
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Compositional dependence of hydrodeoxygenation pathway selectivity for Ni2−xRhxP nanoparticle catalysts†
Nicole J.
LiBretto‡
a,
Sean A.
Tacey§
 a,
Muhammad
Zubair
b,
Tuong V.
Bui¶
a,
Kinga A.
Unocic
c,
Frederick G.
Baddour
a,
Michael B.
Griffin
a,
Joshua A.
Schaidle
a,
Carrie A.
Farberow
*a,
Daniel A.
Ruddy
*a,
Nicholas M.
Bedford
*b and
Susan E.
Habas
*a
a,
Muhammad
Zubair
b,
Tuong V.
Bui¶
a,
Kinga A.
Unocic
c,
Frederick G.
Baddour
a,
Michael B.
Griffin
a,
Joshua A.
Schaidle
a,
Carrie A.
Farberow
*a,
Daniel A.
Ruddy
*a,
Nicholas M.
Bedford
*b and
Susan E.
Habas
*a
aNational Renewable Energy Laboratory, Catalytic Carbon Transformation and Scale-up Center, Golden, CO, USA. E-mail: carrie.farberow@nrel.gov; dan.ruddy@nrel.gov; susan.habas@nrel.gov
bUniversity of New South Wales, School of Chemical Engineering, Sydney, NSW, Australia. E-mail: n.bedford@unsw.edu.au
cOak Ridge National Laboratory, Center for Nanophase Materials Sciences, Oak Ridge, TN, USA
First published on 26th July 2023
Abstract
Transition-metal phosphides (TMPs) are promising materials for biomass conversion processes, where their metal–acid bifunctionality provides active sites for the necessary variety of catalytic reactions (e.g., hydrogenation, hydrogenolysis, decarbonylation, and dehydration). Aiming to understand the catalytic performance of ternary TMP catalysts for the hydrodeoxygenation (HDO) reaction of biomass-derived oxygenates, a synthetic protocol was developed herein to prepare a series of ternary Ni2−xRhxP nanoparticles (NPs), incorporating Rh into a parent Ni2P template. This solution synthesis method allowed for a series of NPs to be prepared having precisely controlled compositions with similar morphology and crystalline structure. Detailed characterization of this series of Ni2−xRhxP (x ≤ 1) NPs revealed that Rh substituted into the parent hexagonal crystal lattice of Ni2P with concomitant expansion of the lattice. The influence of composition on the catalytic performance of silica-supported Ni2−xRhxP (x = 0 to 0.8, and cubic Rh2P) NPs was investigated through the HDO reaction of m-cresol. Whereas Rh2P was more selective for direct deoxygenation relative to Ni2P, increasing concentrations of Rh in Ni2−xRhxP resulted in a decreased selectivity to direct deoxygenation products (i.e., toluene, benzene, xylene), and an associated increased selectivity to hydrogenation products (i.e., methylcyclohexene). Through in situ high energy X-ray diffraction and density functional theory modeling, we identified OH* adsorption energy and surface-P sp-band center as effective descriptors for the observed shift in selectivities across this series of TMPs. Furthermore, the calculated electronic-structure changes were found to exert greater influence over the observed product selectivity than the subtle geometric changes associated with lattice expansion. Identification of this structure–function relationship demonstrates that the controlled synthesis of TMPs enables an understanding of composition-dependent selectivity for the HDO reaction of phenolic molecules and this approach could be extended to other ternary TMP compositions for diverse catalytic applications.
Introduction
Transition-metal phosphides (TMPs) are exceptional catalysts for a variety of biomass upgrading reactions, such as hydrogenation, hydrogenolysis, decarbonylation, and dehydration, due to their metal−acid bifunctionality.1–4 TMPs also exhibit hydrotreating activities similar to, or better than, both traditional transition-metal sulfide-based materials and their parent metals, and can activate hydrogen at low pressures and moderate temperatures.5–7 Tunable reaction rates and product selectivities have been demonstrated through compositional control of the metal identity and phosphorus-to-metal ratio.3,8 Among the synthetically accessible binary TMPs (e.g., MyP, where M represents a single metal), Ni2P nanoparticles (NPs) have shown promise for the hydrodeoxygenation (HDO) reaction of oxygenated compounds derived from biomass.9,10The selective HDO of phenolic molecules to deoxygenated aromatic and cyclic compounds is a key step in the production of renewable chemicals and fuels from biomass, but achieving control over the selectivity requires understanding and control over the catalytic active sites.11 For example, two pathways are typically observed in the HDO of phenolics: (i) direct deoxygenation (DDO) to produce an aromatic product (e.g., phenol to benzene), and (ii) hydrogenation (HYD) followed by dehydration and hydrogenation to produce a cycloalkane (e.g., phenol to cyclohexane).12–15 Coplanar (parallel) orientation of the aromatic ring was proposed to drive selectivity toward HYD products due to the proximity of the ring to adsorbed H* species (* denotes an adsorbed species) at the metallic sites, while nonplanar orientation (perpendicular/tilted) promoted DDO selectivity through interaction of the phenolic OH group (Ph–OH) with the Lewis acid surface sites.16–19 The adsorbate orientation can be influenced by the surface electronic structure, often described by metal Lewis acidity, that can be modulated through changes in catalyst composition.20 Stronger Lewis acidity has been correlated with more favourable interaction of the Ph–OH group with Lewis acid sites on the catalyst surface leading to increased DDO selectivity.20 One approach to control HDO pathway selectivity is through the alloying of binary metal phosphides with another metal to form ternary TMPs (e.g.,  , where M′ represents a second metal).21,22 These more complex compositions were shown to modulate both the electronic and geometric properties of the parent binary TMP, leading to increased HDO reaction rates and composition-dependent selectivity.13,20 Recently, the HDO reaction for a variety of biomass-derived model compounds (e.g., phenol, guaiacol, furfural) was investigated over ternary TMPs containing Mo, Fe, Ni, and Co.13,20,23 One noteworthy result was that the addition of Fe to parent Mo2P NPs increased the Lewis acidity and promoted increased activity and selectivity toward benzene from phenol through effective C–O bond cleavage.20
, where M′ represents a second metal).21,22 These more complex compositions were shown to modulate both the electronic and geometric properties of the parent binary TMP, leading to increased HDO reaction rates and composition-dependent selectivity.13,20 Recently, the HDO reaction for a variety of biomass-derived model compounds (e.g., phenol, guaiacol, furfural) was investigated over ternary TMPs containing Mo, Fe, Ni, and Co.13,20,23 One noteworthy result was that the addition of Fe to parent Mo2P NPs increased the Lewis acidity and promoted increased activity and selectivity toward benzene from phenol through effective C–O bond cleavage.20
Considering the added complexity of ternary TMPs, approaches have been developed to provide a high level of synthetic control to establish structure–function relationships that govern HDO pathway selectivity on TMP catalysts. Traditional synthesis methods for TMPs via impregnation of metal and phosphorus salts on high surface area metal-oxide supports followed by high temperature reduction can lead to broad particle size distributions, overreduction of metal salts to metal NPs, non-uniform phosphorus incorporation, and excess phosphate species that do not participate in TMP formation.24,25 Solution synthesis routes, on the other hand, can enable control over composition, particle morphology, and crystal structure,22,26–32 providing model systems to understand the role of the second metal. Our group has previously established solution synthesis approaches to obtain Ni2P, Rh2P, and a variety of alloyed Ni2P NPs via the thermolysis of low-valent metal triphenylphosphine complexes.33,34 The parent Ni2P and Rh2P NPs prepared through this route were evaluated in the guaiacol HDO reaction,35,36 with Rh2P/SiO2 producing higher yields of the deoxygenated products anisole and benzene due to more effective activation of the Ph–OH bond compared to Ni2P/SiO2 which yielded primarily phenol.37
Herein, the composition-dependent performance of a ternary Ni2−xRhxP NP series was evaluated as a low precious-metal alternative to binary Rh2P for the generation of deoxygenated products from phenolic molecules. The Ni2−xRhxP NPs (x = 0.2, 0.4, 0.6, 0.8, 1.0) were synthesized in a two-step solution synthesis approach via the thermal decomposition of molecular precursors to maintain uniform morphology and crystal structure across the series. The NPs were characterized by X-ray diffraction (XRD), scanning transmission electron microscopy (STEM), and pair distribution function analysis (PDF) of in situ high energy (HE)-XRD patterns. These data revealed that Rh substituted into Ni sites of the hexagonal Ni2P crystal structure while maintaining the bulk structure with only a slight loss of metal–phosphorus bonds in the presence of H2 at 350 °C. Density functional theory (DFT) calculations showed that the increasing concentration of Rh in Ni2−xRhxP was compensated by a decreasing negative charge at the P atom, whereas the charge of Ni and Rh remained constant. Composition-dependent selectivity in the m-cresol HDO reaction was observed, where increasing concentrations of Rh promoted HYD selectivity (up to 72% with Ni1.4Rh0.6P), in contrast to the binary Rh2P catalyst which promoted DDO selectivity (90%). DFT calculations demonstrated that increasing Rh content (x) substituted in the Ni2−xRhxP(0001) surface resulted in weakened OH* adsorption energetics across the series, providing a good descriptor for the experimentally observed decrease in DDO and increase in HYD selectivity with increasing Rh concentration. The surface-P sp-band center was identified as an effective electronic structure descriptor for the OH* binding energy, and thus DDO selectivity. The composition-dependent understanding developed herein demonstrates that the catalytic performance of ternary TMPs is not easily represented by a linear combination of two parent materials, but instead, is intimately related to the crystal structure and electronic structure that dictate surface–substrate interactions.
Experimental methods
Catalyst synthesis
A two-step synthesis route was employed to prepare Ni2−xRhxP (x = 0.2, 0.4, 0.6, 0.8, 1) NPs, as described previously.33 First, amorphous Ni–P NPs were prepared by rapidly heating Ni(CO)2(PPh3)2 (0.639 g, 1.0 mmol) and PPh3 (1.05 g, 4.0 mmol) in dried oleylamine (OAm, 6.5 mL) and dried 1-octadecene (ODE, 6.5 mL) to 250 °C and holding for 15 min before cooling to ambient temperature. After the addition of 0–0.50 molar equivalents of solid RhCl(CO)(PPh3)2 at ambient temperature under N2, the reaction vessel was purged with N2 through a needle inserted through the septum for 2 min. Then, the reaction mixture was heated rapidly to 300 °C under N2 and held at this temperature for 1 h before removing the heat source and allowing the flask to cool to ambient temperature. The reaction mixture was then transferred to a centrifuge tube with the aid of approximately 0.1 mL chloroform. The NPs were flocculated with the addition of 15 mL of 2-propanol and separated via centrifugation at 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 10 min. The NPs were again redispersed in 0.1 mL chloroform and flocculated with 30 mL 2-propanol before recovering by centrifugation.
000 rpm for 10 min. The NPs were again redispersed in 0.1 mL chloroform and flocculated with 30 mL 2-propanol before recovering by centrifugation.
Ni2P NPs were prepared, as described previously,33,34 by heating a mixture of Ni(CO)2(PPh3)2 (0.639 g, 1.0 mmol), PPh3 (1.049 g, 4.0 mmol), dried OAm (6.5 mL), and dried ODE (6.5 mL) rapidly to 300 °C and holding at this temperature for 1 h before the heat source was removed and the reaction mixture allowed to cool to ambient temperature. Similarly, Rh2P was prepared by heating a mixture of RhCl(CO)(PPh3)2 (0.691 g, 1.0 mmol), OAm (4.9 mL), and ODE (8.0 mL) rapidly to 300 °C and holding for 1 h before the heat source was removed and the reaction mixture allowed to cool to ambient temperature. The NPs were purified, as described above.
All NPs were supported at approximately 5 wt% Ni2−xRhxP loading on SiO2. After recovery, the purified NPs were redispersed in 10 mL chloroform. In a separate Erlenmeyer flask containing a 25 mm long stir bar, SiO2 was dispersed in chloroform (10 mL g−1 SiO2) and sonicated for 5 min. The SiO2 mixture was stirred at 400 rpm and the NP suspension was added dropwise. The combined mixture was sonicated again for 5 min and left to stir overnight at ambient temperature. The supported NPs were collected by centrifugation at 10000 rpm for 5 min and dried under flowing N2, before being stored in an N2-filled glovebox. Additional information about the materials and purity used is provided in the ESI.†
Characterization
The metal and phosphorus loadings of both unsupported and SiO2-supported NPs were determined using inductively coupled plasma optical emission spectroscopy (ICP-OES) at Galbraith Laboratories (Knoxville, TN). Scanning transmission electron microscopy (STEM) imaging was performed on an aberration-corrected Nion Co. Ultra-STEM 100 operated at 100 kV and a JEOL 2200FS STEM/TEM instrument equipped with a CEOS GmbH (Heidelberg, Ger) corrector on the illuminating lenses to determine the NP morphology. The AMAG 5C mode was used to achieve a probe with a nominal 150 pA current and associated resolution of a nominal 0.07 nm. The presence and distribution of Ni, Rh, and P in the NPs were confirmed by utilizing energy dispersive X-ray spectroscopy (EDS) analysis and acquiring spectrum images with a Bruker-AXS silicon-drift detector system (SDD) on the JEOL 2200FS STEM/TEM microscope. Powder XRD data were collected using a Rigaku Ultima IV diffractometer with a Cu Kα source (40 kV, 44 mA). Diffraction patterns were collected in the 2θ range of 20–100° at a scan rate of 4° min−1 on both unsupported and SiO2-supported NPs. Theoretical diffraction patterns were simulated at room temperature to aid phase identification. In situ high energy XRD (HE-XRD) experiments were performed at the 6-ID-D beamline of the Advanced Photon Source (APS) at Argonne National Laboratory. HE-XRD patterns were collected at room temperature, 350 °C in 5% H2/Ar, and at 350 °C in guaiacol-saturated 5% H2/Ar. Catalytic site densities of the SiO2-supported Ni2−xRhxP NPs were determined by H2 chemisorption. Volumetric chemisorption experiments were performed using a Quantachrome Instruments Autosorb-1C gas sorption instrument. Additional details about sample preparation and data processing for each characterization technique is available in the ESI.†Catalyst evaluation
Catalytic performance was evaluated in a tubular fixed-bed reactor at 350 ± 1 °C and 0.50 ± 0.03 MPa. The reactor tube was 7.62 mm diameter with 20 mL total volume (Autoclave Engineers). The catalyst bed consisted of 0.2–0.5 g of catalyst diluted with 177–250 μm diameter particles of silicon carbide to a volume of 4 mL. The catalyst bed was loaded into the reactor tube with the remaining reactor volume packed with crushed quartz. Approximately 2 mL of fine-crushed quartz (150–250 μm) was packed above and below the catalyst bed, and the remaining reactor volume (∼12 mL) was packed with coarse-crushed quartz (300–425 μm). The loaded reactor tube was positioned within the isothermal region of the furnace. The temperature of the isothermal zone was measured with a 4-point thermocouple inserted into the center of the catalyst bed. For catalyst evaluation in the guaiacol HDO reaction, the reactor was co-fed with a 95% H2/5% Ar mixture and guaiacol at a molar ratio of 12:1 (i.e., twice the stoichiometric amount of H2 required to reduce guaiacol to cyclohexane, water, and methane). For catalyst evaluation in the m-cresol HDO reaction, the reactor was co-fed with a 95% H2/5% Ar mixture and m-cresol at a molar ratio of 8:1 (i.e., twice the stoichiometric amount of H2 required to reduce m-cresol to methylcyclohexane and water). An Eldex Optos 1 LMP HPLC pump was used to feed guaiacol or m-cresol into the reactor at a rate corresponding to the desired weight hourly space velocity (2–5 h−1), which is the ratio of the mass flow rate of reactant to the catalyst mass per hour. The guaiacol or m-cresol was vaporized immediately upstream of the reactor in heated stainless-steel tubing at 250 °C and mixed with the 95% H2/Ar mixture.
The reactor effluent was fractionated into liquid and gas phases in a condenser operated at 30 °C and samples of each phase were quantified with two different gas chromatography (GC) instruments. The uncondensed reactor effluent was sampled immediately downstream of the condensable trap and analyzed with an on-line Agilent Technologies 7890B gas chromatograph modified by Wasson ECE Instrumentation, Inc. The condensed samples were identified and quantified with an Agilent Technologies 7890A GC equipped with a flame ionization detector (FID) and mass spectrometer (MS). In all cases, the mass balance and carbon balance, based on measured inlet and outlet flowrates were over 90%. The equations used to calculate conversion, selectivity, and site time yield (STY) are presented in the ESI.†
Computational methods
Density functional theory (DFT) calculations were implemented through the Vienna Ab initio Simulation Package (VASP).38,39 Exchange–correlation effects were captured through the spin-polarized generalized gradient approximation (GGA) using the Perdew–Burke–Ernzerhof (PBE) exchange–correlation functional.40 Electron-ion interactions were described using projector augmented-wave (PAW) potentials with valence wavefunctions expanded through a plane-wave basis set with a cutoff energy of 500 eV.41,42 Dispersion corrections were included through the D3 method by Grimme et al.43Density functional theory (DFT) calculations were performed on bulk and surface Ni2−xRhxP models with x values ranging from 0 to 0.83, in increments of 0.17, since phase separation was observed experimentally for x ≥ 1. The preferred sites for Rh substitution into the bulk unit cell were determined for each value of x. Because the primitive bulk Ni2P unit cell contains six metal sites, the bulk unit cell was doubled in the x direction to simulate all compositions. The DFT-calculated lattice parameters for bulk Ni2−xRhxP are summarized in Table S13,† showing good agreement with the experimental lattice parameters for parent Ni2P and Rh2P. Ni2−xRhxP surfaces were modeled as the (0001) facet, due to the prevalence of this facet in previous Ni2P Wulff particle constructions over a range of P chemical potentials (μP).44 The (0001) surface involves repeating Ni3P2 and Ni3P layers, resulting in two possible bulk terminations. Under P-poor conditions, previous work by He and coworkers found that the Ni3P-terminated Ni2P(0001) surface was thermodynamically favoured;44 therefore, this termination was simulated given the absence of a P-source in the reactor feed (i.e., P-poor conditions) during catalyst evaluation. A vacuum layer at least 10 Å thick was employed to separate successive slabs in the z direction. For each surface, a 4-layer slab model was simulated in which all adsorbates and the top half of the slab model could freely relax, while the bottom half was fixed in its bulk-truncated position. The position of Rh atoms in the periodic slabs was optimized using a (1 × 1) unit cell, and the optimal Ni2−xRhxP(0001) surfaces (x = 0, 0.17, 0.33, 0.50, 0.67, and 0.83) are illustrated in Fig. 3. Adsorption of OH* on Ni3P-terminated Ni2−xRhxP(0001) was studied using a (1 × 1) surface unit cell. Due to its large size, adsorption of m-cresol* on Ni2−xRhxP(0001) was studied using a (3 × 3) surface unit cell, with Rh occupying the preferred substitution site for the corresponding (1 × 1) surfaces. A four-layer Rh2P(100) surface was also studied based on the cubic Wulff shape of Rh2P particles;45 a (4 × 4) surface unit cell was studied for all surface intermediates. For Rh2P(100), a P-terminated surface was evaluated due to its stability relative to the Rh-terminated surface across a wide range of μP45 (refer to ESI† for details).
Results and discussion
Synthesis and characterization of Ni2−xRhxP (x = 0, 0.2, 0.4, 0.6, 0.8, 1.0) nanoparticles
Ternary Ni2−xRhxP NPs were synthesized using a two-step approach beginning with the formation of amorphous Ni–P NPs by thermolysis of the molecular precursor Ni(CO)2(PPh3)2 in a mixture of oleylamine and 1-octadecene at 250 °C, according to our previously published procedure.33,34 After cooling the reaction mixture to ambient temperature, 0.10–0.50 molar equivalents of the second molecular precursor, RhCl(CO)(PPh3)2, were added to target specific stoichiometric ratios of Ni2−xRhxP (x = 0.2–1.0). The reaction was then heated to 300 °C for 1 h to promote decomposition of the Rh precursor, incorporation of Rh into the pre-formed amorphous Ni–P NPs, and phosphidation of the NPs. Particle formation was monitored by removing aliquots of the reaction mixture for XRD analysis while heating to the reaction temperature, and at several time points during the 1 h hold at 300 °C in the case of Ni1.6Rh0.4P NP synthesis (Fig. S1†). At 275 °C, the particles were amorphous, as indicated by the lack of reflections in the XRD pattern. Small crystalline particles (crystallite size of ∼1 nm determined by Scherrer analysis of the (111) reflection) with the hexagonal Ni2P structure were observed by XRD analysis of an aliquot taken after the reaction mixture reached 300 °C, with increased crystallite size observed after 60 min at 300 °C (10.1 nm). Binary hexagonal Ni2P (x = 0) and cubic Rh2P (x = 2.0) NPs were prepared similarly to our previous report for comparison to the ternary series of Ni2−xRhxP NPs.34The Ni, Rh, and P composition of the as-prepared Ni2−xRhxP, Ni2P, and Rh2P NPs was determined using inductively coupled plasma optical emission spectroscopy (ICP-OES), and measured x values were in close agreement with the targeted x values (Table 1). The Ni2−xRhxP NPs contained excess P compared to the expected total metal-to-phosphorus (M/P) ratio of 2.0 but maintained similar M/P ratios of 1.6–1.8 throughout the series (Table 1). The excess P may include residual P-containing organic species (e.g., surface ligands) on the NPs, as observed in our previous reports.33,34
| Target x | Measured x | mol% Ni | mol% Rh | mol% P | (Ni + Rh)/P molar ratio | Particle size by TEM (nm) | Volume average particle size by TEM (nm) | Crystallite size by XRD (nm) |
|---|---|---|---|---|---|---|---|---|
| 0 | — | 64.5 | 0 | 35.5 | 1.7 | 10.9 ± 0.4 | 11.1 ± 0.7 | 9.8 |
| 0.2 | 0.21 | 59.3 | 7.3 | 33.4 | 1.6 | 9.2 ± 0.3 | 9.7 ± 0.6 | 8.9 |
| 0.4 | 0.43 | 54.4 | 14.8 | 30.8 | 1.8 | 10.0 ± 0.5 | 10.5 ± 0.8 | 9.9 |
| 0.6 | 0.65 | 47.2 | 22.8 | 30.0 | 1.8 | 10.2 ± 0.4 | 10.8 ± 0.7 | 9.1 |
| 0.8 | 0.82 | 41.0 | 28.7 | 30.3 | 1.8 | 9.5 ± 0.3 | 10.1 ± 0.6 | 9.4 |
| 1.0 | 0.99 | 35.3 | 34.8 | 29.8 | 1.8 | 11.1 ± 0.4 | 11.8 ± 0.7 | 11.2 |
| 2.0 | — | 0 | 66.1 | 33.9 | 1.6 | 10.4 ± 0.7 | 10.7 ± 0.9 | 13.6 |
XRD was performed to identify the crystal phase and crystallite size (Fig. 1a) of the synthesized NPs. The diffraction pattern of the parent Ni2P NPs matches that of the hexagonal Ni2P phase. The hexagonal crystal structure was maintained in the ternary compositions for x between 0.2 and 0.8. However, the (111) reflection systematically shifted from 40.8° to 40.0° with increasing Rh concentration (Fig. 1a and Table S1†), suggesting lattice expansion with the incorporation of Rh into the parent Ni2P material. The computed Ni2P diffraction pattern has two superlattice diffraction peaks at 30.4° and 32.0° corresponding to the (211) and (101) planes, respectively, which decreased in intensity with increasing Rh incorporation, indicating increased atomic disorder.48,49 At x = 1, the appearance of reflections at 32.7° and 47.3° indicated that the cubic Rh2P phase was present in addition to the hexagonal Ni2P phase (Fig. 1a). Scherrer analysis performed on the (111) reflection demonstrated that the crystallite sizes for the phase-pure ternary materials with x of 0.2–0.8 (8.9–9.9 nm) were of comparable size to the parent Ni2P NPs (9.8 nm) (Table 1).
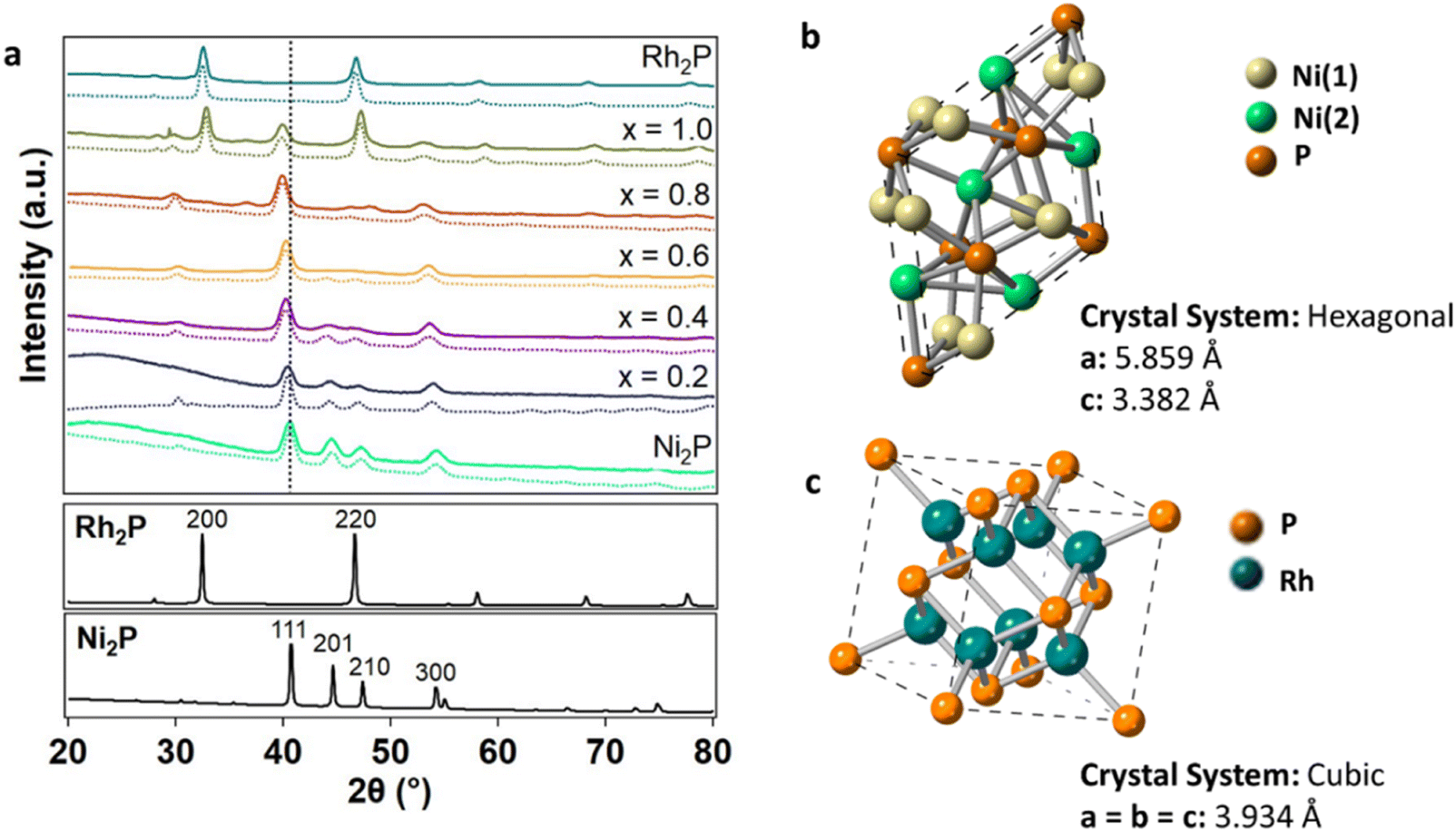 | ||
| Fig. 1 (a) XRD patterns of Ni2−xRhxP NPs (x = 0, 0.2, 0.4, 0.6, 0.8, 1.0, 2.0) with simulated patterns (dashed lines) from Rietveld refinement and the Ni2P (111) reflection highlighted (vertical black dashed line). The corresponding bulk Ni2P (mp-21167)46 and Rh2P (mp-2732)47 simulated patterns are provided below. (b) Hexagonal Ni2P structure with Ni(1) and Ni(2) sites denoted46 and (c) cubic Rh2P structure.47 | ||
In the hexagonal Ni2P crystal structure, Ni atoms are located in two distinct sites, denoted as Ni(1) and Ni(2), that differ based on their local environment (Fig. 1b).26,46 Ni(1) is defined as a near-tetrahedral Ni site with four Ni–P bonds at an average bond distance of 2.24 Å and eight distant Ni neighbours at 2.64 Å. Ni(2) is defined as a square pyramidal Ni site with five P neighbours at 2.43 Å and six Ni neighbours at 2.65 Å. Rh2P has a cubic crystal structure with one distinct Rh site (Fig. 1c). Previously, preferential site occupancy at Ni(2) over Ni(1) sites was observed with the incorporation of Fe atoms.21 Here, substitution of Rh at specific Ni sites in Ni2P was quantified by simulating the diffraction patterns of the Ni2−xRhxP NPs using Rietveld refinement in the MAUD crystal structure software (Table 2).50 The simulations (Fig. 1a, dashed lines) accounted for the specific crystallite sizes determined from Scherrer analysis of each XRD pattern and the measured metal concentrations determined by ICP-OES analysis. Rh was determined to preferentially substitute into the square pyramidal Ni(2) sites at 67%, 63%, 64%, and 53% for x equal to 0.2, 0.4, 0.6, and 0.8, respectively. At higher concentrations, Rh increasingly populated the near-tetrahedral Ni(1) sites with 33%, 37%, 36%, and 47% occupancy at x equal to 0.2, 0.4, 0.6, and 0.8, respectively. This is consistent with literature reports that demonstrated higher concentrations of heterometals leading to increased substitution at the tetrahedral Ni(1) site.51 For x less than 0.8, substitution of Rh atoms into the Ni2P lattice resulted in slight lattice parameter expansion in both a and c (Table 2 and Fig. S2†), consistent with that observed for the preferential substitution of Ni by Fe.15,21 The Rietveld refinement performed here also revealed phase separation in materials with x of 0.8 and 1. For x equal to 0.8, minimal phase separation was observed (∼3 vol%); however, for x equal to 1, more extensive phase separation (50 vol%) was observed, as was apparent in the XRD pattern (Fig. 1a). Phase separation into the hexagonal Ni2P and cubic Rh2P phases was also observed in a recent report by Brock, et al. for Ni2−xRhxP NPs with x ≥ 1.5, where it was determined that Rh incorporation reached a maximum of x equal to 1.25 without separation using a single-step synthesis method.52,53
To identify the distribution of Ni, Rh, and P within the NPs, scanning transmission electron microscopy (STEM) coupled with energy dispersive X-ray spectroscopy (EDS) was performed. Exemplary STEM images and associated analysis is provided in Fig. 2 for Ni1.6Rh0.4P. This analysis was also performed for all compositions of x between 0.2 and 1.0 (Fig. S3–S6†), and co-location of Rh and Ni atoms within the NPs of each composition was confirmed (Fig. 2d–h, S3–S6d–h†). The bright field (BF) STEM image (Fig. 2a, S3–S6a†) indicated that the particles are uniform in size and shape. Average particle diameters for compositions with x ≤ 1 were between 9.2 and 11.1 nm (Table 1 and Fig. S7†), which were comparable or slightly larger than the crystallite sizes calculated from the Scherrer analysis of the (111) reflection in the XRD patterns of the Ni2−xRhxP NPs. There was no obvious trend in particle size observed with addition of Rh. The average particle diameter for the Rh2P NPs (10.4 nm) was smaller than that determined by Scherrer analysis of the XRD pattern (13.6 nm), possibly due to the higher polydispersity of the NPs (standard deviation of 0.7 nm). High resolution (HR) high-angle annular dark-field (HAADF)-STEM (Fig. 2b, S3–S6b†) indicated that the Ni2−xRhxP NPs were highly crystalline. Fast Fourier transform (FFT) analysis of individual particles showed that the Ni1.6Rh0.4P composition had the same hexagonal crystal structure as the parent Ni2P (Table S2†). For all Ni2−xRhxP NPs with x ≤ 1, the FFT patterns were indexed to the hexagonal Ni2P phase (Fig. 2c, S3–S6c, Tables S3–S6†), consistent with the phase determined by XRD (Fig. 1a).
 | ||
Fig. 2 (a) Bright field STEM image of Ni1.6Rh0.4P NPs, (b) high resolution HAADF-STEM image with the corresponding (c) FFT oriented along the [12![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) 3] direction, (d) HAADF-STEM image on a different particle for EDS analysis, and associated EDS elemental maps for (e) Ni, (f) Rh, (g) P, and (h) Ni and Rh overlaid. 3] direction, (d) HAADF-STEM image on a different particle for EDS analysis, and associated EDS elemental maps for (e) Ni, (f) Rh, (g) P, and (h) Ni and Rh overlaid. | ||
The phase-pure Ni2−xRhxP NPs (x ≤ 0.8) were supported on SiO2 with a target loading of 5 wt% NPs (see ESI† for additional experimental details) and further structural characterization was performed. XRD analysis confirmed that the crystal phase did not change upon supporting (Fig. S8†), and ICP-OES analysis confirmed that the molar ratios of Ni, Rh, and P atoms were maintained (Table S7†). High energy (HE)-XRD coupled with atomic pair distribution function (PDF) analysis was performed on as-prepared Ni2P/SiO2, Rh2P/SiO2, and Ni1.6Rh0.4P/SiO2 to understand the local arrangement of metal atoms in the NPs. Ni1.6Rh0.4P/SiO2 was selected as a representative ternary TMP material because 20 mol% provides a high enough Rh concentration to detect structural changes associated with Rh incorporation while maintaining phase homogeneity, as determined by standard XRD analysis (Fig. 1a). The diffraction patterns observed in both laboratory- and HE-XRD (Fig. S9†) align in Q-space, indicating that the structures of each sample are consistent with bulk Ni2P, bulk Rh2P, and Rh-containing Ni2P, respectively. PDF analysis was performed on the HE-XRD patterns (Fig. S10 and S11†) and the first shell average coordination numbers (CNs) (i.e., first nearest neighbor) and bond distances were calculated using the reverse Monte Carlo (RMC) method (Fig. S12–S15 and Table S8†). This analysis revealed that Ni2P/SiO2 was found to have 2.4 Ni–P bonds at 2.30 Å and 3.4 Ni–Ni bonds at 2.63 Å. Rh2P/SiO2 had 2.7 Rh–P bonds at 2.40 Å and 3.0 Rh–Rh bonds at 2.70 Å. The bond distances and CNs of both Ni2P/SiO2 and Rh2P/SiO2 were consistent with those expected for the respective binary TMP phases.46,47 Ni1.6Rh0.4P/SiO2 had 2.2 Ni–P bonds at 2.35 Å and 2.3 Rh–P bonds at 2.42 Å, in addition to 3.1 Ni–Ni bonds at 2.65 Å, 0.4 Ni–Rh bonds at 2.69 Å, 1.5 Rh–Ni bonds at 2.69 Å, and 2.1 Rh–Rh bonds at 2.69 Å. The incorporation of Rh led to bond distance elongation in the Ni–P and Ni–Ni scattering pairs in Ni1.6Rh0.4P/SiO2 relative to Ni2P/SiO2, consistent with the lattice parameter expansion observed by standard XRD (Fig. 1a).
With this detailed understanding of the as-prepared TMP catalyst structures, Ni2P/SiO2, Rh2P/SiO2, and Ni1.6Rh0.4P/SiO2 were evaluated by in situ HE-XRD in H2 and a representative phenolic probe molecule (guaiacol) at 350 °C to understand the structure of the NPs under reaction conditions. During treatment in H2, increased atomic disorder (Fig. S11†) and the loss of local Ni–P (CN = 2.0) and/or local Rh–P (CN = 2.1) coordination is concomitant with an increase in Ni–Ni (CN = 3.4) and/or Rh–Ni (CN = 1.5) coordination in the second coordination sphere for all materials, while all bond distances remained unchanged (Table S8†). The Ni–P, Ni–Ni, and Ni–Rh coordination numbers were within 11% of the as-synthesized values, suggesting that the overall hexagonal crystal structure was maintained. For Ni2P/SiO2, increased atomic disorder in H2 was also observed in the bond angle distributions (Fig. S16†) derived from RMC models, where an increase in the average Ni–P–Ni bond angle in H2 corresponded to an increase of the full width at half maximum (FWHM) of each feature (i.e., peak broadening) (Table S9†). Increased disorder was also observed in Ni1.6Rh0.4P/SiO2 relative to Ni2P/SiO2 (Fig. S17 and Table S9†). It is important to note that the differences in CNs are a result of the increased disorder in the system and not due to a loss of P atoms, as the stoichiometry was maintained for each RMC simulation. No further structural changes were observed upon subsequent exposure to guaiacol, suggesting that the H2-treated structure is relevant for understanding the composition-dependent performance described below.
Electronic structure of Ni2−xRhxP nanoparticles
Models were developed for Ni2−xRhxP NPs (x = 0.2 to 0.8) by determining the most energetically favourable configuration of Rh sites in a Ni2P surface. The lattice parameter of each theoretically determined composition was consistent with that obtained experimentally by XRD (Table S13 and Fig. S2†). DFT calculations, using Bader charge analysis, were performed on bulk Ni2−xRhxP to understand the charge distribution between Ni, Rh, and P atoms with increasing x (Table 3). This analysis was performed for x ranging from 0 to 0.83, as distinct phase separation was observed experimentally at x equal to 1. The Bader charge for both Ni (+0.25 to +0.27e−) and Rh (+0.11 to +0.14e−) in bulk Ni2−xRhxP remained relatively constant across the x range. The bulk P charge, however, became less negative with increasing Rh concentration to compensate for the substitution of more positively charged Ni atoms with less positively charged Rh atoms, increasing from −0.55 to −0.41e− for x increasing from 0 to 0.83.| x | Q (e−) | ||
|---|---|---|---|
| Ni | Rh | P | |
| 0 | +0.27 | — | −0.55 |
| 0.17 | +0.27 | +0.13 | −0.52 |
| 0.33 | +0.27 | +0.13 | −0.49 |
| 0.50 | +0.26 | +0.13 | −0.46 |
| 0.67 | +0.26 | +0.14 | −0.44 |
| 0.83 | +0.25 | +0.14 | −0.41 |
| 2 | — | +0.11 | −0.22 |
A similar analysis was performed to evaluate charge transfer in Ni2−xRhxP surfaces. The (0001) surface is often used in computational studies of Ni2P catalysis,54–56 due to its low surface energy relative to other Ni2P facets and its presence in Wulff particle constructions over a range of P chemical potentials (μP).44 The Ni3P-terminated (0001) surface was considered for this analysis based on work by He et al. demonstrating that this mixed surface termination is thermodynamically favoured under P-poor conditions, which is representative of the HDO reaction environment.44 Likewise, for cubic Rh2P, a P-terminated (100) surface was evaluated due to its stability across a wide range of μP.45 The preferred locations for Rh substitution into Ni2−xRhxP(0001) slab models were calculated for x ranging from 0.17 to 0.83 (Fig. 3). The incorporation of higher Rh concentrations led to varying degrees of Rh substitution into the first and third layers of Ni2−xRhxP(0001). The electronic structure of the individual atoms and average values for the metal sites in the topmost layer for each Ni2−xRhxP(0001) surface was evaluated using Bader charge analyses. As observed for bulk Ni2−xRhxP, the surface-Ni and surface-Rh charges remained relatively constant (Table 4), while the introduction of Rh into the surface layer resulted in a less-negative charge for surface-P, increasing from −0.47e− (x = 0) to −0.32e− (x = 0.83). However, considering the average surface metal (Mavg) charge based on the composition of the topmost surface layer (Fig. 3), the addition of Rh to the surface layer resulted in a decrease in the Mavg charge from +0.25e− (i.e., QNi) at x = 0 and 0.17, to +0.18e− (i.e., (2QNi + QRh)/3) at x = 0.33. 0.50, and 0.67, and to +0.10e− (i.e., (QNi + 2QRh)/3) at x = 0.83. Previous studies on TMP-catalyzed HDO used Bader charge analysis as a proxy for Lewis acidity, suggesting that systems with greater positive charges for surface-metal atoms have stronger Lewis acidities and promote increased DDO selectivity.13,20 In the Ni2−xRhxP series, the surface-P charge became less negative with increasing x while the charge of Ni and Rh surface atoms did not change. Instead, addition of Rh to the surface layer resulted in a decrease to Mavg charge (Table 4). In contrast, Hicks et al. reported increasing average positive charge on the metal atoms (Fe and Mo) and decreasing average negative charge on the P atoms in the FexMo2−xP system with increasing Fe.20 They postulated that electronegativity (EN) differences were a primary driving force for the observed charge transfer between M, M′, and P atoms. Less electronegative metals, like Fe (EN = 1.64) and Mo (EN = 1.30), may have a greater propensity for charge transfer at different molar ratios in FexMo2−xP due to the larger EN difference relative to P (EN = 2.06).20 Conversely, Ni (EN = 1.75) and Rh (EN = 2.28) have EN values closer to that of P, which may limit charge transfer. The lower EN of Ni relative to Rh also accounts for the greater charge transfer between Ni and P atoms in Ni2P compared to Rh and P atoms in Rh2P.
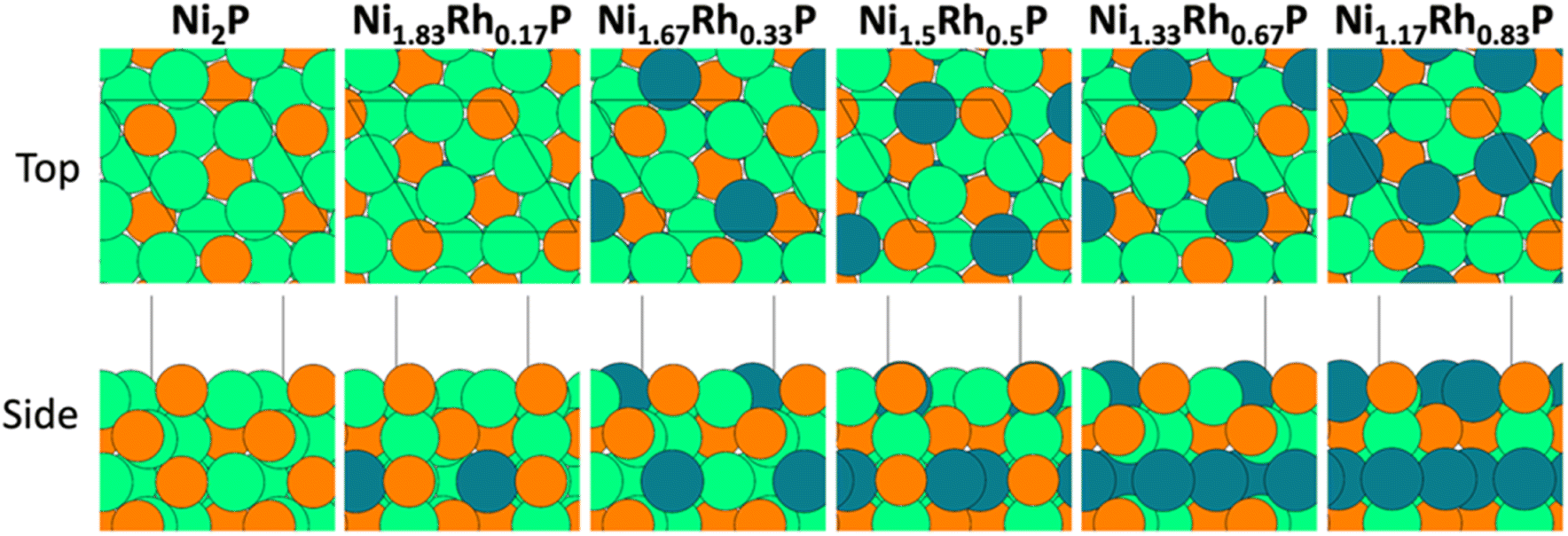 | ||
| Fig. 3 Top and side views of the (1 × 1) Ni3P-terminated Ni2−xRhxP(0001) surface (x = 0, 0.17, 0.33, 0.50, 0.67, and 0.83). Black lines denote the surface unit cell. Atom colors: orange – P, green – Ni, teal – Rh. | ||
| x | Ni | Rh | M avg | P | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Q (e−) | E d (eV) | E sp (eV) | Q (e−) | E d (eV) | E sp (eV) | Q (e−) | E d (eV) | E sp (eV) | Q (e−) | E sp (eV) | |
| 0 | +0.25 | −1.33 | +0.07 | — | — | — | +0.25 | −1.33 | +0.07 | −0.47 | −3.08 |
| 0.17 | +0.25 | −1.35 | +0.06 | — | — | — | +0.25 | −1.35 | +0.06 | −0.47 | −3.11 |
| 0.33 | +0.25 | −1.36 | +0.07 | +0.03 | −1.60 | +0.22 | +0.18 | −1.44 | +0.12 | −0.40 | −3.21 |
| 0.50 | +0.25 | −1.37 | +0.09 | +0.03 | −1.61 | +0.25 | +0.18 | −1.45 | +0.14 | −0.40 | −3.23 |
| 0.67 | +0.25 | −1.38 | +0.12 | +0.03 | −1.61 | +0.28 | +0.18 | −1.46 | +0.17 | −0.40 | −3.24 |
| 0.83 | +0.24 | −1.35 | +0.20 | +0.03 | −1.56 | +0.31 | +0.10 | −1.49 | +0.27 | −0.32 | −3.26 |
| 2.00 | — | — | — | +0.10 | −2.07 | −0.35 | +0.10 | −2.07 | −0.35 | −0.12 | −2.92 |
The metal sp- and d-band centers, as well as the P sp-band centers, were also evaluated to further explore the influence of Rh substitution on the electronic structure of Ni2−xRhxP NPs. The d-band center has been established as a descriptor for both metal reactivity57 and adsorption strength of key surface intermediates, while the sp-band center has shown promise as a descriptor for multi-component materials.58 The calculated average d- and sp-band centers of the Ni, Rh, M, and P atoms in the Ni2−xRhxP(0001) surface are reported in Table 4. Similar to the average metal surface charge, while the d-band centers for Ni and Rh surface atoms do not change appreciably with increasing x, the average d-band values for the surface-metal ensemble decrease as Rh incorporates into the surface layer.
Hydrodeoxygenation of phenolic lignin model compounds
The hydrogen (H*) binding site densities on the Ni2−xRhxP catalysts were determined by H2 chemisorption. When normalized to the metal content on a molar basis, the H* binding site density remained relatively unchanged at 0.010–0.014 molH*/molM across the Ni2−xRhxP series (Table 5) despite changing Rh concentration, and in contrast to a 3–4× increase in H* binding site density observed for Rh2P (0.040 molH*/molM). These values are lower than the ideal values of ca. 0.1 molH*/molM based on a geometric model of 10 nm NPs, where inaccessible sites may be due to a combination of residual ligand decomposition products, lost NP surface area from interaction with the support surface, and the inherent differences in metal phosphide H2 activation compared to that of traditional metals. No trend with Rh content was observed, and therefore, any observed changes in catalytic activity and/or selectivity are not likely due to increased or decreased number of available catalytic sites as a function of Rh content. Indeed, the site-time yield values (STY, calculated as in eqn (4) in the ESI†) from the m-cresol HDO reaction were comparable across the Ni2−xRhxP series (0.064–0.097 s−1) and displayed no trend with H* binding site density.| H2 chemisorption μmolH*/gcat | H2 chemisorption molH*/molM | m-cresol HDO STY (s−1) | |
|---|---|---|---|
| x = 0 | 6.8 | 0.012 | 0.068 |
| x = 0.2 | 7.4 | 0.012 | 0.083 |
| x = 0.4 | 7.1 | 0.014 | 0.064 |
| x = 0.6 | 6.0 | 0.011 | 0.096 |
| x = 0.8 | 5.3 | 0.010 | 0.097 |
| x = 2.0 | 14.3 | 0.040 | 0.038 |
The catalytic performance of SiO2-supported Ni2−xRhxP NPs was evaluated in the HDO reaction of phenolic model compounds. Guaiacol was initially selected as a probe molecule based on our previous study.35 However, initial evaluation of the catalysts in a packed-bed reactor (H2/guaiacol = 12:1, 350 °C, 0.5 MPa) yielded ca. 15% conversion, and only minor changes in product selectivity were observed between Ni2P/SiO2 and Ni1.6Rh0.4P/SiO2, with both favouring phenol at 42–48% (Table S10†), indicating that demethoxylation of guaiacol to phenol was the primary transformation occurring at low conversion. Therefore, m-cresol was selected as a model compound to understand how changes in Ni2−xRhxP composition impacts the selectivity to fully deoxygenated products. HDO of m-cresol over the SiO2-supported materials was evaluated under similar conditions to our previous reports (H2/m-cresol = 8:1, 350 °C, 0.5 MPa)35,59,60 with an m-cresol weight-hourly space velocity (WHSV) of 10 h−1.
The model compound m-cresol enables a comparative study of reaction pathway selectivity for C–O bond scission to produce toluene (DDO pathway) versus ring hydrogenation (HYD pathway) to produce methylcyclohexane, methylcyclohexanol, and subsequently, methylcyclohexene and methylcyclohexane after dehydration and further hydrogenation (Fig. 4a).9,12 The pathway selectivity to HYD and DDO products across the SiO2-supported Ni2−xRhxP NP series were compared to those for binary Ni2P/SiO2 and Rh2P/SiO2 at m-cresol conversions less than 15% (Fig. 4b, Tables S11 and S12†) using grouped product analysis. Across the Ni2−xRhxP NP series, a minor portion of the total products (4.99–13.6%) were light hydrocarbons (e.g., C1−C5 paraffins) likely formed by cracking reactions. No trend was observed in the selectivity to these light hydrocarbons, and they were excluded from our analysis of pathway selectivity (Table S11†). The major products observed over the Ni2−xRhxP NP series were methylcyclohexanone (33.9–40.4%), toluene (21.2–33.7%), and methylcyclohexene (7.99–17.9%), with minor products including methylcyclohexanol (2.3–7.6%), methylcyclohexane (1.6–2.8%), and benzene/xylenes (<1% each). HYD selectivity was determined by the sum of carbon selectivities to methylcyclohexanone, methylcyclohexanol, methylcyclohexene, and methylcyclohexane. DDO selectivity was determined by the sum of carbon selectivities to toluene, benzene, and xylenes, where benzene and xylenes are formed through methyl transfer reactions. Rh2P/SiO2 was highly selective toward DDO products (90%), consistent with previous observations for guaiacol and phenol HDO reactions.35,37 On the other hand, Ni2P/SiO2 slightly favoured HYD (54%) over DDO (46%). We anticipated that incorporation of Rh into the Ni2−xRhxP NP series would invoke Rh2P-like selectivity, leading to enhanced DDO selectivity. However, the introduction of Rh instead increased the HYD pathway selectivity from 58% for Ni1.8Rh0.2P/SiO2 to 66% for Ni1.6Rh0.4P/SiO2 to 72% for Ni1.4Rh0.6P/SiO2 (Fig. 4b and Table S12†), with a corresponding decrease in DDO selectivity. A modest decrease in HYD selectivity to 63% was observed for Ni1.2Rh0.8P/SiO2, attributed to the small degree of phase separation to Rh2P occurring in that composition (3 vol%, determined by Rietveld refinement). These data suggest that the TMPs cannot be treated simply as a linear combination of the performance of their parent metal phosphides.
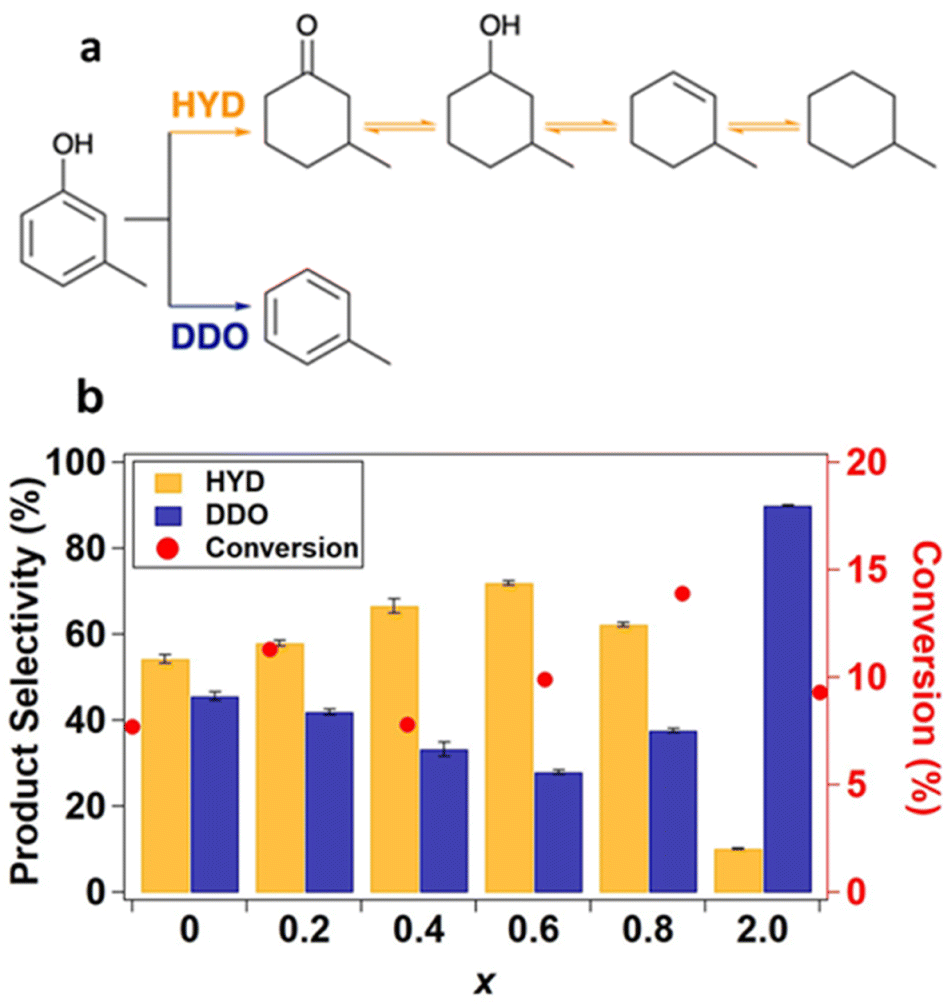 | ||
| Fig. 4 (a) m-cresol HDO reaction pathways.61 (b) Pathway selectivity in m-cresol HDO at 350 °C, 0.5 MPa, WHSV 10 h−1 in an 8:1 molar feed ratio of H2:m-cresol with data averaged over 290 to 460 minutes time-on-stream. | ||
Hydrodeoxygenation selectivity descriptors
DFT calculations were employed to understand the unanticipated selectivity trends for m-cresol HDO on the Ni2−xRhxP NP series in the context of developing computationally tractable descriptors. Previous studies have proposed distinct descriptors for DDO versus HYD selectivity in phenol HDO chemistry, including: Bader charge as a metric for change in Lewis acidity from charge transfer between M, M′, and P,4,20,62 the energy difference between the phenolic reactant adsorption in parallel versus perpendicular/tilted binding modes,23,63 the Ph–OH bond distance in the adsorbed phenolic compound,64–66 and the binding energy of OH*.66 In assessing correlations between possible descriptors and the experimentally measured m-cresol HDO selectivity, the descriptor values on the simulated TMP compositions were compared with the measured selectivity on the most similar Ni2−xRhxP composition (e.g., DFT calculated properties for x equal to 0.17 were compared to experimental data for x equal to 0.2). Due to some phase separation, the measured DDO selectivity for Ni1.2Rh0.8P may be affected by contributions from the cubic Rh2P phase (Fig. 4b). Therefore, x equal to 0.8 was not considered in analyses aiming to understand selectivity trends across the TMPs.As described above, there was not an appreciable change in surface-Ni or -Rh charge across the Ni2−xRhxP series (Table 4), indicating that individual surface-metal charge is not a good descriptor for the observed selectivity changes at varying x for this Ni2−xRhxP series. The m-cresol* adsorption geometry was evaluated on Ni3P-terminated Ni2−xRhxP(0001) and P-terminated Rh2P(100) surfaces. The results, presented in Fig. S18,† indicate that m-cresol* preferentially adsorbs in a parallel orientation (par.) versus a perpendicular/tilted orientation (perp./tilt) on all surfaces by a range of ca. 0.5 eV to 0.7 eV. The difference in binding energy between the parallel and perpendicular adsorption geometries of m-cresol* was not well-correlated to the observed HDO selectivity. The Ph–OH bond distance in the preferred parallel adsorption configuration did not vary significantly or systematically across the TMP series (Fig. S18†) and is thus also not an effective m-cresol HDO selectivity descriptor for this series of TMP catalysts.
For the DDO of phenolic molecules, the thermodynamics associated with Ph–OH bond scission depend on the adsorption energy of OH*, with stronger OH* binding generally making the elementary step more exothermic. Previous work has also implicated the OH* binding energy as a descriptor for phenol DDO selectivity on Ni-based single-atom surface alloys;66 therefore, OH* was evaluated as a descriptor for the observed DDO versus HYD selectivity in the Ni2−xRhxP NP series. The most-stable adsorption sites for OH* binding are provided in Fig. 5 for the Ni3P-terminated Ni2−xRhxP(0001) and P-terminated Rh2P(100) surfaces. The OH* binding energy was observed to decrease with incorporation of additional Rh atoms into the surface layer of Ni3P-terminated Ni2−xRhxP(0001), ranging from −3.44 eV for Ni2P(0001) to −3.10 eV for Ni1.17Rh0.83P(0001). On P-terminated Rh2P(100), OH* binds stronger with a binding energy of −3.73 eV. When the DDO selectivities are plotted against the OH* binding energies across the TMP series, a strong correlation is observed (R2 = 0.96; Fig. 6a). Notably, OH* binds to surface-metal hollow sites on Ni2−xRhxP(0001) and to surface-P atoms on P-terminated Rh2P(100). Thus, the average surface-Rh d-band center is not able to describe the OH* binding energy on P-terminated Rh2P(100) because the Rh sites are in the second layer (i.e., subsurface) and are blocked by surface-P. However, the average surface-metal d-band center is well-correlated with the average surface-P sp-band center for Ni3P-terminated Ni2−xRhxP(0001) (Fig. 6b), and as a result, modulation in the OH* binding strength was observed to be a strong function of the average sp-band center of surface-P atoms (R2 = 0.98; Fig. 6c) across the entire series of these binary and ternary TMP catalysts. In summary, OH* binding energy is well-described by the sp-band center of surface-P atoms, because P atoms are present in the surface layer of the studied parent Ni2P, parent Rh2P, and ternary Ni2−xRhxP surface models. As a result, both OH* binding energy and sp-band center of surface-P atoms may provide computationally tractable descriptors for m-cresol HDO pathway selectivity on TMPs containing P in the surface layer, including TMPs with varying OH* binding sites (i.e., M or P) as found here.
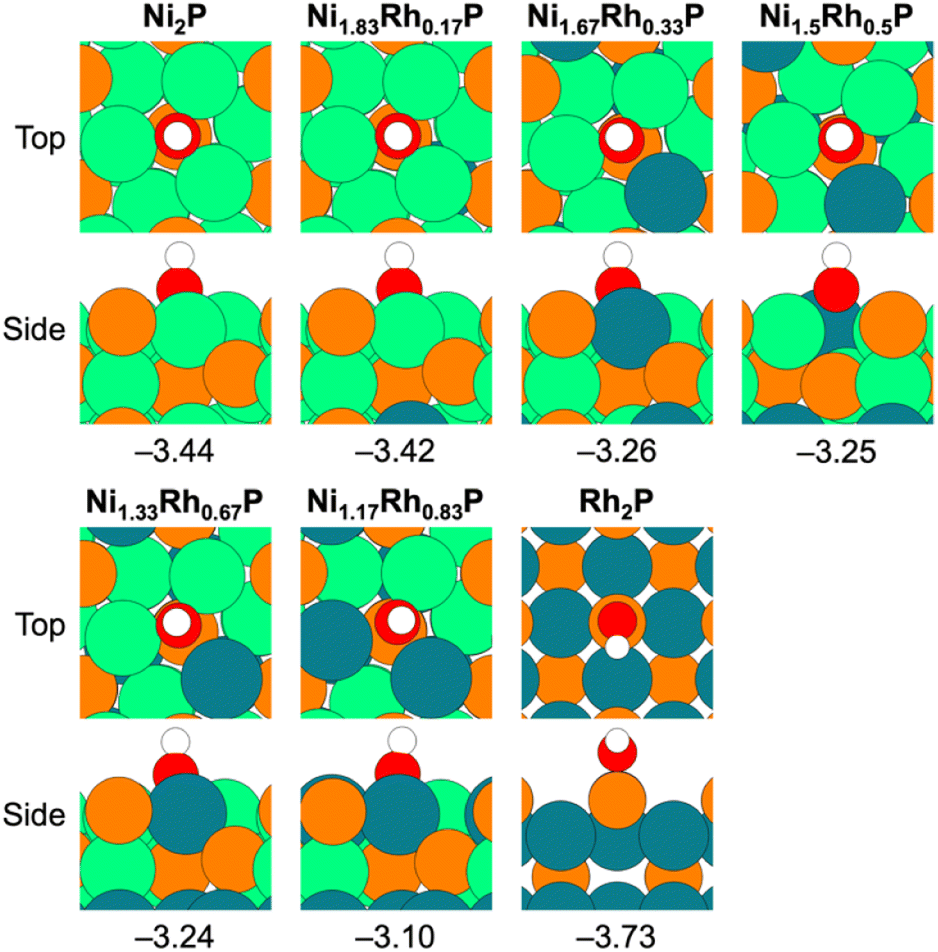 | ||
| Fig. 5 Top and side views for OH* adsorption on Ni3P-terminated Ni2−xRhxP(0001) (x = 0, 0.17, 0.33, 0.5, 0.67, 0.83) and P-terminated Rh2P(100) (x = 2.0). Adsorption energies, in eV, are provided below each snapshot. Atom colors: orange – P, green – Ni, teal – Rh, white – H, red – O. | ||
 | ||
| Fig. 6 (a) Experimental DDO selectivity versus OH* binding energy (EB(OH), in eV). (b) Average d-band center for surface-metal atoms (Ed(surf M), in eV) versus average sp-band center for surface-P atoms (Esp(surf P), in eV) (c) EB(OH) versus sp-band center for surface-P atoms (Esp(surf P), in eV) for Ni3P-terminated Ni2−xRhxP(0001) (x = 0, 0.2, 0.4, 0.6, 0.8) and P-terminated Rh2P(100) (x = 2.0). Data labels correspond to x. Linear fits are marked by the dashed black line alongside the corresponding R2 value. | ||
Given that the charge on the individual Ni and Rh surface sites does not change with varying x, the DFT calculations indicated that it is the formation of Ni–Rh surface ensembles that leads to the modulation of both the OH* adsorption energy and average surface-P sp-band center, and thereby the observed selectivity changes. To assess whether geometric effects (i.e., lattice expansion) from Rh incorporation might also affect the observed DDO versus HYD selectivity, the OH* adsorption energy was evaluated on Ni3P-terminated Ni2P(0001) with the lattice systematically expanded to match the lattice constants corresponding to each studied x value for bulk Ni2−xRhxP (Table S13†). These results, presented in Table S14,† indicate that the lattice expansion resulting from increased incorporation of Rh resulted in only negligible weakening of the OH* binding energy, with values ranging from −3.44 eV to −3.38 eV for Ni2P(0001) with lattice parameters representative of x equal to 0 and 0.83, respectively. Therefore, the calculated OH* binding energies on Ni2−xRhxP(0001) are primarily influenced by the formation of the Ni–Rh surface-ensembles arising from Rh substitution into the surface layer of Ni2−xRhxP(0001).
Previous reports attributed composition-dependent HDO selectivity to either the addition of a metal modifier site that perturbs the adsorption geometry or ligand effects modifying the electronic structure of the parent metal site in the ternary TMP.15,20,64 Though both geometric and electronic consequences occur with varying x values in the Ni2−xRhxP system, DFT analysis suggests a minimal geometric influence on the HYD versus DDO selectivity in the m-cresol HDO reaction (Table S14†). Concomitant electronic-structure changes were imposed with increasing x leading to weaker OH* binding energies at the surface-metal ensemble sites, promoting the formation of HYD products over DDO products. To identify a robust descriptor of catalyst performance, the sp-band center of surface-P atoms was calculated to include the electronic contribution of P atoms, which is not necessarily encompassed in oxophilicity and electronegativity arguments. Although oxophilicity and electronegativity arguments can describe the phenolic molecule HDO pathway selectivity for some ternary TMP systems,13,17 they do not provide universal descriptors for the rational design of novel ternary TMP catalysts, as evidenced by the results for this Ni2−xRhxP NP series. Oxophilicity lacks universality as a descriptor because it describes discrete metal properties, and not necessarily how the metal atoms interact with P atoms in an ordered structure, which can change depending on the crystal structure. We demonstrated that the OH* adsorption energy provides a promising computationally tractable descriptor for the HYD/DDO selectivity on TMP surfaces with varying composition, crystal structure, and surface termination.
In this report, OH* adsorption energy trends well with the catalytic performance observed across the hexagonal Ni2−xRhxP and cubic Rh2P NPs. Further, this study showed that incorporating varying concentrations of Rh into a parent Ni2P framework did not invoke Rh2P-like catalytic properties, highlighting that crystal structure and surface termination are interwoven in ternary TMPs and play important roles in the resultant catalytic selectivity.
Conclusions
A two-step solution synthesis approach was employed to access well-defined ternary Ni2−xRhxP NPs with consistent crystal structure and particle morphology across a range of compositions. Through extensive characterization, the addition of Rh was observed to result in preferential Ni(2) site substitution in binary Ni2P. For all compositions of Ni2−xRhxP (x ≤ 1), Ni and Rh atoms were co-located in a single particle with uniform distribution of both elements. Lattice parameter expansion was observed with increasing Rh concentration. At high Rh concentrations (x ≥ 0.8), phase separation to hexagonal and cubic phases associated with parent Ni2P and Rh2P, respectively, was observed. The H2-pretreated catalyst exhibited an 11% loss in the number of Ni–P bonds with a corresponding increase in metal–metal bonding but maintained a coordination number ratio (Ni–P/Ni–M) consistent with the hexagonal crystal structure observed in the XRD patterns. Subsequent structural changes were not observed after guaiacol exposure at 350 °C, suggesting that the H2-treated structure is relevant for understanding performance in phenolic molecule HDO reactions. The catalytic performance of Ni2−xRhxP NPs was assessed via m-cresol HDO and composition-dependent selectivity was observed. Binary Ni2P had comparable selectivity to HYD and DDO, while binary Rh2P favoured DDO. Increasing Rh concentration in Ni2−xRhxP NPs promoted HYD selectivity until x equal to 0.6, but minor phase separation at x equal to 0.8 likely resulted in the slight increase in DDO selectivity. The composition-dependent understanding developed here demonstrates that the catalytic performance of ternary TMPs is not easily represented by a linear combination of the parent materials.We sought to identify a robust descriptor to explain composition-dependent performance of the Ni2−xRhxP series through DFT calculations. Although the Bader charge analysis is often used as a proxy for Lewis acidity, suggesting that systems with greater positive charges for surface-metal atoms have stronger Lewis acidities and promote increased DDO selectivity, the charge of surface-metal atoms in Ni2−xRhxP remained relatively unchanged and could not describe the observed changes in catalyst performance in this series. DFT calculations revealed that the decreased DDO selectivity with increasing x is described by the OH* adsorption energy, as Rh incorporation into the Ni3P-terminated Ni2−xRhxP(0001) surface resulted in weaker OH* adsorption. Modulation in the OH* binding strength was a strong function of the average sp-band center of surface-P atoms across all evaluated TMPs, regardless of the specific OH* binding site. These studies highlight that (1) the surface-metal and -P atoms in TMPs, not the bulk structures, must be investigated to understand catalytically relevant sites, and (2) the heterogeneity of surface sites in TMPs makes it challenging to find a single descriptor encompassing the role of material composition, crystal structure, and surface termination. The OH* binding energy is a promising descriptor for both the reactivity of hexagonal Ni2P and cubic Rh2P crystal structures during m-cresol HDO and could serve as a general descriptor for novel ternary TMP formulations. Electronic structure descriptors, such as the P sp-band center or the metal d-band center, can also serve as effective HDO selectivity descriptors but their utility is dependent on the presence of each species (i.e., P or M) in the TMP surface. Future work should continue to evaluate these descriptors as general/universal descriptors for DDO versus HYD pathway selectivity in phenolic molecule HDO. Further analysis of the electronic structure of TMPs should also rely on developing fundamental structure–function relationships for catalytically relevant surfaces, rather than solely on the bulk material properties. More broadly, this work highlights the necessity of precisely defined structures to deconvolute geometric and electronic effects, especially in complex reactions, such as phenolic HDO, and the approach undertaken in this work is extensible to a wider set of materials to aid the development of structure–function relationships for rational catalyst design.
Author contributions
Nicole J. LiBretto: conceptualization, methodology, investigation, formal analysis, data curation, validation, writing – original draft, writing – review & editing; Sean A. Tacey: conceptualization, methodology, software, investigation, formal analysis, writing – review & editing, visualization; Muhammad Zubair: methodology, software, formal analysis, data curation, writing – review & editing, visualization; Tuong V. Bui: investigation, writing – review & editing; Kinga A. Unocic: investigation, visualization, writing – review & editing; Frederick G. Baddour: conceptualization, formal analysis, writing – review & editing; Michael B. Griffin: resources, investigation, writing – review & editing; Joshua A. Schaidle: conceptualization, supervision, funding acquisition; Carrie A. Farberow: conceptualization, supervision, methodology, writing – review & editing; Daniel A. Ruddy: conceptualization, supervision, formal analysis, writing – review & editing; Nicholas M. Bedford: methodology, investigation, resources, supervision, writing – review & editing, project administration; Susan E. Habas: conceptualization, investigation, supervision, writing – review & editing, funding acquisition.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was authored in part by the National Renewable Energy Laboratory, operated by Alliance for Sustainable Energy, LLC, and in part by Oak Ridge National Laboratory, operated by UT-Battelle, LLC, for the U.S. Department of Energy (DOE) under Contract no. DE-AC36-08GO28308 and DE-AC05-00OR22725, respectively. Funding provided by the U.S. DOE Office of Energy Efficiency and Renewable Energy, Bioenergy Technologies Office in collaboration with the Chemical Catalysis for Bioenergy (ChemCatBio) Consortium, a member of the Energy Materials Network (EMN). In situ HE-XRD experiments were performed at the 6-ID-D beamline of the Advanced Photon Source, a U.S. DOE Office of Science User Facility operated by Argonne National Laboratory under Contract no. DE-AC02-06CH11357. M. Z. acknowledges scholarship support from the UNSW Scientia PhD Scholarship scheme. We would like to thank Doug Robinson for assistance at 6-ID-D and Yang Ren for providing the capillary furnace cell for HE-XRD measurements, as well as Evan C. Wegener for contributions in understanding the initial material structures using X-ray absorption spectroscopy. We would also like to thank Elisa M. Miller for X-ray photoelectron spectroscopy and helpful discussions. The microscopy was performed as part of a user project at the Center for Nanophase Materials Sciences (CNMS), which is a U.S. DOE Office of Science User Facility. The authors thank Kimberley S. Reeves for assistance with TEM sample preparation. A portion of the research was performed using computational resources sponsored by the U.S. DOE Office of Energy Efficiency and Renewable Energy and located at the National Renewable Energy Laboratory. This work also used computational resources at the Extreme Science and Engineering Discovery Environment (XSEDE), which is supported by National Science Foundation grant number ACI-1548562. The authors thank Vassili Vorotnikov for collaboration on initial computational models.Notes and references
- S. Kim, E. E. Kwon, Y. T. Kim, S. Jung, H. J. Kim, G. W. Huber and J. Lee, Recent advances in hydrodeoxygenation of biomass-derived oxygenates over heterogeneous catalysts, Green Chem., 2019, 21, 3715–3743 RSC.
- X. Lan, R. Pestman, E. J. M. Hensen and T. Weber, Furfural hydrodeoxygenation (HDO) over silica-supported metal phosphides – The influence of metal–phosphorus stoichiometry on catalytic properties, J. Catal., 2021, 403, 181–193 CrossRef CAS.
- S. T. Oyama, T. Gott, H. Zhao and Y.-K. Lee, Transition metal phosphide hydroprocessing catalysts: A review, Catal. Today, 2009, 143, 94–107 CrossRef CAS.
- V. Jain, Y. Bonita, A. Brown, A. Taconi, J. C. Hicks and N. Rai, Mechanistic insights into hydrodeoxygenation of phenol on bimetallic phosphide catalysts, Catal. Sci. Technol., 2018, 8, 4083–4096 RSC.
- S. Boullosa-Eiras, et al., Potential for metal-carbide, -nitride, and -phosphide as future hydrotreating (HT) catalysts for processing of bio-oils, in Catalysis, ed. Spivey J. J. and Han Y-F., Dooley K. M., RSC, 2014, vol. 26, pp. 29–71 Search PubMed.
- R. Prins and M. E. Bussell, Metal phosphides: preparation, characterization and catalytic reactivity, Catal. Lett., 2012, 142, 1413–1436 CrossRef CAS.
- H. Y. Zhao, D. Li, P. Bui and S. T. Oyama, Hydrodeoxygenation of guaiacol as model compound for pyrolysis oil on transition metal phosphide hydroprocessing catalysts, Appl. Catal., A, 2011, 391, 305–310 CrossRef CAS.
- L. Song and S. Zhang, A versatile route to synthesizing bulk and supported nickel phosphides by thermal treatment of a mechanical mixing of nickel chloride and sodium hypophosphite, Powder Technol., 2011, 208, 713–716 CrossRef CAS.
- V. O. O. Gonçalves, P. M. de Souza, V. T. da Silva, F. B. Noronha and F. Richard, Kinetics of the hydrodeoxygenation of cresol isomers over Ni2P/SiO2: Proposals of nature of deoxygenation active sites based on an experimental study, Appl. Catal., B, 2017, 205, 357–367 CrossRef.
- A. Berenguer, J. A. Bennet, J. Hunns, I. Moreno, J. M. Coronado, A. F. Lee, P. Pizarro, K. Wilson and D. P. Serrano, Catalytic hydrodeoxygenation of m-cresol over Ni2P/hierarchical ZSM-5, Catal. Today, 2018, 304, 72–79 CrossRef CAS.
- X. Wu, Q. Ge and X. Zhu, Vapor phase hydrodeoxygenation of phenolic compounds on group 10 metal-based catalysts: reaction mechanism and product selectivity control, Catal. Today, 2021, 365, 143–161 CrossRef CAS.
- D. A. Ruddy, J. A. Schaidle, J. R. Ferrell, J. Wang, L. Moens and J. E. Hensley, Recent advances in heterogeneous catalysts for bio-oil upgrading via “ex situ catalytic fast pyrolysis”: catalyst development through the study of model compounds, Green Chem., 2014, 16, 454–490 RSC.
- Y. Bonita and J. C. Hicks, Periodic trends from metal substitution in bimetallic Mo-based phosphides for hydrodeoxygenation and hydrogenation reactions, J. Phys. Chem. C, 2018, 122, 13322–13332 CrossRef CAS.
- L. Nie and D. E. Resasco, Kinetics and mechanism of m-cresol hydrodeoxygenation on a Pt/SiO2 catalyst, J. Catal., 2014, 317, 22–29 CrossRef CAS.
- L. Nie, P. de Souza, F. B. Noronha, W. An, T. Sooknoi and D. E. Resasco, Selective conversion of m-cresol to toluene over bimetallic Ni–Fe catalysts, J. Mol. Catal. A: Chem., 2014, 388, 47–55 CrossRef.
- A. M. Robinson, L. Mark, M. J. Rasmussen, J. E. Hensley and J. W. Medlin, Surface chemistry of aromatic reactants on Pt- and Mo-modified Pt catalysts, J. Phys. Chem. C, 2016, 120(47), 26824–26833 CrossRef CAS.
- G. Held, Adsorption and dissociation of benzene on bimetallic surfaces—the influence of surface geometry and electronic structure, J. Phys.: Condens. Matter, 2003, 15, R1501–R1516 CrossRef CAS.
- S. J. Jenkins, Aromatic adsorption on metals via first-principles density functional theory, Proc. R. Soc. A, 2009, 465, 2949–2976 CrossRef CAS.
- J. Zhong, J. Chen and L. Chen, Selective hydrogenation of phenol and related derivatives, Catal. Sci. Technol., 2014, 4, 3555–3569 RSC.
- D. J. Rensel, J. Kim, V. Jain, Y. Bonita, N. Rai and J. C. Hicks, Composition-directed FexMo2−xP bimetallic catalysts for hydrodeoxygenation reactions, Catal. Sci. Technol., 2017, 7, 1857–1867 RSC.
- A. Hitihami-Mudiyanselage, M. P. Arachchige, T. Seda, G. Lawes and S. L. Brock, Synthesis and characterization of discrete FexNi2–xP nanocrystals (0 < x < 2): compositional effects on magnetic properties, Chem. Mater., 2015, 27, 6592–6600 CrossRef CAS.
- D. R. Liyanage, S. J. Danforth, Y. Liu, M. E. Bussell and S. L. Brock, Simultaneous control of composition, size, and morphology in discrete Ni2–xCoxP nanoparticles, Chem. Mater., 2015, 27, 4349–4357 CrossRef CAS.
- Y. He and S. Laursen, The surface and catalytic chemistry of the first row transition metal phosphides in deoxygenation, Catal. Sci. Technol., 2018, 8, 5302–5314 RSC.
- D. Li, H. Baydoun, B. Kulikowski and S. L. Brock, Boosting the catalytic performance of iron phosphide nanorods for the oxygen evolution reaction by incorporation of manganese, Chem. Mater., 2017, 29, 3048–3054 CrossRef CAS.
- A. Parra-Puerto, K. L. Ng, K. Fahy, A. E. Goode, M. P. Ryan and A. Kucernak, Supported transition metal phosphides: Activity survey for HER, ORR, OER, and corrosion resistance in acid and alkaline electrolytes, ACS Catal., 2019, 9, 11515–11529 CrossRef CAS.
- S. J. Danforth, D. R. Liyanage, A. Hitihami-Mudiyanselage, B. Illic, S. L. Brock and M. E. Bussell, Probing hydrodesulfurization over bimetallic phosphides using monodisperse Ni2−xMxP nanoparticles encapsulated in mesoporous silica, Surf. Sci., 2016, 648, 126–135 CrossRef CAS.
- T. Su’a, M. N. Poli and S. L. Brock, Homogeneous nanoparticles of multimetallic phosphides via precursor tuning: Ternary and quaternary M2P phases (M = Fe, Co, Ni), ACS Nanosci. Au, 2022, 2, 503–519 CrossRef.
- S. Carenco, D. Portehault, C. Boissière, N. Mézailles and C. Sanchez, Nanoscaled metal borides and phosphides: Recent developments and perspectives, Chem. Rev., 2013, 113, 7981–8065 CrossRef CAS PubMed.
- A. E. Henkes, Y. Vasquez and R. E. Schaak, Converting metals into phosphides: A general strategy for the synthesis of metal phosphide nanocrystals, J. Am. Chem. Soc., 2007, 129, 1896–1897 CrossRef CAS PubMed.
- S. Carenco, Y. Hu, I. Florea, O. Ersen, C. Boissiere, N. Mezeilles and C. Sanchez, Metal-dependent interplay between crystallization and phosphorus diffusion during the synthesis of metal phosphide nanoparticles, Chem. Mater., 2012, 24, 4134–4145 CrossRef CAS.
- M. E. Mundy, D. Ung, N. L. Lai, E. P. Jahrman, G. T. Seidler and B. M. Cossairt, Aminophosphines as versatile precursors for the synthesis of metal phosphide nanocrystals, Chem. Mater., 2018, 30, 5373–5379 CrossRef CAS.
- F. D'Accriscio, E. Schrader, C. Sassoye, M. Selmane, R. F. Andre, S. Lamaison, D. Wakerley, M. Fontecave, V. Mougel, G. LeCorre, H. Grutzmacher, C. Sanchez and S. Carenco, A single molecular stoichiometric P-source for phase-selective synthesis of crystalline and amorphous iron phosphide nanocatalysts, ChemNanoMat, 2020, 6, 1208–1219 CrossRef.
- C. A. Downes, K. Van Allsburg, S. Tacey, K. A. Unocic, F. G. Baddour, D. A. Ruddy, N. J. LiBretto, M. M. O'Connor, C. A. Farberow, J. A. Schaidle and S. E. Habas, Controlled synthesis of transition metal phosphide nanoparticles to establish composition-dependent trends in electrocatalytic activity, Chem. Mater., 2022, 34(14), 6255–6267 CrossRef CAS.
- S. E. Habas, F. G. Baddour, D. A. Ruddy, C. P. Nash, J. Wang, M. Pan, J. E. Hensley and J. A. Schaidle, A facile molecular precursor route to metal phosphide nanoparticles and their evaluation as hydrodeoxygenation catalysts, Chem. Mater., 2015, 27, 7580–7592 CrossRef CAS.
- M. B. Griffin, F. G. Baddour, S. E. Habas, D. A. Ruddy and J. A. Schaidle, Evaluation of silica-supported metal and metal phosphide nanoparticle catalysts for the hydrodeoxygenation of guaiacol under ex situ catalytic fast pyrolysis conditions, Top. Catal., 2016, 59, 124–137 CrossRef CAS.
- M. B. Griffin, F. G. Baddour, S. E. Habas, C. P. Nash, D. A. Ruddy and J. A. Schaidle, An investigation into support cooperativity for the deoxygenation of guaiacol over nanoparticle Ni and Rh2P, Catal. Sci. Technol., 2017, 7, 2954–2966 RSC.
- Y. Kanda, T. Chiba, R. Aranai, T. Yasuzawa, R. Ueno, T. Tayao, K. Kato, Y. Obora, K.-i. Shimizu and Y. Uemichi, Catalytic activity of rhodium phosphide for selective hydrodeoxygenation of phenol, Chem. Lett., 2019, 48, 471–474 CrossRef CAS.
- G. Kresse and J. Furthmüller, Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed.
- G. Kresse and J. Furthmüller, Efficiency of ab initio total energy calculations for metals and semiconductors using a plane-wave basis set, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Generalized gradient approximation made simple, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- P. E. Blöchl, Projector augmented-wave method, Phys. Rev., 1994, 50, 17953–17979 Search PubMed.
- G. Kresse and D. Joubert, From ultrasoft pseudopotentials to the projector augmented-wave method, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef CAS.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- J. He, Á. Morales-García, O. Bludský and P. Nachtigall, The surface stability and equilibrium crystal morphology of Ni2P nanoparticles and nanowires from an ab initio atomistic thermodynamic approach, CrystEngComm, 2016, 18, 3808–3818 RSC.
- H. Duan, D. Li, Y. Tang, Y. He, S. Ji, R. Wang, H. Lv, P. P. Lopes, A. P. Paulikas, H. Li, S. X. Mao, C. Wang, N. M. Markovic, J. Li, V. R. Stamenkovic and Y. Li, High-performance Rh2P electrocatalyst for efficient water splitting, J. Am. Chem. Soc., 2017, 139, 5494–5502 CrossRef CAS PubMed.
- K. Persson, Materials Data on Ni2P (SG:189) by Materials Project, 2014 Search PubMed.
- K. Persson, Materials Data on Rh2P (SG:225) by Materials Project, 2014 Search PubMed.
- D. Kim, C. Xie, N. Becknell, Y. Yu, M. Karamad, K. Chan, E. J. Crumlin, J. K. Korskov and P. Yang, Electrochemical activation of CO2 through atomic ordering transformations of AuCu nanoparticles, J. Am. Chem. Soc., 2017, 139, 8329–8336 CrossRef CAS PubMed.
- J. J. Choi, C. R. Bealing, K. J. Hughes, E. Zhang, D.-M. Smilgies, R. G. Hennig, J. R. Engstrom and T. Hanrath, Controlling nanocrystal superlattice symmetry and shape-anisotropic interactions through variable ligand surface coverage, J. Am. Chem. Soc., 2011, 133, 3131–3138 CrossRef CAS PubMed.
- L. Lutterotti and M. Bortolotti, Object-oriented programming and fast computation techniques in Maud, a program for powder diffraction analysis written in Java, Compcomm. Newslett., 2003, 1, 43–50 Search PubMed.
- P. E. R. Blanchard, A. P. Grosvenor, R. G. Cavell and A. Mar, Effects of metal substitution in transition-metal phosphides (Ni1−xMx)2P (M = Cr, Fe, Co) studied by X-ray photoelectron and absorption spectroscopy, J. Mater. Chem., 2009, 19, 6015–6022 RSC.
- D. R. Liyanage, D. Li, Q. B. Cheek, H. Baydoun and S. L. Brock, Synthesis and oxygen evolution reaction (OER) catalytic performance of Ni2−xRuxP nanocrystals: enhancing activity by dilution of the noble metal, J. Mater. Chem. A, 2017, 5, 17609–17618 RSC.
- T. N. Batugedara and S. L. Brock, Role of noble- and base-metal speciation and surface segregation in Ni2−xRhxP nanocrystals on electrocatalytic water splitting reactions in alkaline media, Chem. Mater., 2022, 34, 4414–4427 CrossRef CAS.
- C. Wei, R. R. Rao, J. Peng, B. Huang, I. E. L. Stephens, M. Risch, Z. J. Xu and Y. Shao-Horn, Recommended practices and benchmark activity for hydrogen and oxygen electrocatalysis in water splitting and fuel cells, Adv. Mater., 2019, 31, 1806296 CrossRef.
- R. B. Wexler, J. M. P. Martirez and A. M. Rappe, Active role of phosphorus in the hydrogen evolving activity of nickel phosphide (0001) surfaces, ACS Catal., 2017, 7, 7718–7725 CrossRef CAS.
- L. Partanen, M. Hakala and K. Laasonen, Hydrogen adsorption trends on various metal-doped Ni2P surfaces for optimal catalyst design, Phys. Chem. Chem. Phys., 2018, 21, 184–191 RSC.
- J. K. Nørskov, F. Abild-Pedersen, F. Studt and T. Bligaard, Density functional theory in surface chemistry and catalysis, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 937–943 CrossRef PubMed.
- R. Michalsky, Y.-J. Zhang, A. J. Medford and A. A. Peterson, Departures from the adsorption energy scaling relations for metal carbide catalysts, J. Phys. Chem. C, 2014, 118, 13026–13034 CrossRef CAS.
- F. G. Baddour, V. A. Witte, C. P. Nash, M. B. Griffin, D. A. Ruddy and J. A. Schaidle, Late-transition-metal-modified β-Mo2C catalysts for enhanced hydrogenation during guaiacol deoxygenation, ACS Sustainable Chem. Eng., 2017, 5, 11433–11439 CrossRef CAS.
- M. B. Griffin, G. A. Ferguson, D. A. Ruddy, M. J. Biddy, G. T. Beckham and J. A. Schaidle, Role of the support and reaction conditions on the vapor-phase deoxygenation of m-cresol over Pt/C and Pt/TiO2, Catalysts, 2016, 6(4), 2715–2727 CAS.
- W. Jin, L. Pastor-Perez, D. Shen, A. Sepulveda-Escribano, A. Gu and T. R. Reina, Catalytic upgrading of biomass model compounds: novel approaches and lessons learnt from traditional hydrodeoxygenation – a review, ChemCatChem, 2019, 11, 924–960 CrossRef CAS.
- H. He, S. Xia and D. Luo, Sn-assisted nickel synergistically catalyzes the direct cleavage of CArO bond in lignin-derived m-cresol: theoretical and experimental analysis, J. Catal., 2022, 410, 180–193 CrossRef CAS.
- Y. He and S. Laursen, Trends in the surface and catalytic chemistry of transition-metal ceramics in the deoxygenation of a woody biomass pyrolysis model compound, ACS Catal., 2017, 7, 3169–3180 CrossRef CAS.
- X. Liu, W. An, C. H. Turner and D. E. Resasco, Hydrodeoxygenation of m-cresol over bimetallic NiFe alloys: kinetics and thermodynamics insight into reaction mechanism, J. Catal., 2018, 359, 272–286 CrossRef CAS.
- X. Liu, W. An, Y. Wang, C. H. Turner and D. E. Resasco, Hydrodeoxygenation of guaiacol over bimetallic Fe-alloyed (Ni, Pt) surfaces: reaction mechanism, transition-state scaling relations and descriptor for predicting C–O bond scission reactivity, Catal. Sci. Technol., 2018, 8, 2146–2158 RSC.
- J. Zhou, W. An, Z. Wang and X. Jia, Hydrodeoxygenation of phenol over Ni-based bimetallic single-atom surface alloys: mechanism, kinetics and descriptor, Catal. Sci. Technol., 2019, 9, 4314–4326 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental methods, XRD patterns for aliquots taken during synthesis, major XRD reflections and Rietveld refinement data, calculated and experimental lattice parameters, detailed electron microscopy data and analysis, particle size distributions, XRD patterns of supported catalysts, physical properties of supported catalysts, in situ HE-XRD reciprocal space raw data, long- and short-range PDFs, RMC simulation fits, summary of structural parameters from RMC simulations, RMC models and associated bond angle distributions, catalytic performance data from guaiacol and m-cresol HDO, HYD and DDO pathway selectivities, DFT-calculated and experimental lattice parameters, average d-band center for surface-metal atoms versus average sp-band center for surface-P atoms, adsorption energetics for OH* with lattice constant. See DOI: https://doi.org/10.1039/d3ta02071a |
| ‡ Present address for NJL, Honeywell UOP, 8400 Joliet Road, McCook, IL 60525. |
| § Present address for SAT, AbbVie Inc., 1 North Waukegan Road, North Chicago, IL 60064. |
| ¶ Present address for TVB, 821 South Johnstone Avenue, Bartlesville, OK 74003. |
| This journal is © The Royal Society of Chemistry 2023 |