
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSintering resistance of Pd single atoms on steam-modified ceria: deciphering the role of hydroxyl groups†
Yuanyuan
An
a,
Sheng-Yu
Chen
 a,
Li
Zhou
a,
Beibei
Wang
b,
Guoxiu
Hao
a,
Junchen
Chen
a,
Yanli
Wang
a,
Hui
Zhang
c,
Zheng
Peng
b,
Tsung-Cheng
Yang
d,
Chia-Min
Yang
d,
Jeng-Lung
Chen
e,
Chia-Kuang
Tsung‡
f,
Zhi
Liu
*abg and
Lien-Yang
Chou
*a
a,
Li
Zhou
a,
Beibei
Wang
b,
Guoxiu
Hao
a,
Junchen
Chen
a,
Yanli
Wang
a,
Hui
Zhang
c,
Zheng
Peng
b,
Tsung-Cheng
Yang
d,
Chia-Min
Yang
d,
Jeng-Lung
Chen
e,
Chia-Kuang
Tsung‡
f,
Zhi
Liu
*abg and
Lien-Yang
Chou
*a
aSchool of Physical Science and Technology, ShanghaiTech University, Shanghai 201210, China. E-mail: liuzhi@shanghaitech.edu.cn; zhuoly@shanghaitech.edu.cn
bCenter for Transformative Science, ShanghaiTech University, Shanghai 201210, China
cState Key Laboratory of Functional Materials for Informatics, Shanghai Institute of Microsystem and Information Technology, Chinese Academy of Sciences, Shanghai 200050, China
dDepartment of Chemistry, National Tsing Hua University, Hsinchu 300044, Taiwan
eNational Synchrotron Radiation Research Center, Hsinchu 30076, Taiwan
fDepartment of Chemistry, Boston College, Merkert Chemistry, Chestnut Hill, Massachusetts 02467, USA
gShanghai High Repetition Rate XFEL and Extreme Light Facility (SHINE), ShanghaiTech University, Shanghai 201210, China
First published on 22nd September 2023
Abstract
It remains a significant challenge to achieve good stability for single-atom catalysts while maintaining high activity. On the basis of observing the sintering resistance up to 800 °C of Pd single atoms dispersed on OH-modified CeO2 and their sustainable activity for CO oxidation at low temperature, the relationship between the unique structure and catalytic performance of the single-atom catalyst was deeply investigated. The structural characterization revealed that the OH groups on the ceria can anchor the Pd-loaded sites and promote the formation of a stable OH-Pd1O4-doped structure, leading to its high-temperature stability. Moreover, low-temperature activity retention was attributed to the multiple active palladium species (Pd2+, Pdδ+, and Pd0). This study highlights the crucial role of structural regulation in determining catalyst performance, and provides guidance for the rational design of high-performance single-atom catalysts.
1. Introduction
For precious metal catalysts, reducing the metal size is a highly effective approach for raising the active sites and material usage of the loaded metals.1 Compared to the reported catalysts with nanoscale particles, single-atom catalysts (SACs) have recently gained increasing attention.1,2 SACs offer significant advantages over metal nanoparticles, such as unique electronic structures, high atomic dispersion, and a great many unsaturated coordination atoms,1,3,4 due to the atomic metal sites.5,6 However, the surface energy increases rapidly along with the decreasing metal size, which leads to a tendency for atomic scale metal aggregation.3 Therefore, it remains a significant challenge to improve the stability of SACs while maintaining high activity.Reducible supports, such as cerium oxide and titanium oxide, are often used to stabilize single-atom metals because of their strong interaction with transition metals.4,7 This interaction, known as metal–support interaction (MSI), can be constructed by forming covalent bonds between metals and supports to achieve high stability for the loaded metals.8,9 However, strong MSI also potentially weakens the catalytic activity of active metals. Taking CO oxidation, the most commonly studied probe reaction to explore the structure–property relationship of catalysts,10 as an example, the catalytic activity of CeO2-based Pt SACs for CO oxidation was found to be greatly influenced by the coordination status of the Pt structure.11 The study showed that Pt species with higher coordination numbers usually exhibit better stability, but it also results in the loss of some effective active sites. Besides Pt, a highly stable Pd-doped ceria catalyst for CO oxidation was also reported.12 The oxidized Pd single atoms as the active sites were well-stabilized for CO oxidation because the doping structure facilitated the transfer of electrons from the Pd to the CeO2 supports.13,14 Nevertheless, some of the Pd single atoms were embedded in the supports and were not able to participate in the surface reaction. Therefore, although it is not difficult to prepare a highly stable SAC using a ceria support for CO oxidation, it remains a great challenge to develop a SAC that can simultaneously maintain high activity and stability. Furthermore, the lack of systematic research on the relationship between catalyst structure and high stability/activity for CO oxidation limits the development of efficient catalysts. In our previous study,15 we observed that OH-modified CeO2 can effectively disperse Pd metal and exhibits high activity for CO oxidation at room temperature (9.72 molcoh−1 gPd−1, 27 °C). The strong MSI of the Pd/OH-CeO2 catalyst implies the potential for reusability. Therefore, studying the stability and figuring out the relationship between structure and stability/activity of the catalyst may provide valuable insights towards achieving precise regulation of high-performance catalysts.
Herein, we comprehensively studied the stability of Pd/OH-CeO2 towards CO oxidation, including high-temperature sintering resistance and low-temperature catalytic stability. The experimental results demonstrated that the Pd/OH-CeO2 catalyst can effectively operate over a wide temperature range (22–800 °C) and exhibits excellent cyclability (>10 cycles), highlighting its remarkable sintering resistance. Furthermore, the catalytic activity was stably maintained at room temperature for at least 60 min, suggesting that the OH-modified CeO2 can efficiently stabilize the loaded Pd single atoms. We investigated the structure–activity relationship of the oxidized Pd on OH-modified CeO2 through operando characterization techniques and explained the steady high activity at low temperature. It was found that the OH-modified CeO2 not only anchors the Pd loading site to form a stable OH-Pd1O4 structure but also creates new active Pd species under the low-temperature CO oxidation conditions. These results highlight the dual benefits of the OH-modified CeO2 support for both the stability and activity of Pd single-atom catalysts.
2. Results and discussion
2.1 Sintering resistance and catalytic stability of Pd on the OH-modified CeO2
A strategy of steam-modifying the ceria support before Pd loading has been reported to boost Pd SACs for low-temperature CO oxidation.15 The preparation process for the ceria-based palladium catalysts is depicted in Fig. 1a. The as-synthesized ceria (A-CeO2) was treated with steam (2 vol%) to obtain the OH-modified ceria (OH-CeO2). Pd species were loaded onto the OH-CeO2 and then dispersed by calcination in oxygen at 800 °C (ref. 16–18) to form the atomically dispersed Pd/OH-CeO2 composite catalyst. As shown in Fig. S1,† no Pd nanoparticles were observed in the high-resolution transmission electron microscope (HRTEM) image, and energy-dispersive spectroscopy (EDS) revealed that the Pd species was in a highly dispersed state. To comprehensively assess the stability of Pd/OH-CeO2, CO oxidation was selected as a probe reaction to evaluate its sintering resistance performance in cyclic catalysis tests from room temperature (RT) to high temperature (800 °C) and to evaluate its sustainable stability at low temperature. As shown in Fig. 1b and S2,† the CO conversion of Pd/OH-CeO2 consistently remained around 53% at RT (∼25 °C) for at least 10 cycles, suggesting the excellent sintering resistance of Pd/OH-CeO2 in a high-temperature catalytic environment. In contrast, the catalytic performance of the highly dispersed Pd catalyst loaded onto ceria without modified OH groups (Pd/D-CeO2, Fig. S3†) was significantly deactivated in the second cycle (Fig. S4†). These results indicate that the OH-modified CeO2 plays an important role in the formation of a stable structure in the composite catalyst. To measure the temperature limit of the thermal stability of Pd/OH-CeO2, we tested Pd/OH-CeO2 over a wider temperature range from RT to 900 °C (Fig. S5†). Pd/OH-CeO2 displayed a consistent and stable catalytic performance toward CO conversion, even after three cycling runs. Notably, the high activity of the Pd/OH-CeO2 catalyst was even sustained in the second cycle within the RT – 1000 °C cyclic range, despite the loss of RT activity that occurred in the third cycle. To evaluate the thermal stability of Pd/OH-CeO2 in comparison to the reported state-of-the-art Pd- or Pt-catalysts that exhibit good catalytic performance in CO oxidation, we compiled a list of their working temperature ranges and cycle times.12,14,16,18–25T50 (temperature of 50% conversion) and Tupper (the upper limit of operating temperature) represent the starting and ending temperatures of the working range (Fig. 1c), respectively, and more details are shown in Table S1.† Pd/OH-CeO2 exhibited not only an ultra-wide working temperature range (22–800 °C) over a long cycle time (10 cycles) but also a remarkably low T50 at RT (22 °C), whereas the Tupper and T50 of other samples were mainly below 300 °C and around 50–120 °C, respectively. It is obvious that the OH-modified CeO2 greatly regulates the sintering resistance of the loaded active Pd metals at high temperatures. Furthermore, Pd/OH-CeO2 also exhibited sustainable CO oxidation activity at RT and 50 °C for at least 60 min (Fig. 1d), indicating good resistance to surface poisoning by CO at low temperatures. However, compared to the catalytic activity at 50 °C, a slight attenuation in the activity was observed at RT, which may be attributed to the accumulation of the product (CO2). To confirm this speculation, Pd/OH-CeO2 was reacted at RT for 50 min, then purged with He to remove the reaction gas and subjected to the temperature-programmed test. The changes in CO2 during the whole process were detected by a mass spectrometer (Fig. 1e). When the temperature was raised to 45 °C, an obvious CO2 signal was detected, which was ascribed to the remaining product on the Pd/OH-CeO2 at RT and was responsible for the observed decrease of activity at RT. The catalytic test at 50 °C showed a more stable activity curve because CO2 could be desorbed. In summary, Pd displayed excellent high-temperature sintering resistance and low-temperature catalytic stability on OH-modified CeO2 supports. Undoubtedly, the OH-modified CeO2 plays a critical role in stabilizing the Pd, but the mechanism of the stability improvement of the loaded metal remains unclear. To verify whether the presence of the OH group improves the stability of Pd or not, CeO2 with the same rod-like morphology as OH-CeO2 but with fewer defects was treated by steam to prepare a fewer-OH-modified CeO2, which was then loaded with Pd and denoted as Pd_f-ST. The activity of Pd_f-ST was comparable to that of Pd/OH-CeO2 only in the first run and then decreased significantly in the following two cycles (Fig. S6a†). In addition, an obvious activity drop of Pd_f-ST was observed within 15 min during the sustainability test at RT (Fig. S6b†). These catalytic data preliminarily proved that OH groups contributed to the formation of a stable catalyst structure between Pd and CeO2.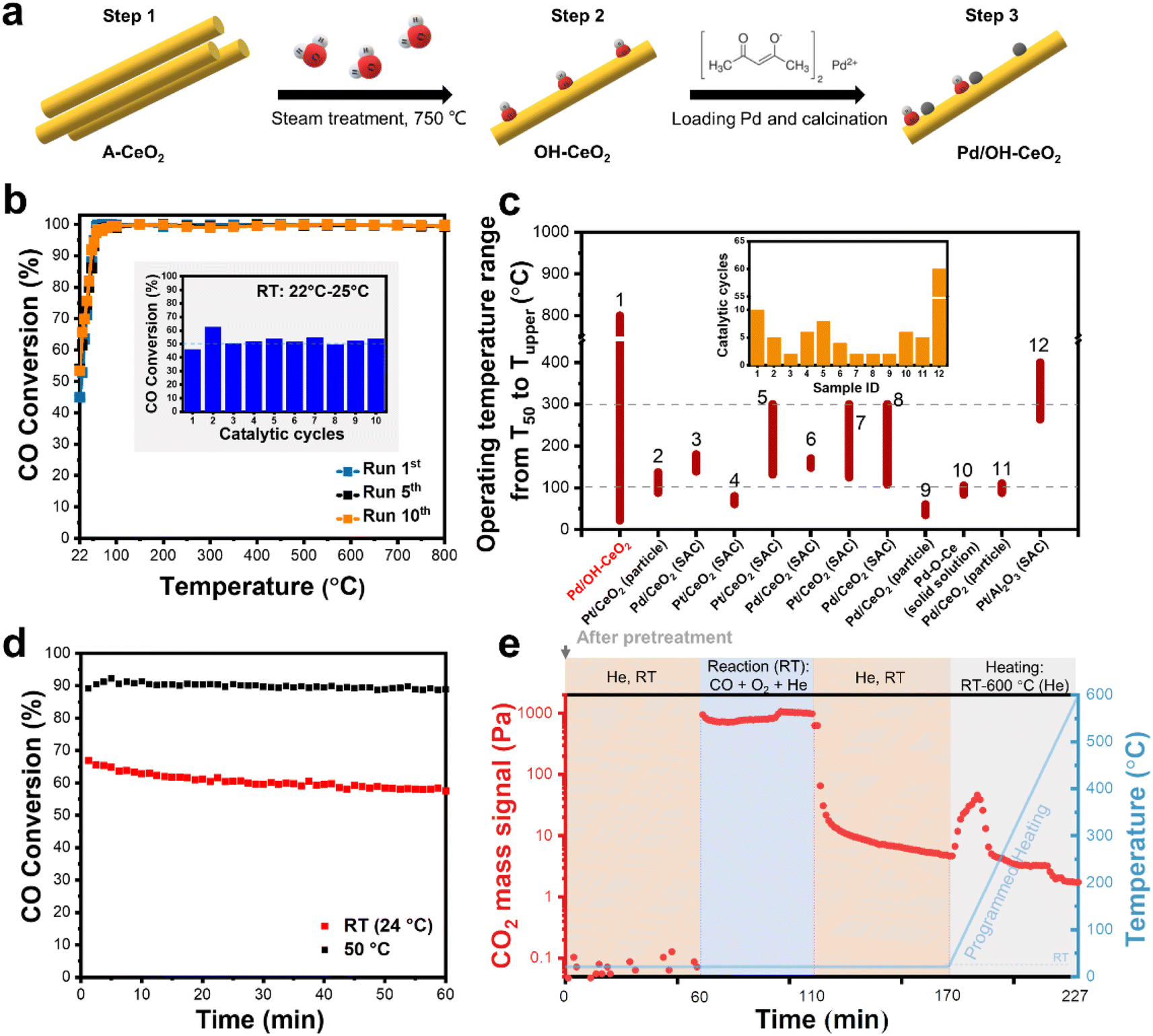 | ||
| Fig. 1 (a) The preparation process for Pd/OH-CeO2 includes a steam treatment and a metal-loading step. (b) Catalytic cycling performance of Pd/OH-CeO2 (Run 1–10) toward CO oxidation in the range of RT-800 °C. The inset of the bar graph shows the room-temperature activity for each cycle. (c) A summary of the temperature ranges of the Pd- and Pt-catalysts for CO oxidation in the cyclic reaction and the number of cycles (inset of the bar graph). (d) Sustainability test of Pd/OH-CeO2 at RT and 50 °C for 60 min. (e) Mass signal of CO2 obtained from the detection of Pd/OH-CeO2 under different atmosphere and temperature conditions. | ||
2.2 The effect of OH groups on the formation of the stable structure
To gain a deep understanding of the possible role of OH groups in forming the stable structure of Pd/OH-CeO2, Pd K-edge extended X-ray absorption fine structure (EXAFS) analysis was performed to analyze the structure of Pd formed in Pd/OH-CeO2. The Pd structure in Pd/D-CeO2 was also analyzed as a control sample to exclude the influence of other factors. To fit the EXAFS data of Pd/OH-CeO2 well, a structure model with OH groups (OH-Pddoped, Fig. 2a) was built based on the reported Pd-doped CeO2 structure,26,27 and the fitting parameters are listed in Table S2.† As shown in Fig. 2b, the absence of Pd–Pd (second shell) in Pd/OH-CeO2 indicated that all the Pd species were atomically dispersed, whereas Pd–Pd (second shell) from Pd clusters was found in Pd/D-CeO2. The average coordination number of O bonded to Pd in both Pd/OH-CeO2 and Pd/D-CeO2 samples (CNPd–O: ∼3.7) was less than the highest coordination number of 4, indicating that both samples possessed Pd open sites for CO adsorption. Density functional theory (DFT) calculations were applied to further study the contribution of OH groups to the formation of the Pddoped structure, which was built by replacing a Ce atom with a Pd atom and then forming a square planar structure with four oxygen atoms.26,28 Two ideal CeO2 surface structures, with OH groups (Str.1_OH) and without OH groups (Str.2_w/o), were constructed to calculate the Gibbs free energy at each stage of the substitution process. The initial structural state of CeO2 without OH (step I in Pd-Str.2_w/o) was set to 0 kJ mol−1, and the corresponding energy differences are shown in Fig. 2c and Table S3.† The first step energy of Pd-Str.1_OH was 168.8 kJ mol−1, which was lower than that of Pd-Str.2_w/o (I), indicating that the incorporation of the OH group was beneficial for decreasing the energy of the cerium oxide system. The formation of Ce vacancy (ΔEI→II) is a decisive step for Pd substitution in the substitution process. ΔEI→II of Pd-Str.1_OH was calculated as 1617.3 kJ mol−1, which was much lower than that of Pd-Str.2_w/o (2071.5 kJ mol−1). After the Pd atom was replaced with the Ce atom, the relaxation energy of the Pd atom in Str.1_OH further reduced the energy by 1.9 kJ mol−1 (ΔEIII→IV), while the energy increased by 71.4 kJ mol−1 in Str.2_w/o. These results indicated that the presence of OH groups was favorable for the formation of a Pddoped structure on ceria, which has been considered an energetically stable state. In general, OH-functionalized ceria can anchor the loaded Pd to form a stable OH-Pd1O4-doped structure (Fig. 2d), greatly enabling the catalyst to achieve good sintering resistance performance in catalytic cycle tests (>10) over a wide temperature range (RT–800 °C). In contrast, the calcined ceria support of Pd/D-CeO2 lacks OH modification and the Pd species tend to form clusters due to weaker interaction with the support, eventually leading to catalyst deactivation during repeated catalytic cycles.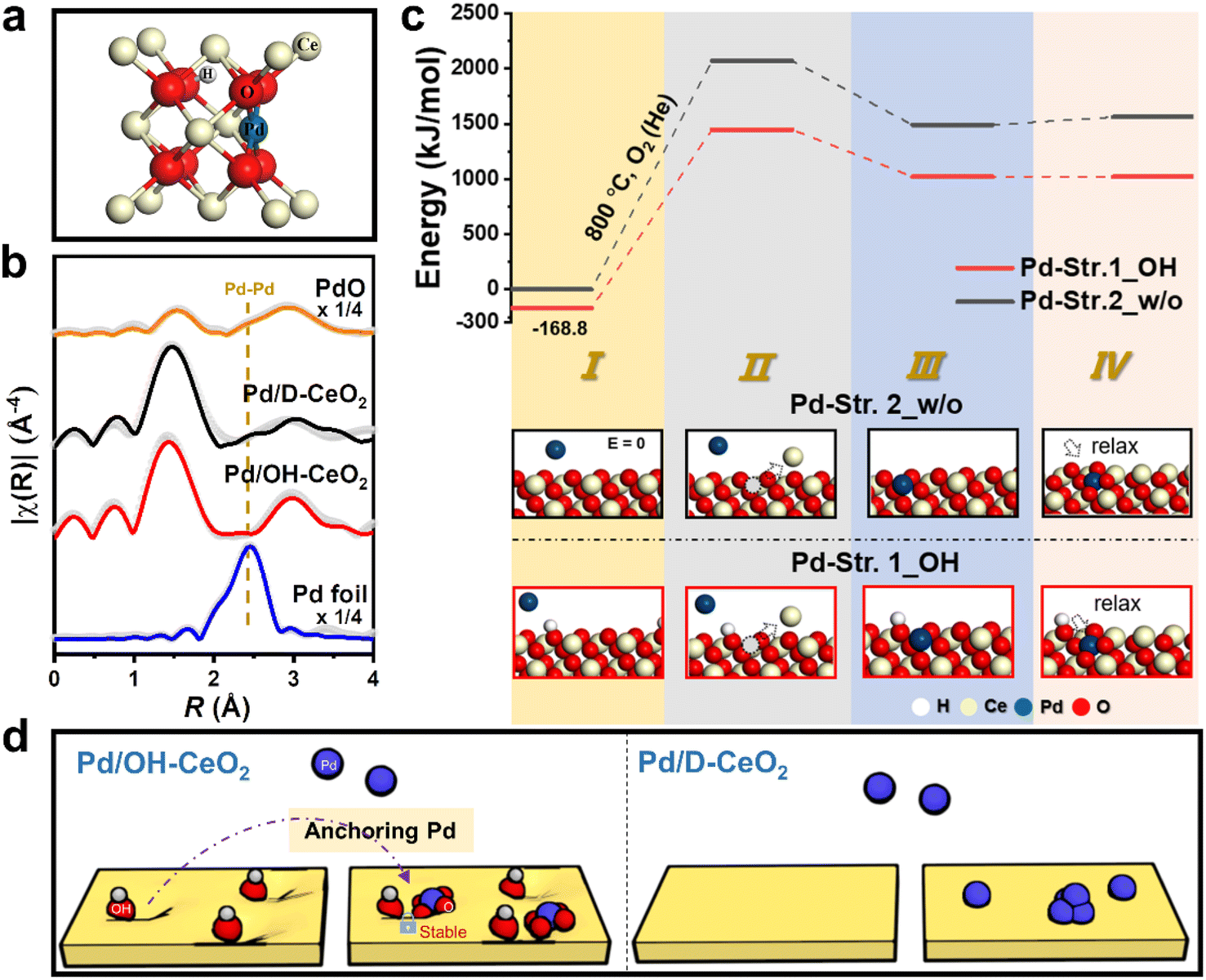 | ||
| Fig. 2 (a) Model of the Pd/OH-CeO2 (OH-Pddoped) structure. (b) The k3-weighted Fourier transforms of EXAFS of the Pd K-edge of Pd/OH-CeO2 and Pd/D-CeO2. The data for PdO and Pd foil were obtained from standard samples. Light-colored dots represent experimental data and the dark solid lines represent the fitting results. (c) DFT calculations of the process of replacing a Ce atom with a Pd atom and then relaxing to form a square planar structure on Str. 1_OH model and Str. 2_w/o model. (d) Schematic diagram of structure formation on OH-modified CeO2 and non-modified CeO2. | ||
2.3 Active species of Pd for CO oxidation
To have sustainable low-temperature performance, catalysts should not only have good resistance to CO poisoning but also require stable valence states of the active metals.13 To further confirm this point of view, we employed quasi in situ XPS to investigate the changes in the valence state of Pd species on different ceria supports before and after CO oxidation (Fig. 3). The XPS spectrum of the as-synthesized Pd/OH-CeO2 exhibited only one Pd oxidation state at 337.8 eV (Fig. 3a), which was attributed to the binding energy of the covalent linkage between the Pd single atom and CeO2 in the Pd–O–Ce structure.12,29,30 We inferred the Pd species to be Pd2+ since the absorption threshold energy in the Pd K-edge XANES spectrum of this sample was similar to that of PdO (Fig. S7†). In contrast to Pd/OH-CeO2, Pd/D-CeO2 contained another oxidation state of Pd species, Pdδ+ (6%, 336 eV), in addition to Pd2+ (94%, 337.5 eV). Furthermore, the binding energy of Pd2+ in Pd/OH-CeO2 is 0.3 eV higher than that in Pd/D-CeO2, suggesting that Pd/OH-CeO2 possesses a stronger MSI than Pd/D-CeO2.15,31 After reacting with CO and O2 at room temperature (22 °C) for 20 min (Fig. 3b), the Pd species in Pd/OH-CeO2 still possessed more than 65% Pd2+ state (337.8 eV), approximately 10% reduced metallic Pd0 (334.8 eV), and nearly 25% semi-oxidized state Pdδ+ (335.8 eV). However, Pd/D-CeO2 had less than 20% of the Pd2+ remaining after the same reaction, while most Pd2+ was reduced to the semi-oxidized Pdδ+ state (67%) and metallic Pd0 state (14%). The quasi situ XPS analysis results indicated that the Pd2+ species formed in Pd/OH-CeO2 were more stable than those in Pd/D-CeO2, which could be attributed to the stronger MSI via insertion of the precious metal into the support lattice.8,12,32 To further confirm the difference in MSI, H2-temperature-programmed reduction (H2-TPR) testing was performed. The pure ceria support has only one H2 consumption peak at ∼450 °C below the 600 °C region (Fig. S8†), which is attributed to the reduction of cerium on the CeO2 surface.33 This peak disappeared in the composite catalysts, and a new peak showed up at lower temperature (∼90 °C) assigned to Pd–oxo species,34 which was the result of MSI influence.35 The reduction temperature of Pd–oxo species in Pd/OH-CeO2 (96 °C) was higher than that in Pd/D-CeO2 (90 °C), suggesting that Pd/OH-CeO2 possessed a higher MSI.34 We further repeated the same quasi in situ XPS test for both samples at 100 °C (Fig. S9†) and obtained similar results to those at RT, suggesting that the original stable oxidized Pd species in both samples still exhibit stability in the low-temperature reaction region (22–100 °C).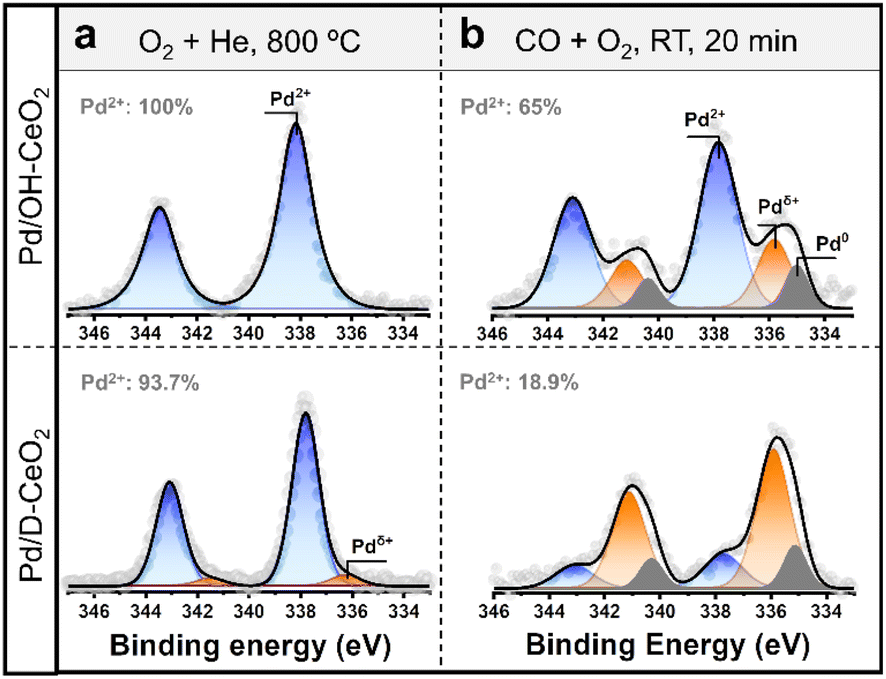 | ||
| Fig. 3 (a and b) Quasi in situ XPS spectra of the Pd 3d core level of Pd/OH-CeO2 and Pd/D-CeO2 after (a) as synthesized, and (b) reacting at RT for 20 min. | ||
Furthermore, a certain percentage (35%) of the oxidized Pd species in Pd/OH-CeO2 was reduced under the catalytic conditions, however, the activity of Pd/OH-CeO2 did not show a clear drop in the catalysis test (Fig. 1). Such an interesting phenomenon is different from the cognition in the reported studies, in which Pd2+ single-atom species were generally considered as the active sites for low-temperature CO oxidation.12–14 Considering the relationship between the structure and activity, we speculated that some changes might occur in the metal active sites for CO oxidation in Pd/OH-CeO2. To explore the relationship between Pd/OH-CeO2 active sites and correlated catalytic performance, in situ analysis of diffuse reflectance infrared Fourier transform spectroscopy (in situ DRIFTS) for Pd/OH-CeO2 and Pd/D-CeO2 was performed at RT using CO as a probe agent. With the successive four gas exchange flows (CO–N2–CO–O2) introduced, IR spectra were simultaneously collected to monitor the adsorption and oxidation behavior of CO on both catalysts (Fig. 4). The stretching band at 2090 cm−1 in the spectra represented the linear-bonded CO (COlinear) on the atomically dispersed Pd2+, while the band at 1970–1920 cm−1 was attributed to bridge-bonded CO (CObridge) on Pd particles.14,36,37 Specifically, when CObridge mode is on the edge/corner or facet of Pd particles, the corresponding stretching bands are at 1970 cm−1 and 1920 cm−1, respectively.38 As shown in Fig. 4a, when CO was fed at the beginning, Pd/OH-CeO2 exhibited only one COlinear peak centered around 2090 cm−1, confirming the highly dispersed single-atomic state of the Pd species. With increasing feeding time, the peak for CObridge (∼1970 cm−1) appeared, which was derived from the partial reduction and agglomeration of Pd2+ single atoms under the CO atmosphere. This result also indicated the existence of some unstable Pd species on the surface of Pd/OH-CeO2 besides the OH-anchored Pd metal (Fig. 4i-1), consistent with the analysis of the quasi in situ XPS experiment. Unlike Pd/OH-CeO2, the control sample Pd/D-CeO2 had a CObridge peak at the first recording time point (Fig. 4e), showing the existence of Pd clusters, which agreed with the EXAFS analysis (Fig. 4i-4). The adsorbed CO on both samples was not completely removed after purging N2 for 12 min (Fig. 4b and f) due to the strong interaction between CO and the surface Pd sites. CO was then reintroduced to both samples for 10 min (Fig. 4c, g, i-2, i-5), following the injection of O2 to test the active sites for CO oxidation at low temperatures (Fig. 4d and h). For Pd/OH-CeO2, the COlinear peak (∼2090 cm−1) disappeared rapidly after the introduction of O2 (within 0 to 3.5 min), and the CObridge band (1970–1920 cm−1) subsequently started to decrease from the 4th min and completely vanished after 10 min (Fig. 4d). The phenomenon revealed that the CO adsorbed on both Pd2+ single atoms and Pd clusters (Pdδ+ and Pd0) in Pd/OH-CeO2 can be oxidized at RT (Fig. 4i-3). However, for Pd/D-CeO2, the CObridge band persisted even after treatment with O2 for 20 min (Fig. 4h), revealing the lack of RT activity of the reduced Pd clusters at low temperature (Fig. 4i-6).
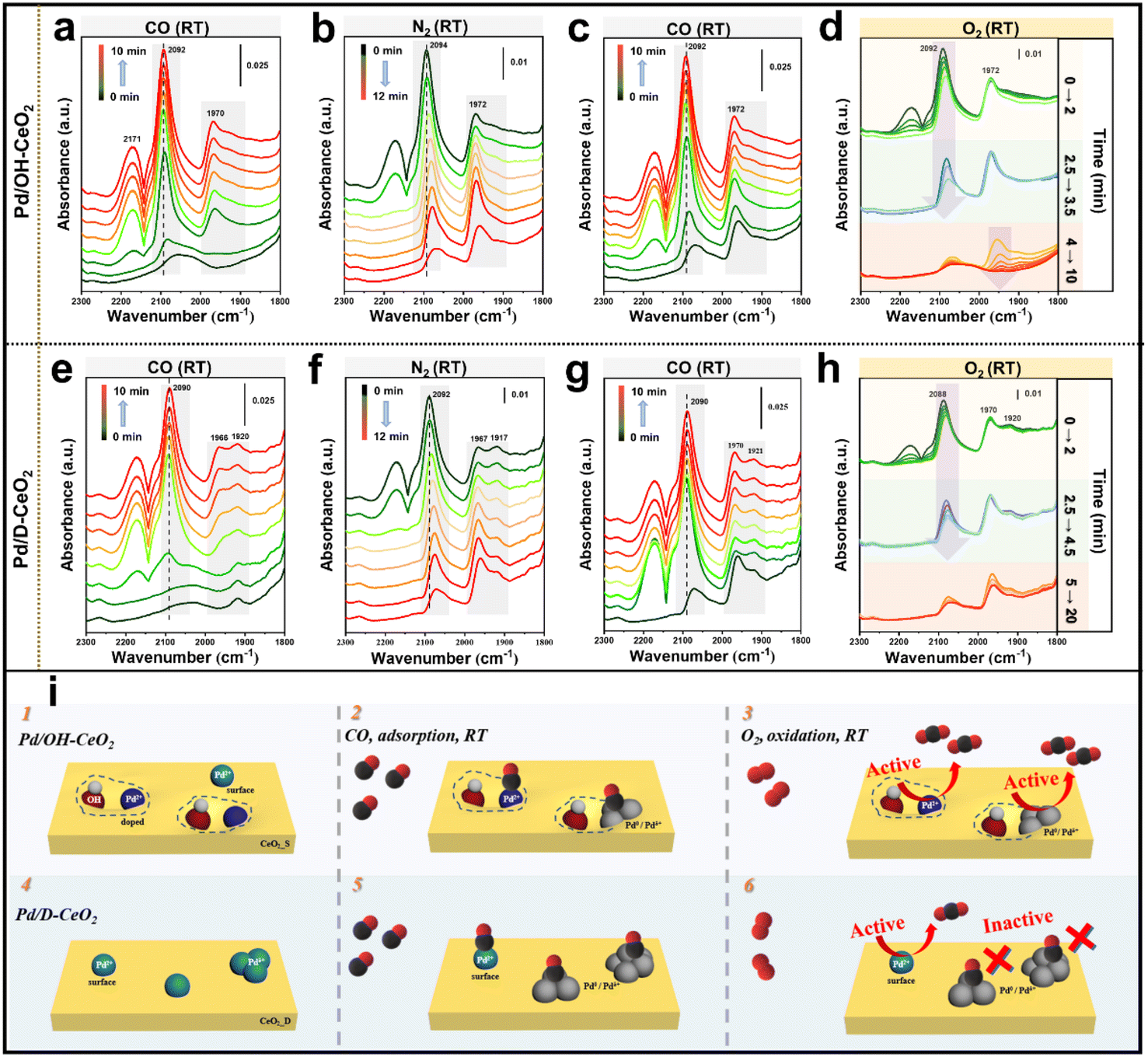 | ||
| Fig. 4 Characterization of the active Pd species. (a–h) In situ DRIFTS experiments of Pd/OH-CeO2 (a–d) and Pd/D-CeO2 (e–h) at RT with a gas feeding cycle (CO–N2–CO–O2). (i) Schematic illustration of the initial dispersion of Pd species on different CeO2 supports and the changes after reacting with CO and O2 in Pd/OH-CeO2 and Pd/D-CeO2. | ||
The increase in active Pd species (Pd2+, Pdδ+ and Pd0) on the Pd/OH-CeO2 sample is undoubtedly the primary reason for its high performance towards CO oxidation in the low-temperature region. The unusual activity of Pd0 at low temperature (even at RT) on Pd/OH-CeO2 may be attributed to the low reaction energy barrier (11.6 kJ mol−1)15 of CO oxidation, which is related to the transition of reacting molecules to the active status on the catalyst surface. Generally speaking, CO oxidation catalyzed by palladium-ceria systems follows the Mars–van Krevelen mechanism,12,13,39 which means that there is no competitive adsorption between O2 and CO molecules on the Pd sites. CO and O2 molecules are adsorbed and activated on the Pd surface and CeO2 support, respectively. However, the low-temperature performance of Pd catalysts is often limited by CO poisoning on the active metals or difficult oxygen activation.5,22,40,41 In the case of the Pd/OH-CeO2 sample, on the one hand, OH-modified CeO2 can reduce the adsorption strength of Pd-CO and promote the activation of CO molecules.15 On the other hand, the activation of oxygen is related to the surface charge of the catalyst,41,42 and the increased charge density of the catalyst promotes electron transfer to the anti-bonding π* orbital of oxygen, weakening the molecular bonds of oxygen, and then forming active oxygen species.43 Based on these insights, DFT was used to calculate the effect of OH groups on the local charge concentration of the CeO2 surface. As shown in Fig. S10a,† when the OH group was introduced into the CeO2 supercell model (Str. 2_w/o), the Fermi level of CeO2 (Str. 1_OH) shifted close to the conduction band and the total density area increased, indicating the increased charge density of CeO2 with an OH group. The charge density contour plots projected from the cut plane visually revealed that the contour line of the charge density of the Ce atom next to OH was distorted (Fig. S10b†). The charge density of Ce with an OH group was clearly higher than that of the pristine CeO2, implying that oxygen molecules would be more easily activated near OH. To sum up, OH not only anchors the metal loading sites, but also facilitates the activation of the reactants, resulting in the enhanced reactivity of different Pd species on the OH-modified CeO2.
3. Conclusions
In summary, the CeO2 support modified by OH groups is beneficial for stabilizing the loaded Pd and increasing the active species for CO oxidation. Pd/OH-CeO2 maintained excellent sintering resistance over a wide operating temperature range (RT – 800 °C) during a 10-cycle test and exhibited stable activity for 60 min under low-temperature catalytic conditions. The relationship between the excellent activity and structure is shown in Fig. 5. After correlating the results of XAFS, quasi in situ XPS and DFT calculations, it was found that functionalized ceria (OH-CeO2) can disperse and stabilize Pd species much better than non-modified ceria because of the formation of the stable OH-Pd1O4 structure, which was resistant to sintering in high-temperature cycling tests. In situ DRIFTS measurements indicated that both Pd single atoms and clusters on Pd/OH-CeO2 were active sites for CO oxidation at low temperature, as a result of property regulations of the metal and support promoting the activation process of the reactants, and the increased Pd active species also explained the sustainable low-temperature activity. Pd/OH-CeO2 with excellent properties towards CO oxidation also has potential applications in several important heterogeneous reactions that require low-temperature catalytic oxidation performance, including the purification of exhaust emissions, the removal of CO from gas streams, and water–gas shift reactions for fuel cells.4,10,44 In addition, these studies revealed that modifying the support to regulate the properties can have a substantial impact on both metal loading and catalytic performance. We believe that the research on the relationship between unique structure and excellent performance in this manuscript provides a certain reference for regulating more efficient single-atom catalysts.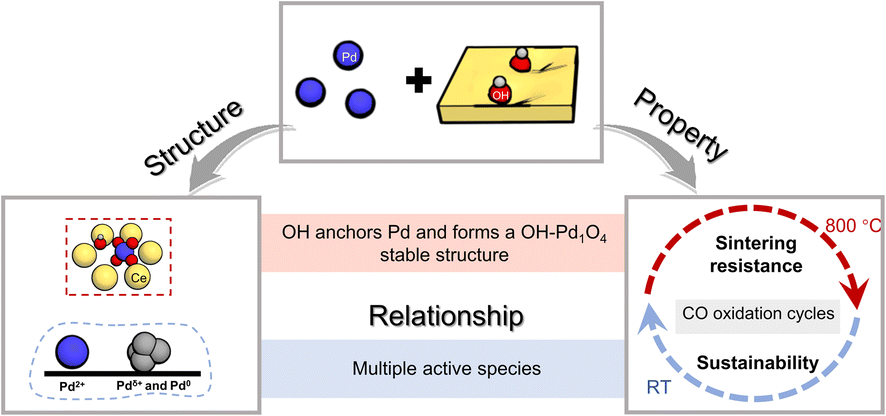 | ||
| Fig. 5 Schematic summary of the relationship between the structure and property of Pd/OH-CeO2. | ||
Author contributions
Y. A. performed most sample synthesis, catalytic tests, material characterization, data analysis, and article writing. S.-Y. C. participated in the construction of the catalytic system, data analysis, and article revision. L. Z. performed all the DFT calculations in this article. B. W. performed the quasi in situ XPS experiments. G. H. performed supplementation of the catalysis data and analysis. J. C. participated in article revision. Y. W. performed supplementation of the catalysis data. H. Z. helped the data analysis of XAFS. Z. P. participated in the discussion of data analysis. T.-C. Y., C.-M. Y. and J.-L. C. provided assistance for data collection and analysis. Z. L. and L.-Y. C. provided major guidance, construction of the research idea, and article revision. C.-K. T. initiated and led this project. The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the start-up funding from ShanghaiTech University. This work was partially supported by the Center of high-resolution Electron Microscopy (CℏEM) of the SPST at ShanghaiTech under grant EM02161943. Z. L. is partially supported by Shanghai high repetition rate XFEL and extreme light facility (SHINE). The authors thank BLO2B01 of the Shanghai Synchrotron Radiation Facility and lab-based SPECS AP-XPS instrument supported by the National Natural Science Foundation of China, No. 11227902. The authors thank Dr Peihong Cheng, Haiyin Zhu, Qing Zhang, Lili Du, Liuliu Long and Na Yu for their assistance in characterization. The authors thank Dr Xiangwen Liu for assistance with sample synthesis.Notes and references
- W. Zhang and W. Zheng, Adv. Funct. Mater., 2016, 26, 2988–2993 CrossRef CAS.
- J. Wu, L. Xiong, B. Zhao, M. Liu and L. Huang, Small Methods, 2020, 4, 1900540 CrossRef CAS.
- X.-F. Yang, A. Wang, B. Qiao, J. Li, J. Liu and T. Zhang, Acc. Chem. Res., 2013, 46, 1740–1748 CrossRef CAS PubMed.
- A. Beniya and S. Higashi, Nat. Catal., 2019, 2, 590–602 CrossRef.
- K. Ding, A. Gulec, A. M. Johnson, N. M. Schweitzer, G. D. Stucky, L. D. Marks and P. C. Stair, Science, 2015, 350, 189–192 CrossRef CAS PubMed.
- Y. Lykhach, A. Bruix, S. Fabris, V. Potin, I. Matolínová, V. Matolín, J. Libuda and K. M. Neyman, Catal. Sci. Technol., 2017, 7, 4315–4345 RSC.
- Y. Nagai, T. Hirabayashi, K. Dohmae, N. Takagi, T. Minami, H. Shinjoh and S. i. Matsumoto, J. Catal., 2006, 242, 103–109 CrossRef CAS.
- P. Hu, Z. Huang, Z. Amghouz, M. Makkee, F. Xu, F. Kapteijn, A. Dikhtiarenko, Y. Chen, X. Gu and X. Tang, Angew. Chem., Int. Ed., 2014, 126, 3486–3489 CrossRef.
- N. Acerbi, S. E. Tsang, G. Jones, S. Golunski and P. Collier, Angew. Chem., Int. Ed., 2013, 125, 7891–7895 CrossRef.
- H. J. Freund, G. Meijer, M. Scheffler, R. Schlögl and M. Wolf, Angew. Chem., Int. Ed., 2011, 50, 10064–10094 CrossRef CAS PubMed.
- J. Ke, W. Zhu, Y. Jiang, R. Si, Y.-J. Wang, S.-C. Li, C. Jin, H. Liu, W.-G. Song, C.-H. Yan and Y.-w. Zhang, ACS Catal., 2015, 5, 5164–5173 CrossRef CAS.
- V. Muravev, G. Spezzati, Y.-Q. Su, A. Parastaev, F.-K. Chiang, A. Longo, C. Escudero, N. Kosinov and E. J. Hensen, Nat. Catal., 2021, 4, 469–478 CrossRef CAS.
- E. J. Peterson, A. T. DeLaRiva, S. Lin, R. S. Johnson, H. Guo, J. T. Miller, J. Hun Kwak, C. H. Peden, B. Kiefer, L. F. Allard, F. H. Ribeiro and A. K. Datye, Nat. Commun., 2014, 5, 4885 CrossRef CAS PubMed.
- G. Spezzati, Y. Su, J. P. Hofmann, A. D. Benavidez, A. T. DeLaRiva, J. McCabe, A. K. Datye and E. J. Hensen, ACS Catal., 2017, 7, 6887–6891 CrossRef CAS PubMed.
- Y. An, S.-Y. Chen, B. Wang, L. Zhou, G. Hao, Y. Wang, J. Chen, C.-K. Tsung, Z. Liu and L.-Y. Chou, J. Mater. Chem. A, 2023, 11, 16838–16845 RSC.
- X. I. Pereira-Hernández, A. DeLaRiva, V. Muravev, D. Kunwar, H. Xiong, B. Sudduth, M. Engelhard, L. Kovarik, E. J. Hensen, Y. Wang and A. K. Datye, Nat. Commun., 2019, 10, 1–10 CrossRef PubMed.
- J. Jones, H. Xiong, A. T. DeLaRiva, E. J. Peterson, H. Pham, S. R. Challa, G. Qi, S. Oh, M. H. Wiebenga, X. I. Pereira Hernández, Y. Wang and A. K. Datye, Science, 2016, 353, 150–154 CrossRef CAS PubMed.
- D. Jiang, G. Wan, C. E. Garcia-Vargas, L. Li, X. I. Pereira-Hernandez, C. Wang and Y. Wang, ACS Catal., 2020, 10, 11356–11364 CrossRef CAS.
- Y. Xia, J. Ye, D.-g. Cheng, F. Chen and X. Zhan, Catal. Sci. Technol., 2018, 8, 5137–5147 RSC.
- K. Tang, D. Zeng, F. Lin, Y. Yang and L. Wu, CrystEngComm, 2020, 22, 1251–1260 RSC.
- M. Farrag, M. K. Das, M. Moody and M. Samy El-Shall, ChemPhysChem, 2021, 22, 312–322 CrossRef CAS PubMed.
- L. Nie, D. Mei, H. Xiong, B. Peng, Z. Ren, X. I. P. Hernandez, A. Delariva, M. Wang, M. H. Engelhard, L. Kovarik, A. K. Datye and Y. Wang, Science, 2017, 358, 1419–1423 CrossRef CAS PubMed.
- J. Chen, Y. Wanyan, J. Zeng, H. Fang, Z. Li, Y. Dong, R. Qin, C. Wu, D. Liu, M. Wang, Q. Kuang, Z. Xie and L. s. Zheng, ACS Sustainable Chem. Eng., 2018, 6, 14054–14062 CrossRef CAS.
- C. Wang, X.-K. Gu, H. Yan, Y. Lin, J. Li, D. Liu, W.-X. Li and J. Lu, ACS Catal., 2017, 7, 887–891 CrossRef CAS.
- Z. Zhang, Y. Zhu, H. Asakura, B. Zhang, J. Zhang, M. Zhou, Y. Han, T. Tanaka, A. Wang, T. Zhang and N. Yan, Nat. Commun., 2017, 8, 1–10 CrossRef PubMed.
- C. I. Hiley, J. M. Fisher, D. Thompsett, R. J. Kashtiban, J. Sloan and R. I. Walton, J. Mater. Chem. A, 2015, 3, 13072–13079 RSC.
- Y.-Q. Su, I. A. Filot, J.-X. Liu and E. J. Hensen, ACS Catal., 2018, 8, 75–80 CrossRef CAS PubMed.
- D. O. Scanlon, B. J. Morgan and G. W. Watson, Phys. Chem. Chem. Phys., 2011, 13, 4279–4284 RSC.
- A. I. Boronin, E. M. Slavinskaya, I. G. Danilova, R. V. Gulyaev, Y. I. Amosov, P. A. Kuznetsov, I. A. Polukhina, S. V. Koscheev, V. I. Zaikovskii and A. S. Noskov, Catal. Today, 2009, 144, 201–211 CrossRef CAS.
- Y. Chen, J. Chen, W. Qu, C. George, M. Aouine, P. Vernoux and X. Tang, Chem. Commun., 2018, 54, 10140–10143 RSC.
- X.-C. Sun, K. Yuan, W.-D. Hua, Z.-R. Gao, Q. Zhang, C.-Y. Yuan, H.-C. Liu and Y.-W. Zhang, ACS Catal., 2022, 12, 11942–11954 CrossRef CAS.
- S. Colussi, A. Gayen, M. Farnesi Camellone, M. Boaro, J. Llorca, S. Fabris and A. Trovarelli, Angew. Chem., Int. Ed., 2009, 48, 8481–8484 CrossRef CAS PubMed.
- Z. Hu, X. Liu, D. Meng, Y. Guo, Y. Guo and G. Lu, ACS Catal., 2016, 6, 2265–2279 CrossRef CAS.
- V. Muravev, A. Parastaev, Y. van den Bosch, B. Ligt, N. Claes, S. Bals, N. Kosinov and E. J. Hensen, Science, 2023, 380, 1174–1179 CrossRef CAS PubMed.
- F. Jiang, S. Wang, B. Liu, J. Liu, L. Wang, Y. Xiao, Y. Xu and X. Liu, ACS Catal., 2020, 10, 11493–11509 CrossRef CAS.
- H. Jeong, J. Bae, J. W. Han and H. Lee, ACS Catal., 2017, 7, 7097–7105 CrossRef CAS.
- J. Xu, L. Ouyang, W. Mao, X.-J. Yang, X.-C. Xu, J.-J. Su, T.-Z. Zhuang, H. Li and Y.-F. Han, ACS Catal., 2012, 2, 261–269 CrossRef CAS.
- H. Wang, X.-K. Gu, X. Zheng, H. Pan, J. Zhu, S. Chen, L. Cao, W.-X. Li and J. Lu, Sci. Adv., 2019, 5, eaat6413 CrossRef PubMed.
- J.-X. Liu, Y. Su, I. A. Filot and E. J. Hensen, J. Am. Chem. Soc., 2018, 140, 4580–4587 CrossRef CAS PubMed.
- H. Wang, J.-X. Liu, L. F. Allard, S. Lee, J. Liu, H. Li, J. Wang, J. Wang, S. H. Oh, W. Li, M. Flytzani-Stephanopoulos, M. Shen, B. R. Goldsmith and M. Yang, Nat. Commun., 2019, 10, 1–12 CrossRef PubMed.
- M. M. Montemore, M. A. van Spronsen, R. J. Madix and C. M. Friend, Chem. Rev., 2017, 118, 2816–2862 CrossRef PubMed.
- R. Long, K. Mao, M. Gong, S. Zhou, J. Hu, M. Zhi, Y. You, S. Bai, J. Jiang, Q. Zhang, X. Wu and Y. Xiong, Angew. Chem., Int. Ed., 2014, 53, 3205–3209 CrossRef CAS PubMed.
- Y. Bai, H. Huang, C. Wang, R. Long and Y. Xiong, Mater. Chem. Front., 2017, 1, 1951–1964 RSC.
- C. Ratnasamy and J. P. Wagner, Catal. Rev., 2009, 51, 325–440 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ta04200f |
| ‡ Deceased, January 5, 2021. |
| This journal is © The Royal Society of Chemistry 2023 |