
The structure-dependent mechanism of single-atom cobalt on macroporous carbon nitride in (photo-)Fenton-like reactions†
Jingkai
Lin
,
Lin
Jiang
,
Wenjie
Tian
 ,
Yangyang
Yang
,
Xiaoguang
Duan
,
Yan
Jiao
*,
Huayang
Zhang
* and
Shaobin
Wang
*
,
Yangyang
Yang
,
Xiaoguang
Duan
,
Yan
Jiao
*,
Huayang
Zhang
* and
Shaobin
Wang
*
School of Chemical Engineering, The University of Adelaide, North Terrace, Adelaide, SA 5005, Australia. E-mail: huayang.zhang@adelaide.edu.au; yan.jiao@adelaide.edu.au; shaobin.wang@adelaide.edu.au
First published on 18th May 2023
Abstract
Single-atom catalysts have been believed to be ideal materials for achieving maximum utilization of metal active sites in Fenton-like catalysis for eliminating organic pollutants. However, the relationship between the single-atom structure and catalytic activity remains largely uninvestigated. Herein, a spatial confinement strategy to anchor Co single atoms (0.6–10.2 wt%) on macroporous carbon nitride (MCN) was developed, and the single atom catalysts were tested in peroxymonosulfate activation for (photo-)Fenton-like reactions. Single-atom Co-MCN was discovered to show different molecular structures, and a light-dependent mechanism in Fenton-like catalysis was revealed. Co atoms in Co–N4 configuration present Co–N1+3/Co–N2+2 geometric structures, dependent on a Co load. Co–N1+3 is thermodynamically favorable to form, serving as the main active site. Co–N2+2 possesses an inferior catalytic activity and induces negative effects on the adjacent Co–N1+3 site. Moreover, experimental and theoretical investigations reveal a 100% nonradical reaction pathway that can be photo-switched to a nonradical/radical process by visible light. This work enriches the fundamentals of single-atom catalysis by providing new insights into the atomic metal structure, reaction pathways and mechanisms, and structure–activity relationships in organic degradation.
 Huayang Zhang | Huayang Zhang is currently a research fellow at the School of Chemical Engineering and Advanced Materials, The University of Adelaide, Australia. In 2018, he received his Ph.D. degree in chemical engineering from Curtin University, Australia. His current research focuses on the design and synthesis of novel artificial and semiartificial nanostructured materials for solar-light-driven energy and environmental applications. |
1. Introduction
Downsizing bulk or nanosized metal species to an atomic scale endows catalysts with enormous merits, including unique electronic structures, controllable coordination environments, and ultrahigh atom utilization efficiency.1,2 Thus, single-atom catalysts (SACs) with isolated metal atoms on support surfaces have shown remarkable performances of reactions in diverse fields, such as energy conversion,3,4 environmental remediation,5,6 and biomedical applications.7,8 Extensive studies suggest enhancing the catalytic performance of SACs by maximizing single-atom capacity.9,10 However, in many cases, increasing metal loads can lead to the formation of metal nanoclusters/nanoparticles, making it challenging to prepare SACs with a high atomic meal load.4,11,12 Although SACs with high atomic metal loads were reported recently,6,13 few investigations have focused on the structural variations and activity. Variations in the local coordination environment may affect the geometric and electronic structures of SACs, thereby influencing their catalytic behaviours.14,15 The unclear understanding of the metal load-induced structural variations and catalytic performances limits the catalyst structure design for performance optimization in single-atom catalysis. Therefore, more work is desired to fundamentally understand the impact of atomic metal loading on its coordination structure and resulting catalytic properties.In environmental remediation, peroxymonosulfate (PMS)-based advanced oxidation processes are considered a powerful Fenton-like technique for eliminating persistent organic pollutants (POPs) in water. The emerging SACs serve as efficient and stable catalysts for PMS activation.6,16 One or multiple reactive radicals (superoxide anion (O2˙−), hydroxyl (˙OH), and sulfate (SO4˙−)) and nonradicals (electron transfer regime or singlet oxygen (1O2)) can be involved in PMS-related Fenton-like catalysis.17,18 Compared to radical-based processes with high oxidation potentials,16,19 nonradical processes usually possess mild oxidation potentials and strong electrophilicity, endowing them with high selectivity and reactivity in oxidizing electron-rich organic pollutants in water treatment or green organic synthesis.20,21 Therefore, nonradical systems can maintain high reactivity in treating an actual water matrix with complex surroundings, e.g., varied pH conditions, natural organic matter, and inorganic ions.6,17,21 Some efforts have been made to regulate nonradical and radical processes in Fenton-like catalysis,17,22–24 whereas the principles remain ambiguous. Light irradiation can enhance the catalytic efficiency by establishing a photo-Fenton-like system,16 but there are few in-depth studies on comparing reaction mechanisms under dark and visible light irradiation conditions.
To address the above issues, we develop a spatial confinement strategy to enable the controllable immobilization of single-atom Co on macroporous carbon nitride (MCN), with a Co load in a wide range (0.6–10.2 wt%). Carbon nitride (CN) was recognized as a promising support for anchoring single-atom metals, owing to its easy synthesis and ideal electron-rich ‘nitrogen pot’ for metal incorporation.25–27 As a visible-light-active semiconductor, functional CN provides great potential in catalytic photo-Fenton-like environmental remediation.28,29
In this work, we found the formation of different geometric structures of a Co SAC in Co–N1+3/Co–N2+2 and the structure-dependent catalytic behaviour of Co-MCN SACs. Co-MCN SACs effectively activate PMS for a selective 100% nonradical pathway in the dark. Interestingly, the nonradical pathway is photo-switched to a radical/nonradical process under visible light irradiation, inducing O2˙− formation. Density functional theory (DFT) calculations revealed that the Co–N1+3 geometric structure tends to form preferentially with its lower formation energy. Moreover, Co–N1+3 functions as the primary active site for PMS adsorption and activation. However, an isomeric Co–N2+2 site that possesses low PMS adsorption/activation ability could co-exist with Co–N1+3 at a higher Co load, lowering the catalytic activity of the adjacent Co–N1+3 site. This study provides new insights into the single-atom structure and mechanism in catalysis and nonradical/radical pathway regulation for different chemical synthesis and environmental remediation applications.
2. Experimental section
2.1. Chemicals
All the chemicals in this work are of an analytical grade without further purification. Acyclovir (ACV), acetone, ammonium hydroxide (NH3·H2O), ammonium hydrogen difluoride (NH4HF2), bisphenol A (99%, BPA), cobalt(II) acetylacetonate (Co(C5H7O2)2), cobalt(II,III) oxide (Co3O4), di-ammonium oxalate monohydrate (AMO), dicyandiamide (DCD), ethanol, furfuryl alcohol (FFA), hydrochloric acid (37%, HCl), isopropanol, methanol (MeOH), methyl phenyl sulfoxide (PMSO), Nafion, naproxen (NPX), p-benzoquinone (p-BQ), phenol, p-hydroxybenzoic acid (HBA), potassium iodide (KI), sodium bicarbonate (NaHCO3), sodium sulfate (Na2SO4), sulfamethoxazole (SMX), tert-butanol (TBA), 2,2,6,6-tetramethyl-4-piperidinol (TEMP) and tetraethyl orthosilicate (TEOS) were purchased from Sigma-Aldrich. Ultra-pure water (18.2 mΩ cm−1) was used in all the experiments.2.2. Fabrication of ordered silica
SiO2 microspheres with a size of approximately 300 nm were prepared using a modified Stöber-like strategy.30 Specifically, 8 mL of TEOS and 192 mL of ethanol were added to a 400 mL beaker as solution A and stirred at 500 rpm and 25 °C. Solution B was prepared by mixing 14 mL of NH3·H2O, 56.6 mL of ethanol and 29.4 mL of H2O and stirred at 500 rpm and 25 °C for 20 min. Then, solution B was added to solution A drop by drop at 800 rpm and 25 °C. The mixture was kept stirring at 800 rpm and 25 °C for 24 h. After that, the prepared SiO2 was collected by centrifuging at 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500 rpm and 20 °C and rinsed with ultrapure water to remove unreacted residues. The as-obtained silica solids were then dried in an oven at 60 °C overnight. The white solids were then collected, ground, and dispersed in ultrapure water (5 wt%). The resulting solution was then dispensed into 10 mL vials for ultrasonication. The mixed solution was placed in an oven for evaporation at 110 °C. Finally, aligned SiO2 microspheres were obtained on the wall of the vials.
500 rpm and 20 °C and rinsed with ultrapure water to remove unreacted residues. The as-obtained silica solids were then dried in an oven at 60 °C overnight. The white solids were then collected, ground, and dispersed in ultrapure water (5 wt%). The resulting solution was then dispensed into 10 mL vials for ultrasonication. The mixed solution was placed in an oven for evaporation at 110 °C. Finally, aligned SiO2 microspheres were obtained on the wall of the vials.
2.3. Fabrication of Co-MCN SACs
DCD (0.6 g) was dissolved in ethanol/acetone solution (100 mL for each) with Co(C5H7O2)2 at certain amounts: 25.7, 257.1, 771.3, and 1799.7 mg. The solution was stirred at 200 rpm and 70 °C for evaporation. After that, the collected solids were thoroughly ground in a mortar for further use. The resulting solid (0.6 g) was uniformly tiled on the surface of silica templates (1.0 g) and calcined in nitrogen gas at 520 °C with a ramp of 2 °C min−1 for 2 h and further to 550 °C with a ramp of 4 °C min−1 for 2 h. The resulting product was etched with 4 M NH4HF2 aqueous solution (50 mL) for 48 h to remove the silica template completely and washed with a mixture of HCl, ethanol, and water (20 mL for each) for 24 h to remove surface metal species. After that, the as-obtained solid was washed with ultrapure water several times and dried at 60 °C overnight. The sample collected was denoted as Co-MCN SACs, varying with a different Co load (0.6, 2.8, 5.6, and 10.2 wt%). The fabrication of MCN and CN is provided in the Experimental procedures in the ESI.†2.4. Characterization
Fourier transform infrared (FTIR) spectroscopy was carried out using a Nicolet 6700 Thermo Fisher. X-ray diffraction (XRD) was recorded on a Rigaku MiniFlex 600 X-ray diffractometer. The morphology of catalysts was investigated by scanning electron microscopy (SEM, FEI Quanta 450). High-angle annular dark-field scanning TEM (HAADF-STEM) with energy-dispersive X-ray spectroscopy (EDX) elemental mapping images were acquired on a FEI Titan Themis 80-200. The Brunauer–Emmett–Teller (BET) surface area was determined by nitrogen adsorption/desorption on an ASAP 2460 analyzer. The surface compositions of samples were recorded by X-ray photoelectron spectroscopy (XPS, Thermo Scientific™ Nexsa™). The cobalt weighting percentage was examined by using a thermogravimetric analysis-differential scanning calorimeter (air, 800 °C, TGA-DSC, Mettler Toledo). X-ray absorption near-edge structure (XANES) spectra for the C, N K-edge and Co L-edge were obtained using the Soft X-ray Spectroscopy beamline at the Australian Synchrotron in Melbourne. All the spectral data were processed and analyzed using the QANT software program developed by the Australian Synchrotron.31 The XANES of the Co K-edge and extended X-ray absorption fine structure (EXAFS) spectra were recorded in a transmission mode at the BL14W1 station in the Shanghai Synchrotron Radiation Facility. Co foil, CoO, Co3O4, and cobalt phthalocyanine (CoPc) were regarded as the standard references. All the spectra were collected under ambient conditions; data were processed and analyzed using standard methods. Here, the resulting spectra were energy calibrated, background corrected and normalized at the height of the edge step using the ATHENA module in the IFEFFIT packages.32 UV-vis diffuse reflectance spectra were recorded on a Cary 100 UV-vis spectrophotometer (Agilent, US). An RF-5301PC spectrofluorophotometer (Shimadzu, Japan) was used to obtain steady-state photoluminescence (PL) spectra at room temperature (25 °C). The free radicals (such as ˙OH, SO4˙−, and O2˙−) and nonradical single oxygen (1O2) were investigated on an electron paramagnetic resonance (EPR) spectrometer (Bruker EMXplus-6/1, Germany) with 60.0 mM of DMPO and TEMP as the spin-trapper reagents, respectively. The Co leaching concentration in the reaction solution was analyzed by using an Agilent 8900x triple quad inductively coupled plasma mass spectrometry instrument (QQQ-ICP-MS). The illuminator intensity (415 mW cm−2) was measured with a 340 solar meter (OAI, USA).2.5. Electrochemical and photoelectrochemical tests
Electrochemical impedance spectroscopy (EIS) measurements and Mott–Schottky plot analysis were performed with an identical three-electrode system using a Zennium electrochemical workstation (Zahner, Germany). Photocurrent measurements were performed using a CHI760E electrochemical workstation. A 300 W xenon lamp (Aulight CEL-PF300-T8) was used as the light source. These measurements used a three-electrode configuration that included a working electrode, a counter electrode (Pt ring), and a reference electrode (Ag/AgCl electrode). An electrolyte solution of 0.5 M Na2SO4 (pH = 6.8, at room temperature) was used. The working electrodes were prepared as follows: fluorine-doped tin oxide (FTO) glasses were firstly ultrasonicated in ultrapure water, acetone and ethanol for 15 min in sequence and then dried at 60 °C. The sample film was then fabricated on the clean FTO glasses. Specifically, 3 mg catalysts were blended with 250 μL isopropanol and 5 μL of Nafion® 117 solution to form a suspension by sonication. The obtained slurry was then drop-cast onto a predefined area (d = 0.50 cm, geometrical area: ∼0.196 cm2) on an FTO glass substrate (1 × 1.5 cm2) three times to prepare the electrode (5 μL each time).Open-circuit potential (OCP) and cyclic voltammetry (CV) measurements were carried out using a CHI760E electrochemical workstation. The CV tests were performed in a three-electrode configuration that included a working electrode (1 ×1 cm2 graphite plate), a counter electrode (Pt ring), and a reference electrode (Ag/AgCl electrode) in solution (i.e., 0.5 M Na2SO4 (pH = 6.8, at room temperature) with/without 10 ppm BPA). The reversible hydrogen electrode (RHE) was calibrated through eqn (1).
| ERHE = EAg/AgCl + 0.059pH + 0.1976 | (1) |
The half-wave potential (φ1/2) of BPA oxidation was thus calculated using the Nernst formula (eqn (2)).
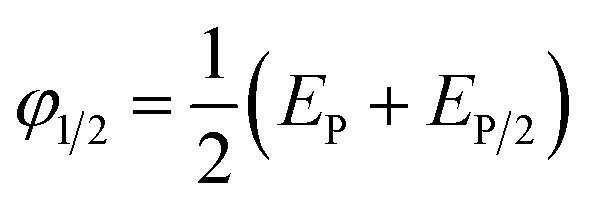 | (2) |
For the OCP measurement, the working electrode was prepared following a similar procedure described above. Precisely, the sample film was fabricated on the clean FTO glasses, and 3 mg catalysts were blended with 250 μL isopropanol and 5 μL of Nafion® 117 solution to form a suspension by sonication. The obtained slurry was then added dropwise onto the pre-treated FTO glass via a spin-coating method. The prepared electrode was dried at 50 °C for 24 h. Then, the working electrode was dipped in H2O overnight, maintaining a stable potential before the electrochemical and photoelectrochemical analysis. Then, the OCPs were monitored by chronopotentiometry analysis using Ag/AgCl as the reference electrode and a Pt ring as the counter electrode in different solutions, including H2O and 10 ppm BPA, respectively, with or without the presence of 0.5 mM PMS. For the OCP measurement under visible light irradiation, a 300 W xenon lamp (Aulight CEL-PF300-T8) was used as the light source with a cut-off filter (λ ≥ 420 nm).
2.6. Activity evaluation
The degradation test was performed on an ultrahigh performance liquid chromatograph (UHPLC, Thermo Scientific) with a C-18 column and a UV detector set at 270 nm. A 100 mL reaction system was established in a 150 mL reactor with BPA (10 ppm), catalyst (0.2 g L−1) and PMS (0.5 mM) with a circulating water system to control the temperature at 25 °C. A solution sample was periodically withdrawn by using a syringe and quickly filtered through a 0.22 μm polyether sulfone filter and then injected into a HPLC vial. Acetonitrile (60%) and ultrapure water (40%) were applied as the mobile phase at a flow rate of 1.0 mL min−1. Degradation experiments in the dark were conducted in a blackout box. Photodegradation experiments were carried out using a 300 W xenon lamp (Aulight CEL-PF300-T8) with a cut-off filter (λ ≥ 420 nm). Pseudo-first-order kinetic rate constants were calculated according to eqn (3), where C0 is the initial concentration of the pollutant; C is the concentration of the pollutant at the time (t); k is the first-order reaction rate constant. The model was evaluated by plotting ln(C/C0) versus the reaction time (t).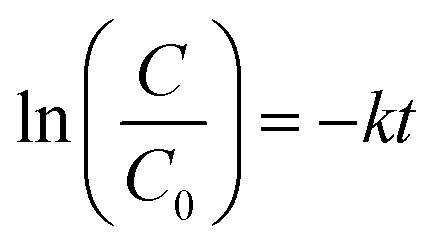 | (3) |
The details of quenching experiments, activity evaluation of different pollutants, pH effect study, cycling test, PMS adsorption evaluation, and theoretical calculation methods are provided in the Experimental procedures in the ESI.†
3. Results and discussion
3.1. Material synthesis and characterization
As illustrated in Scheme 1, Co-MCN SACs were prepared by a hard-templating-assisted pyrolysis method using silica nanoparticles as the templates (Fig. S1 and S2, ESI†). The pyrolysis of dicyandiamide at 550 °C allows the formation of hydrogen-bonded polymeric melon-structured carbon nitride with the maximum NH and NH2 functional groups (Fig. S3†).33 The spatial confinement effect between the silica templates and surrounding precursors facilitates the immobilization of single-atom Co between melon polymer chains (details are illustrated in Fig. S4†). After template and excessive Co removal by acid etching, Co-MCN SACs with a macroporous structure were produced. For comparison, MCN and CN were also synthesized. | ||
| Scheme 1 Schematic illustration of the synthesis route for Co-MCN SACs. | ||
The scanning electron microscopy (SEM) image (Fig. 1a and S5b†) indicated the macroporous structure of Co-MCN SAC and MCN. In comparison, the SEM images of pristine CN are shown in Fig. S5a,† which displayed a dense bulky structure. The mesoporous features of MCN and Co-MCN SACs were further revealed by N2 sorption isotherms and pore size distribution curves (Fig. S6†). As shown in Table S1,† compared with CN, MCN and Co-MCN SACs possessed a larger specific surface area (SSA). The interconnected macro/mesoporous architecture will boost the reaction kinetics of single-atom Co sites to reactants during catalytic reactions.34,35 X-ray photoelectron spectroscopy (XPS) confirms Co introduction on MCN (Table S1†). Co weight ratios in Co-MCN SACs were evaluated as 0.6, 2.8, 5.6 and 10.2 wt%, respectively, based on thermogravimetric analysis (TGA, Fig. S7, Table S1†).36 The uniform dispersion of atomic Co (0.6–10.2 wt%) on Co-MCN SACs was verified by aberration-correction high-angle annular dark-field scanning transmission electron microscopy (AC-HAADF-STEM) images (Fig. 1b and S8†). The energy dispersive spectroscopy (EDS) elemental mapping images (Fig. 1c and S8†) reveal the homogeneous distributions of C, N, and Co. X-ray diffraction (XRD) patterns confirm the atomic form of Co as no metal peak emerged (Fig. S9†).
C K-edge X-ray absorption near edge structure (XANES) spectra in Fig. 1d indicate the existence of π* C–C or C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C at around 286 eV and π* C–N–C at approximately 289 eV in CN, MCN, and Co-MCN SACs.37,38 The N K-edge XANES spectra (Fig. 1e) show two typical π* resonances at 399.5 and 402.4 eV, corresponding to π* C–N–C and π* N–C (i.e., N–C–2H and 2C–N–H), respectively.39 π* C–N–C or π* C–C/CC remains almost unchanged after loading Co atoms. The N–C peak tapered with a higher Co load in Co-MCN SACs, suggesting the loss of H-bonding interactions between melon chains and the breaking of intralayer long-range atomic order patterns after bonding with Co single atoms. XRD and Fourier transform infrared spectroscopy (FTIR) spectra also confirm the variation of C–N groups and gradual destruction of the long-range order of the carbon nitride structure after coordinating with increasing Co single atoms (Fig. S9 and S10†).
C at around 286 eV and π* C–N–C at approximately 289 eV in CN, MCN, and Co-MCN SACs.37,38 The N K-edge XANES spectra (Fig. 1e) show two typical π* resonances at 399.5 and 402.4 eV, corresponding to π* C–N–C and π* N–C (i.e., N–C–2H and 2C–N–H), respectively.39 π* C–N–C or π* C–C/CC remains almost unchanged after loading Co atoms. The N–C peak tapered with a higher Co load in Co-MCN SACs, suggesting the loss of H-bonding interactions between melon chains and the breaking of intralayer long-range atomic order patterns after bonding with Co single atoms. XRD and Fourier transform infrared spectroscopy (FTIR) spectra also confirm the variation of C–N groups and gradual destruction of the long-range order of the carbon nitride structure after coordinating with increasing Co single atoms (Fig. S9 and S10†).
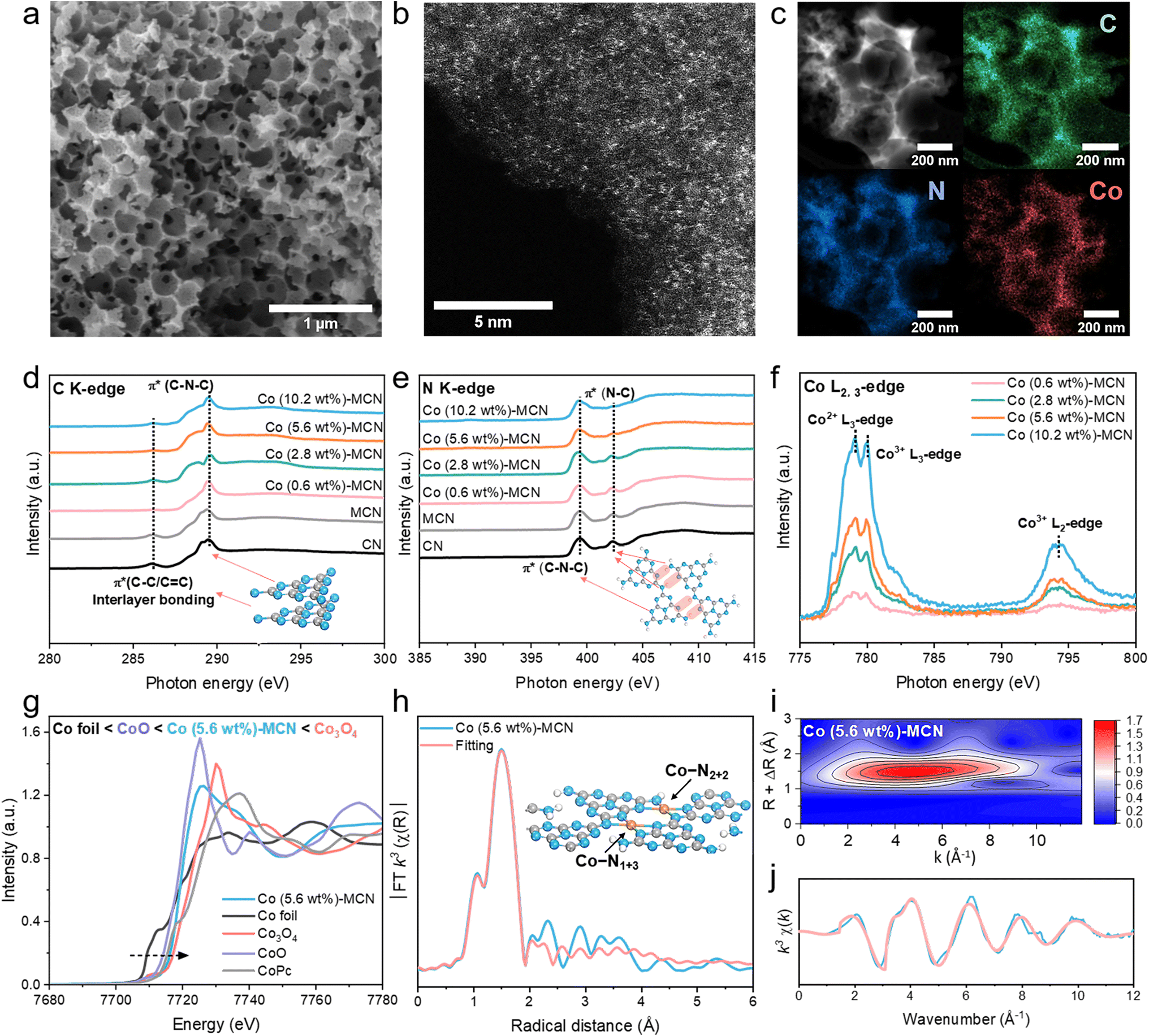 | ||
| Fig. 1 (a) SEM, (b) AC-HAADF-STEM, and (c) HAADF-STEM and elemental mapping images of Co (5.6 wt%)-MCN. (d) C K-edge, (e) N K-edge, and (f) Co L-edge XANES spectra of Co-MCN SACs, MCN and CN. (g) Normalized Co K-edge XANES spectra of Co (5.6 wt%)-MCN, Co foil, Co3O4, CoO and CoPc. (h) FT k3-weighted EXAFS and fitted EXAFS spectra in R-space. Inset: Co–N1+3 and Co–N2+2 structures on the MCN matrix. (i) Wavelet transform EXAFS plot and (j) k3-weighted EXAFS in k-space of Co (5.6 wt%)-MCN. | ||
The chemical state of Co in Co-MCN SACs was examined by Co L-edge XANES. Multiple peaks at 779.1, 780.1, and 794.3 eV corresponding to Co2+ and Co3+ in the L3-edge and Co3+ in the L2-edge have a relatively higher intensity (Fig. 1f),40 suggesting the co-existence of Co2+ and Co3+ in Co-MCN SACs. For Co K-edge XANES spectra, the normalized Co adsorption spectrum of Co (5.6 wt%)-MCN is situated between those of CoO and Co3O4, confirming the Co valence state between +2 and +3 (Fig. 1g).
Extended X-ray absorption fine structure spectra (EXAFS) verify the atomic dispersion of Co single atoms and their coordination structure. The k3-weighted Fourier-transformed (FT) EXAFS of the Co K-edge in Fig. 1h shows the prominent FT peak at approximately 1.53 Å, ascribed to the first-shell coordination between Co and C/N elements. The FT peak of Co–Co at 2.2 Å was undetectable,23,41 suggesting no metallic Co. Wavelet transform (WT) formation of the Co K-edge EXAFS oscillation analysis in Fig. 1i shows only the WT maximum at 4.5 Å for Co–C/N coordination. By contrast, no WT intensity maximum related to Co–Co coordination (intensity maxima at about 7.0 Å)41 was observed.
N in a CN structure is well recognized as the electron-abundant site, which can provide rich electron lone pairs to incorporate with single metal ions.42 According to the quantitative EXAFS fitting results (Fig. 1h, j), the Co–N coordination number of a Co site in Co (5.6 wt%)-MCN is about 4 (Co–N4) with an average coordination distance of 1.97 ± 0.04 Å (Table S2†).43 Isomeric Co–N1+3 and Co–N2+2 sites for Co–N4 configurations will exist, depending on the difference of coordinating sp hybridized N atoms (sp3 N or sp2 N atoms, Fig. S11a†). Optimized MCN/Co–N1+3/2+2 models with Co–N1+3 and Co–N2+2 coordination geometries were established (average Co–N bond distance of 1.94 Å, Fig. 1h and S11b†). Considering individual Co–N1+3 and Co–N2+2 sites, we also established MCN/Co–N1+3 and MCN/Co–N2+2 models with average Co–N bonds of 1.96 and 2.08 Å, respectively (Fig. S11c and d†). Considering the similar XRD, FTIR, and XANES results, it is speculated that Co-MCN SACs have analogous Co–N1+3 or/and Co–N2+2 geometric structures but in different proportions. The formation tendency of the two geometric Co–N4 configurations will be investigated thermodynamically in the subsequent theoretical study.
3.2. Performances of Co-MCN SACs in (photo-)Fenton-like catalysis
The catalytic activity of Co-MCN SACs was first evaluated for bisphenol A (BPA) removal via PMS activation in the dark (Fig. 2a). Pristine CN and MCN were also investigated, showing negligible BPA removal. After Co loading on MCN, the degradation rate of BPA was significantly enhanced. All catalysts displayed poor adsorption of BPA; thus, BPA removal was related to catalytic PMS activation. With a relatively low PMS addition (0.5 mM) compared to most reported AOP systems (e.g., 2 mM PMS addition for SAC/PMS systems44), our system can greatly reduce the usage of persulfates, as well as the generation of by-product sulfate ions that may threaten aquatic life.45 The activity of Co-MCN SACs increased with a higher Co load from 0.6 to 5.6 wt%, yet it decreased at a Co of 10.2 wt%. The optimum degradation efficiency was achieved at a Co load of 5.6 wt%, almost completely removing BPA in 90 min, with a first-order kinetic constant (k) of 0.045 min−1. Considering the same amount of Co, Co in SACs is much better in BPA removal than in Co3O4 (Fig. S12a†). Many early studies pointed out that elevating the metal load is critical to acquiring abundant active sites to improve the catalytic ability.12,16,46 However, our findings indicate that excessive atomic metal loads do not necessarily signify a high catalytic performance.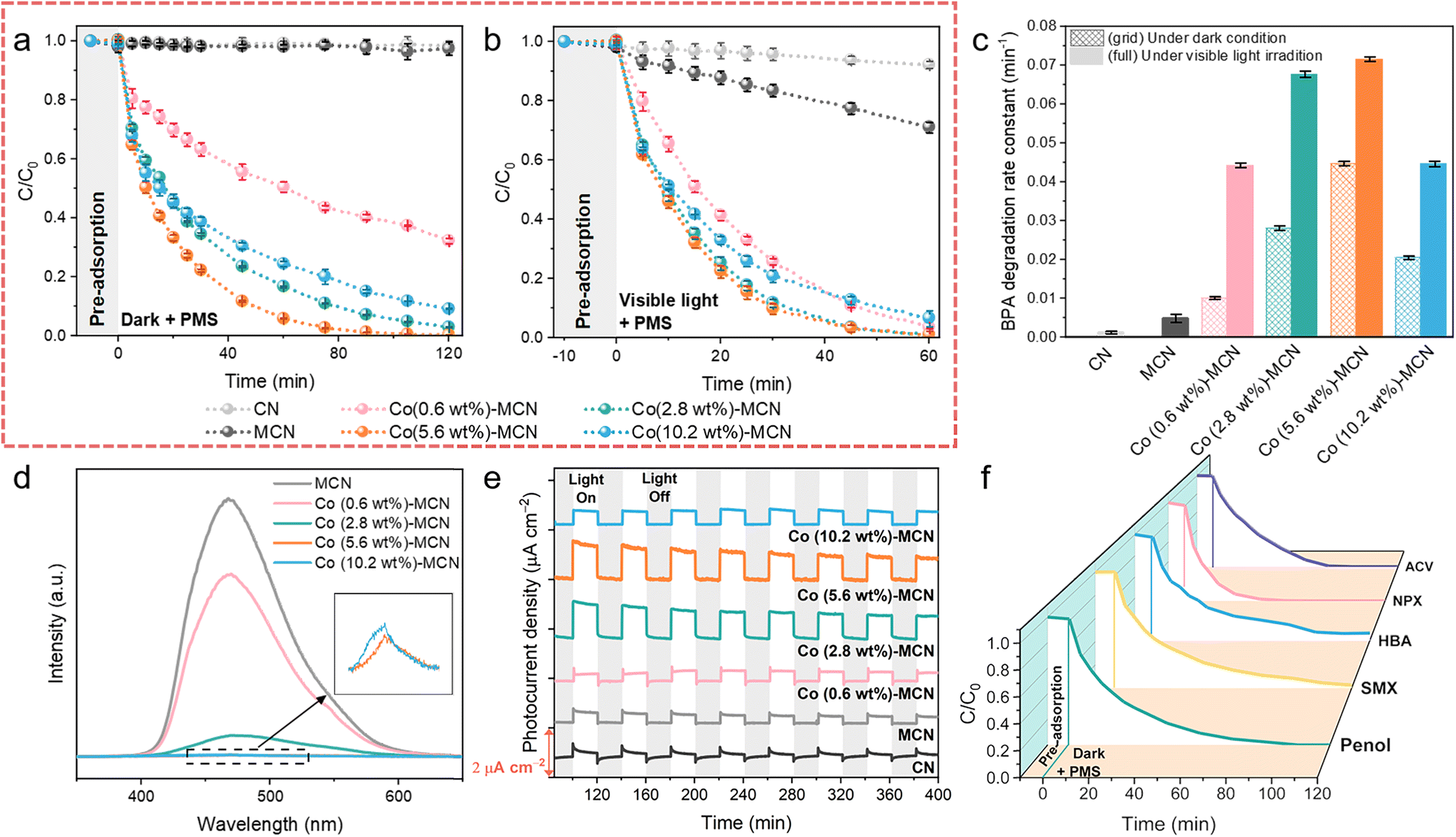 | ||
| Fig. 2 (a–c) Catalytic performances of Co-MCN SACs in PMS activation for BPA removal at neutral pH (6.8). BPA removal kinetics (a) in the dark and (b) under visible light irradiation. (c) Corresponding degradation rate constants. (d–f) Applicability study of Co (5.6 wt%)-MCN SAC/PMS in different systems. (d) PL spectra and (e) photocurrent responses of Co-MCN SACs. (f) Removal of other POPs (phenol, SMX, HBA, NPX, and ACV) under darkness. Reaction conditions: [POP] = 10 mg L−1, [PMS] = 0.5 mM, [catalyst] = 0.2 g L−1, and T = 25 °C. A 300 W xenon lamp (>420 nm) was used for visible light irradiation (illuminator intensity of 415 mW cm−2). | ||
The as-prepared samples were further investigated for photo-Fenton-like catalysis. In the presence of visible light and PMS, pristine CN presented less than 8% BPA photodegradation in 60 min (Fig. 2b), while MCN achieved a BPA removal rate of 3.8 times higher than CN (Fig. 2c). After atomic Co introduction, all Co-MCN SACs exhibited fast BPA photodegradation in 60 min, with Co (2.8 wt%)-MCN and Co (5.6 wt%)-MCN being superior to Co (0.6 wt%)-MCN and Co (10.2 wt%)-MCN. Meanwhile, catalysts alone had negligible photocatalytic performance (Fig. S12b†).
A series of photochemical characterization techniques were performed to unveil the origin of the improved photoactivity of the SACs. Compared to MCN, the steady-state photoluminescence (PL) spectra of Co-MCN SACs were markedly reduced, especially for SACs with a higher Co load, indicating the effectively retarded recombination of electron–hole pairs (Fig. 2d). Co (5.6 wt%)-MCN exhibited the lowest PL intensity, suggesting the best charge separation ability. The photocurrent responses of the samples (Fig. 2e) followed a similar trend to their photocatalytic performances in BPA removal. UV-Vis diffuse reflectance spectra (Fig. S13a†) showed that Co-MCN SACs exhibited gradually enhanced absorption in the visible-light region (420–800 nm) with higher Co loads. With increasing Co loads, the absorption edge of Co-MCN SACs showed progressive redshifts, indicating a narrowed bandgap (Eg) (Fig. S13b†). Moreover, Co-MCN SACs showed gradually increased Urbach tail absorption in the visible light region, associated with the formation of a mid-gap state (represented by transition energy (ET))47 within the bandgap (Fig. S13c†). Although higher light absorption and photon capture capabilities were achieved in Co-MCN SACs with a higher Co load, the mid-gap state position was positively shifted toward the valence band (VB), resulting in the excitation of hot electrons with low and ineffective energy,47,48 leading to weakened photoactivity with 10.2 wt% Co.
Based on the above analysis, we can derive that the photocatalytic activity of Co-MCN SACs is determined by active Co sites, charge carrier separation, and high-energy electron generation. Increasing atomic Co loading will introduce active sites for PMS activation and facilitate charge carrier separation but weaken the high-energy electron photoactivation process. Under the combined action of these factors, the photocatalytic performances of Co-MCN SACs increase when Co loading rises from 0.6 to 5.6 wt%, whereas the Co (10.2 wt%)-MCN SAC with excessive Co immobilization showed an impaired catalytic ability. Overall, the catalytic performance of Co-MCN SACs is not proportional to the atomic Co load, and Co (5.6%)-MCN was determined to have a robust catalytic PMS activation activity under both light and dark conditions.
Co (5.6 wt%)-MCN also exhibited robust reactivity and pH compatibility (5–11) for PMS activation under both dark and visible light irradiation (Fig. S14†). Cycling tests demonstrated excellent stability and reusability in the dark or under visible light irradiation (Fig. S15†). XANES spectroscopy was used to probe the structural destruction of Co (5.6 wt%)-MCN during the process (Fig. S15†). As shown in the C K-edge XANES spectra (Fig. S16a†), a wider and stronger π* C–C or CC peak at 286 eV and a shifted π* C–N–C/O–CO peak at approximately 289 eV indicate the coverage by reaction intermediates.19,34 N K-edge XANES spectra show that the reclaimed Co (5.6 wt%)-MCN SAC had no discrepancy compared to the fresh sample, suggesting its structural stability (Fig. S16b†). No obvious changes were observed by employing characterization techniques including SEM, FTIR, and XRD on fresh and used Co (5.6 wt%)-MCN SACs, confirming its structural stability (Fig. S17–S19†). ICP-MS measurements demonstrate that there is no Co leaching in the Co (5.6 wt%)-MCN/PMS system after BPA degradation (Table S3†). Co (5.6 wt%)-MCN also shows broad applicability for the degradation of different contaminants, e.g., phenol, sulfamethoxazole (SMX), p-hydroxybenzoic acid (HBA), naproxen (NPX), and acyclovir (ACV) in PMS-related Fenton-like catalysis (Fig. 2f).
3.3. Mechanisms in the dark and under visible light irradiation
We conducted ROS quenching experiments (Fig. 3a–d and S20–S25†), electron paramagnetic resonance (EPR), and electrochemical measurements to study the reaction mechanism of Co-MCN SACs/PMS under darkness and visible light irradiation. Methanol (MeOH, a radical scavenger for both ˙OH and SO4˙−), tert-butanol (TBA, a radical scavenger for ˙OH), p-benzoquinone (p-BQ, a radical scavenger for O2˙−), 2,2,6,6-tetramethyl-4-piperidinol (TEMP, a scavenger for 1O2) and furfuryl alcohol (FFA, a scavenger for 1O2) were employed as quenching reagents. Taking Co (5.6 wt%)-MCN/PMS as an example (Fig. 3a and c), the quenching effects of the scavengers under darkness followed the sequence of p-BQ + FFA + TBA = p-BQ + FFA = FFA > TEMP > TBA > MeOH ≈ p-BQ. This suggests that 1O2 contributed significantly to BPA degradation in the dark; SO4˙− contributes little; a small quantity of ˙OH, and O2˙− was generated but not directly involved in BPA degradation.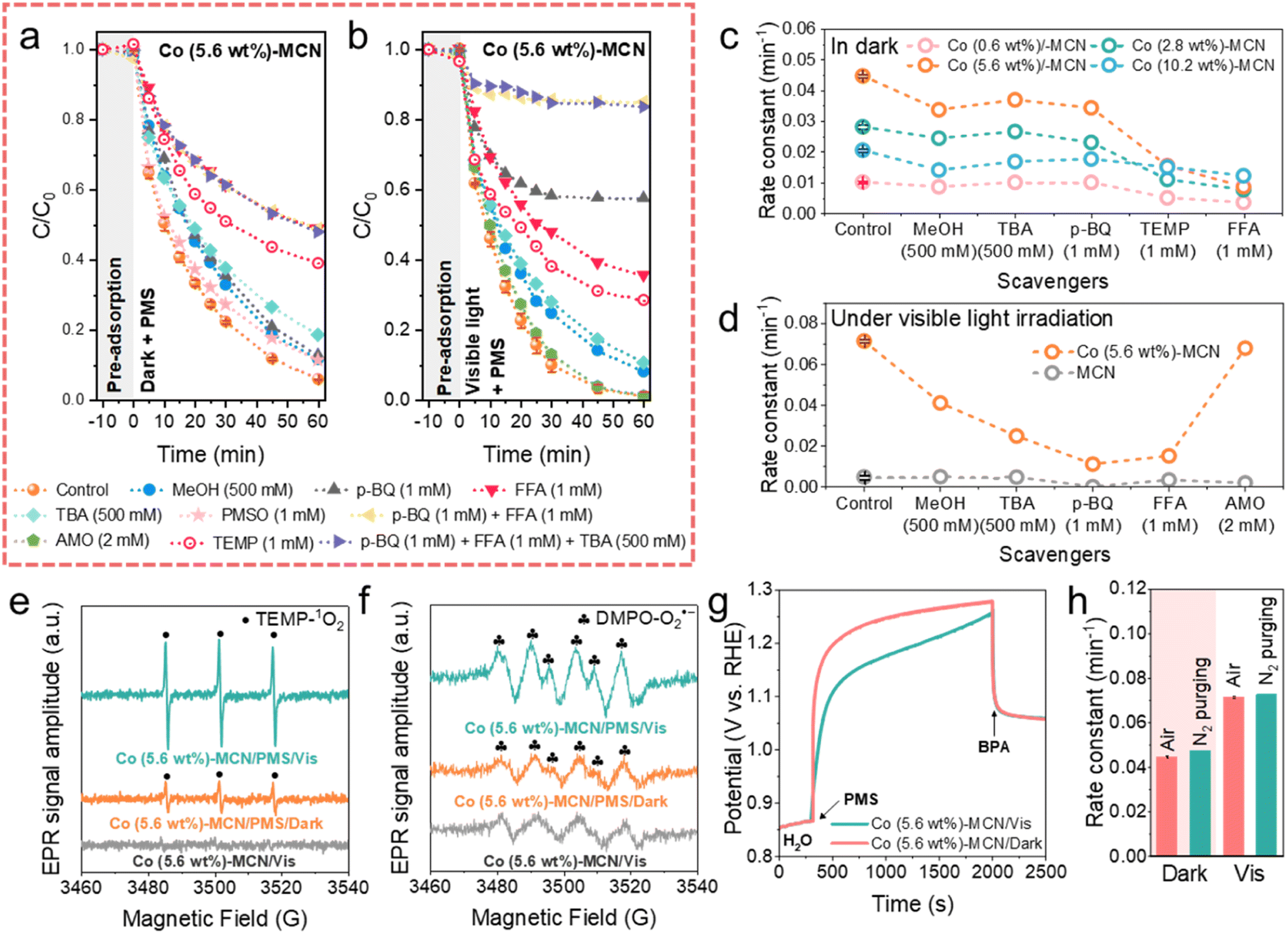 | ||
| Fig. 3 BPA degradation kinetics by Co (5.6 wt%)-MCN/PMS using different scavengers (a) in the dark and (b) under visible light irradiation. (c) Degradation rate constant comparison using different scavengers for Co (0.6, 2.8, 5.6, 10.2 wt%)-MCN/PMS in the dark. (d) Photodegradation rate constants using different scavengers for MCN/PMS and Co (5.6 wt%)-MCN/PMS under visible light irradiation. EPR spectra for detecting (e) 1O2 and (f) O2˙− in the dark and under visible light irradiation. (g) Open-circuit potential measurements in the dark and under visible light irradiation. (h) Degradation rate by Co (5.6 wt%)-MCN under different atmospheres. Reaction conditions: [BPA] = 10 mg L−1, [PMS] = 0.5 mM, [catalyst] = 0.2 g L−1, T = 25 °C, and initial solution pH = 6.8. 300 W xenon lamp (>420 nm) was used for visible light irradiation (illuminator intensity of 415 mW cm−2). | ||
Moreover, the negligible quenching effect of methyl phenyl sulfoxide (PMSO) indicates that high-valent cobalt species were not involved in BPA oxidation. ROS quenching tests were also conducted for Co-MCN SACs with different Co loads (Co = 0.6, 2.8, 5.6, 10.2 wt%, Fig. S20†-23) and showed similar trends for a conclusion of 1O2 as the main ROS for BPA removal (Fig. 3c), except for the Co (10.2 wt%)-MCN/PMS system, where the contribution of 1O2 obviously declined.
ROS quenching tests for the Co (5.6 wt%)-MCN/PMS/Vis system (Fig. 3b, d, and S24†) showed the quenching effects of different scavengers in the sequence of p-BQ + FFA + TBA = p-BQ + FFA > p-BQ > FFA > TEMP > TBA > MeOH > di-ammonium oxalate monohydrate (AMO, an h+ scavenger). This result indicates that visible light irradiation induced O2˙− as another critical ROS for BPA photodegradation, in addition to 1O2. The influence of h+ was excluded by AMO. In the MCN/PMS/Vis system, p-BQ almost completely quenched the reaction, and other scavengers displayed negligible effects (Fig. 3d and S25†), strongly suggesting the dominant role of O2˙−.
The ROS generation in the Co (5.6 wt%)-MCN/PMS system was further validated by EPR measurements under darkness and light irradiation. We used 2,2,6,6-tetramethyl-4-piperidinyloxyl (TEMP) to trap 1O2 and 5,5-dimethyl-1-pyrroline-N-oxide (DMPO) to capture SO4˙−, ˙OH, and O2˙− (Fig. 3e, f, and S26†). Consistent with ROS quenching results, SO4˙− was not detected in the dark and light. The signals of TEMP-1O2, DMPO-˙OH and -O2˙− were captured in the dark, which was enhanced under visible light illumination. Control EPR measurements in the Co (5.6 wt%)-MCN/Vis system demonstrated that ROS was generated from PMS activation. Combining EPR with ROS quenching experiments, 1O2 was produced and dominantly contributed to BPA degradation under darkness. A trace amount of ˙OH and O2˙− was produced in the dark, but not directly contributing to the BPA degradation, which may in situ form 1O2. Visible light promoted the generation of O2˙− for BPA removal.
Nonradical electron transfer regimes in Co-MCN SACs/PMS systems were further monitored by electrochemical measurements (Fig. S27†). The open-circuit potential (OCP) under dark conditions verified that Co-MCN SACs could donate electrons to PMS, forming complexes of Co-MCN SACs/PMS* for BPA oxidation.34,49 Co (5.6 wt%)-MCN/PMS* complexes presented the highest OCP value, consistent with the best catalytic performance. In contrast, the direct electron transfer oxidation process could not occur in the MCN/PMS system. In addition, FTIR spectra in Fig. S28† further confirmed the structural stability of Co (5.6 wt%)-MCN after the electrochemical test (i.e., OCP measurement).
A comparative study under visible light irradiation showed that the OCP of Co (5.6 wt%)-MCN/PMS* under visible light irradiation was lower than that in the dark, suggesting the weakened oxidation ability of the electron transfer regime (Fig. 3g). We speculate that PMS molecules adsorbed on Co (5.6 wt%)-MCN could trap photoinduced high-energy electrons for decomposition toward O2˙− generation, weakening the direct electron transfer regime.
Thus, the selective nonradical pathway (1O2 and electron transfer) in the dark is photo-switched to nonradical (1O2 and electron transfer)/radical (O2˙−) mixed pathways. Fig. 3h and S29† show that BPA degradation kinetics in the Co (5.6 wt%)-MCN/PMS system was not influenced by the atmosphere (N2 and air) under both dark and light conditions, verifying that O2˙− and 1O2 were not derived from dissolved O2, but from PMS decomposition. Specifically, 1O2 can be produced during the loss of H atoms and e− from PMS to a catalyst or during the electron transfer from the catalyst to PMS. The latter reaction pathway (eqn (S3)–(S9)†)17,22 aligns with the OCP tests (Fig. 3g and S27b†), confirming electron transfer from Co-MCN to PMS. In the related mechanism, ˙OH and O2˙− can also be generated in the dark (eqn (S10)–(S14)†),50 which can be further transformed to 1O2 (eqn (S16)†). However, quenching and EPR experiments suggest that this contribution was insignificant. Under visible light irradiation, the as-generated photoelectrons can facilitate PMS activation, especially for O2˙− formation via eqn (S11)–(S14),† as suggested by the experimental results.
3.4. Theoretical simulation
To get an insight into the relationship between Co load and catalytic activity, we performed DFT calculations based on the established MCN, MCN/Co–N1+3, MCN/Co–N2+2, and MCN/Co–N1+3/2+2 models in Fig. S11a–d.† As shown in Fig. S11b,† MCN/Co–N1+3 exhibited much lower formation energy (1.65 eV) than MCN/Co–N2+2 (2.03 eV). Hence, the formation of the geometric Co–N1+3 structure is favourable, especially at a low Co load, owing to the sufficient space and uncoordinated sites on MCN. At a high Co load, the formation of Co–N2+2 is inevitable when considering the spatial confinement effect. However, it is unlikely to exist solely on Co-MCN SACs. For Co-MCN SACs with low or medium atomic Co density, the ratio of Co–N1+3/Co–N2+2 is relatively high. An excessive Co load will introduce more Co–N2+2 sites and reduce the ratio of Co–N1+3/Co–N2+2.PMS adsorption ability was evaluated on MCN, MCN/Co–N1+3, MCN/Co–N2+2, and MCN/Co–N1+3/2+2 (on both Co–N1+3 and Co–N2+2 moieties) with the adsorption energy (Eads) of PMS as an indicator. MCN possesses very weak PMS adsorption ability (|Eads| = 0.46 eV, Fig. 4a). Such weak adsorption agrees with a previous report on the weak van der Waals interaction between the triazine ring in carbon nitride and O–S bond in PMS.18 For PMS adsorption on single-atom Co sites, the calculation results in Fig. S30–S32† show that the adsorption tends to occur between the single-atom Co site and PMS site a (O connecting with –OH), rather than site b (O connecting to S). It is noteworthy that Co–N1+3 exhibits much higher |Eads| (3.05 eV) than Co–N2+2 (1.92 eV), indicating a higher affinity to PMS (Fig. 4a). In addition, the |Eads| for PMS on Co–N1+3 and Co–N2+2 sites in MCN/Co–N1+3/2+2 was 2.88 and 2.67 eV, respectively, which are lower than that on the single MCN/Co–N1+3 site (3.05 eV). Co–N2+2 weakens the PMS adsorption ability of the adjacent Co–N1+3 site, which reduces the overall PMS adsorption capability. Experimentally, PMS adsorption is stronger when the Co load increased from 0.6 to 5.6 wt% and slightly decreased when the Co load reached 10.2 wt% (Fig. S33†). This confirms that excessive Co loads negatively affect the PMS adsorption due to lowered Co–N1+3/Co–N2+2 ratios.
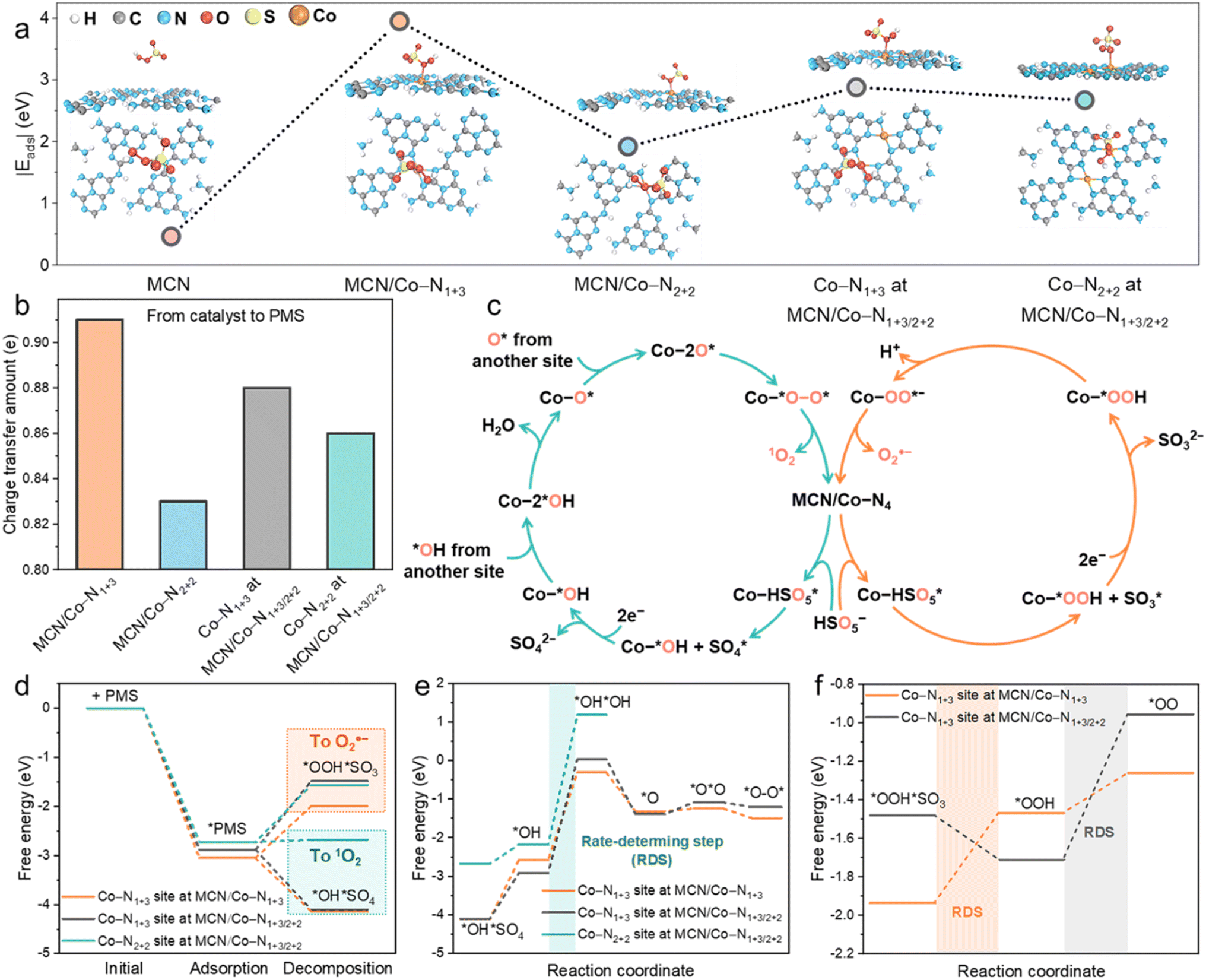 | ||
| Fig. 4 (a) Optimized model for PMS adsorption on MCN, MCN/Co–N1+3, MCN/Co–N2+2, and MCN/Co–N1+3/2+2. (b) Charge transfer from the catalyst to PMS. (c) Proposed catalytic cycles for PMS activation toward 1O2 and O2˙− generation. (d) Bader charge analysis of key reaction intermediates (*OH + SO4* for 1O2 generation and *OOH + SO3* for O2˙− generation). (e) Fee energy diagram for the reaction pathway of 1O2 generation based on *OH and SO4* moieties. (f) Free energy diagram for the reaction pathway of O2˙− generation based on *OOH and SO3* moieties. | ||
We further investigated the charge transfer numbers between PMS and different Co sites (Fig. 4b). Individual Co–N1+3 possessed the highest electron transfer ability (0.91 e) to favour electron accumulation on Co–O bonding. This robust electron transfer ability allows direct electron transfer to PMS and activation of PMS for ROS generation. In comparison, Co–N1+3 on MCN/Co–N1+3/2+2 delivers 0.88 e electron transfer to PMS. This suggests that Co–N2+2 decreased the electron donation ability of the adjacent Co–N1+3 site. Generally, the experimental PMS adsorption (Fig. S33†) and electron transfer tests (Fig. S27b and c†) are consistent with the DFT calculations.
As shown in Fig. 4c, the catalytic cycles of involved ROS, i.e., 1O2 and O2˙− generation, were calculated over different Co–N4 sites. The free energy change of intermediates in each elementary step was calculated based on the 1O2 (eqn (S3)–(S9)†) and O2˙− (eqn (S11)–(S14)†) formation pathway. Specifically, 1O2 generation mainly proceeds by HSO5− → HSO5* → *OH + SO4* → *OH → 2*OH → O* → 2O* → *O–O* → 1O2, as shown in the optimized models in Fig. S34 and S35† (* represents the adsorption of the intermediates on catalytic Co sites). In 1O2 generation, *OH and SO4* formation in the first step was highly critical. The O2˙− generation pathway was driven by the generation of *OOH and SO3* in the initial step, namely, HSO5− → HSO5* → *OOH + SO3* → *OOH → OO* → O2˙−, as shown in Fig. S36.† For Co–N1+3 sites on MCN/Co–N1+3 and MCN/Co–N1+3/2+2 structures, the downward free energy change for *OH + SO4* formation indicates that this transformation is thermodynamically more favourable and eventually leads to the formation of 1O2 (Fig. 4d). As for the initial step for O2˙− generation, the formation of *OOH and SO3* shows a highly positive intermediate free energy, which unfavourably occurred. Under light irradiation, external energy helps overcome the free energy changes for *OOH + SO3* generation, allowing for the simultaneous O2˙− pathway, confirmed by our experimental results. We also evaluated *OH + SO4* and *OOH + SO3* formation on the Co–N2+2 site in MCN/Co–N1+3/2+2. With nearly zero free energy changes, *OH + SO4* formation is still preferable due to the lower free energy changes than *OOH + SO3*. The above results uncovered the nature of the switched nonradical/radical reaction mechanism in the dark and under visible light irradiation.
Free energy changes in each step for the formation of 1O2 on different moieties are provided in Fig. 4e. As is shown, the rate-determining step (RDS) is the formation of a 2*OH intermediate. The positive free energy of the intermediate for RDS follows the order of Co–N1+3 in MCN/Co–N1+3 (2.26 eV) < Co–N1+3 in MCN/Co–N1+3/2+2 (2.94 eV) < Co–N2+2 in MCN/Co–N1+3/2+2 (3.36 eV). Individual Co–N1+3 possessed the highest PMS activation activity toward 1O2 generation. With higher Co loads, the co-existence of Co–N2+2 will reduce the PMS activation activity of the adjacent Co–N1+3 site, which can decrease the overall performance of the catalysts. We also evaluated the free energy changes during the formation of O2˙−, as shown in Fig. 4f. Co–N1+3 in MCN/Co–N1+3 and MCN/Co–N1+3/2+2 has different RDSs, i.e., the formation of *OOH (0.47 eV) and *OO (0.75 eV), respectively, and the free energy changes on individual Co–N1+3 were much lower than that on Co–N1+3/2+2. Based on the theoretical study of the single-atom site-to-site interaction, we can demonstrate the significance of Co loads and geometric Co–N4 configuration in Co-MCN SACs for Fenton-like catalysis, as summarized in Scheme S1.†
The detailed mechanism in Co-MCN/PMS for (photo)Fenton-like catalysis is illustrated in Scheme 2. The catalytic performance of Co-MCN SACs is not proportional to the increased atomic Co load due to the structural change (Scheme 2a), and visible light can switch the reaction pathway from a nonradical to a radical/nonradical mixed route (Scheme 2b). Experimentally and theoretically, we demonstrate that geometric Co–N1+3 will be formed preferentially for Co–N4 configuration and serve as the primary active sites. Isomeric Co–N2+2 sites with a lower activity could co-exist to decrease the catalytic activity of adjacent Co–N1+3. The catalytic activity of Co-MCN SACs is closely related to the amount and proportion of Co–N1+3/Co–N2+2. An excessive Co load to generate Co–N2+2 sites will reduce the ratio of Co–N1+3/Co–N2+2, weakening the intrinsic activity of active sites and reducing the reaction kinetics. The nature of the switched nonradical/radical reaction mechanism was uncovered by DFT simulation. In the dark, it is thermodynamically favourable to generate nonradical 1O2via direct PMS decomposition. In contrast, light irradiation can help overcome the free energy changes for generating the initial-step intermediates for O2˙− generation. Combining both experiment and DFT simulation, the catalytic performance of Co-MCN SACs in the dark is highly dependent on the intrinsic activity of active sites, which is determined by the atomic Co loads and Co–N1+3/Co–N2+2 ratios. In addition to this factor, the photocatalytic activity of Co-MCN SACs is also determined by the charge carrier separation and high-energy electron, which are significantly affected at a high atomic Co load.
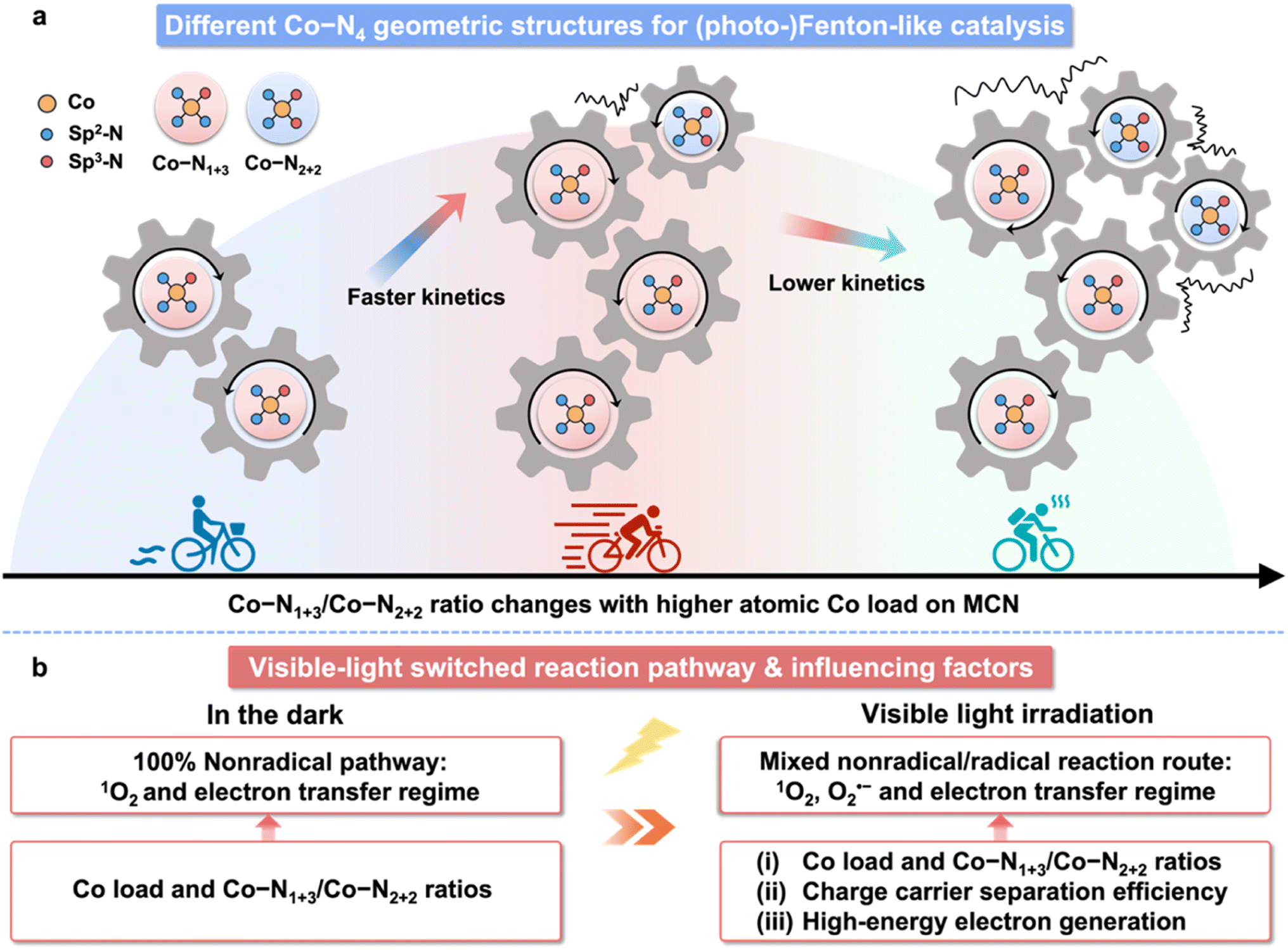 | ||
| Scheme 2 (a) Schematic illustration of different Co–N4 geometric configurations for (photo-)Fenton-like catalysis. (b) Visible-light switched reaction pathway and factors related to catalysis in (photo-)Fenton-like reactions with Co-MCN SACs. | ||
4. Conclusion
This study reports an efficient confinement strategy to fabricate high metal-loaded SACs for environmental remediation and reveals a photo-switched nonradical/radical Fenton-like process. Notably, with an optimum Co load of 5.6 wt%, the Co-MCN SAC exhibits an excellent PMS activation activity for bisphenol A removal via a 100% nonradical pathway in the dark. Embedding visible light irradiation switches the PMS activation pathway from a mild nonradical reaction to a radical/nonradical mixed route. Co–N1+3 for Co–N4 geometric structures was thermodynamically more favourable to form and serve as the main active site. Co–N2+2 with inferior PMS adsorption/activation abilities could co-exist and induce negative effects on the adjacent Co–N1+3 site. Overall, an excessive Co load will significantly reduce the proportion of Co–N1+3/Co–N2+2, resulting in a weakened PMS activation performance. Both nonradical and radical pathways were studied in-depth to elucidate the visible light-switched reaction mechanism. This work not only reveals the effect of single-atom metal load on the atomic structure and catalytic pollutant degradation performance but also enriches the understanding of the selective regulation of nonradical and radical pathways in (photo-)Fenton-like catalysis for practical applications.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The authors acknowledge the financial support from the Australian Research Council (DP200103206). W. Tian would like to acknowledge the ARC Discovery Early Career Researcher Award (DE220101074). The authors gratefully thank Dr Anton Tadich for conducting the X-ray absorption measurements at the Australian Synchrotron and also acknowledge the technical assistance from Dr Ashley Slattery at Adelaide Microscopy.References
- H. Zhang, W. Tian, X. Duan, H. Sun, S. Liu and S. Wang, Adv. Mater., 2020, 32, 1904037 CrossRef CAS PubMed.
- C. Gao, J. Low, R. Long, T. Kong, J. Zhu and Y. Xiong, Chem. Rev., 2020, 120, 12175–12216 CrossRef CAS PubMed.
- H. Yang, S. Hung, S. Liu, K. Yuan, S. Miao, L. Zhang, X. Huang, H. Wang, W. Cai, R. Chen, J. Gao, X. Yang, W. Chen, Y. Huang, H. Chen, C. Li, T. Zhang and B. Liu, Nat. Energy, 2018, 3, 140–147 CrossRef CAS.
- X. Li, S. Zhao, X. Duan, H. Zhang, S. ze Yang, P. Zhang, S. P. Jiang, S. Liu, H. Sun and S. Wang, Appl. Catal., B, 2021, 283, 119660 CrossRef CAS.
- J. Xu, X. Zheng, Z. Feng, Z. Lu, Z. Zhang, W. Huang, Y. Li, D. Vuckovic, Y. Li, S. Dai, G. Chen, K. Wang, H. Wang, J. K. Chen, W. Mitch and Y. Cui, Nat. Sustain., 2021, 4, 233–241 CrossRef PubMed.
- L. S. Zhang, X. H. Jiang, Z. A. Zhong, L. Tian, Q. Sun, Y. T. Cui, X. Lu, J. P. Zou and S. L. Luo, Angew. Chem., Int. Ed., 2021, 60, 21751–21755 CrossRef CAS PubMed.
- S. Ji, B. Jiang, H. Hao, Y. Chen, J. Dong, Y. Mao, Z. Zhang, R. Gao, W. Chen, R. Zhang, Q. Liang, H. Li, S. Liu, Y. Wang, Q. Zhang, L. Gu, D. Duan, M. Liang, D. Wang, X. Yan and Y. Li, Nat. Catal., 2021, 4, 407–417 CrossRef CAS.
- J. Tang, Y. Wu, X. Li, L. Bu and B. Chang, Prog. Mater. Sci., 2022, 128, 100959 CrossRef CAS.
- J. Wang, Z. Li, Y. Wu and Y. Li, Adv. Mater., 2018, 30, 1801649 CrossRef PubMed.
- S. Ji, Y. Qu, T. Wang, Y. Chen, G. Wang, X. Li, J. Dong, Q. Chen, W. Zhang, Z. Zhang, S. Liang, R. Yu, Y. Wang, D. Wang and Y. Li, Angew. Chem., Int. Ed., 2020, 59, 10651–10657 CrossRef CAS PubMed.
- L. Liu and A. Corma, Chem. Rev., 2018, 118, 4981–5079 CrossRef CAS PubMed.
- S. An, G. Zhang, T. Wang, W. Zhang, K. Li, C. Song, J. T. Miller, S. Miao, J. Wang and X. Guo, ACS Nano, 2018, 12, 9441–9450 CrossRef CAS PubMed.
- Y. Cheng, S. Zhao, B. Johannessen, J. P. Veder, M. Saunders, M. R. Rowles, M. Cheng, C. Liu, M. F. Chisholm, R. De Marco, H. M. Cheng, S. Z. Yang and S. P. Jiang, Adv. Mater., 2018, 30, 1706287 CrossRef PubMed.
- Y. Xiong, H. Li, C. Liu, L. Zheng, C. Liu, J. Wang, S. Liu, Y. Han, L. Gu, J. Qian and D. Wang, Adv. Mater., 2022, 34, 2110653 CrossRef CAS PubMed.
- X. Liang, D. Wang, Z. Zhao, T. Li, Y. Gao and C. Hu, Adv. Funct. Mater., 2022, 32, 2203001 CrossRef CAS.
- Y. Wang, X. Zhao, D. Cao, Y. Wang and Y. Zhu, Appl. Catal., B, 2017, 211, 79–88 CrossRef CAS.
- Z. Wang, E. Almatrafi, H. Wang, H. Qin, W. Wang, L. Du, S. Chen, G. Zeng and P. Xu, Angew. Chem., Int. Ed., 2022, 134, e202202338 Search PubMed.
- P. Duan, J. Pan, W. Du, Q. Yue, B. Gao and X. Xu, Appl. Catal., B, 2021, 299, 120714 CrossRef CAS.
- W. Tian, J. Lin, H. Zhang, X. Duan, H. Sun, H. Wang and S. Wang, J. Hazard. Mater., 2021, 408, 124459 CrossRef CAS PubMed.
- Y. Zhao, L. Yu, C. Song, Z. Chen, F. Meng and M. Song, Environ. Sci. Technol., 2022, 56, 10710–10720 CrossRef CAS PubMed.
- W. Ren, L. Xiong, G. Nie, H. Zhang, X. Duan and S. Wang, Environ. Sci. Technol., 2020, 54, 1267–1275 CrossRef CAS PubMed.
- Y. Gao, T. Wu, C. Yang, C. Ma, Z. Zhao, Z. Wu, S. Cao, W. Geng, Y. Wang, Y. Yao, Y. Zhang and C. Cheng, Angew. Chem., Int. Ed., 2021, 60, 22513–22521 CrossRef CAS PubMed.
- X. Mi, P. Wang, S. Xu, L. Su, H. Zhong, H. Wang, Y. Li and S. Zhan, Angew. Chem., Int. Ed., 2021, 60, 4588–4593 CrossRef CAS PubMed.
- B. Liu, W. Guo, Q. Si, W. Jia, S. Zheng, H. Wang, Q. Zhao, H. Luo, J. Jiang and N. Ren, Chem. Eng. J., 2022, 446, 137277 CrossRef CAS.
- H. Zhang, W. Tian, X. Duan, H. Sun, Y. Shen, G. Shao and S. Wang, Nanoscale, 2020, 12, 6937–6952 RSC.
- J. Lin, W. Tian, H. Zhang, X. Duan, H. Sun and S. Wang, Energy Fuels, 2021, 35, 7–24 CrossRef CAS.
- J. Lin, W. Tian, Z. Guan, H. Zhang, X. Duan, H. Wang, H. Sun, Y. Fang, Y. Huang and S. Wang, Adv. Funct. Mater., 2022, 32, 2201743 CrossRef CAS.
- Y. Yang, X. Li, C. Zhou, W. Xiong, G. Zeng, D. Huang, C. Zhang, W. Wang, B. Song, X. Tang, X. Li and H. Guo, Water Res., 2020, 184, 116200 CrossRef CAS PubMed.
- X. Wang, J. Meng, X. Zhang, Y. Liu, M. Ren, Y. Yang and Y. Guo, Adv. Funct. Mater., 2021, 31, 2010763 CrossRef CAS.
- W. Stöber, A. Fink and E. Bohn, J. Colloid Interface Sci., 1968, 26, 62–69 CrossRef.
- E. Gann, C. R. McNeill, A. Tadich, B. C. C. Cowie and L. Thomsen, J. Synchrotron Radiat., 2016, 23, 374–380 CrossRef CAS PubMed.
- B. Ravel and M. Newville, J. Synchrotron Radiat., 2005, 12, 537–541 CrossRef CAS PubMed.
- P. Niu, L. Zhang, G. Liu and H. M. Cheng, Adv. Funct. Mater., 2012, 22, 4763–4770 CrossRef CAS.
- W. Tian, J. Lin, H. Zhang, X. Duan, H. Wang, H. Sun and S. Wang, J. Hazard. Mater., 2022, 423, 127083 CrossRef CAS PubMed.
- J. Lin, W. Tian, H. Zhang, X. Duan, H. Sun, H. Wang, Y. Fang, Y. Huang and S. Wang, J. Hazard. Mater., 2022, 434, 128866 CrossRef CAS PubMed.
- C. Lu, Y. Chen, Y. Yang and X. Chen, Nano Lett., 2020, 20, 5522–5530 CrossRef CAS PubMed.
- G. Zhang, Q. Ji, Z. Wu, G. Wang, H. Liu, J. Qu and J. Li, Adv. Funct. Mater., 2018, 28, 1706462 CrossRef.
- Z. Mo, J. Di, P. Yan, C. Lv, X. Zhu, D. Liu, Y. Song, C. Liu, Q. Yu, H. Li, Y. Lei, H. Xu and Q. Yan, Small, 2020, 16, 2003914 CrossRef CAS PubMed.
- J. Ran, W. Guo, H. Wang, B. Zhu, J. Yu and S. Z. Qiao, Adv. Mater., 2018, 30, 1800128 CrossRef PubMed.
- J. Wang, J. Zhou, Y. Hu and T. Regier, Energy Environ. Sci., 2013, 6, 926–934 RSC.
- W. Liu, W. Hu, L. Yang and J. Liu, Nano Energy, 2020, 73, 104750 CrossRef CAS.
- X. H. Jiang, L. S. Zhang, H. Y. Liu, D. S. Wu, F. Y. Wu, L. Tian, L. L. Liu, J. P. Zou, S. L. Luo and B. B. Chen, Angew. Chem., Int. Ed., 2020, 59, 23312–23316 CrossRef.
- C. Chu, Q. Zhu, Z. Pan, S. Gupta, D. Huang, Y. Du, S. Weon, Y. Wu, C. Muhich, E. Stavitski, K. Domen and J. H. Kim, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 6376–6382 CrossRef CAS PubMed.
- Y. Gao, Y. Zhu, T. Li, Z. Chen, Q. Jiang, Z. Zhao, X. Liang and C. Hu, Environ. Sci. Technol., 2021, 55, 8318–8328 CrossRef CAS PubMed.
- X. Duan, S. Yang, S. Wacławek, G. Fang, R. Xiao and D. D. Dionysiou, J. Environ. Chem. Eng., 2020, 8, 103849 CrossRef CAS.
- Y. Yang, G. Zeng, D. Huang, C. Zhang, D. He, C. Zhou, W. Wang, W. Xiong, B. Song, H. Yi, S. Ye and X. Ren, Small, 2020, 16, 2001634 CrossRef CAS PubMed.
- R. A. Fernandes, M. J. Sampaio, E. S. Da Silva, H. Boumeriame, T. Lopes, L. Andrade, A. Mendes, J. L. Faria and C. G. Silva, Appl. Catal., A, 2021, 609, 117912 CrossRef CAS.
- M. L. Brongersma, N. J. Halas and P. Nordlander, Nat. Nanotechnol., 2015, 10, 25–34 CrossRef CAS PubMed.
- W. Ren, C. Cheng, P. Shao, X. Luo, H. Zhang, S. Wang and X. Duan, Environ. Sci. Technol., 2022, 56, 78–97 CrossRef CAS PubMed.
- W. Han, D. Li, M. Zhang, H. Ximin, X. Duan, S. Liu and S. Wang, J. Hazard. Mater., 2020, 395, 122695 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ta01926h |
| This journal is © The Royal Society of Chemistry 2023 |