
Multi-functional photocatalytic systems for solar fuel production
Young Hyun
Hong
a,
Yong-Min
Lee
 *ab,
Wonwoo
Nam
*a and
Shunichi
Fukuzumi
*ac
*ab,
Wonwoo
Nam
*a and
Shunichi
Fukuzumi
*ac
aDepartment of Chemistry and Nano Science, Ewha Womans University, Seoul 03760, Korea. E-mail: fukuzumi@chem.eng.osaka-u.ac.jp; yomlee@ewha.ac.kr; wwnam@ewha.ac.kr
bResearch Institute for Basic Sciences, Ewha Womans University, Seoul 03760, Korea
cDepartment of Chemistry, University of Tsukuba, Tennodai, Tsukuba, Ibaraki 305-8571, Japan
First published on 9th June 2023
Abstract
Solar fuel production from water has received increasing attention due to increasing energy and environmental concerns. Herein we highlight dual- and multi-functional photocatalytic systems, which have both photosensitizing and thermal redox functions for solar fuel production. For example, polymeric cyano (CN)-bridged heteronuclear metal complexes act as dual functional photoredox catalysts that incorporate both a light harvesting photosensitizer and redox catalytic sites for the photocatalytic oxidation of water and reduction of dioxygen to produce H2O2. On the other hand, metal–organic frameworks (MOFs) contain both organic parts which act as photosensitizers and inorganic metal parts which act as redox catalysts, catalyzing water oxidation, H2 evolution, CO2 reduction and H2O2 production. An organic photoredox catalyst can be incorporated into nano-sized mesoporous cellular silica foams together with a redox catalyst for water oxidation, H2 production, CO2 reduction and H2O2 production. Finally, construction of multi-functional catalytic systems is discussed for solar-driven production of H2 as molecular models of photosystem I (PSI) and photosystem II (PSII) in photosynthesis.
 Young Hyun Hong | Young Hyun Hong received her B.S. degree from the Department of Chemistry and Nano Science at Ewha Womans University, Korea, and Ph.D. degree from Ewha Womans University, Korea, under the supervision of Professor Wonwoo Nam and Professor Shunichi Fukuzumi in 2022. She is currently a postdoctoral fellow at Ewha Womans University. |
 Yong-Min Lee | Yong-Min Lee received his M.S. and Ph.D. degrees in Inorganic Chemistry from Pusan National University, Republic of Korea, under the supervision of Professor Sung-Nak Choi in 1999. After he worked in the Magnetic Resonance Centre (CERM) at the University of Florence, Italy, as a Postdoctoral fellow and Researcher under the direction of Professors Ivano Bertini and Claudio Luchinat from 1999 to 2005, he joined the Centre for Biomimetic Systems at Ewha Womans University, as a Research Professor in 2006. He is currently a Special Appointment Professor at Ewha Womans University since 2009. |
 Wonwoo Nam | Wonwoo Nam earned his B.S. (Honours) degree in Chemistry from California State University, Los Angeles, and his Ph.D. degree in Inorganic Chemistry from the University of California, Los Angeles (UCLA) under the supervision of Professor Joan S. Valentine in 1990. After working as a postdoctoral fellow at UCLA for one year, he became an Assistant Professor at Hong Ik University in 1991. In 1994, he moved to Ewha Womans University, where he is currently a Distinguished Professor. His current research has been focused on dioxygen activation, water oxidation, and important roles of metal ions in bioinorganic chemistry. |
 Shunichi Fukuzumi | Shunichi Fukuzumi earned a bachelor's degree and Ph.D. degree in applied chemistry at the Tokyo Institute of Technology in 1973 and 1978, respectively. After working as a postdoctoral fellow from 1978 to 1981 at Indiana University in the USA, he became an Assistant Professor in 1981 at Osaka University where he was promoted to a Full Professor in 1994. His research has been focused on electron transfer chemistry, particularly artificial photosynthesis. He is currently a Professor at Ewha Womans University and a Professor Emeritus at Osaka University. |
1. Introduction
Fossil fuels that had been accumulated as products of photosynthesis on our planet have been consumed at a much faster rate than the current production of solar fuels by natural photosynthesis.1 Such rapid consumption of fossil fuels has created concerns about global warming and climate change as well as their depletion in the near future.2 Thus, an extensive effort has so far been devoted to produce solar fuels as renewable energy and material sources by artificial photosynthesis.2–11 Artificial photosynthesis consists of light-harvesting and charge-separation units, combined with catalytic units for water oxidation and proton or CO2 reduction, requiring multi-catalytic functions.12–19 Semiconductor-based photocatalysts have been developed to realize multi-functional catalysis, which involves three steps:20–32 (i) photoexcitation by photons with an energy greater than that of their band gap excites electrons to the conduction band, leaving holes in the valence band. (ii) The photogenerated electrons and holes are separated by overcoming coulombic attraction, followed by their diffusion and transfer. (iii) The surviving photogenerated electrons and holes react with adsorbed species on the surface of the photocatalyst for oxidation of water and reduction of protons or CO2 with the use of cocatalysts combined with photocatalysts.20–33 In the case of heterogeneous semiconductor-based photocatalysts, however, the detailed photocatalytic mechanisms have yet to be clarified because of the difficulty of identification and detection of reaction intermediates in heterogeneous systems in precluding rational molecular design for improvement of the catalytic efficiency and selectivity.34In contrast, an extensive effort has been devoted to clarifying catalytic mechanisms of homogeneous water oxidation, proton or CO2 reduction.9–19,35–48 Dual functional homogeneous catalysts have been developed for photochemical synthesis.49–54 However, multi-functional homogeneous photocatalysts have been less developed for photocatalytic water oxidation combined with proton or CO2 reduction.
This review focuses on multi-functional photocatalytic systems for solar fuel production by combining photocatalytic water oxidation and proton or CO2 reduction. Synthetic coordination polymer catalysts will be described, having dual catalytic functions, long-term stability, tolerance to experimental conditions, and good product selectivity including mechanistic investigations through in situ spectroscopy and kinetics. Metal–organic frameworks (MOFs) and mesoporous cellular silica foams containing both organic parts and inorganic metal parts will also be described inducing various photoredox reactions such as water oxidation, H2 production, CO2 reduction and H2O2 production. Then, immobilization of different molecular catalysts on solid-state supports for solar fuel production will be described to provide a system displaying dual catalytic function, enhanced selectivity and simplified identification of the active species, while also facilitating product isolation and catalyst recycling.14,40,41,55 Finally, construction of multi-functional catalytic systems is discussed for solar-driven fuel production from water.27,41–57
2. Coordination polymeric cyano (CN)-bridged heteronuclear catalysts
Hydrogen peroxide has attracted increasing interest as a solar fuel that can be generated from H2O and O2 using solar energy and used in H2O2 fuel cells.4,58–70 CN-bridged heteronuclear metal complex polymers (M(II)[(bpy)Ru(II)(CN)4]: M(II) = Mn(II), Fe(II) and Ni(II); bpy = 2,2′-bipyridine: see Fig. 1) were reported to act as a dual functional photoredox catalyst for generation of H2O2 from H2O and O2 under visible light irradiation, catalyzing both photo-reduction of O2 to H2O2 and photo-oxidation of H2O to O2.71 The highest turnover number (TON) was obtained as 247 based on the monomer unit of Ni(II)[(bpy)Ru(II)(CN)4] in the presence of Sc3+ under visible light irradiation.71 As shown in Scheme 1, in the photocatalytic cycle it was proposed that electron transfer from {Ni(II)[(bpy)Ru(II)(CN)4]}*, which is the excited state of Ni(II)[(bpy)Ru(II)(CN)4], to S2O82− occurs to afford {Ni(II)[(bpy)Ru(III)(CN)4]}+, SO4˙− and SO42−.71 Then, {Ni(II)[(bpy)-Ru(III)(CN)4]}+ is converted to {Ni(III)[(bpy)Ru(II)(CN)4]}+.71 Ni(II)[(bpy)Ru(II)(CN)4] is also oxidized by SO4˙− to form {Ni(III)[(bpy)Ru(II)(CN)4]}+, which oxidizes H2O to evolve O2.71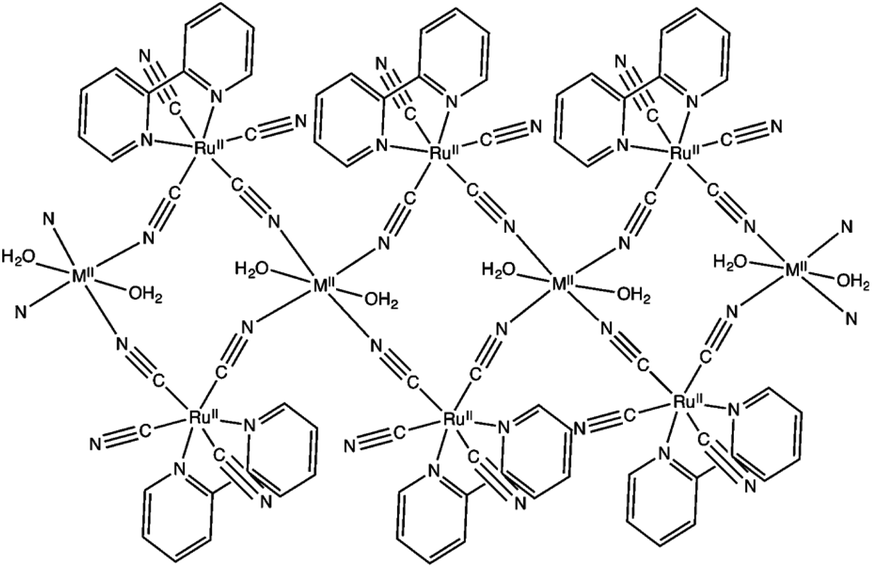 | ||
| Fig. 1 Schematic structure of M(II)[(bpy)Ru(II)(CN)4]. Reprinted from ref. 71 with permission from the Royal Society of Chemistry (Copyright 2017). | ||
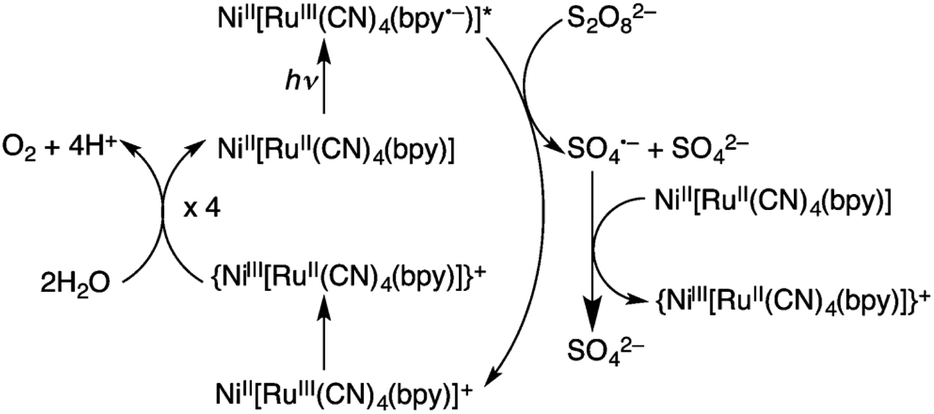 | ||
| Scheme 1 Photocatalytic cycle of water oxidation by S2O82− with Ni(II)[(bpy)Ru(II)(CN)4]. Reprinted from ref. 71 with permission from the Royal Society of Chemistry (Copyright 2017). | ||
The photocatalytic oxidation of water with Ni(II)[(bpy)Ru(II)(CN)4] is combined with the two-electron/two-proton reduction of O2 to produce H2O2 from H2O and O2 in the presence of Sc3+ (Scheme 2).71 Electron transfer from the photoexcited state of Ni(II)[(bpy)Ru(II)(CN)4] to O2 occurs in the presence of Sc3+ to afford {Ni(II)[(bpy)Ru(III)(CN)4]}+ and an O2˙−–Sc3+ complex that disproportionates in the presence of protons to form H2O2.72–75 The dual-functional photocatalysis of a single catalyst M(II)[(bpy)Ru(II)(CN)4] provides an efficient way to produce H2O2 from H2O and O2 as a promising solar fuel using sunlight.71 The yields of H2O2 with M(II)[(bpy)Ru(II)(CN)4] are summarized in Table 1, in which the maximum concentration of H2O2 obtained with the use of NiII[RuII(CN)4(bpy)] was 2.2 mM after 70 h of photoirradiation.71
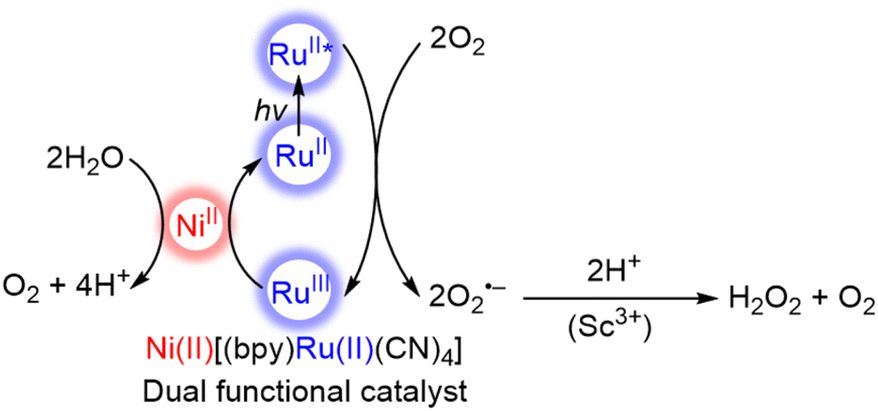 | ||
| Scheme 2 Catalytic mechanism for the formation of hydrogen peroxide from water and dioxygen with a dual functional catalyst, Ni(II)[(bpy)Ru(II)(CN)4], under visible light irradiation.71 | ||
| Entry | Catalyst | Irradiated wavelength (nm) | Reaction conditions | H2O2 yield (h) | Ref |
|---|---|---|---|---|---|
| 1 | FeII[RuII(CN)4(bpy)] | λ > 390 nm | O2-saturated H2O in the presence of Sc(NO3)3 (0.10 M) at 298 K | 1.0 mM (300 h) | 71 |
| 2 | NiII[RuII(CN)4(bpy)] | λ > 390 nm | O2-saturated CH3OH/H2O (5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v) in the presence of Sc(NO3)3 (67 mM) at 298 K :1, v/v) in the presence of Sc(NO3)3 (67 mM) at 298 K |
2.2 mM (70 h) | 71 |
| 3 | [Fe(H2O)3]2[Ru(CN)6]@sAl-MCM-41 | λ > 390 nm | O2-saturated CH3CN (2.9 mL) and H2O (0.40 mL) in the presence of Sc(NO3)3 (0.10 M) at 298 K | 3.0 mM (114 h) | 76 |
| 4 | [RuII(Me2phen)3]2+/[Ir(Cp*)(H2O3)]2+ | λ > 420 nm | O2-saturated H2O (3.0 mL) in the presence of Sc(NO3)3 (0.10 M) at 298 K | 2.0 mM (23 h) | 77 |
| 5 | [RuII(Me2phen)3]2+/[Co(Cp*)(bpy)(H2O)]2+ | λ > 390 nm | O2-saturated H2O (3.0 mL) in the presence of Sc(NO3)3 (0.10 M) at 298 K | 1.1 mM (3 h) | 58 |
| 6 | MIL-125-NH2 MOF | λ > 420 nm | O2-saturated CH3CN (5.0 mL)/H2O (1.0 mL) | 0.16 mM (20 h) | 96 |
| 7 | TTF–BT–COF | λ > 420 nm | O2-saturated H2O (10 mL) at 298 K | 1.38 mM (1 h) | 99 |
A CN-bridged polynuclear metal complex containing iron(II) and ruthenium(II) incorporated in mesoporous silica–alumina {[Fe(II)(H2O)3]2[Ru(II)(CN)6]@sAl-MCM-41} was also reported to act as a dual-functional photocatalyst for the photocatalytic formation of H2O2 from H2O and O2 and oxidation of benzene (C6H6) to PhOH using O2 as an oxygen source and as an oxidant under photoirradiation at 298 K.76 [Fe(II)(H2O)3]2[Ru(II)(CN)6] exhibited catalytic activity in thermal hydroxylation of C6H6 by H2O2, where the apparent TON of PhOH formation reached ∼390 for 60 h.76 The TON increased to 2.5 × 103 for 114 h by incorporation of [Fe(II)(H2O)3]2[Ru(II)(CN)6] with sAl-MCM-41.76 The iron ions in [Fe(II)(H2O)3]2[Ru(II)(CN)6] have extra H2O ligands to form an octahedral coordination (Fig. 2).76 The iron(II) ions may react with H2O2 to form hydroxyl radicals or iron-oxo species, which oxidize C6H6 to form PhOH.76 [Fe(II)(H2O)3]2[Ru(II)(CN)6] acts as both an excellent catalyst for C6H6 hydroxylation with H2O2 to produce PhOH and also an effective photocatalyst for the H2O oxidation with O2 to generate hydrogen peroxide.76
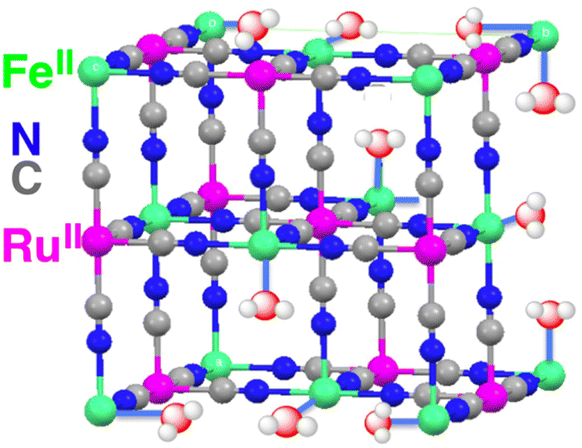 | ||
| Fig. 2 Partial structure of [Fe(II)(H2O)3]2[Ru(II)(CN)6]. Reprinted from ref. 76 with permission from American Chemical Society (Copyright 2016). | ||
The photocatalytic mechanism for generating H2O2 from H2O and O2 and hydroxylating C6H6 using a composite photocatalyst, [Fe(II)(H2O)3]2[Ru(II)(CN)6]@sAl-MCM-41, under irradiation is shown in Scheme 3.76 Electron transfer from the photoexcited state of [Fe(II)(H2O)3]2[Ru(II)(CN)6] to O2 occurs in the presence of Sc3+ to form {[Fe(II)(H2O)3]2[Ru(III)(CN)6]}+ and O2˙−–Sc3+ species, which disproportionates in the presence of protons to yield H2O2.76 The maximum concentration of H2O2 was 3.0 mM after 114 h of photoirradiation (entry no. 3 in Table 1).76 The H2O2 concentration was higher than those obtained with the use of a combination of [RuII(Me2phen)3]2+ with water oxidation catalysts (entry no. 3 and no. 4 in Table 1). H2O2 produced thus is used as an oxidant for hydroxylation of C6H6.77,78 The combination of the photocatalytic H2O2 formation from H2O and O2 and the catalytic C6H6 oxidation to PhOH with H2O2 provides a new strategy to achieve single-pot C6H6 oxidation using O2, which is an oxidant as well as an ideal oxygen source.76
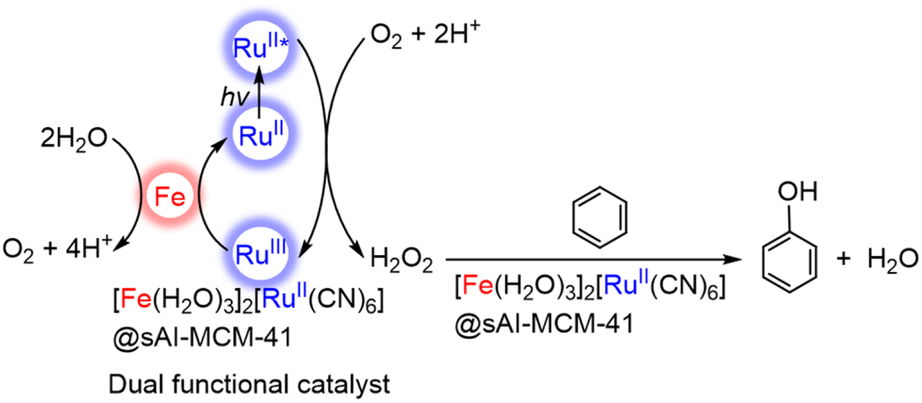 | ||
| Scheme 3 Photocatalytic cycle for PhOH production from C6H6 and O2 with [Fe(II)(H2O)3]2[Ru(II)(CN)6]@sAl-MCM-41.76 | ||
A semiconductor–catalyst hybrid assembly has recently been synthesized for photocatalytic H2O oxidation by forming CoFe Prussian blue (PB) particles on Dion–Jacobson type niobate (NB) nanosheets (KCa2Nb3O10), which results in a p–n junction shown by the Mott–Schottky plot.79 The assembly consisted of NB nanosheets and PB nanostructures with a particle size of about 50 nm.79 Photocatalytic oxidation of H2O was investigated on both bare NB nanosheets and the hybrid assembly in the presence of S2O82−, which is an electron scavenger, showing that the hybrid assembly exhibited greater photocatalytic activity (89.5 μmol g−1 h−1) than NB nanosheets.79 Various spectroscopic measurements, such as IR, XRD, XPS, SEM, and TEM, carried out on both raw and post-catalytic samples, showed that the hybrid assembly exhibited an appropriate band energy alignment for the photocatalytic H2O oxidation process and was stable even after 12 h of photocatalytic experiments.79 This implies that the hybrid assembly not only retained its structural integrity, but also enabled electron transfer between the NB nanosheets and PB units through charge transfer during the photocatalytic process.79 As the hybrid assembly exhibited approximately 6.5 times higher catalytic activity than pure NB nanosheets, it can therefore be considered one of the first efficient p–n junction type NB hybrid assemblies with an earth-abundant cocatalyst.79 The p–n junction feature allowed the hybrid assembly to be active in the UV as well as the visible region.79 The facile restructuring of both NB nanosheets and PB particles with metal doping also allowed for several facile strategies to further explore NB/PB interaction.79
The moldability, wide composition, and compatibility with other substrates of coordination polymer materials as described above may enable achieving unique multi-catalytic functions for various photoredox reactions in the future.
3. Metal–organic frameworks
Metal–organic frameworks (MOFs) have been adopted to fabricate homogeneous polyoxometallate (POM) water oxidation catalysts (WOCs) as well as heterogeneous photosystems for recyclable utilization.80–82 POM can be incorporated into MOF cavities to construct POM@MOF composites as a heterogeneous photocatalyst for photocatalytic water oxidation, where the MOF acts as a light harvester (Scheme 4).15,83 It has been reported that a tetranuclear cobalt porphyrin (Porp–Co4) is immobilized in the pores of porphyrin-linked zirconium MOF-545 to produce a Porp–Co4 @MOF-545 complex for the photocatalytic oxidation of water.84 The proposed mechanism is shown in Scheme 5.84 First, the porphyrin captured the light and electron transfer from the excited state to the sacrificial electron acceptor occurred to produce the one-electron oxidized porphyrin.84 Then, Co4–POM was oxidized and after 4 oxidizing equivalents on Co4–POM, water was oxidized to evolve O2.84 The excellent performance of this photocatalytic system was attributed to the increase in the oxidative power of the MOF by the porphyrin ligand and the stabilization of the active site by confining Co4–POM inside the pore of MOF-545.84 Enhanced efficiency was achieved using Porp–Co4 thin films on a TiO2 support, which overcame the problems of the random orientation of the active sites and hindrance of light penetration.85 Absorption capacity and charge separation at the Co4–POM@MOF junction were increased by transferring electrons from the MOF to Co4–POM to increase the efficiency.85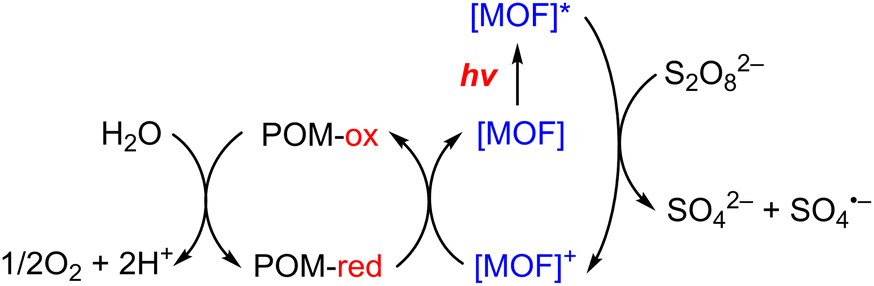 | ||
| Scheme 4 Photocatalytic cycle of a Co4–POM@MOF photosystem.15 | ||
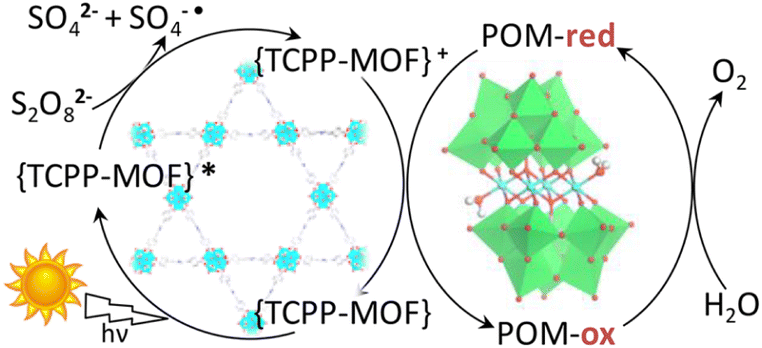 | ||
| Scheme 5 Proposed mechanism for the light-driven water oxidation by Porp–Co4@MOF-545. Reprinted from ref. 84 with permission from American Chemical Society (Copyright 2018). | ||
A Pt-loaded phosphorescent MOF was also utilized for efficient H2 generation with higher turnover frequencies (TOFs) and an improved TON (7.0 × 103) compared to similar homogeneous catalysts.73 [(bpy)Ir(III)(ppy)2]Cl-derived dicarboxylic acids (H2Lx in Fig. 3) were initially utilized for the successful synthesis of PtNP@MOFs (PtNP = Pt nanoparticle).73 Internally grown octahedral nanocrystals were used to achieve H2 evolution by electron injection into PtNPs and synergistic photoexcitation of the MOFs.73
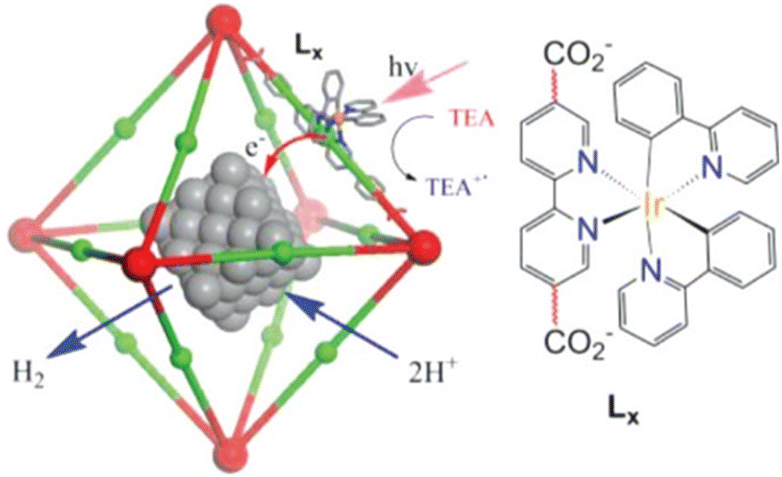 | ||
| Fig. 3 Reaction scheme for the photocatalytic H2 evolution via photoinjection of electrons from the light-harvesting MOFs into platinum NPs (PtNPs). The red and green balls represent Zr6(O)4(OH)4(carboxylate)12 cores and the Ir–phosphor ligand of the MOFs, respectively. Reprinted from ref. 73 with permission from American Chemical Society (Copyright 2012). | ||
A dinuclear iron complex, [FeFe](dcbdt)(CO)6 (dcbdt = 1,4-dicarboxylbenzene-2,3-dithiolate), was incorporated into a highly robust Zr(IV)-based metal–organic framework (MOF), where postsynthetic ligand and metal ion exchange (PSE) occurred to construct UiO-66–[FeFe](dcbdt)(CO)6, which is an organometallic, multinuclear, and nonprecious-metal-based proton reduction catalyst (Fig. 4).86 UiO-66–[FeFe](dcbdt)(CO)6 is a hybrid material that combines the highly stable and ordered inorganic support with the advantages of molecular catalysts.86 The molecular integrity of the organometallic site in the MOF was confirmed by a variety of spectroscopic methods, including XAS.86 Photocatalytic H2 evolution occurs using ascorbate (Asc) as an electron donor, [RuII(bpy)3]2+ as a photosensitizer and MOF–[FeFe](dcbdt)(CO)6 as a bifunctional photoredox catalyst (Scheme 6).86 Incorporation of the Fe2 complex in MOF–[FeFe](dcbdt)(CO)6 resulted in higher stability under the photocatalytic conditions, protected the reduced species from charge recombination, and promoted the disproportionation reactions that generated catalytically active dianions.86 Electron transfer from Asc to the photoexcited [RuII(bpy)3]2+ occurs to undergo photocatalytic H2 evolution with UiO-66–[FeFe](dcbdt)(CO)6.86
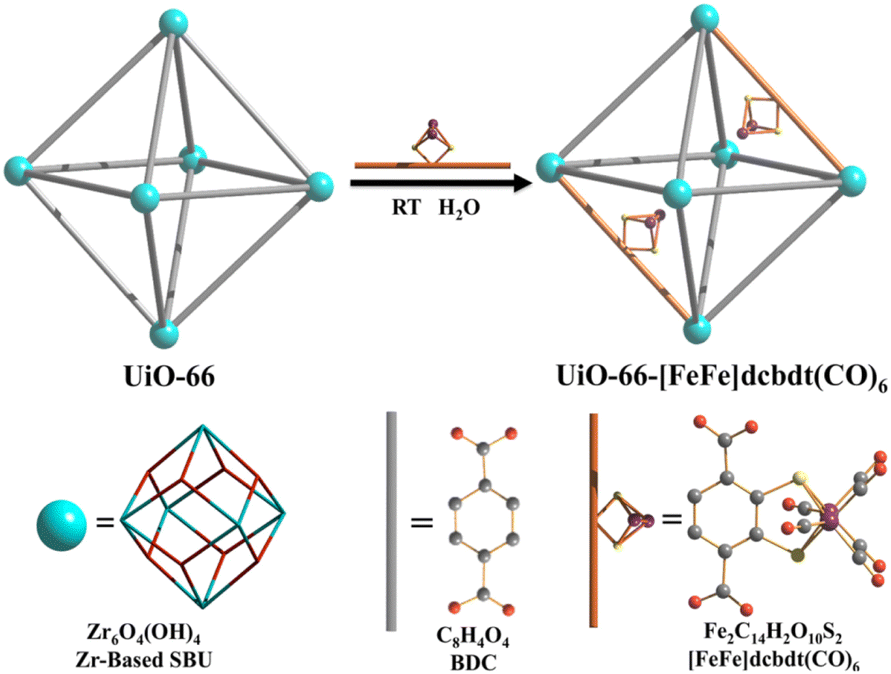 | ||
| Fig. 4 Schematic representation of PSE of [FeFe](dcbdt)(CO)6 into UiO-66. Reprinted from ref. 86 with permission from American Chemical Society (Copyright 2013). | ||
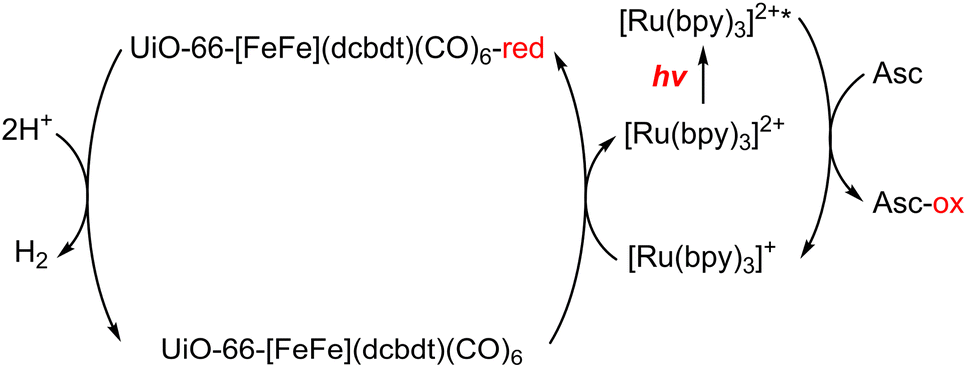 | ||
| Scheme 6 Scheme for the photocatalytic H+ reduction.86 | ||
A POM-based TBA5[P2Mo(V)16Mo(VI)8O71(OH)9Zn8(L)4] MOF (NNU-29), which exhibits strong chemical stability, can maintain the advantageous electronic properties of POM as well as keep the complex stable in aqueous solution.87 In addition, the sensitive photocurrent response of this MOF enables a high separation efficiency of light-induced electron–hole pairs for the efficient heterogeneous photocatalytic conversion of CO2 to HCO2H in H2O, reducing competitive H2 generation (TON = 28).88 Based on the analysis of the experimental results and theoretical calculations, the proposed mechanism for the photocatalytic conversion of CO2 to HCOOH over NNU-29 is shown in Scheme 7.88 When the photosensitizer [RuII(bpy)3]2+ is irradiated with visible light, an electron of [RuII(bpy)3]2+ is excited from HOMO to LUMO, and then transferred to NNU-29, resulting in reduction of NNU-29 and formation of [RuIII(bpy)3]3+.88 This is because NNU-29, which is like an electron sponge, can accept photoelectrons due to the valence change of several Mo atoms.88 The POM moiety of reduced NNU-29 captures and reduces CO2 and then returns to its original state.88 Meanwhile, [RuIII(bpy)3]3+ accepts an electron from TEOA as an electron donor and returns to [RuII(bpy)3]2+ while oxidizing TEOA.88
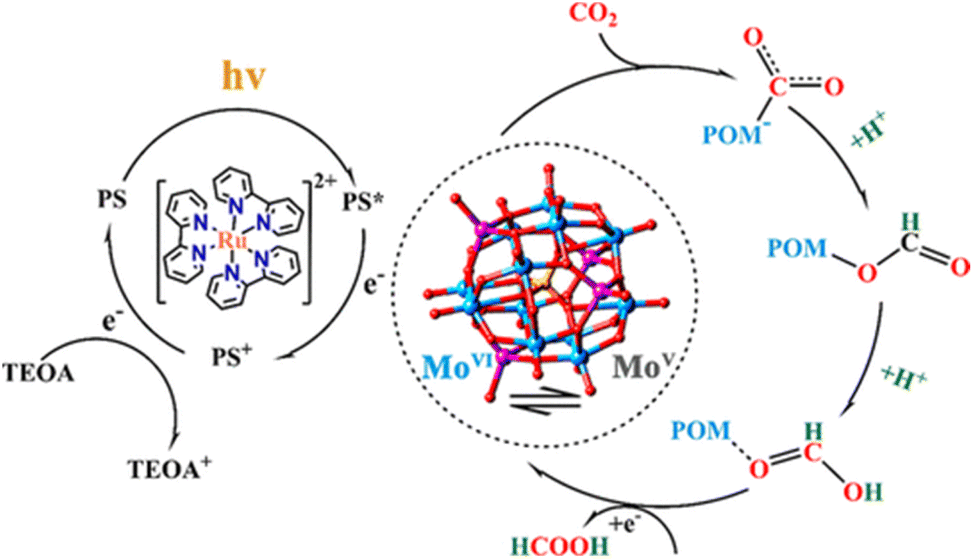 | ||
| Scheme 7 Proposed mechanism for the photocatalytic CO2 reduction to HCO2H catalyzed by NNU-29. Reprinted from ref. 88 with permission from American Chemical Society (Copyright 2019). | ||
In the latest study, the Keggin-type polyoxometalate (POM) PW12O403− and an Rh catalyst Cp*Rh(bpydc)Cl2 (bpydc = 2,2′-bipyridine-5,5′-dicarboxylic acid) in the UiO MOF (PW12,Cp*Rh)@UiO-67 demonstrated high photocatalytic activity for CO2 reduction to produce formate (Scheme 8).89 Compared to that observed for the Cp*Rh@UiO-67 catalyst without POM, the production of formate was doubled and achieved a maximum TON of 175 when fabricated as thin films, demonstrating the beneficial effect of POM.89 However, PW12 has a minimal effect on the electronic structure of the composite but acts as a proton relay.89
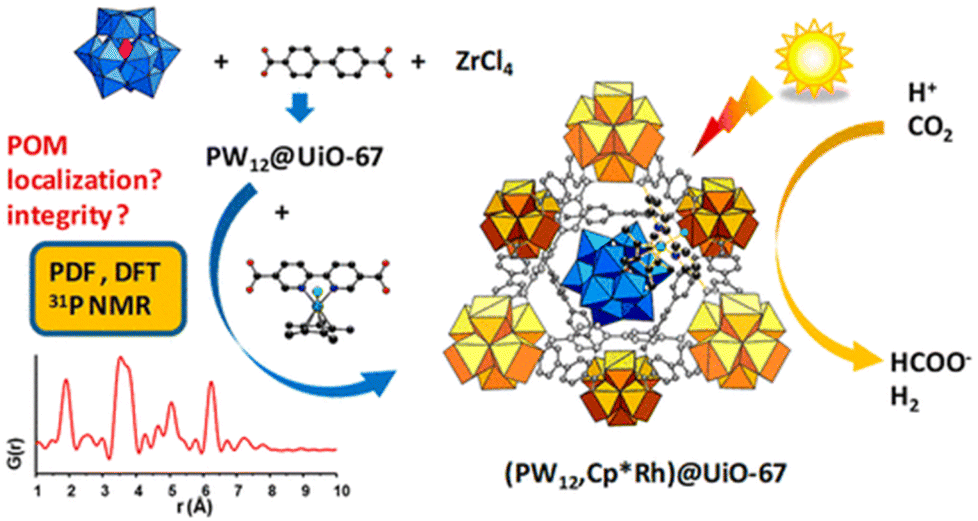 | ||
| Scheme 8 Scheme for photocatalytic reduction of CO2 to produce formate catalyzed by (PW12,Cp*Rh)@UiO-67. Reprinted from ref. 89 with permission from American Chemical Society (Copyright 2020). | ||
Photocatalytic reduction of CO2 into value added fuel (i.e., CO) together with reduction of H+ to H2 using an Fe(III)(TCPP)@NU-1000 catalyst was reported by Hupp and co-workers.90 The Fe(III)(TCPP)@NU-1000 catalyst was prepared by grafting an iron(III)–porphyrin (Fe(III)(TCPP); TCPP = tetrakis(4-carboxy-phenyl)porphyrin) onto the hexagonal portion of the NU-1000 MOF mesopore.90 This catalyst was available in the 8-connected nodes, giving a TON of ∼20 for both products, CO and H2, under light for 2 h.90 Hupp and co-workers also addressed the problem of catalyst leaching from the chromophoric MOF after the TON reached 20 and discussed the need to design better linkers or binding sites to improve catalyst robustness as well as increase TON.90 The mechanism of the photocatalytic reduction of CO2 with Fe(III)(TCPP)@NU-1000 was proposed as shown in Scheme 9.90,91 In the presence of TEOA as a sacrificial reductant, the photoexcited electrons were transferred initially from NU-1000* to Fe(III)(TCPP) to generate Fe(I)(TCPP), followed by reformation of NU-1000 and production of TEOA+.90 In the presence of a second molecule of TEOA, spontaneous decomposition of TEOA+ generated a strongly reducing radical species with TEOAH+.92–94 The resulting radical species could donate the electron required to reach the catalytically active Fe(0)(TCPP) species.90 Alternatively, disproportionation of two Fe(I)(TCPP) molecules to Fe(II)(TCPP) and Fe(0)(TCPP) could effectively double the number of available reducing equivalents, when the radical species that is the decomposition product of TEOA+ reduces Fe(II)(TCPP) to Fe(I)(TCPP).90 Then, CO2 is bound to Fe(0)(TCPP) species to produce a Fe(0)(TCPP)–CO2 adduct species, which is susceptible to further reaction to generate Fe(II)(TCPP)–CO.90 Released O2− reacts with protons from TEOA (or contingent sources) to produce H2O.90 When CO was released from Fe(II)(TCPP)–CO, Fe(II)(TCPP) was regenerated and the next catalytic cycle started again.90 As a competing process, Fe(0)(TCCP) may convert H+ to H− to form an iron(II)-hydride complex that reacts with an additional proton to give H2.90
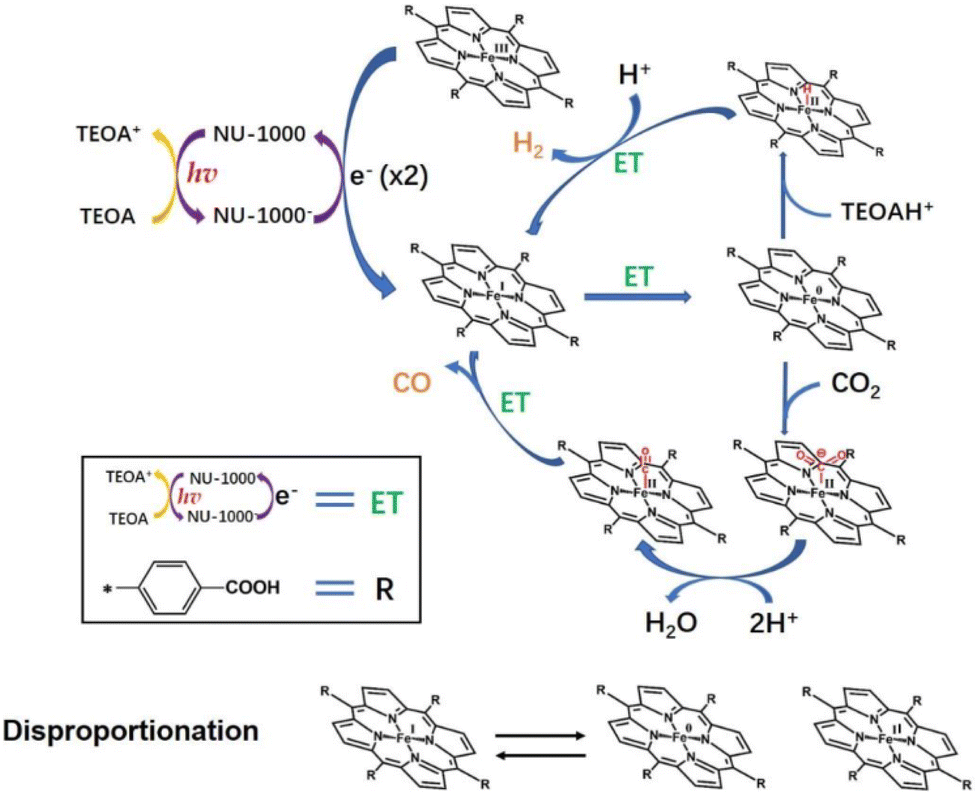 | ||
| Scheme 9 Proposed mechanism for photocatalytic reduction of CO2 using an Fe–TCPP@NU-1000 heterogeneous catalyst with TEOA as a sacrificial agent. Reprinted from ref. 90 with permission from Elsevier (Copyright 2022). | ||
A microporous anthracene-functionalized Zr-MOF (NNU28) was prepared as a dual functional photoredox catalyst for reduction of CO2 to produce formate.95 Zr-MOF demonstrated efficient photoinduced charge generation, broadband visible light absorption, and high CO2 absorption, along with excellent chemical and thermal stability.95 NNU28 exhibited the highest rate of formation of formate (∼180 μmol h−1 mmol(MOF)−1) among Zr-based MOF photocatalysts.95 The Zr6 oxo cluster and anthracene-based organic ligand both act as photocatalytic centers for CO2 reduction under visible irradiation.95 The dual photocatalytic pathways for photoreduction of CO2 with NNU28 were demonstrated to be generally more efficient for visible-light-driven CO2 reduction than that typically relying on a ligand-to-metal charge transfer process, illustrating a new strategy to design and synthesize novel visible-light photocatalysts for CO2 reduction with high efficiency.95 A photocatalytic mechanism of dual catalytic pathways for photoreduction of CO2 with NNU28 is shown in Scheme 10.95 Upon photoirradiation, the anthracene-based ligand in NNU28 acts as an efficient visible light harvester for the sensitized Zr6 oxo cluster via the ligand-to-metal charge transfer (LMCT) process, undergoing photoinduced electron transfer from TEOA.95 The two catalytic pathways in Scheme 10 contribute to the photoreduction of CO2 to formate, accounting for the high catalytic performance of NNU28.95 The large conjugation of the anthracene-based ligand resulted in unexpected optical performance for the MOFs.95
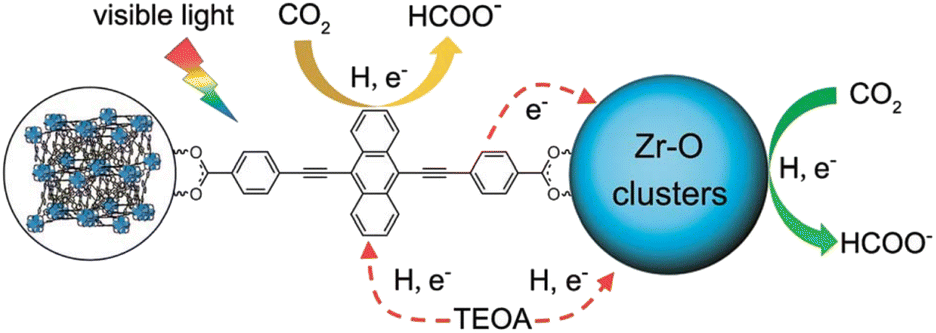 | ||
| Scheme 10 A photocatalytic scheme for CO2 reduction by TEOA with NNU28 under visible irradiation. Reprinted from ref. 95 with permission from the Royal Society of Chemistry (Copyright 2016). | ||
Visible light-driven 2e− O2 reduction to H2O2 with TEOA or benzyl alcohol was also catalyzed by a metal–organic framework (MIL-125-NH2).96 When NiO nanoparticles (NPs) were deposited onto MIL-125-NH2, the photocatalytic reduction of O2 to H2O2 was dramatically enhanced through disproportionation of O2˙− (Scheme 11).96 NiO acted as a WOC when water was used as a source of electrons.96 The H2O2 concentration was 0.16 mM after 20 h photoirradiation (entry no. 6 in Table 1). The photocatalytic mechanism of generation of H2O2 by 2e− O2 reduction with TEOA catalyzed by NiO/MIL-125-NH2 is shown in Scheme 11.96 Photoexcitation of MIL-125-NH2 results in production of Ti8O8(OH)4˙− and a hole at the linker.97,98 The produced hole is quenched by electron transfer from TEOA, followed by O2 reduction with Ti8O8(OH)4˙− to produce O2˙− that disproportionates with H+ to yield H2O2.96
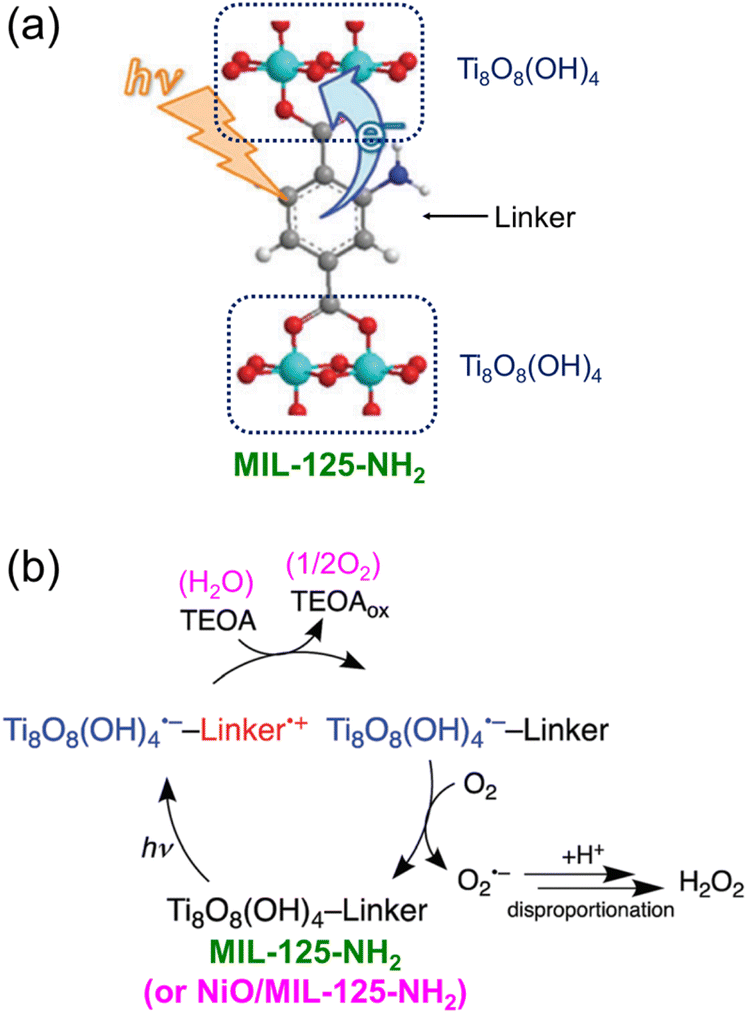 | ||
| Scheme 11 Visible light-driven 2e− O2 reduction to H2O2 with TEOA catalyzed by Ni/MIL-125-NH2. Reproduced from ref. 96 with permission from the Royal Society of Chemistry (Copyright 2018). | ||
Photocatalytic production of H2O2 by combination of H2O oxidation and O2 reduction to H2O2 without sacrificial agents is highly required (vide supra).99 A redox molecular-conjugated COF (TTF–BT–COF) was synthesized through the covalent bonding of tetrathiafulvalene (TTF) as a photooxidation site and benzothiazole (BT) as a photoreduction site.99 This photocatalyst was applied to the generation of H2O2 without sacrificial agents (Scheme 12).99 The concentration of H2O2 reached 1.38 mM after 1 h of photoirradiation (entry no. 7 in Table 1).99 The covalent linkage between them creates a redox junction, providing appropriate photo-redox ability, effective electron–hole separation efficiency, and visible light adsorption, allowing effective visible light-driven electron transfer from BT to TTF, resulting in the simultaneous WOR and ORR.99 It is worth noting that the production rate (276000 μM h−1 g−1) for H2O2 photoproduction without sacrificial donors can be record-high among porous crystalline photocatalysts, approximately 10 times higher than that with single linkers or physical mixtures.99 In addition, theoretical calculations have demonstrated redox molecular junctions that can enhance the performance by facilitating charge transfer and significantly reducing the energy barriers of both the WOR and ORR.99 This is the first research capable of designing redox molecular junction COF for production of H2O2, potentially shedding new light on the exploration of porous crystalline materials in industrial scale H2O2 production.99
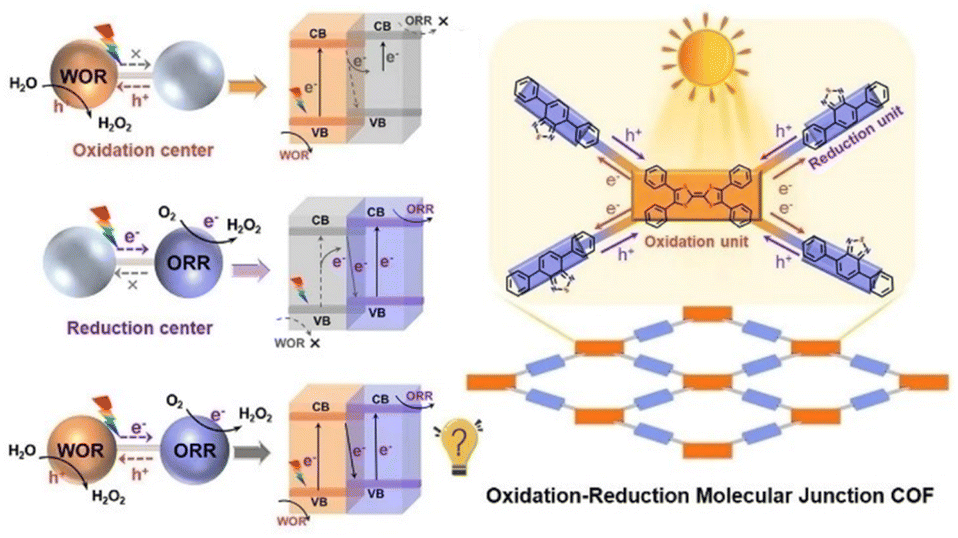 | ||
| Scheme 12 Schematic diagram for H2O2 formation with a redox molecular-conjugated COF photocatalyst. Reprinted from ref. 99 with permission from John Wiley and Sons (Copyright 2023). | ||
Carbon-based semiconductors and graphic carbon nitride (g-C3N4) and graphene oxide systems have also been frequently used for photocatalytic production of H2O2 from H2O and O2.67,100–102 These heterogeneous materials may not be suitable to combine molecular catalysts to develop multi-catalytic functions. In contrast, the multi-catalytic functions of the COF and MOF can be further expanded because the COF and MOF have not only the intrinsic catalytic activity but also the capability to encapsulate additional catalytic sites.
4. Mesoporous cellular silica foams
Numerous photocatalytic systems of interest have been developed by utilizing mesoporous silica, which has high stability due to its robust structural framework, confinement effects inside the channels with controllable hydrophobicity/hydrophilicity, and high specific surface area with uniform pore size.28,103–115Recently, Inagaki and co-workers reported photocatalytic water splitting for the oxygen evolution reaction (OER) with the use of periodic mesoporous organosilica (PMO) grafted with a [Ru(II)(bpy)3]2+ sensitizer.116 The unique pore-wall structure (bpy-PMO) in the PMO pores is responsible for the generation of photoredox-active metal complexes on the outer surface of the material (Scheme 13).116
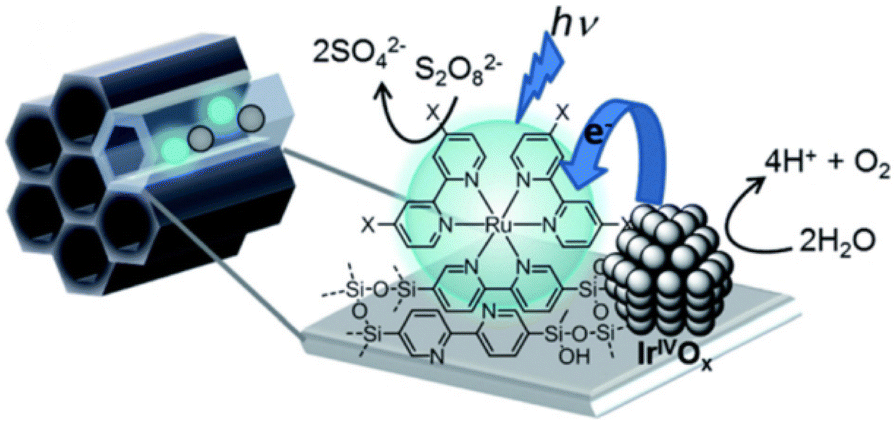 | ||
| Scheme 13 Photocatalytic water oxidation by Ru complexes supported on BPy-PMO. Reprinted from ref. 116 with permission from the Royal Society of Chemistry (Copyright 2020). | ||
Inagaki and co-workers also reported fine-tuning of the photocatalyst to increase the photocatalytic rate up to 20 times under visible light irradiation.116 The photochemical properties of the catalyst (Ru(II)(X2bpy)3/PMO) were controlled by changing the 4,4′-di-X substituents of the bpy ligand (X2bpy = 4,4′-X-2,2′-bipyridine) in the PMO framework, where X = H, Me, tBu, and C(O)OMe.116
The mesospaces uniformly distributed among Al–SiO2NPs were utilized to construct a self-assembled and active catalytic structure of 2-phenyl-4-(1-naphthyl)quinolinium ion (QuPh+-NA) and PtNPs for photocatalytic H2 evolution in artificial photosynthetic systems at room temperature.117 This composite catalyst (PtNPs/QuPh+-NA/Al–SiO2NPs) was synthesized using an aqueous suspension of PVP-capped PtNPs and an MeCN solution of QuPh+-NA by sequential addition to the aqueous dispersion of Al–SiO2NPs and then by placing it in an ultrasonicator for 30 min.117 The mesospaces allowed the controlled incorporation of PtNPs surrounded by several QuPh+-NA ions and the surface of the Al–SiO2NPs was covered with QuPh+-NA ions as shown in Fig. 5.117 This mesoporous structure allowed the PtNP to receive multiple electrons from QuPh˙-NA species, which were produced by electron transfer from β-dihydronicotinamide adenine dinucleotide (NADH) to the QuPh˙-NA˙+ (photoinduced ET state of QuPh+-NA ions). As a result, H2 was efficiently produced photocatalytically, and the catalyst was recycled multiple times.117 In contrast to the case of the PtNPs/QuPh+-NA/Al–SiO2NPs catalyst, almost no H2 formation was observed in the conventional mesoporous silica–alumina photocatalytic reaction system containing QuPh+-NA ions together with PtNPs instead of the PtNPs/QuPh+-NA/Al–SiO2NPs catalyst under the same reaction conditions.114 PtNPs with a size of 2 nm were too large to interact with QuPh+-NA ions immobilized inside the cylindrical mesopores, resulting in no photocatalytic H2 formation.114 Self-assembled structures of such metal oxide NPs are a promising platform for the assembly of functional organic compounds and NPs.114
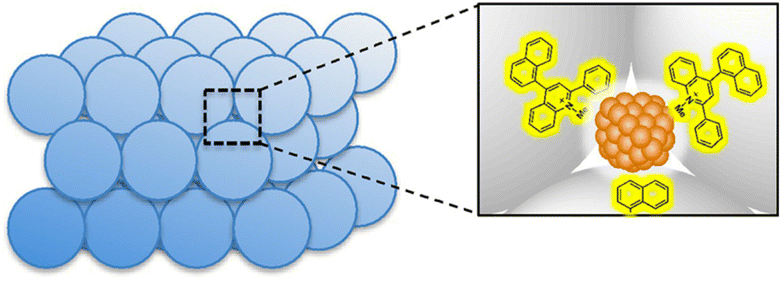 | ||
| Fig. 5 Structure of the catalyst prepared with silica–alumina NPs (Al–SiO2NPs) supporting QuPh+-NA on silica–alumina and PtNPs. Reproduced from ref. 117 with permission from John Wiley and Sons (Copyright 2016). | ||
Liu and co-workers reported that a [(Cp*)IrIIICl(μ-Cl)]2 complex was immobilized on bipyridine-based organosilica NTs (bpy/ENT) to obtain an organic–inorganic hybrid solid photocatalyst (Ir-bpy/ENT) for photocatalytic H2 evolution from both formate and aldehyde (Scheme 14).118 Ir-bpy/ENT has high surface area, stable nanotube structure, and confining effects inside its channels due to its large pore diameter, reducing diffusion inhibition and facilitating the facile transport of substrates, which is an important function to enhance the catalytic reactivity (Scheme 14).118 In addition, Ir-bpy/ENT has controllable hydrophobicity/hydrophilicity and isolated active sites.118 Ir-bpy/ENT can also provide high efficiency in the pH-dependent production of pure H2 (TOF = 16.2 h−1 and QY = ∼8.0%) from formate.118 The Ir-bpy/ENT catalyst can be reused up to 6 cycles.118
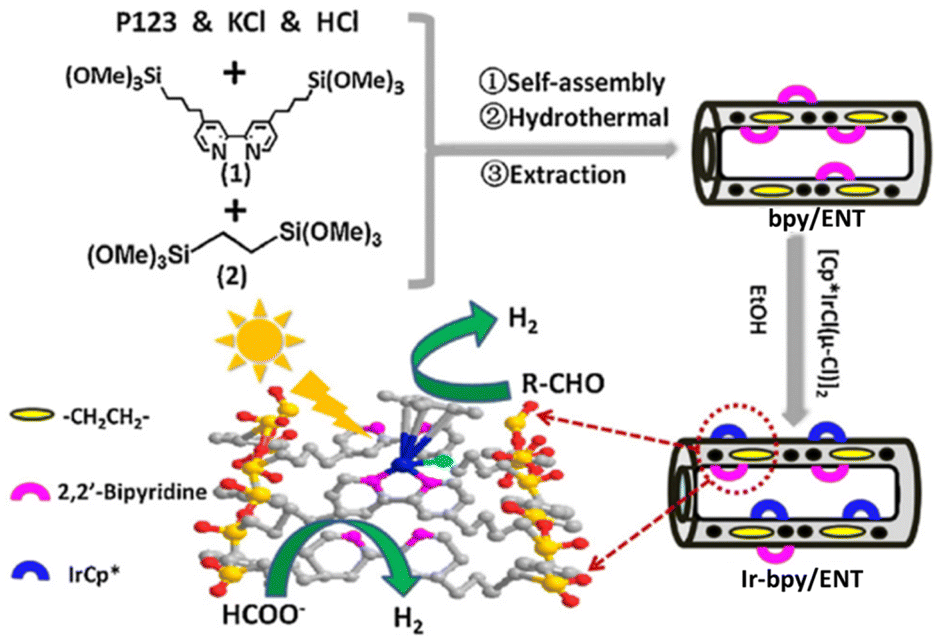 | ||
| Scheme 14 Synthesis of Ir-bpy/ENT and photocatalysis with Ir-bpy/ENT, where ENT is an ethenyl-bridged organosilica nanotube. Reprinted from ref. 118 with permission from Elsevier (Copyright 2018). | ||
Li and co-workers immobilized a Co(III) cyclam complex (cyclam = 1,4,8,11-tetraazacyclotetradecane) on the surface of mesoporous silica with the use of two different covalent linkages (Fig. 6).119 Li and co-workers also reported a heterogeneous photocatalyst designed for the photocatalytic reduction of CO2 to CO in the presence of p-terphenyl as a molecular photosensitizer.119 A TON of 12.5 for CO formation was achieved using Co(L)/SiO2 after CO2 reduction. Using one of the modified CoIII(cyclam)X/SiO2, the TON of CO formation reached 160 in the photocatalytic CO2 reduction.119
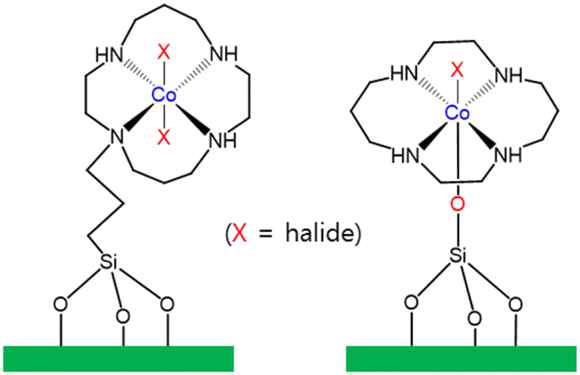 | ||
| Fig. 6 Co(L)/SiO2 bearing a cyclam ligand, showing two different covalent linkages.119 | ||
Various photocatalysts were embedded with PMO to afford abundant organic molecules (R) immobilized to the silica framework as light harvesters (Fig. 7a).120,121 10-Me-acridone groups able to absorb visible light were used as R in the PMO framework.121 The silica units interact strongly with the phosphonic acid groups of the PRu-Re photocatalyst, trapping PRu-Re inside PMO, resulting in a hybrid catalyst (Fig. 7b).121 The photons harvested by about 40 molecules of 10-Me-acridone were transferred to a Ru(II) unit of a PRu-Re photocatalyst in the hybrid catalyst, and then the photocatalytic CO2 reduction was initiated.121 The PMO hybrid with a light-harvesting function showed a 10-fold increase in the photocatalytic CO release rate compared to the PRu-Re-adsorbed PMO without a light harvester.121 When a benzimidazoline derivative was used as an electron donor in the photocatalytic CO2 reduction, the apparent TON for CO formation was more than 600.121
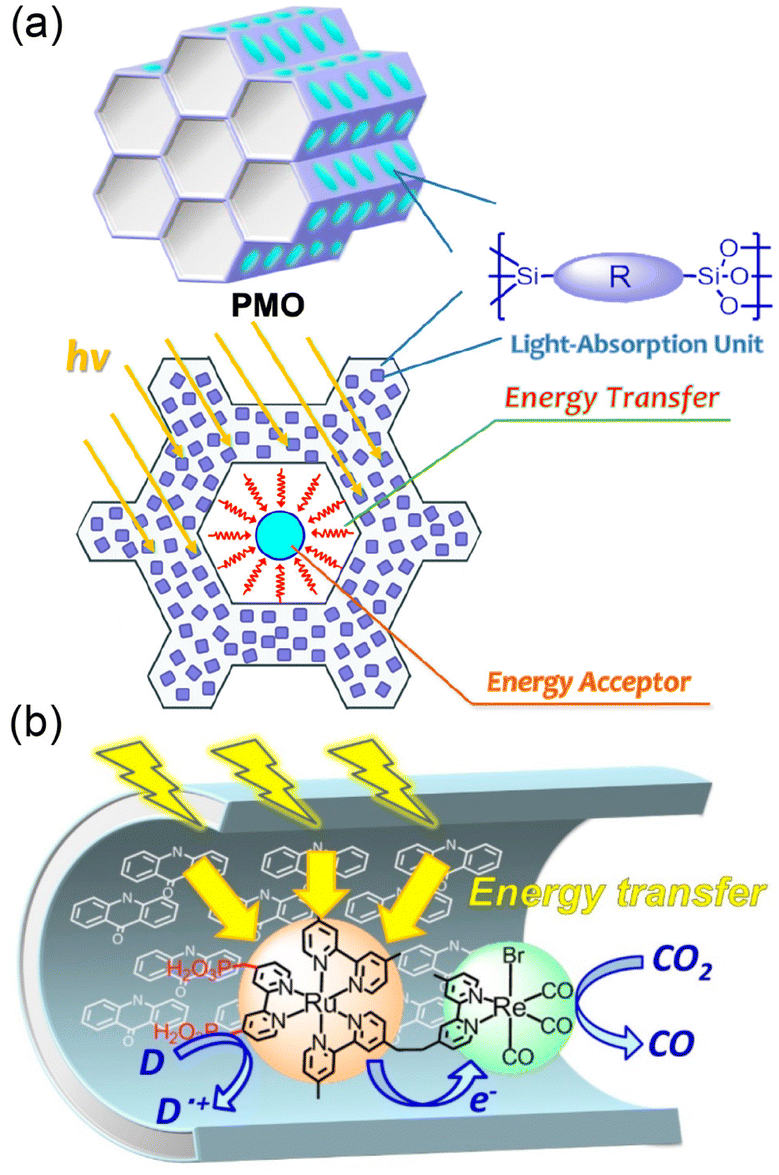 | ||
| Fig. 7 (a) Structural arrangement of each part of the PMO. (b) Photocatalytic CO2 reduction by light harvesting hybrids of PRu-Re and 10-Me-acridone in PMO. Reprinted from ref. 121 with permission from American Chemical Society (Copyright 2022). | ||
On the other hand, a multi-functional hybrid catalyst material was obtained by incorporating a 9-mesityl-10-methylacridinium ion (Acr+-Mes)47 into mesoporous silica–alumina, in which a copper complex [Cu(II)(TPA)](ClO4)2 (TPA = tris(2-pyridylmethyl)amine) was embedded by cation exchange.122 The hybrid composite catalyst [Cu(II)(TPA)]2+/Acr+-Mes@AlMCM-41 acted as an efficient and robust photocatalyst in the oxidation of p-xylene by O2 to yield 4-Me-benzaldehyde selectively and H2O2.122 A nano-sized mesoporous silica–alumina (sAlMCM-41 and tAlMCM-41) also stabilizes the electron-transfer state of Acr+-Mes, which is an electron donor–acceptor dyad cation.122 Acr+-Mes was easily incorporated into nano-sized mesoporous silica–alumina to produce a composite by a cation-exchange reaction.122 Photoexcitation of sAlMCM-41 and tAlMCM-41 resulted in generation of an extremely long-lived Acr˙-Mes˙+, which is an electron-transfer state, as detected by EPR and laser induced transient absorption measurements.122 Acr˙-Mes˙+ incorporated into tAlMCM-41 also has a very long lifetime lasting over 1 s at ambient temperature and decays via intramolecular back electron transfer as opposed to the diffusion-limited intermolecular back electron transfer between two Acr˙-Mes˙+ molecules in solution.122
The photocatalytic mechanism for the oxidation of p-xylene with [Cu(II)(TPA)]2+/Acr+-Mes@AlMCM-41 is shown in Scheme 15.122 First, photoexcitation of Acr+-Mes@AlMCM-41 results in formation of the long-lived Acr˙-Mes˙+, followed by electron transfer from p-xylene to the Mes˙+ moiety of Acr˙-Mes˙+.122 The radical cation, which is 1 e− oxidized species of p-xylene, thus deprotonates to form a 4-Me-benzyl radical, which reacts rapidly with O2 to produce a 4-Me-benzylperoxyl radical.122 Then, 4-Me-benzylperoxyl radical disproportionates to form 4-Me-benzyl alcohol, 4-Me-benzaldehyde and O2.122 4-Me-benzyl alcohol was further oxidized via electron transfer from 4-Me-benzyl alcohol to the Mes˙+ moiety of Acr˙-Mes˙+ to produce 4-Me-benzaldehyde selectively as the final oxygenated product of p-xylene.123 On the other hand, the reduction of O2 by Acr˙-Mes is catalyzed by [Cu(II)(TPA)]2+.122 The incorporation of Acr+-Mes into nano-sized mesoporous silica–alumina coupled with [Cu(II)(TPA)]2+ as an O2 reduction catalyst presented a promising way to develop efficient and robust organic photocatalysts combined with a metal complex catalyst, which acts as a multi-functional catalytic material for the oxidation of the substrate by O2 that is the ultimate environmentally benign oxidant in artificial photosynthesis to produce solar fuels.123
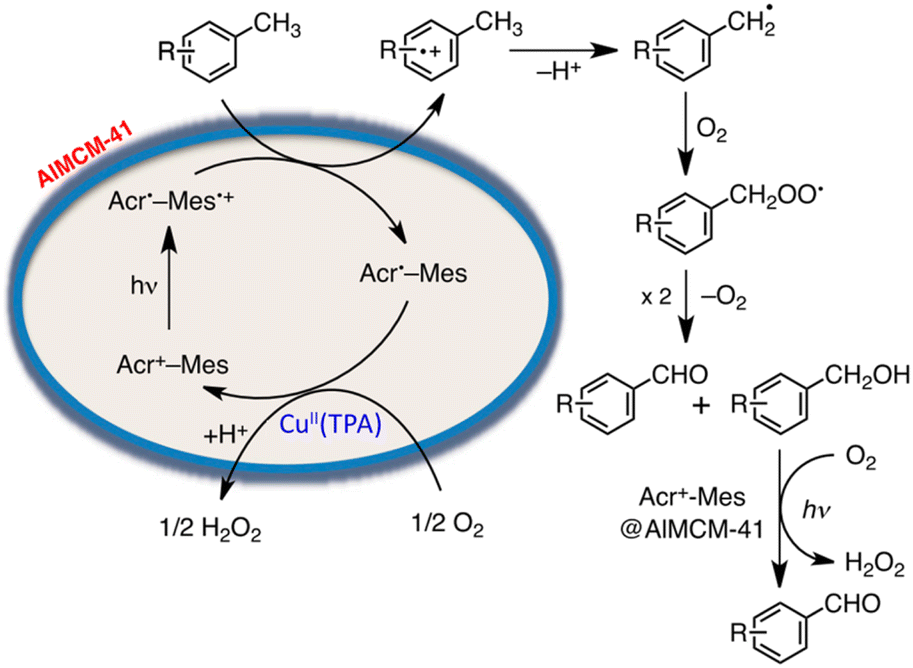 | ||
| Scheme 15 Photocatalytic mechanism for the oxidation of p-xylene selectively by O2 with [Cu(II)(TPA)]2+/Acr+-Mes@AlMCM-41. Reprinted from ref. 122 with permission from the National Academy of Sciences (Copyright 2012). | ||
Immobilization of different functional catalysts in mesoporous silica–alumina may enable the development of more efficient multifunctional catalytic systems in the future.
5. Hybrid photoelectrochemical cell
Hybrid photoelectrochemical cells composed of metal complexes and semiconductor materials with relatively high photochemical oxidation power have recently been developed for visible-light-driven multi-functional solar fuel production.124–131 Photocatalysts with semiconductor particles afford both high selectivity and strong oxidation power by photoexcitation of both the redox photosensitizer and the semiconductor via the so-called Z-scheme mechanism.124–131A Z-scheme configuration of a hybrid photoelectrochemical cell composed of a RuRe/CuGaO2 photocathode and a CoOx/TaON photoanode has been constructed to perform CO2 reduction by H2O without bias potential as shown in Scheme 16.132,133 Two compartment cells that contain a CO2-saturated electrolyte solution were separated using a Nafion membrane.132 Visible light irradiation of the RuRe/CuGaO2 photocathode and the CoOx/TaON photoanode in the wavelength region of λex > 460 nm and λex > 400 nm, respectively, resulted in formation of reduced products (CO and H2) and O2 (the four-electron oxidized product of water) in the cathode and anode cells, respectively.132 The amount of CO produced was 232 nmol and the TONCO reached 22 based on the amount of immobilized RuRe.132 The hybrid photoelectrochemical cell made it possible to reduce CO2 to CO as well as to oxidize water to O2 by photoexcitation of both RuRe and TaON via a Z-scheme-type electron transfer under visible-light irradiation.132
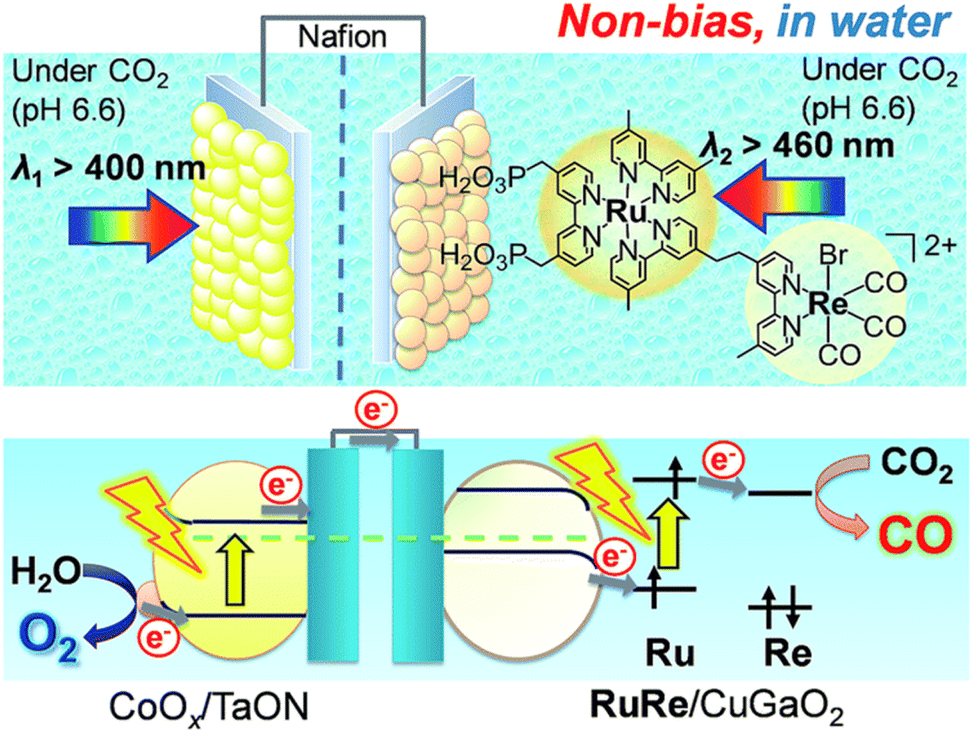 | ||
| Scheme 16 Schematic image of a hybrid photoelectrochemical cell in a Z-scheme configuration. Reprinted from ref. 132 with permission from the Royal Society of Chemistry (Copyright 2017). | ||
On the other hand, a p-type nickel oxide (NiO) photocathode was modified with a hexaphosphonated Ru(bpy)3-based dye (RuP3) that is linked with a tetraphosphonated molecular [Ni(P2N2)2]2+ type proton reduction catalyst (NiP) to be employed for the photocatalytic H2 evolution in water (Scheme 17).134 By using a layer-by-layer deposition method, Zr4+-phosphonates were linked to the phosphonate units of RuP3 and NiP in a supramolecular assembly on the p-type semiconductor NiO photocathode.134 Such a layer-by-layer method made it possible to control spatial arrangement of individual units without elaborate chemical synthesis of the dye–linker–catalyst dyad,134 keeping the dye located close to the catalyst and semiconductor surface to facilitate the photocatalytic activity, but spatially separating NiP from NiO to avoid charge recombination.134 The NiO|RuP3–Zr4+–NiP electrodes generate higher and more stable photocurrents as compared to photocathodes with RuP3 and NiP co-immobilized on the NiO surface in the absence of Zr4+ cations linking the dye and catalyst.134 Directed electron transfer from the excited dye to the catalyst occurs efficiently, whereas charge recombination was slowed down in the dye–linker–catalyst dyad.134 Thus, photoelectrocatalytic generation of H2 occurred at the NiO|RuP3–Zr4+–NiP hybrid electrode at an electrochemical underpotential, thereby demonstrating the potential of this photoelectrocatalytic system to store light in the chemical bonds of H2.134
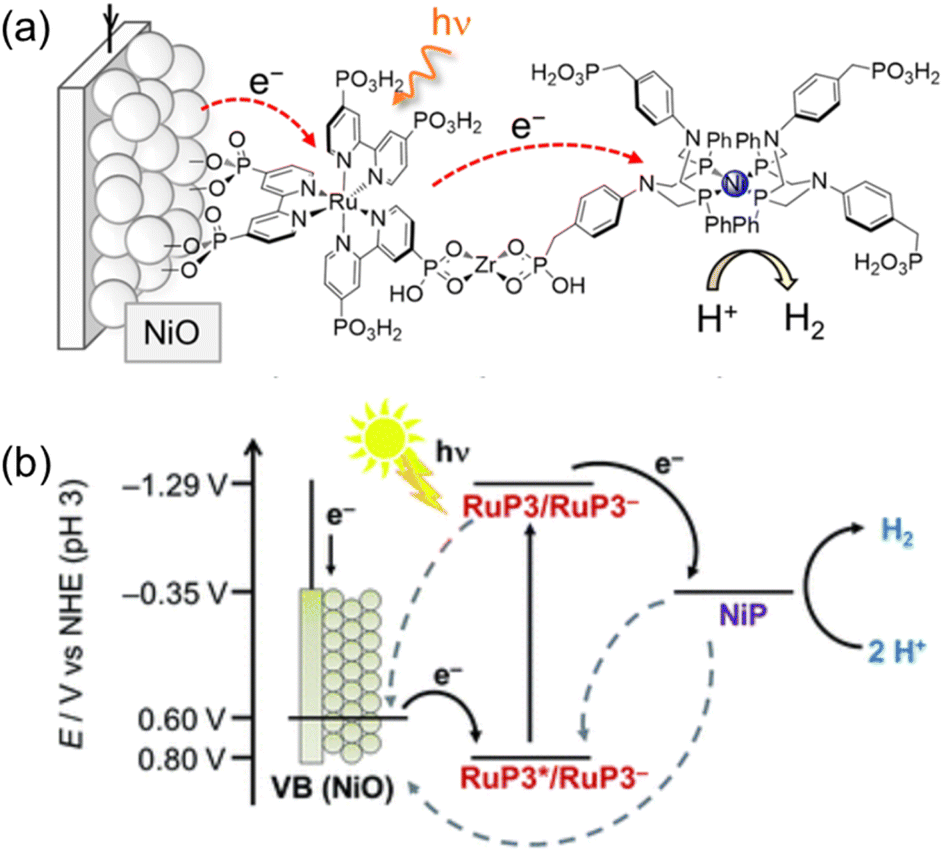 | ||
| Scheme 17 (a) Schematic procedure of sequential immersion of Zr4+ bridge-based photocathodes (NiO|RuP3–Zr4+–NiP). (b) Energy diagram of photoinduced electron injection from a NiO photocathode to the excited state of RuP3. Back solid arrows show forward electron transfer and grey dashed arrows show possible recombination pathways between p-NiO, RuP3 and NiP. Reprinted from ref. 134 with permission from the Royal Society of Chemistry (Copyright 2016). | ||
The efficiency of the NiO|RuP3–Zr4+–NiP electrodes may be improved by developing phosphonated push–pull dyes optimized for the photoelectrocatalytic H2 evolution.135 Further work may also include the synthesis and investigation of dyes, which are more suitable for injecting holes into the NiO valence band.135 Thus, the multifunctions of light-harvesting, electron transfer and H2 evolution catalytic functions can be further optimized.
6. Multi-functional molecular models of photosystems in photosynthesis
Much attention has been paid to the design of direct Z-scheme photocatalysts for solar energy conversion due to their effectiveness in spatially separating photogenerated electron–hole pairs and optimizing the oxidation and reduction capabilities of the photocatalytic system.136–145 In natural photosynthesis, water oxidation is a major reaction using a Mn4CaO5 cluster under solar illumination in photosystem II (PSII).146–149 In PS II, photocatalytic water oxidation evolves O2 with the supply of electrons and protons for solar fuel production.150 Photoexcited chlorophyll aggregates, P680*, undergo charge separation and electrons are transferred to quinones (QA and QB).151 Finally plastoquinone (PQ) accepted electrons to produce plastoquinol (PQH2) by taking two protons from its surroundings.152,153 As one molecule of plastoquinone is oxidized, two electrons are transferred to P700. Photosystem I (PSI) has the photooxidation and reduction cycle of P700.154 In this cycle, the electron flows to NADP+ to produce NADPH through electron carriers.155 In photosynthetic linear electron flow, the so-called Z-scheme, electrons that came from H2O oxidation in photosystem II are transferred to NADP+ to produce NADPH. On the other hand, in the dark reaction, driven by the energy stored in ATP, CO2 is finally stepwise reduced into carbohydrates.156 Light energy is stored in the form of NADPH, while some of the light energy is stored in the form of ATP through the phosphorylation of ADP.157,158 That is, the overall process of photosynthesis is the process of producing organic compounds such as carbohydrates by the reduction of CO2 along with the oxidation of water.157,158Inspired by the Z-scheme in photosynthesis, photocatalytic water splitting was achieved with the use of molecular catalysts.159 The combination of PSI and PSII models achieved photocatalytic H2O splitting for the first time, allowing the production of O2 and H2 in a ratio of 1:2 (Scheme 18).159 As shown in Fig. 8, each molecular model of photosystem, PSI or PSII, contains two solvent phases, toluene (PhCH3) and water/trifluoroethanol mixture (H2O/TFE, 3:1) phases, which are separated using a liquid membrane.159 The PhCH3 and H2O/TFE mixture phases of the PSII functional model were each connected to the same phase of the PSI functional model using two glass membranes.159 The PSII functional model contains the p-benzoquinone derivative (X-Q) as a plastoquinone analog in PhCH3 and a nonheme iron(II) complex ([FeII(N4Py)]2+; N4Py = N,N-bis(2-pyridylmethyl)-N-bis(2-pyridyl)methylamine) as a H2O oxidation catalyst in H2O/TFE.159 Photoirradiation of the PSII model cell resulted in production of the stoichiometric amount of O2 and formation of a hydroquinone derivative (X-QH2) that is a plastoquinol analog.159 On the other hand, the PSI model cell contains nothing in PhCH3, but X-QH2, which is the product of the photocatalytic H2O oxidation in the PSII model, Acr+-Mes as a photoredox catalyst (a simple model of the photosynthetic reaction center for the charge separation),47,160–162 and a cobalt(III) complex (CoIII(dmgH)2ClPy) as an H2 evolution catalyst in H2O/TFE (3:1).159 Photoirradiation of the PSI model cell resulted in evolution of the stoichiometric amount of H2 and the formation of X-Q.159 These PSI and PSII molecular models were combined through glass membranes to achieve overall water splitting to O2 and H2 in a 1:2 stoichiometric ratio under photoirradiation.159 A TON of >100 has been achieved for the formation of H2 by combining the PSI and PSII models, where X-Q acted as a photoredox catalyst in the photocatalytic H2O splitting system as in the case of plastoquinone in photosynthesis.159
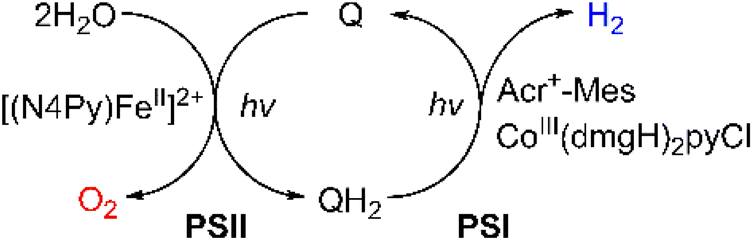 | ||
| Scheme 18 Schematic diagram of light-driven H2O splitting with the combination of PSI and PSII models. Reprinted from ref. 159 with permission from American Chemical Society (Copyright 2022). | ||
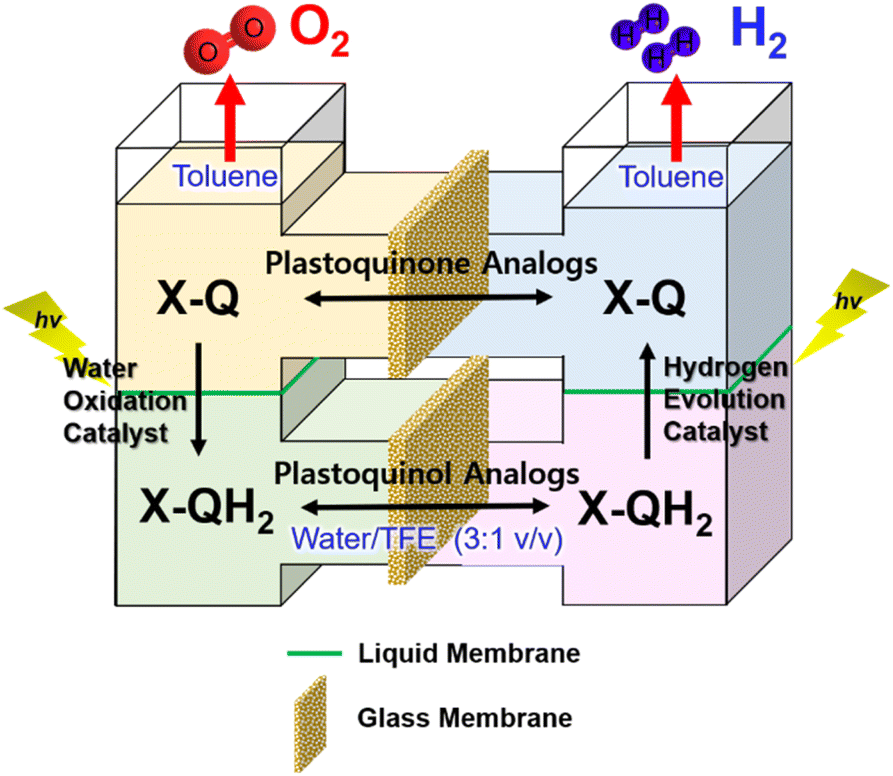 | ||
| Fig. 8 An O-type photocatalytic cell for photocatalytic H2O oxidation and reduction with a combination of PSI and PSII models. Reprinted from ref. 159 with permission from American Chemical Society (Copyright 2022). | ||
In the functional model of PSII, three steps of electron transfer from [FeII(N4Py)]2+ to X-Q*, which is the excited state of X-Q, generate an active intermediate ([FeV(O)(N4Py)]3+) which is able to oxidize H2O to produce O2 (Scheme 19).163 In the functional model of PSI, electron transfer from X-QH2 to 3(Acr˙-Mes˙+)*, which is the triplet ET state of Acr+-Mes, arises to form Acr˙-Mes and QH2˙+ (Scheme 20).164 QH2˙+ rapidly deprotonates to form QH˙, accompanied by electron transfer from Acr˙-Mes to CoIII(dmgH)2ClPy which produces [CoII(dmgH)2ClPy]− and regenerates Acr+-Mes.164 On the other hand, H-atom transfer from QH˙ to [CoII(dmgH)2ClPy]− occurs to yield a cobalt(III)-hydride complex ([CoIII(H)(dmgH)2ClPy]−) that reacts with protons to produce H2 and regenerate CoIII(dmgH)2ClPy (Scheme 20).164 This is the first example of combining PSI and PSII functional models in solution to obtain photocatalytic H2O splitting using a homogeneous molecular photocatalyst that produces O2 and H2 in a ratio of 1:2. Thus, this combinatorial system provides valuable mechanistic insights and reaction intermediates that are not possible using heterogeneous photocatalyts.159
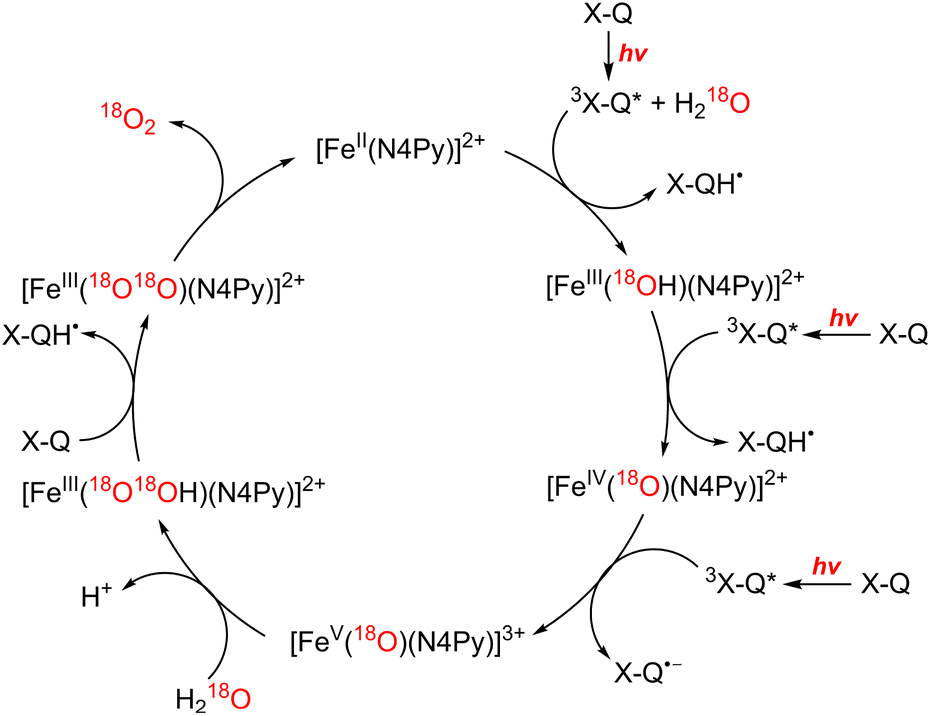 | ||
| Scheme 19 Proposed mechanism of photocatalytic oxidation of water by X-Q with [FeII(N4Py)]2+. Reprinted from ref. 163 with permission from American Chemical Society (Copyright 2019). | ||
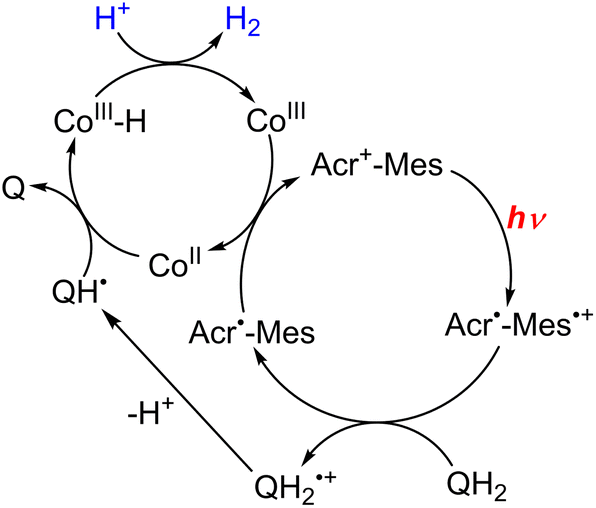 | ||
| Scheme 20 Proposed mechanism of photocatalytic evolution of H2 from X-QH2 with Acr+-Mes and CoIII(dmgH)2ClPy. Reprinted from ref. 164 with permission from American Chemical Society (Copyright 2020). | ||
Hydrogen produced in the PSI model can reduce NAD+ to NADH as well as CO2 to HCOOH with an Ir complex catalyst.165 Therefore, the molecular photocatalytic H2O splitting system (Fig. 8) can be applied to the generation of liquid solar fuels from H2O and CO2 by combining various catalysts reported for CO2 reduction with H2.166–171 Combination of PSI and PSII models will enable the development of a variety of catalytic reduction reactions using water as an earth-abundant reductant.
7. Conclusion
Production of solar fuels from water requires multi-functional catalysts for photosensitization, water oxidation and reduction of O2 to H2O2 or reduction of CO2 to HCOOH, HCHO, CH3OH etc. Coordination polymeric cyano (CN)-bridged heteronuclear metal complexes act as a dual functional photocatalyst for the photocatalytic production of H2O2 under visible light irradiation, catalyzing both photocatalytic oxidation of H2O to O2 and photocatalytic reduction of O2 to H2O2. In particular, the combination of semiconductor photocatalysts and Prussian blue analogues provides a composite photocatalyst that exhibited improved photocatalytic performance for water oxidation. On the other hand, metal–organic frameworks (MOFs) and mesoporous cellular silica foams (MCFs) have provided effective scaffolds to accommodate photocatalysts and redox catalysts for visible-light-driven water oxidation, H2 production, CO2 reduction and H2O2 production by incorporating both organic parts and inorganic metal parts. New visible-light photocatalysts of MOFs and MCFs can be designed and synthesized by incorporating optically functionalized organic chromophores which is a feasible approach with high efficiency. Construction of a hybrid photoelectrochemical cell demonstrated solar fuel production from water and CO2 with energy inputs from sunlight. Finally molecular models of PSII and PSII in photosynthesis were developed and they were combined with the use of liquid membranes to produce H2 from H2O under visible light photoirradiation through a quinone/hydroquinone redox couple as in the case of the plastoquinone/plastoquinone redox couple in photosynthesis. Such multi-functional molecular models of photosynthesis may hopefully pave a new and promising way for production of solar fuels more effectively than natural photosynthesis. In this review, bimetals play important roles in most multi-functional photocatalytic systems. Combination of coordination polymers, MOFs, COFs and MCFs with the use of multi-metals may enable the construction of more efficient model systems of photosynthesis.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge the contributions of their collaborators and coworkers cited in the listed references and support from JSPS (No. 16H02268 and 23K04686 to S. F.) and NRF of Korea (NRF-2023R1A2C1007668 to Y.-M. L. and NRF-2021R1A3B1076539 to W. N.).Notes and references
- W. W. Fischer, J. Hemp and J. E. Johnson, Annu. Rev. Earth Planet. Sci., 2016, 44, 647–683 CrossRef CAS.
- N. S. Lewis, Science, 2016, 351, aad1920 CrossRef PubMed.
- T. Ahmad and D. Zhang, Energy Rep., 2020, 6, 1973–1991 CrossRef.
- S. Fukuzumi, Joule, 2017, 1, 689–738 CrossRef CAS.
- X. Tao, Y. Zhao, S. Wang, C. Li and R. Li, Chem. Soc. Rev., 2022, 51, 3561–3608 RSC.
- Z. Wang, Y. Hu, S. Zhang and Y. Sun, Chem. Soc. Rev., 2022, 51, 6704–6737 RSC.
- M. Thangamuthu, Q. Ruan, P. O. Ohemeng, B. Luo, D. Jing, R. Godin and J. Tang, Chem. Rev., 2022, 122, 11778–11829 CrossRef CAS PubMed.
- T. A. Faunce, W. Lubitz, A. W. Rutherford, D. MacFarlane, G. F. Moore, P. Yang, D. G. Nocera, T. A. Moore, D. H. Gregory, S. Fukuzumi, K. B. Yoon, F. A. Armstrong, M. R. Wasielewski and S. Styring, Energy Environ. Sci., 2013, 6, 695–698 RSC.
- A. M. Appel, J. E. Bercaw, A. B. Bocarsly, H. Dobbek, D. L. DuBois, M. Dupuis, J. G. Ferry, E. Fujita, R. Hille, P. J. A. Kenis, C. A. Kerfeld, R. H. Morris, C. H. F. Peden, A. R. Portis, S. W. Ragsdale, T. B. Rauchfuss, J. N. H. Reek, L. C. Seefeldt, R. K. Thauer and G. L. Waldrop, Chem. Rev., 2013, 113, 6621–6658 CrossRef CAS.
- M. Aresta, A. Dibenedetto and A. Angelini, Chem. Rev., 2014, 114, 1709–1742 CrossRef CAS PubMed.
- K. A. Grice, Coord. Chem. Rev., 2017, 336, 78–95 CrossRef CAS.
- E. A. R. Cruz, D. Nishiori, B. L. Wadsworth, N. P. Nguyen, L. K. Hensleigh, D. Khusnutdinova, A. M. Beiler and G. F. Moore, Chem. Rev., 2022, 122, 16051–16109 CrossRef PubMed.
- M. E. El-Khouly, E. El-Mohsnawy and S. Fukuzumi, J. Photochem. Photobiol., C, 2017, 31, 36–83 CrossRef CAS.
- S. Fukuzumi, Y.-M. Lee and W. Nam, Tetrahedron, 2020, 76, 131024 CrossRef CAS.
- A. Paul, S. D. Adhikary, S. Kapurwan and S. Konar, J. Mater. Chem. A, 2022, 10, 13152–13169 RSC.
- B. Zhang and L. Sun, Chem. Soc. Rev., 2019, 48, 2216–2264 RSC.
- M. D. Kärkäs, O. Verho, E. V. Johnston and B. Åkermark, Chem. Rev., 2014, 114, 11863–12001 CrossRef PubMed.
- J. S. O'Neill, L. Kearney, M. P. Brandon and M. T. Pryce, Coord. Chem. Rev., 2022, 467, 214599 CrossRef.
- H. Sun, W. Ou, L. Sun, B. Wang and C. Su, Nano Res., 2022, 15, 10292–10315 CrossRef CAS.
- Q. Wang and K. Domen, Chem. Rev., 2020, 120, 919–985 CrossRef CAS PubMed.
- S. Chen, T. Takata and K. Domen, Nat. Rev. Mater., 2017, 2, 1–17 Search PubMed.
- U. V. Ghorpade, M. P. Suryawanshi, M. A. Green, T. Wu, X. Hao and K. M. Ryan, Chem. Rev., 2023, 123, 327–378 CrossRef CAS.
- Y. Fang, Y. Hou, X. Fu and X. Wang, Chem. Rev., 2022, 122, 4204–4256 CrossRef CAS PubMed.
- Z. Wang, C. Li and K. Domen, Chem. Soc. Rev., 2019, 48, 2109–2125 RSC.
- X. Chen, S. Shen, L. Guo and S. S. Mao, Chem. Rev., 2010, 110, 6503–6570 CrossRef CAS.
- S. Yoshino, T. Takayama, Y. Yamaguchi, A. Iwase and A. Kudo, Acc. Chem. Res., 2022, 55, 966–977 CrossRef CAS PubMed.
- A. Kudo and Y. Miseki, Chem. Soc. Rev., 2009, 38, 253–278 RSC.
- H. Yamashita, K. Mori, Y. Kuwahara, T. Kamegawa, M. Wen, P. Verma and M. Che, Chem. Soc. Rev., 2018, 47, 8072–8096 RSC.
- J. Ran, J. Zhang, J. Yu, M. Jaroniec and S. Z. Qiao, Chem. Soc. Rev., 2014, 43, 7787–7812 RSC.
- P. Niu, J. Dai, X. Zhi, Z. Xia, S. Wang and L. Li, InfoMat, 2021, 3, 931–961 CrossRef CAS.
- Y. Guo, H. Li, W. Ma, W. Shi, Y. Zhu and W. Choi, Carbon Energy, 2020, 2, 308–349 CrossRef CAS.
- A.-Y. Lo and F. Taghipour, J. Mater. Chem. A, 2021, 9, 26430–26453 RSC.
- A. Nakada, H. Kumagai, M. Robert, O. Ishitani and K. Maeda, Acc. Mater. Res., 2021, 2, 458–470 CrossRef CAS.
- F. Zaera, Chem. Rev., 2022, 122, 8594–8757 CrossRef CAS PubMed.
- S. Fukuzumi, Y.-M. Lee and W. Nam, Coord. Chem. Rev., 2018, 355, 54–73 CrossRef CAS.
- S. Fukuzumi, D. Hong and Y. Yamada, J. Phys. Chem. Lett., 2013, 4, 3458–3467 CrossRef CAS.
- S. Fukuzumi, Y.-M. Lee, H. S. Ahn and W. Nam, Chem. Sci., 2018, 9, 6017–6034 RSC.
- R. Cauwenbergh and S. Das, Green Chem., 2021, 23, 2553–2574 RSC.
- Y. Yamazaki, M. Miyaji and O. Ishitani, J. Am. Chem. Soc., 2022, 144, 6640–6660 CrossRef CAS PubMed.
- L. Wang and L. Wang, Front. Chem., 2022, 10, 996383 CrossRef CAS PubMed.
- Y. H. Hong, Y.-M. Lee, W. Nam and S. Fukuzumi, ACS Catal., 2023, 13, 308–341 CrossRef CAS.
- X. Li, H. Lei, L. Xie, N. Wang, W. Zhang and R. Cao, Acc. Chem. Res., 2022, 55, 878–892 CrossRef CAS PubMed.
- H. Lei, X. Li, J. Meng, H. Zheng, W. Zhang and R. Cao, ACS Catal., 2019, 9, 4320–4344 CrossRef CAS.
- D. L. Ashford, M. K. Gish, A. K. Vannucci, M. K. Brennaman, J. L. Templeton, J. M. Papanikolas and T. J. Meyer, Chem. Rev., 2015, 115, 13006–13049 CrossRef CAS PubMed.
- T. R. Cook, D. K. Dogutan, S. Y. Reece, Y. Surendranath, T. S. Teets and D. G. Nocera, Chem. Rev., 2010, 110, 6474–6502 CrossRef CAS PubMed.
- N. Armaroli and V. Balzani, Chem.–Eur. J., 2016, 22, 32–57 CrossRef CAS PubMed.
- S. Fukuzumi, K. Ohkubo and T. Suenobu, Acc. Chem. Res., 2014, 47, 1455–1464 CrossRef CAS PubMed.
- S. Fukuzumi, Curr. Opin. Chem. Biol., 2015, 25, 18–26 CrossRef CAS PubMed.
- K. Sakai and H. Ozawa, Coord. Chem. Rev., 2007, 251, 2753–2766 CrossRef CAS.
- H. Ozawa and K. Sakai, Chem. Commun., 2011, 47, 2227–2242 RSC.
- K. L. Skubi, T. R. Blum and T. P. Yoon, Chem. Rev., 2016, 116, 10035–10074 CrossRef CAS PubMed.
- K. R. Bajya and S. Selvakumar, Eur. J. Org. Chem., 2022, 2022, e202200229 Search PubMed.
- L. Qin, R. Wang, X. Xin, M. Zhang, T. Liu, H. Lv and G.-Y. Yang, Appl. Catal., B, 2022, 312, 121386 CrossRef CAS.
- Q. Zhang and C. Wang, Eur. J. Org. Chem., 2022, 2022, e202200431 CAS.
- T. E. Schirmer and B. König, J. Am. Chem. Soc., 2022, 144, 19207–19218 CrossRef CAS PubMed.
- E. B. McLean and A.-L. Lee, Tetrahedron, 2018, 74, 4881–4902 CrossRef CAS.
- R. Ham, C. J. Nielsen, S. Pullen and J. N. H. Reek, Chem. Rev., 2023, 123, 5225–5261 CrossRef CAS PubMed.
- S. Kato, J. Jung, T. Suenobu and S. Fukuzumi, Energy Environ. Sci., 2013, 6, 3756–3764 RSC.
- S. Fukuzumi, Y.-M. Lee and W. Nam, Chem.–Eur. J., 2018, 24, 5016–5031 CrossRef CAS PubMed.
- C. J. McDonnell-Worth and D. R. MacFarlane, Aust. J. Chem., 2018, 71, 781–788 CrossRef CAS.
- S. Fukuzumi, Biochim. Biophys. Acta, 2016, 1857, 604–611 CrossRef CAS PubMed.
- Y. Yamada, M. Yoneda and S. Fukuzumi, Energy Environ. Sci., 2015, 8, 1698–1701 RSC.
- S. Fukuzumi and Y. Yamada, ChemElectroChem, 2016, 3, 1978–1989 CrossRef CAS.
- L. An, T. Zhao, X. Yan, X. Zhou and P. Tan, Sci. Bull., 2015, 60, 55–64 CrossRef CAS.
- S. Fukuzumi and Y. Yamada, Aust. J. Chem., 2014, 67, 354–364 CrossRef CAS.
- S. Fukuzumi, Y. Yamada and K. D. Karlin, Electrochim. Acta, 2012, 82, 493–511 CrossRef CAS PubMed.
- H. Hou, X. Zeng and X. Zhang, Angew. Chem., Int. Ed., 2020, 59, 17356–17376 CrossRef CAS PubMed.
- Y. Sun, L. Han and P. Strasser, Chem. Soc. Rev., 2020, 49, 6605–6631 RSC.
- X. Hu, X. Zeng, Y. Liu, J. Lu and X. Zhang, Nanoscale, 2020, 12, 16008–16027 RSC.
- Y.-X. Ye, C. Wen, J. Pan, J.-W. Wang, Y.-J. Tong, S. Wei, Z. Ke, L. Jiang, F. Zhu, N. Zhou, M. Zhou, J. Xu and G. Ouyang, Appl. Catal., B, 2021, 285, 119726 CrossRef CAS.
- Y. Aratani, T. Suenobu, K. Ohkubo, Y. Yamada and S. Fukuzumi, Chem. Commun., 2017, 53, 3473–3476 RSC.
- T. Zhang and W. Lin, Chem. Soc. Rev., 2014, 43, 5982–5993 RSC.
- C. Wang, K. E. deKrafft and W. Lin, J. Am. Chem. Soc., 2012, 134, 7211–7214 CrossRef CAS PubMed.
- Y. Horiuchi, T. Toyao, M. Saito, K. Mochizuki, M. Iwata, H. Higashimura, M. Anpo and M. Matsuoka, J. Phys. Chem. C, 2012, 116, 20848–20853 CrossRef CAS.
- T. Toyao, M. Saito, S. Dohshi, K. Mochizuki, M. Iwata, H. Higashimura, Y. Horiuchi and M. Matsuoka, Chem. Commun., 2014, 50, 6779–6781 RSC.
- Y. Aratani, K. Oyama, T. Suenobu, Y. Yamada and S. Fukuzumi, Inorg. Chem., 2016, 55, 5780–5786 CrossRef CAS PubMed.
- Y. Isaka, S. Kato, D. Hong, T. Suenobu, Y. Yamada and S. Fukuzumi, J. Mater. Chem. A, 2015, 3, 12404–12412 RSC.
- Y. Isaka, K. Oyama, Y. Yamada, T. Suenobu and S. Fukuzumi, Catal. Sci. Technol., 2016, 6, 681–684 RSC.
- S. S. Akbari, U. Unal and F. Karadas, ACS Appl. Energy Mater., 2021, 4, 12383–12390 CrossRef CAS.
- J. Zhou, W. Chen, C. Sun, L. Han, C. Qin, M. Chen, X. Wang, E. Wang and Z. Su, ACS Appl. Mater. Interfaces, 2017, 9, 11689–11695 CrossRef CAS PubMed.
- D. Zhou and B.-H. Han, Adv. Funct. Mater., 2010, 20, 2717–2722 CrossRef CAS.
- J. Li, W. Chen, L. Chen, X. Zheng, G. Zhu and E. Wang, Adv. Opt. Mater., 2018, 6, 1800225 CrossRef.
- Y. Liu, C. Tang, M. Cheng, M. Chen, S. Chen, L. Lei, Y. Chen, H. Yi, Y. Fu and L. Li, ACS Catal., 2021, 11, 13374–13396 CrossRef CAS.
- G. Paille, M. Gomez-Mingot, C. Roch-Marchal, B. Lassalle-Kaiser, P. Mialane, M. Fontecave, C. Mellot-Draznieks and A. Dolbecq, J. Am. Chem. Soc., 2018, 140, 3613–3618 CrossRef CAS PubMed.
- G. Paille, M. Gomez-Mingot, C. Roch-Marchal, M. Haouas, Y. Benseghir, T. Pino, M.-H. Ha-Thi, G. Landrot, P. Mialane, M. Fontecave, A. Dolbecq and C. Mellot-Draznieks, ACS Appl. Mater. Interfaces, 2019, 11, 47837–47845 CrossRef CAS PubMed.
- S. Pullen, H. Fei, A. Orthaber, S. M. Cohen and S. Ott, J. Am. Chem. Soc., 2013, 135, 16997–17003 CrossRef CAS PubMed.
- D.-Y. Du, J.-S. Qin, S.-L. Li, Z.-M. Su and Y.-Q. Lan, Chem. Soc. Rev., 2014, 43, 4615–4632 RSC.
- X.-X. Li, J. Liu, L. Zhang, L.-Z. Dong, Z.-F. Xin, S.-L. Li, X.-Q. Huang-Fu, K. Huang and Y.-Q. Lan, ACS Appl. Mater. Interfaces, 2019, 11, 25790–25795 CrossRef CAS PubMed.
- Y. Benseghir, A. Lemarchand, M. Duguet, P. Mialane, M. Gomez-Mingot, C. Roch-Marchal, T. Pino, M.-H. Ha-Thi, M. Haouas, M. Fontecave, A. Dolbecq, C. Sassoye and C. Mellot-Draznieks, J. Am. Chem. Soc., 2020, 142, 9428–9438 CrossRef CAS PubMed.
- K. Zhang, S. Goswami, H. Noh, Z. Lu, T. Sheridan, J. Duan, W. Dong and J. T. Hupp, J. Photochem. Photobiol., 2022, 10, 100111 CrossRef.
- J. Bonin, M. Chaussemier, M. Robert and M. Routier, ChemCatChem, 2014, 6, 3200–3207 CrossRef CAS.
- B. Probst, A. Rodenberg, M. Guttentag, P. Hamm and R. Alberto, Inorg. Chem., 2010, 49, 6453–6460 CrossRef CAS PubMed.
- H. Zhang, J. Wei, J. Dong, G. Liu, L. Shi, P. An, G. Zhao, J. Kong, X. Wang, X. Meng, J. Zhang and J. Ye, Angew. Chem., Int. Ed. Engl., 2016, 55, 14310–14314 CrossRef CAS PubMed.
- P. Du, J. Schneider, P. Jarosz and R. Eisenberg, J. Am. Chem. Soc., 2006, 128, 7726–7727 CrossRef CAS PubMed.
- D. Chen, H. Xing, C. Wang and Z. Su, J. Mater. Chem. A, 2016, 4, 2657–2662 RSC.
- Y. Isaka, Y. Kondo, Y. Kawase, Y. Kuwahara, K. Mori and H. Yamashita, Chem. Commun., 2018, 54, 9270–9273 RSC.
- Y. Fu, D. Sun, Y. Chen, R. Huang, Z. Ding, X. Fu and Z. Li, Angew. Chem., Int. Ed., 2012, 51, 3364–3367 CrossRef CAS PubMed.
- L. Shen, M. Luo, L. Huang, P. Feng and L. Wu, Inorg. Chem., 2015, 54, 1191–1193 CrossRef CAS PubMed.
- J.-N. Chang, Q. Li, J.-W. Shi, M. Zhang, L. Zhang, S. Li, Y. Chen, S.-L. Li and Y.-Q. Lan, Angew. Chem., Int. Ed., 2023, 62, e202218868 CrossRef CAS PubMed.
- S. Porcu, F. Secci and P. C. Ricci, Molecules, 2022, 27, 6828 CrossRef CAS PubMed.
- Y. Wu, T. Sakurai, T. Adachi and Q. Wang, Nanoscale, 2023, 15, 6521–6535 RSC.
- H. Song, L. Wei, L. Chen, H. Zhang and J. Su, Top. Catal., 2020, 63, 895–912 CrossRef CAS.
- C. Gérardin, J. Reboul, M. Bonne and B. Lebeau, Chem. Soc. Rev., 2013, 42, 4217–4255 RSC.
- I. Sierra and D. Pérez-Quintanilla, Chem. Soc. Rev., 2013, 42, 3792–3807 RSC.
- P. Yang, S. Gai and J. Lin, Chem. Soc. Rev., 2012, 41, 3679–3698 RSC.
- S.-H. Wu, C.-Y. Mou and H.-P. Lin, Chem. Soc. Rev., 2013, 42, 3862–3875 RSC.
- H. Wang, Q. Tang, Z. Chen, T. Li and J. Wang, Sci. China Mater., 2020, 63, 2189–2205 CrossRef CAS.
- H. Jung, Y. Whang and S. W. Han, Bull. Korean Chem. Soc., 2021, 42, 806–809 CrossRef CAS.
- X. Qian, K. Fuku, Y. Kuwahara, T. Kamegawa, K. Mori and H. Yamashita, ChemSusChem, 2014, 7, 1528–1536 CrossRef CAS PubMed.
- C. Cheng, D. Lu, B. Shen, Y. Liu, J. Lei, L. Wang, J. Zhang and M. Matsuoka, Microporous Mesoporous Mater., 2016, 226, 79–87 CrossRef CAS.
- H. Y. Kim, J. Y. Kim and S. H. Joo, Bull. Korean Chem. Soc., 2021, 42, 724–736 CrossRef CAS.
- J. G. Mahy, C. A. Paez, C. Carcel, C. Bied, A. S. Tatton, C. Damblon, B. Heinrichs, M. Wong Chi Man and S. D. Lambert, J. Photochem. Photobiol., A, 2019, 373, 66–76 CrossRef CAS.
- P. Rana, B. Kaushik, K. Solanki, K. Mohan Saini and R. K. Sharma, Chem. Commun., 2022, 58, 11354–11377 RSC.
- A. A. Yakushev, A. S. Abel, A. D. Averin, I. P. Beletskaya, A. V. Cheprakov, I. S. Ziankou, L. Bonneviot and A. Bessmertnykh-Lemeune, Coord. Chem. Rev., 2022, 458, 214331 CrossRef CAS.
- S. Lin, H. Huang, T. Ma and Y. Zhang, Adv. Sci., 2021, 8, 2002458 CrossRef CAS PubMed.
- M. Waki, S. Shirai, K.-I. Yamanaka, Y. Maegawa and S. Inagaki, RSC Adv., 2020, 10, 13960–13967 RSC.
- Y. Yamada, H. Tadokoro, M. Naqshbandi, J. Canning, M. J. Crossley, T. Suenobu and S. Fukuzumi, ChemPlusChem, 2016, 81, 521–525 CrossRef CAS PubMed.
- S. Zhang, M. Li, Q. Wu, H. Yang, J. Han, H. Wang and X. Liu, Appl. Catal., B, 2018, 236, 466–474 CrossRef CAS.
- T. Jin, C. Liu and G. Li, J. Coord. Chem., 2016, 69, 1748–1758 CrossRef CAS.
- Y. Ueda, H. Takeda, T. Yui, K. Koike, Y. Goto, S. Inagaki and O. Ishitani, ChemSusChem, 2015, 8, 439–442 CrossRef CAS PubMed.
- H. Kumagai, Y. Tamaki and O. Ishitani, Acc. Chem. Res., 2022, 55, 978–990 CrossRef CAS PubMed.
- S. Fukuzumi, K. Doi, A. Itoh, T. Suenobu, K. Ohkubo, Y. Yamada and K. D. Karlin, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 15572–15577 CrossRef CAS PubMed.
- K. Ohkubo, K. Mizushima, R. Iwata, K. Souma, N. Suzuki and S. Fukuzumi, Chem. Commun., 2010, 46, 601–603 RSC.
- T. Arai, S. Sato and T. Morikawa, Energy Environ. Sci., 2015, 8, 1998–2002 RSC.
- T. Arai, S. Sato, K. Uemura, T. Morikawa, T. Kajino and T. Motohiro, Chem. Commun., 2010, 46, 6944–6946 RSC.
- S. Sato, T. Arai, T. Morikawa, K. Uemura, T. M. Suzuki, H. Tanaka and T. Kajino, J. Am. Chem. Soc., 2011, 133, 15240–15243 CrossRef CAS PubMed.
- K. Maeda, R. Kuriki, M. Zhang, X. Wang and O. Ishitani, J. Mater. Chem. A, 2014, 2, 15146–15151 RSC.
- R. Kuriki, O. Ishitani and K. Maeda, ACS Appl. Mater. Interfaces, 2016, 8, 6011–6018 CrossRef CAS PubMed.
- A. Nakada, T. Nakashima, K. Sekizawa, K. Maeda and O. Ishitani, Chem. Sci., 2016, 7, 4364–4371 RSC.
- K. Wada, M. Eguchi, O. Ishitani and K. Maeda, ChemSusChem, 2017, 10, 287–295 CrossRef CAS PubMed.
- R. Kuriki, H. Matsunaga, T. Nakashima, K. Wada, A. Yamakata, O. Ishitani and K. Maeda, J. Am. Chem. Soc., 2016, 138, 5159–5170 CrossRef CAS PubMed.
- H. Kumagai, G. Sahara, K. Maeda, M. Higashi, R. Abe and O. Ishitani, Chem. Sci., 2017, 8, 4242–4249 RSC.
- M. Shizuno, K. Kato, S. Nishioka, T. Kanazawa, D. Saito, S. Nozawa, A. Yamakata, O. Ishitani and K. Maeda, ACS Appl. Energy Mater., 2022, 5, 9479–9486 CrossRef CAS.
- M. A. Gross, C. E. Creissen, K. L. Orchard and E. Reisner, Chem. Sci., 2016, 7, 5537–5546 RSC.
- J. Massin, M. Bräutigam, N. Kaeffer, N. Queyriaux, M. J. Field, F. H. Schacher, J. Popp, M. Chavarot-Kerlidou, B. Dietzek and V. Artero, Interface Focus, 2015, 5, 20140083 CrossRef PubMed.
- B. Samanta, Á. Morales-García, F. Illas, N. Goga, J. A. Anta, S. Calero, A. Bieberle-Hütter, F. Libisch, A. B. Muños-García, M. Pavone and M. C. Toroker, Chem. Soc. Rev., 2022, 51, 3794–3818 RSC.
- M. Grätzel, Acc. Chem. Res., 1981, 14, 376–384 CrossRef.
- A. J. Bard and M. A. Fox, Acc. Chem. Res., 1995, 28, 141–145 CrossRef CAS.
- D. Gust, T. A. Moore and A. L. Moore, Acc. Chem. Res., 2009, 42, 1890–1898 CrossRef CAS PubMed.
- Y. Tachibana, L. Vayssieres and J. R. Durrant, Nat. Photonics, 2012, 6, 511–518 CrossRef CAS.
- F. E. Osterloh, Chem. Soc. Rev., 2013, 42, 2294–2320 RSC.
- Y. Qu and X. Duan, Chem. Soc. Rev., 2013, 42, 2568–2580 RSC.
- T. Hisatomi, J. Kubota and K. Domen, Chem. Soc. Rev., 2014, 43, 7520–7535 RSC.
- C. Jiang, S. J. A. Moniz, A. Wang, T. Zhang and J. Tang, Chem. Soc. Rev., 2017, 46, 4645–4660 RSC.
- S. Chen, T. Takata and K. Domen, Nat. Rev. Mater., 2017, 2, 17050 CrossRef CAS.
- J. Yano and V. Yachandra, Chem. Rev., 2014, 114, 4175–4205 CrossRef CAS PubMed.
- J. Barber, Chem. Soc. Rev., 2009, 38, 185–196 RSC.
- J. P. McEvoy and G. W. Brudvig, Chem. Rev., 2006, 106, 4455–4483 CrossRef CAS PubMed.
- H. Dau and I. Zaharieva, Acc. Chem. Res., 2009, 42, 1861–1870 CrossRef CAS PubMed.
- N. Cox, D. A. Pantazis, F. Neese and W. Lubitz, Acc. Chem. Res., 2013, 46, 1588–1596 CrossRef CAS PubMed.
- F. Rappaport, M. Guergova-Kuras, P. J. Nixon, B. A. Diner and J. Lavergne, Biochemistry, 2002, 41, 8518–8527 CrossRef CAS PubMed.
- J. M. Keough, A. N. Zuniga, D. L. Jenson and B. A. Barry, J. Phys. Chem. B, 2013, 117, 1296–1307 CrossRef CAS PubMed.
- L. A. Malone, M. S. Proctor, A. Hitchcock, C. N. Hunter and M. P. Johnson, Biochim. Biophys. Acta, Bioenerg., 2021, 1862, 148380 CrossRef CAS PubMed.
- S. Wada, Y. Suzuki and C. Miyake, Plants, 2020, 9, 319 CrossRef CAS PubMed.
- C. Miyake, Antioxidants, 2020, 9, 230 CrossRef CAS PubMed.
- T. G. Laughlin, A. N. Bayne, J.-F. Trempe, D. F. Savage and K. M. Davies, Nature, 2019, 566, 411–414 CrossRef PubMed.
- J. Z. Zhang and E. Reisner, Nat. Rev. Chem., 2020, 4, 6–21 CrossRef CAS.
- H. Kubota-Kawai, R. Mutoh, K. Shinmura, P. Sétif, M. M. Nowaczyk, M. Rögner, T. Ikegami, H. Tanaka and G. Kurisu, Nat. Plants, 2018, 4, 218–224 CrossRef CAS PubMed.
- Y. H. Hong, Y.-M. Lee, W. Nam and S. Fukuzumi, J. Am. Chem. Soc., 2022, 144, 695–700 CrossRef CAS PubMed.
- S. Fukuzumi, H. Kotani, K. Ohkubo, S. Ogo, N. V. Tkachenko and H. Lemmetyinen, J. Am. Chem. Soc., 2004, 126, 1600–1601 CrossRef CAS PubMed.
- T. Tsudaka, H. Kotani, K. Ohkubo, T. Nakagawa, N. V. Tkachenko, H. Lemmetyinen and S. Fukuzumi, Chem.–Eur. J., 2017, 23, 1306–1317 CrossRef CAS PubMed.
- S. Fukuzumi, Y.-M. Lee and W. Nam, Biochem. Soc. Trans., 2018, 46, 1279–1288 CrossRef CAS PubMed.
- Y. H. Hong, J. Jung, T. Nakagawa, N. Sharma, Y.-M. Lee, W. Nam and S. Fukuzumi, J. Am. Chem. Soc., 2019, 141, 6748–6754 CrossRef CAS PubMed.
- Y. H. Hong, Y.-M. Lee, W. Nam and S. Fukuzumi, Inorg. Chem., 2020, 59, 14838–14846 CrossRef CAS PubMed.
- Y. Maenaka, T. Suenobu and S. Fukuzumi, J. Am. Chem. Soc., 2012, 134, 367–374 CrossRef CAS PubMed.
- C. Liu, B. C. Colón, M. Ziesack, P. A. Silver and D. G. Nocera, Science, 2016, 352, 1210–1213 CrossRef CAS PubMed.
- W. Tu, Y. Zhou and Z. Zou, Adv. Mater., 2014, 26, 4607–4626 CrossRef CAS PubMed.
- E. Nikoloudakis, I. López-Duarte, G. Charalambidis, K. Ladomenou, M. Ince and A. G. Coutsolelos, Chem. Soc. Rev., 2022, 51, 6965–7045 RSC.
- H. Hayashi, S. Ogo and S. Fukuzumi, Chem. Commun., 2004, 2714–2715 RSC.
- Y. Maenaka, T. Suenobu and S. Fukuzumi, Energy Environ. Sci., 2012, 5, 7360–7367 RSC.
- K. Kamada, J. Jung, T. Wakabayashi, K. Sekizawa, S. Sato, T. Morikawa, S. Fukuzumi and S. Saito, J. Am. Chem. Soc., 2020, 142, 10261–10266 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2023 |