
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTuning and understanding the solvent ratios of localized saturated electrolytes for lithium-metal batteries†
Michael
Yi
,
Laisuo
Su
and
Arumugam
Manthiram
 *
*
McKetta Department of Chemical Engineering & Texas Materials Institute, The University of Texas at Austin, Austin, TX 78712, USA. E-mail: manth@austin.utexas.edu
First published on 10th May 2023
Abstract
LiPF6-based localized saturated electrolytes (LSEs) have been shown to greatly stabilize lithium-metal batteries with high-Ni cathodes to attain high energy densities for commercial feasibility. A mixture of fluoroethylene carbonate (FEC) and ethyl methyl carbonate (EMC) has been proven to be a promising solvent, but regulating the solvent ratios in order to understand the changes in the electrolyte properties and the implications for cycling performance warrants investigation. Herein, four different LSEs with varying FEC/EMC solvent ratios are comprehensively screened through several electrochemical and materials characterization techniques. It is found that the ideal FEC/EMC (v/v) ratio of 1/1 results in remarkable Li-metal half-cell cycling with the cobalt-free LiNiO2 cathode, achieving an exceptional 80.6% retention over 600 cycles with a high cutoff voltage of 4.4 V. The largely improved cycling stability comes from the formation of inorganic-rich and robust cathode–electrolyte interphase and anode solid-electrolyte interphase development along with the best Li plating and stripping efficiency. The formation of beneficial interphase species is further correlated to the unique Li+ solvation structure in the electrolyte. Therefore, a careful balance of FEC and EMC solvents is found to be critical for achieving the best overall performance in lithium-metal batteries.
Introduction
Lithium-metal batteries (LMBs) with high-nickel layered oxide cathodes serve as the ideal battery chemistry for commercial applications due to the very low reduction potential of Li metal (−3.04 V vs. the standard hydrogen electrode) and the high oxidation potential of Ni-layered oxides (≥4.5 V vs. Li+/Li). The large operation voltage coupled with the high specific capacities of high-Ni cathodes allows the LMBs to achieve very high energy densities, even above the targeted metric of 235 W h kg−1 at the pack level set by the US Department of Energy.1–3 However, LMBs are plagued by the extreme reactivity of the Li-metal anode with the electrolyte that causes surface corrosion and unwanted side reactions, leading to low Li utilization from dead Li formation, poor coulombic efficiency (CE), and overall fast capacity fade. Moreover, Li dendrite growth is another key issue associated with LMBs as it can exacerbate all the above problems and cause short circuiting, which in large-scale settings can lead to battery fire and explosions.4–6Intensive research efforts to stabilize LMBs in the past few years have led to the development of advanced electrolytes, known as localized high-concentration electrolytes (LHCEs) composed of ether-based solvents with highly soluble lithium salts, such as the expensive lithium bis(fluorosulfonyl)imide (LiFSI) salt, and a diluent, such as 1,1,2,2-tetrafluoroethyl 2,2,3,3-tetra-fluoropropyl ether (TTE), to reduce the viscosity of the electrolyte. LHCEs have been shown to greatly prolong LMB cycling performance and safety, attributed to their unique solvation structures with localized clusters of coordinated Li+ and FSI− ions in solvents with high salt-to-solvent ratios, which widen the electrochemical stability window of the electrolyte. This correspondingly forms robust inorganic-rich electrode–electrolyte interphases that protect the Li-metal anode and corresponding cathode during cycling.7–16
Recently, our group introduced a new family of electrolytes named localized saturated electrolytes (LSEs), which utilize lithium salts with low to medium solubilities, such as the low-cost lithium hexafluorophosphate (LiPF6). LiPF6 also has lower solubility in carbonate solvents (<4–5 M) compared to LiFSI in ether solvents (>10 M), which makes it easier to concentrate the solvent system while adding less salt, resulting in a cheaper overall electrolyte to be made. It has been shown that these LiPF6-based LSEs bring incredible cycling stability in LMBs even with the highly unstable cobalt-free LiNiO2 (LNO) cathode.17
In typical low-concentration electrolytes, such as commercial LiPF6-based LP57, most of the solvents and salt anions remain as free, uncoordinated molecules. In this case, the free uncoordinated solvent molecules mainly get reduced onto the Li-metal anode, as the lowest unoccupied molecular orbital (LUMO) of free solvent molecules lies at the lowest energy among the other electrolyte components. Such free solvent reduction is largely unfavorable as the reduction products form organic-rich species that dominate the solid-electrolyte interphase (SEI), which is harmful to the Li-metal anode during cycling.4 In LSEs and LHCEs, the increased salt concentration greatly reduces the presence of free solvent as nearly all the solvent molecules will get coordinated with the abundant presence of Li+ cations.8,10 The high amounts of Li+ per solvent molecule also induce joint coordination of the PF6− anion and form contact-ion pairs (CIPs) or cation–anion aggregates (AGGs), all of which can get carried and reduced onto the anode. The unique solvation sheath formed by these CIPs and AGGs allows for the LUMO to shift from the solvent to the anion, which will allow for beneficial F-based inorganic species to form onto the SEI, which is known to greatly stabilize the cycling. Furthermore, the addition of a diluent forms localized clusters of the CIPs and AGGs without largely disrupting the solvation sheath and reduces the viscosity to improve the overall electrolyte wettability.15,16,18–20
Following the promising results attained with the development of the first LSE study,17 our group has carried out a comprehensive screening analysis of various solvents and diluents to incorporate into LSEs and find the combination that yields the optimal cycling performance for LMBs.21 We found that the best result came from a saturated fluoroethylene carbonate (FEC)/ethyl methyl carbonate (EMC) solvent mixture with the TTE diluent. However, the choice of the FEC/EMC solvent ratio was not intensely investigated, which prompts further exploration into understanding how altering the solvent ratio affects the LSE solvation structure and impacts the overall performance of LMBs.
Herein, we investigate the effects of FEC/EMC solvent ratios on the cyclability in LMBs with the LNO cathode. X-ray photoelectron spectroscopy (XPS) analyses indicate that both the LNO cathode and Li-metal anode interphases can be largely stabilized by choosing the appropriate ratio of solvents. Bulk electrolyte characterization experiments with Fourier-transform infrared spectroscopy (FTIR) reveal that at either low or high FEC/EMC ratios, the solvent in greater proportion will favor Li+ and PF6− ion coordination and will force the lesser solvent to de-coordinate and remain as free solvent molecules. Such a de-coordination is unfavorable, as free solvent molecule reduction onto the Li-metal anode forms adverse and unstable organic-rich interphases. Our work provides insights into tailoring solvent components in advanced electrolytes for LMBs.
Experimental
Electrolyte preparation
LiPF6 salt, ethylene carbonate (EC), fluoroethylene carbonate (FEC), and ethyl methyl carbonate (EMC) solvents were all purchased from Gotion. TTE diluent was purchased from SynQuest Labs. All electrolytes were made following the molar ratios of LiPF6 salt, EC/FEC/EMC solvents, and TTE diluent laid out in Table 1. The solvents and diluents were stored with molecular sieves before use. The electrolyte components were all added into glass vials and stirred until all the salt was visibly dissolved into the solution.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 for all LSEs in D1–D7
:2 for all LSEs in D1–D7
| Electrolyte identification | FEC:EMC solvent ratio |
LiPF6 concentration in solvent (M) | Overall composition (mol) | ||
|---|---|---|---|---|---|
| Vol. | Weight | Mol. | |||
| LP57 | N/A | N/A | N/A | 1.0 | LiPF6:EC:EMC = 1:3.7:7.3 |
| D1 | 1/9 | 0.16 | 0.16 | 3.85 | LiPF6:FEC:EMC:TTE = 1:0.36:2.27:3.43 |
| D3 | 3/7 | 0.62 | 0.61 | 3.55 | LiPF6:FEC:EMC:TTE = 1:1.16:1.91:3.72 |
| D5 | 5/5 | 1.44 | 1.41 | 3.25 | LiPF6:FEC:EMC:TTE = 1:2.11:1.49:4.07 |
| D7 | 7/3 | 3.36 | 3.30 | 2.95 | LiPF6:FEC:EMC:TTE = 1:3.25:0.99:4.48 |
Cathode preparation
The Ni(OH)2 precursor was synthesized in-house by transition-metal hydroxide coprecipitation, which is further detailed in our previous studies.22,23 The synthesized hydroxide precursor was mixed with LiOH·H2O at a molar ratio of 1:1.03 and calcined at 655 °C for 12 h under flowing oxygen to yield LiNiO2 (LNO). Cathode slurries were then made by mixing the calcined LiNiO2 with poly(vinylidene fluoride) (PVDF) binder and conductive carbon (Super P) in a 90:5:5 weight ratio in N-methyl-2-pyrrolidone (NMP) solvent. The slurries were cast onto Al foil current collectors with active material loadings of ∼2 mA h cm−2 and were then dried overnight in a 120 °C vacuum oven. The electrodes were then punched out into 1.2 cm disks and paired with Li-metal anodes (400 μm) and the Celgard 2325 separator to make CR2032-type coin half cells. 100 μL of electrolyte was used for all half cells.
Electrochemical testing
All half cells underwent three C/10 rate formation cycles, followed by C/2 charge and 1C discharge cycling between 2.8 and 4.4 V at 25 °C (1C = 180 mA g−1). Rate capability tests were done at a C/5 charge rate with varying discharge rates. Electrochemical impedance spectroscopy measurements were carried out on cells that were charged to 3.8 V with an impedance spectrometer (Solartron 1260A) by applying a 5 mV potential perturbation in the frequency range of 1 MHz to 1 mHz. Linear sweep voltammetry tests were carried out with Li|Al coin half cells with a scan rate of 0.05 mV s−1.Materials characterization
The morphologies of the Ni(OH)2 cathode precursor, calcined LiNiO2 sample, Li-metal, and Li-plated Cu foil were examined with an FEI Quanta 650 field emission scanning electron microscope. The ionic conductivities of the electrolytes were measured with a conductivity probe (Mettler-Toledo SevenCompact S230). For the Li|Cu cells, the cells were disassembled inside a glovebox to extract the plated Cu foils, rinsed with dimethyl carbonate (DMC), and dried before SEM imaging. NMR spectroscopy was performed via a Bruker Avance III 500 MHz NMR spectrometer. FTIR spectra of the various solvents, diluents, and electrolytes were collected with a Thermofisher FTIR spectrometer equipped with an attenuated total reflection attachment and a germanium crystal. XPS characterization of the cycled Li-metal anodes and LiNiO2 cathodes was carried out on an Axis Ultra DLD spectrometer (Kratos) with an Al Kα radiation source (1486.6 eV). The electrodes were disassembled inside a glovebox, rinsed with DMC, and dried before they were transferred to the instrument via an air-free capsule. CasaXPS software was used to fit the collected XPS data with Gaussian–Lorentzian functions after applying a Shirley background correction. All spectra were calibrated with the adventitious carbon peak at 284.8 eV.Results and discussion
Electrolyte formulation
Four different variations of the FEC/EMC solvent LSEs were prepared and compared to the standard LP57 electrolyte. Specifically, LSEs with FEC/EMC (v/v) ratios of 1/9, 3/7, 5/5, and 7/3 were prepared with the full electrolyte formulations as further detailed in Table 1. The LSEs with ratios of 1/9, 3/7, 5/5, and 7/3 will hereafter be referred to as, respectively, D1, D3, D5, and D7 for convenience. The total FEC + EMC solvent to TTE diluent volume ratio was kept constant at 1:2 for all LSEs from D1 to D7. Table 1 interestingly shows that the molar concentration of LiPF6 linearly decreases with increasing FEC/EMC ratio. This is because FEC solvent has a lower LiPF6 salt saturation concentration (∼2.5 M) compared to that of EMC (∼4.0 M), in which a higher proportion of FEC will induce an overall lower salt concentration. As a result of this lowered salt concentration, the LiPF6 to TTE diluent ratio also correspondingly decreases from D1 to D7 when normalized to the moles of LiPF6. Ionic conductivities of the electrolytes were measured and shown in Table S1.† LP57 presents the highest ionic conductivity of 8.80 mS cm−1 while the D1–D7 LSEs have lower conductivities ranging from 1.46 to 2.19 mS cm−1. The conductivities of the LSEs appear to linearly increase from D1 to D7, which is opposite to the trend seen for the LiPF6 salt concentrations as they linearly decrease from D1 to D7. These trends corroborate what is known with higher salt concentrations decreasing the ionic conductivities in electrolytes. The higher conductivity of LP57 is expected as it has the smallest LiPF6 salt concentration of only 1.0 M compared to the LSEs. Additionally, LP57 does not contain any TTE unlike the LSEs, which is known to decrease the conductivity even though it improves electrolyte wettability and viscosity. Further exploration into the electrolyte solvation properties will be discussed later.
Electrochemical performance
All half cells in this study were paired with the LNO cathode, as it serves as the ideal cobalt-free Ni-based layered oxide cathode with the highest specific capacity among all layered oxides. It is also plagued with the most surface and bulk lattice instabilities, all of which will detrimentally impact cycling performance.24 Utilizing the most unstable layered-oxide cathode LNO, therefore, allows us to make very strict and rigorous comparisons of our LSEs in half cells. Fig. S1a and b† show the scanning electron microscopy (SEM) images of the calcined LNO cathode particles used in this study, which have a homogeneous size distribution along with good secondary particle morphologies. Fig. 1 displays the electrochemical data of the LNO half cells with the various electrolytes tested in this study. Long-term half-cell cycling performances are presented in Fig. 1a with the initial 1C discharge capacities of all the LSE samples in D1–D7 having similar values of around 224 mA h g−1, all higher than that of the LP57 cell, which displays a capacity of 218 mA h g−1. After 400 cycles, the LP57 cell decays to 53.5% of its initial capacity. The D3 and D7 samples, however, show significantly better cycling improvements with, respectively, 81.5% and 80.8% retentions. Moreover, the D5 sample further extends the cycling stability up to 600 cycles with an incredible 80.6% retention. This is, by far, the best performing LNO half-cell data among currently published results in the literature on par with our previous LSE work.17 The D1 sample, on the other hand, shows severe capacity decline, reaching only 7.7% retention after just 200 cycles. The overall cycling data fit well in accordance with the CE evolutions shown in Fig. S2.† The LP57 cell has an average CE of 99.89% while the D1 cell has a value of 94.54%. Both cells have the lowest average CE compared to those of the D3–D7 cell, which boasts average CEs of essentially 100% throughout the course of cycling. The vast differences seen with cycling among the LSE samples highlight the great impact altering the FEC/EMC solvent ratios can have.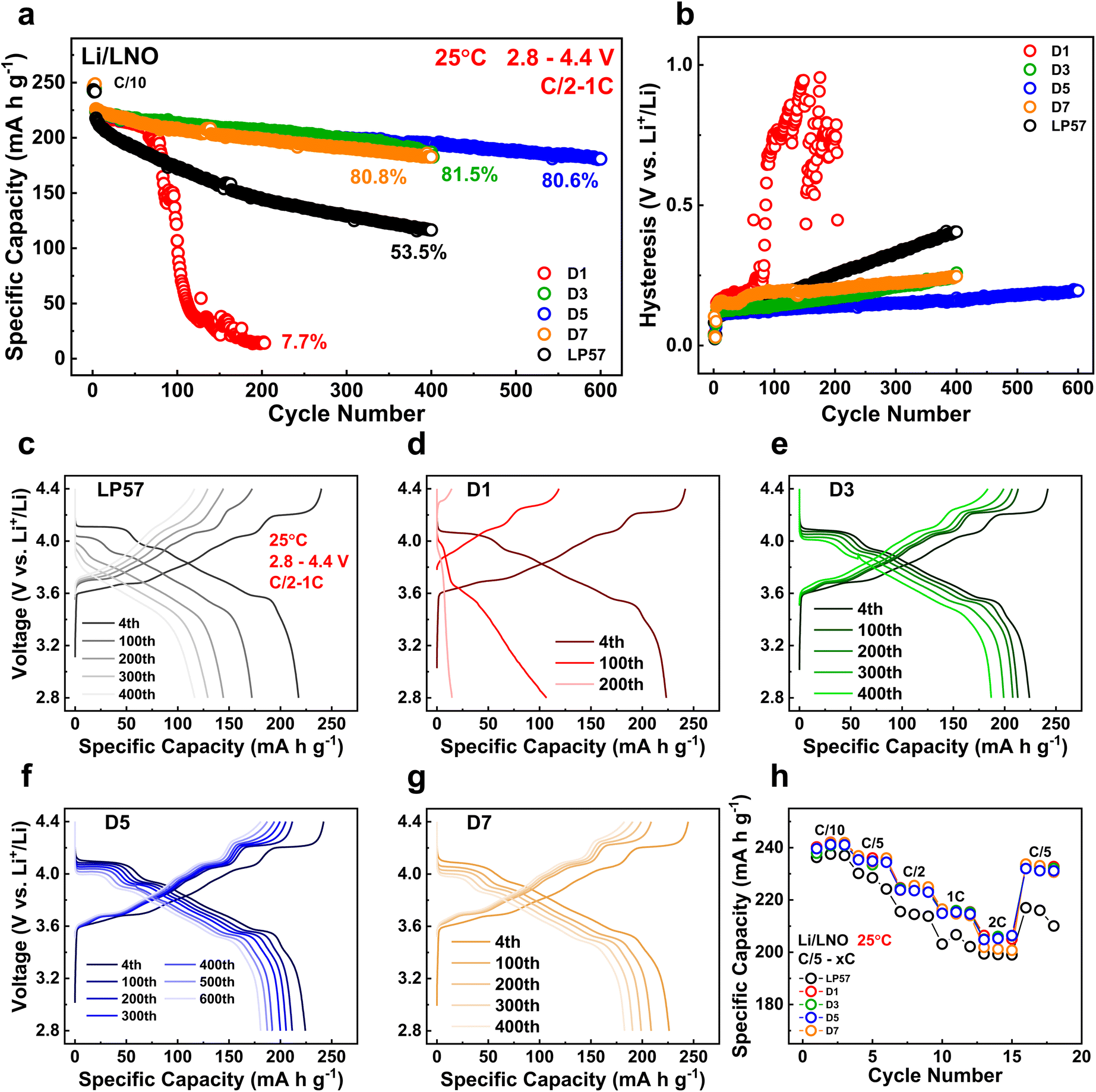 | ||
| Fig. 1 (a) Long-term cycling performances of LNO in Li-metal coin half cells with the various electrolytes of interest at C/2 charge and 1C discharge rates. (b) Voltage hysteresis growth of the average charge and discharge of the cells for each cycle. Charge–discharge curves of the coin cells cycled with (c) LP57, (d) D1, (e) D3, (f) D5, and (g) D7 electrolytes. (h) Rate capabilities of the coin cells with a C/5 charge rate and varying discharge rates. | ||
Fig. 1b additionally portrays the voltage hysteresis of the half cells after long-term cycling with the growth in alignment with the cycling data. D5 shows the lowest polarization growth of all cells at 600 cycles, while D3 and D7 show similar growth at 400 cycles, both lower than that of LP57. D1 expectedly has the largest hysteresis among all cells, indicative of severe impedance growth because of the electrolyte composition impacting the surface and structural stabilities. This is additionally shown in Fig. S3† from the average charge and discharge voltages during the long-term cycling for all cells, as the hysteresis is the difference taken between the charge and discharge voltages. Fig. 1c–g show the voltage curve evolutions of the LP57 and D1–D7 cells throughout long-term C/2 – 1C cycling. The LP57 cell in Fig. 1c shows worsened voltage polarization as highlighted in Fig. 1b, with the voltage curves essentially losing all noticeable plateaus after 200 cycles compared to their 4th 1C cycle curves. These curves in LNO refer to the H1-M, M-H2, and H2–H3 phase transitions with each plateau upon increasing the voltage.25 The loss of these plateaus indicates large voltage polarization stemming from impedance growth and structural degradation and will restrict the cell from reaching the same initial state-of-charge (SOC), resulting in considerable capacity loss. The D1 cell in Fig. 1d evidently suffers from the most extreme voltage polarization among all cells, losing more than half of its 4th cycle capacity after 100 cycles, and essentially losing all capacity after 200 cycles with no visible voltage plateaus whatsoever. Compared to these two, the D3–D7 cells in Fig. 1e–g show significantly more robust voltage profiles with mitigated polarization after cycling, particularly the D5 sample after 600 cycles. For all three samples, there is minimal plateau loss, indicating that these cells are successfully able to retain the phase transitions and thus reach similar SOCs throughout the entirety of their respective cycling, hence explaining their excellent capacity retentions.
The rate capabilities of the cells were assessed and are shown in Fig. 1h. All the LSE samples from D1 to D7 display good rate performance comparable to one another and have notable improvements at all the cycled discharge rates to that of the LP57 sample, further showcasing the improved electrochemical qualities of the LSEs. It is interesting to note that the rate performance of the D1 sample was similar to those of the other LSE samples despite the extremely poor cyclability seen earlier. This most likely implies that the poor performance stems from detrimental interphase species that evolve over the course of longer, continual cycling in this LSE.
The applicability of the D5 LSE is further demonstrated with more practical cell conditions. Fig. S4† shows the cycling performances of LNO paired with thin Li-plated Cu foil with lean electrolyte amounts of LP57 and D5 LSE in coin cells. 5 mA h cm−2 of Li was plated onto a Cu foil substrate to control the thickness and capacity of the Li metal. The plating was carried out with the same respective LP57 and D5 LSE electrolytes that were later used to make half cells with the LNO cathode, which allowed for a more practical N/P ratio of 2.5 to be achieved. Additionally, 30 μL of electrolyte was added for each cell to simulate lean electrolyte conditions. The cell with the D5 LSE shows significantly more stable cycling compared to the LP57 cell. The LP57 cell degrades to 80% capacity retention while the D5 LSE cell maintains 92% retention after 60 cycles. This suggests that the D5 LSE can better protect the thin-Li plated Cu and lean electrolyte cell compared to LP57 and further supports the overall electrochemical performance in Fig. 1.
Electrochemical impedance spectroscopy was carried out with the LNO half cells in the five examined electrolytes with the results laid out in Fig. S5.† The EIS plots after the formation cycles in Fig. S5a† indicate that both the LP57 and D1 samples have the largest total impedance compared to the D3–D7 samples. After 30 cycles shown in Fig. S5b,† the D3 sample intriguingly has the largest impedance, followed by the D1, D7, and LP57 samples. The D5 sample results in the lowest overall impedance among all the samples, in agreement with the cycling data. This change in the total impedance growth between the samples alludes that there are disparities among the formed electrode–electrolyte interphases that result from the different FEC/EMC contents in the LSEs which play as important factors that contribute to cycling degradation. Linear-sweep voltammetry (LSV) scans were also conducted on the electrolytes with Li|Al cells, as illustrated in Fig. S6a and b.† All four LSEs in this study compared to LP57 and LHCE show essentially no oxidative instability up to 5 V and overlap with each other. The LHCE and LP57 electrolytes, however, begin to show large leaking currents at, respectively, 4.5 V and 4.7 V. The LSV results suggest that varying the FEC/EMC ratios does not affect the oxidative stabilities of the LSEs. Therefore, the LSEs showing analogous electrochemical stabilities further support the notion that the differences seen in the cell cycling performances and EIS plots must stem from differences in the chemical and surface stabilities that arise and develop over the course of cycling. Additional EIS was conducted with LNO|LNO and Li|Li symmetric cells harvested from half cells cycled in LP57 and D5 LSE, as shown in Fig. S7a and b,† to deconvolute impedance contributions from the cathode and anode. Both symmetric cells cycled in the LP57 electrolyte exhibit a larger overall resistance increase than those cycled in the D5 LSE. This suggests the growth of more resistive CEIs and SEIs on the respective electrodes cycled with the LP57 electrolyte compared to those cycled in the D5 LSE and helps explain the better long-term cyclability observed for the D5 LSE in half cells. Further discussion on the symmetric cell EIS is given in the ESI.†
Cathode–electrolyte interphase (CEI) and bulk characterization
The striking differences seen in the LNO half-cell cycling results exemplify the degree to which electrolytes can affect the overall performance. As discussed above, the changes that occur in each cell must be dictated by the distinctive interphases that form, driven by reactions between the electrolyte and electrode surface, starting with the cathode–electrolyte interphase (CEI) species that passivate the LNO cathode. In order to deduce the different surface chemistries formed on the cathodes, X-ray photoelectron spectroscopy (XPS) was carried out for the cycled LNO cathodes in the LP57 and D1–D7 electrolytes. Fig. 2 depicts the XPS plots of the C 1s (Fig. 2a), O 1s (Fig. 2b), and F 1s spectra (Fig. 2c) for the cycled LNO cathodes in the five electrolytes and summarizes the overall atomic compositions of the different fragments from the plots (Fig. 2d). The XPS results show slightly reduced carbon concentrations for all the LSE samples, which range from 51.6% to 53.9% compared to the LP57 sample with 57.4%, suggesting the preferential formation of more inorganic species on the CEIs for the LSE samples, as seen in both Fig. 2a and d. Additionally, the C 1s spectra show slightly lowered peak intensities of the C–C/C–H peak at 284.8 eV, which corresponds to the conductive carbon black in the cathode. It has been shown that the lower this peak intensity, the thicker the CEI is, as the carbon black is less exposed, indicating extraneous degradation components that are passivating over the bare cathode.26,27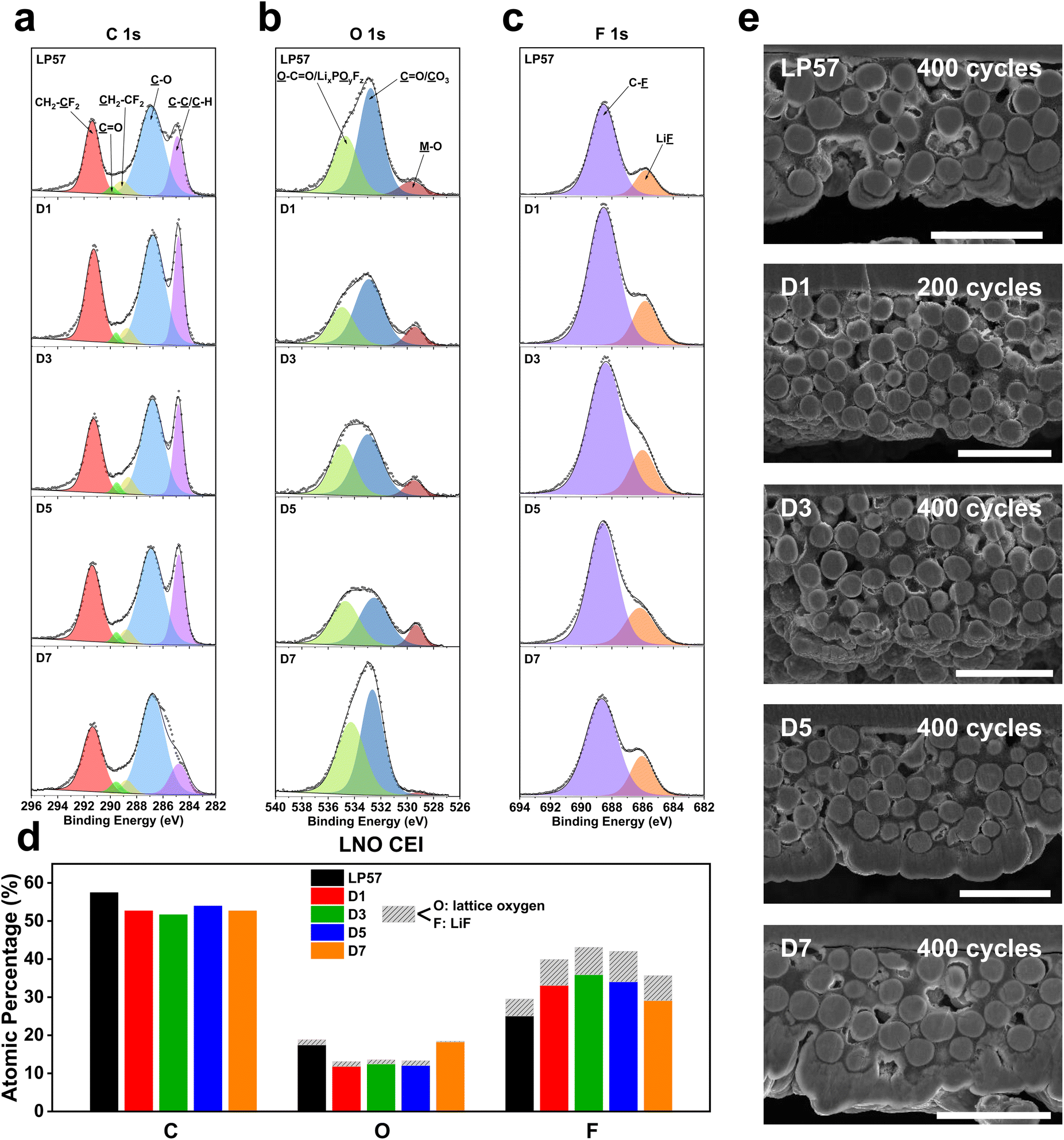 | ||
| Fig. 2 XPS characterization of cycled LNO cathodes harvested after 30 cycles with Li-metal anodes in half cells at C/2–1C charge–discharge rates. XPS plots of the (a) C 1s, (b) O 1s, and (c) F 1s spectra of the cycled LNO cathodes in the various electrolytes tested. (d) Overall XPS quantitative analysis of atomic compositions and components comprising the LNO CEI. (e) Cross-sectional SEM images of LNO cathodes recovered from half cells in the various electrolytes after long-term cycling. All scale bars are 40 μm. See Table S2† for tabulated atomic composition data. | ||
For the O concentrations, the bulk lattice oxygen denoted as M–O in the O 1s spectra in Fig. 2b was distinguished from the rest of the O-based species shown in Fig. 2d in grey to signify it being a beneficial CEI component over the other unfavorable organic based species. The LP57 and D1–D5 samples all have M–O lattice concentrations of around 1.2%, while the D7 sample presents a much smaller amount of 0.2%. By additionally referencing the M–O lattice concentration to that of the overall O composition as a ratio for a more proper comparison, we see that the D1–D5 samples possess the largest M–O lattice ratios over both LP57 and D7. These lowered M–O lattice ratios indicate slightly thicker CEI formations for the LP57 and D7 LNO cathodes and are also indicated by their lower M–O peak intensities compared to that of the other samples, particularly with the D7 M–O peak being barely visible.
The F 1s spectra in Fig. 2c show that there are varying LiF contents among the different electrolyte samples. LiF is the favored species in the F 1s spectra, as it is known to be a beneficial CEI component.28,29 The LP57 sample depicts the smallest LiF composition with a concentration of 4.4%. Also, the overall F atomic composition of the LP57 sample is notably smaller than those of the LSE samples. This directly corresponds to the higher C and O atomic compositions of LP57 over the LSE samples and indicates more organic carbonate-based species decomposition onto the CEI. This additionally highlights the difference discussed earlier between low-concentration electrolytes and LSEs, with the latter favoring inorganic F-based decomposition products that benefit the CEI as opposed to the former creating adverse organic components due to the presence of a large amount of uncoordinated free solvent molecules. Among the LSE samples, D5 presents the largest LiF composition of 8% compared to those of D1, D3, and D7, which have, respectively, 6.8%, 7.2%, and 6.6%. D7 interestingly also has a larger LiF to C–F ratio among the other LSEs as it contains the smallest C–F composition, which may be correlated to the fact that it contains the highest FEC content, which is known to be a beneficial CEI and SEI additive by promoting LiF formation.30–32 However, with the smaller M–O lattice composition over the other organic O species as discussed, it is difficult to conclude that this larger LiF ratio for the D7 LSE indicates a thinner and inorganic-rich CEI. The D5 LSE cathode, on the other hand, yields the largest LiF composition as well as the largest M–O lattice to overall O species ratio, which is indicative of the most robust CEI passivation and correlates well to the best overall cycling performance among all LSEs tested.
The bulk structures of the LNO cathodes were also examined through cross-sectional SEM imaging after long-term cycling, exhibited in Fig. 2e. Based on the images, all cathode secondary particles overall surprisingly do not show much noticeable detection of cracking and have little variations in the overall particle integrities. Upon closer inspection, the D3, D5, and D7 LSE cathodes show slightly more crack propagations from the surface to the center of the particles over the LP57 and D1 samples. This may be because of the significantly improved cycling performance of the D3–D7 cells, as they deliver higher capacities at the same cycles compared to the LP57 and D1 cells, indicating that they undergo larger degrees of the H2–H3 phase transitions, which are known to be the main cause of particle cracking.33 D1 does not show much cracking as it was only cycled to 200 cycles due to its quick capacity fade to nearly zero, and would not have made much impact by keeping it cycling all the way to 400 cycles since it would not have undergone any major phase transitions whatsoever. However, the fact that the D3–D7 cathodes clearly far outperform the LP57 and D1 cathodes despite slightly more noticeable cracking generated suggests that such particle cracking may not be the main cause of capacity fade in cathodes as previous notions have stated. Instead, the rather different CEI formations seen and discussed with the XPS results hint that the surface chemistry and reactivity are the main factors that influence overall cyclability.34
Anode solid-electrolyte interphase (SEI) characterization
LMB stabilization is well assessed by understanding the CEI growth phenomena. However, the huge set of instabilities associated with the Li-metal anode solid-electrolyte interphase (SEI) often dominate LMB performance and must be critically characterized. Thus, the corresponding Li-metal anodes that were paired with the LNO cathodes for XPS cycling were also examined as depicted in Fig. 3, which shows the C 1s (Fig. 3a), O 1s (Fig. 3b), and F 1s (Fig. 3c) peaks along with the quantitative analysis of the specific atomic compositions present in each sample (Fig. 3d). Fig. 3a and d show that there is a notably higher total C atomic composition for the LP57 sample of 47.9% compared to all other LSE samples, which range from 39.8% to 43.8% like that of the cathode XPS results, indicating more organic species formation for LP57 over all other LSEs.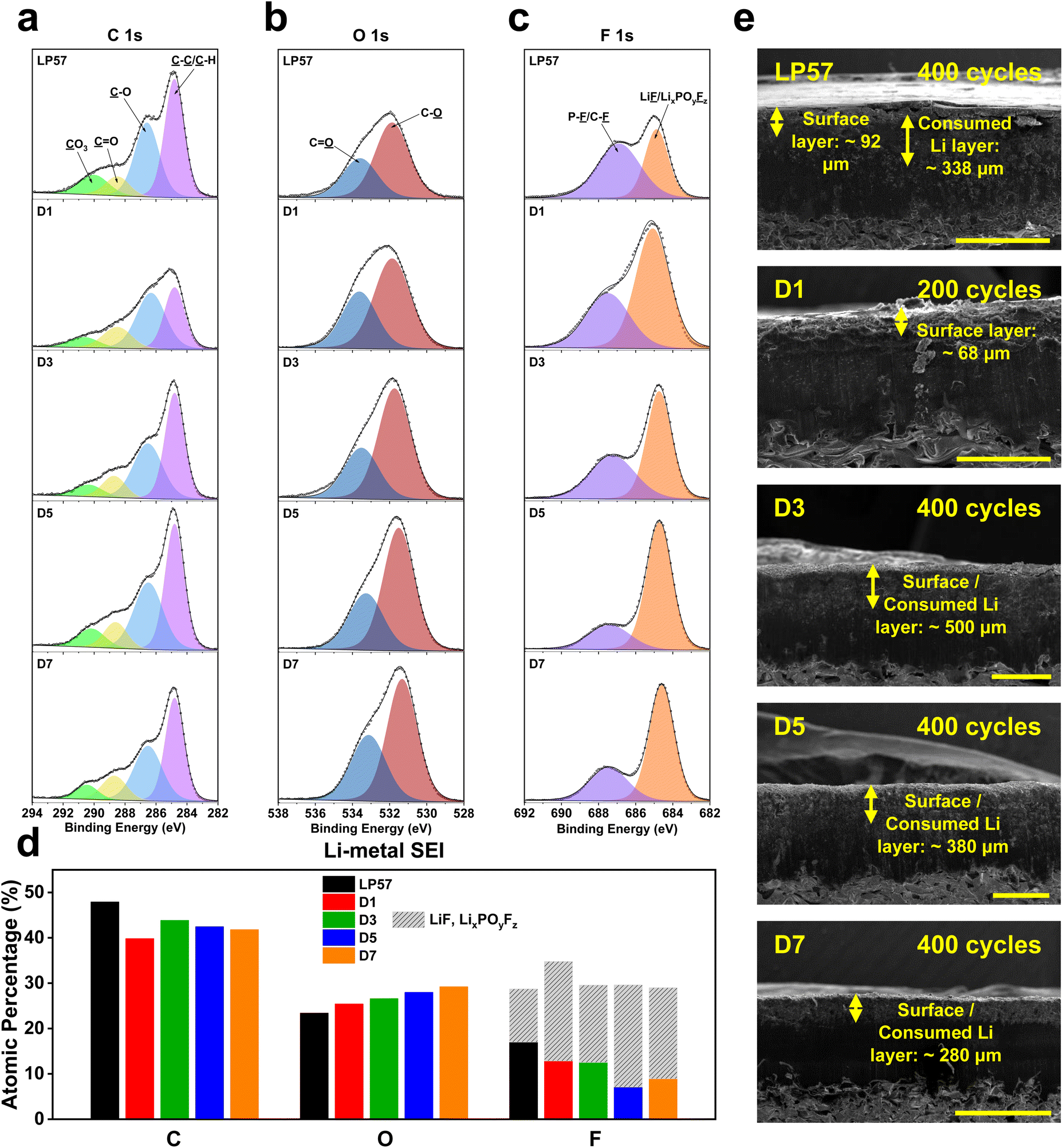 | ||
| Fig. 3 (a) XPS characterization of cycled Li-metal anodes harvested after 30 cycles with LNO cathodes in half cells at C/2–1C charge–discharge rates. XPS plots of the (a) C 1s, (b) O 1s, and (c) F 1s spectra of the cycled Li-metal anodes in the various electrolytes tested. (d) Overall XPS quantitative analysis of atomic compositions and components comprising the Li-metal anode SEI. (e) Cross-sectional SEM images of Li-metal anodes recovered from half cells in the various electrolytes after long-term cycling. LP57 and D7 image scale bars are at 1 mm, while D1–D5 image scale bars are at 500 μm. See Table S3† for tabulated atomic composition data. | ||
Looking at the F 1s spectra in Fig. 3c, we also see a general decrease in the organic-based C–F species from the LP57 to LSE samples. LP57 yields the lowest LiF/LixPOyFz concentration at 11.7% compared to the other LSEs, which have compositions ranging from 17% to 22.5%, further indicating more organic species formation on LP57. In addition to LiF, LixPOyFz is recognized as a favorable inorganic SEI/CEI compound that acts as a scavenging agent for transition-metal dissolution from the cathode to prevent ensuing crossover to the anode.35 Among the LSEs samples, D5 has the largest inorganic LiF/LixPOyFz composition with 22.5%, and correspondingly presents the smallest organic F-based species composition in the C–F/P–F peak, which overall yields the highest inorganic to organic F species ratio. This indicates that the SEI formation for the D5 anode has the largest inorganic composition, which is deemed to be favorable over all other SEI chemistries. These results further help explain the greatest overall cycling performance for the D5 LSE in half cells, as the XPS results indicated that it had the best overall inorganic species formation for both the LNO cathode and Li-metal anode. More importantly, this further asserts the fact that LSEs can simultaneously stabilize and promote the robust formation of CEI and SEI chemistries with a careful ratio of FEC and EMC solvents.
The Li-metal anodes were also examined through cross-sectional SEM like that of the LNO cathodes following long-term cycling to view the deposited layer formed on the surface after cycling, as shown in Fig. 3e. LP57 shows the most apparent cycled surface layer formation with a thickness of 92 μm. The formed layer appears to be very brittle with noticeable cracks spawning across the surface, indicating inadequate layer formation and deposition in terms of mechanical rigidity. The LP57 sample also presents a sublayer underneath the surface layer with a thickness of 338 μm, which here is labeled as the consumed Li layer that hosts the active Li ion deposits from the cathode after intercalating through the SEI. The D1 sample also shows a thick surface layer of 68 μm, but does not really have a noticeable consumed Li layer. This may be because the cell was only cycled to half of the number of cycles the other cells went through so there may not have been enough cycles to form a visually detectable layer. The overall layer, however, also appears quite porous and has an uneven surface formation, which may help explain the poor cycling performance of this sample. D1 has the lowest amount of FEC over EMC, which could indicate the drawbacks associated with inadequate FEC content in the solvent composition, which will be discussed later.
D3–D7 samples overall show rather different surface structures compared to the LP57 and D1 samples, as the surface and consumed Li layers seemed to be condensed into one single indistinguishable layer. This may indicate a very compact and thin formation of both layers, which well highlights their superb performance in half cells. This also may imply thinner and more robust SEI formation for the anodes cycled in these LSEs. One notable trend from D3 to D7 is the decrease in the total apparent layer thickness as the saturated FEC content is increased. However, based on the better cycling performance of D5 over D7, a thinner layer may not necessarily imply better cycling performance. It is also the respective SEI chemistries that are the key factors to determine how robust the formation is and is a better implication for cycling performance. Upon further inspection, we see an increase in the overall P–F/C–F to LiF/LixPOyFz ratio among the F species from D5 to D7, indicating that D7, albeit with higher saturated FEC content, interestingly induces less inorganic F-based species compared to D5. Also, given the similar overall C and O atomic compositions between the two, the differences in the F species among D5 and D7 appear to be the key factor in understanding how varying FEC/EMC ratios affect SEI formation and performance.
Lithium plating morphology and efficiency characterization
Li plating morphologies were characterized in Li|Cu cells to better understand the effects of different electrolytes in stabilizing Li-metal anodes during cycling. Fig. 4 presents SEM images of the Li plating morphologies on Cu foil in the different electrolytes. The plating for the LP57 sample in Fig. 4a shows porous, strain-like morphologies, which indicates poor Li deposition.36 The LSE samples from Fig. 4b–e show thicker and denser morphologies displaying compact aggregates of the deposited Li, with the D5 LSE sample displaying the densest plating, correlating well to its best cyclability in the half cells. While the D1 plating morphology does appear to show thicker Li deposition on the Cu foil compared to the LP57 sample, the sample suffers from the worst overpotential during the plating process as displayed in the voltage profiles in Fig. 4f, even more so than that of the LP57 sample. This large overpotential indicates that the Li+ de-solvating process in this electrolyte severely limits the overall kinetics of the cell and reflects the poor cyclability in half cells.21 With increasing FEC content in the D3 and D5 samples, the plating becomes noticeably thicker with larger Li strands and smaller void spaces. However, the morphology of the D7 sample in Fig. 4e slightly deviates from what was seen with the previous samples and rather shows a more loosely packed deposition. The D7 voltage profile during plating still interestingly shows low overpotential comparable to the D3 and D5 samples. This may suggest that the larger FEC content in D7 alters the Li deposition morphology in half cells albeit having Li+ transport kinetics similar to the D3 and D5 LSEs and shows the unique differences that occur during plating between low and high FEC contents in D1 and D7 LSEs. | ||
| Fig. 4 SEM images of Li plating morphologies on bare Cu foil carried out in Li|Cu cells with (a) LP57, (b) D1, (c) D3, (d) D5, and (e) D7 electrolytes. Insets are photos of the freshly plated Cu foil after cell disassembly. (f) Voltage profiles of the cells during the plating process. A 1 mA cm−2 current density was applied for 20 minutes for the plating process. All scale bars are 10 μm. | ||
To further evaluate the electrolytes on influencing the long-term cycling stability of the Li-metal anodes, the CE evolutions of Li|Cu cells after continual plating/stripping cycles were examined as shown in Fig. 5a. Such long-term cycling of Li|Cu cells has been established as a good protocol to assess CEs for Li-metal anodes and LMBs.37 At first glance, the LP57 and D1 cells display extremely poor stabilities, with both samples having their CEs drop below 70% within 15 cycles. The CEs continue to drastically decline past 20% within 25 cycles as seen in Fig. S8,† which displays the full view of the CE cycling of the electrolytes. Such inadequate CE stabilities of these two electrolytes correspond well to their inferior cycling in LNO half cells. The associated voltage curve evolutions of the plating/stripping cycles are also shown in Fig. 5b and c for, respectively, LP57 and D1 samples over 100 cycles. The poor CEs for both samples are accompanied by very large overpotentials seen in the voltage curves that worsen over continual cycling.
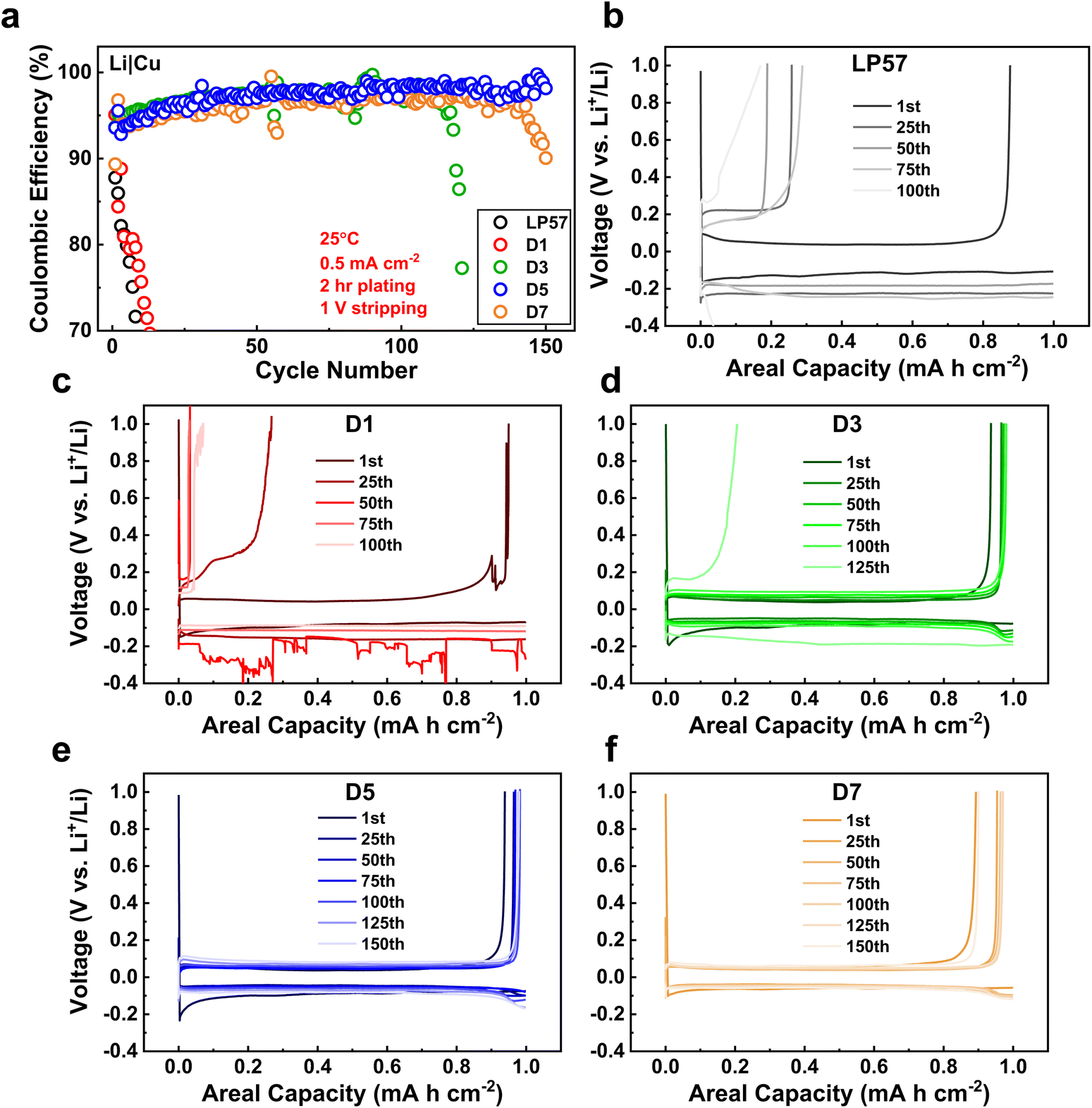 | ||
| Fig. 5 (a) Coulombic efficiencies of Li|Cu cells in the various electrolytes over extended cycling. Corresponding voltage profile evolutions of the cells throughout the cycling period of (b) LP57, (c) D1, (d) D3, (e) D5, and (f) D7. The Li|Cu cells utilized working areas of 1.91 cm2. Each cycle consisted of a two-hour plating step followed by a stripping step to 1 V with the same current densities of 0.5 mA cm−2 for both steps. | ||
In stark contrast, the D3–D7 LSE cells clearly indicate superior plating/stripping efficiencies with high initial CEs of around 93% that gradually increase past 100 cycles. Table S4† presents the average CEs of the cells after their respective cycling ranges for the D3–D7 cells. Note that the cycling ranges to determine the averages are based on the most stable cycling region for each cell with little variation in the CE. The D5 cell shows the highest averaged CE of 97.97%. The striking improvements in these samples match well with the thick and dense Li plating morphologies as seen in Fig. 4. Such formation of high density and low surface area plating greatly reduces the number of interfacial reactions that occur on the Li-metal anode, which helps attain a higher CE throughout cycling.10 The D7 LSE also displays great CE cycling despite its loosely packed plating, which may indicate that the CE stability is not entirely dependent on the Li plating morphology. Furthermore, despite the D3 sample having a good initial plating morphology, the CE rapidly drops to zero after around 120 cycles, which is suggestive of severe FEC consumption.38Fig. 5d shows the large overpotential that forms at the 125th cycle for the D3 cell, which well matches this rapid CE fading. This may also explain the rapid drop of the D1 cell, which occurred immediately upon cycling as the FEC content was notably lower. The D5 and D7 cells show the most stable CE cycling among all samples, particularly D5, with it having retained a high CE up to 150 cycles. The D7 cell displays similar stability, but begins to decline earlier around 140 cycles. Fig. 5e and f illustrate the stable cycling of the D5 and D7 cells as both display the best CE retention coupled with minimal overpotential growth over 150 cycles. All cells were left to cycle until failure as again shown in Fig. S8† where the D5 and D7 cells, interestingly, do not display the same rapid CE drop profile as the other three cells upon failure. Rather, their CEs erratically fluctuate before dropping gradually, which may imply a slightly different degradation mechanism for these LSEs. The fading of the cells, specifically that of the D7 sample, suggests that excessive FEC amounts in LSEs are also unsatisfactory, possibly due to the disruption of the LSE solvation structure, which will be further discussed in the next section. The overall cycling stability of Li-metal anodes is thus heavily regulated by multiple factors, as discussed above in the SEI/CEI compositions and depositions, Li plating morphologies, and the overpotentials associated with plating that is governed by Li+ transport processes during cycling.
Bulk electrolyte characterization
The differences seen across all LSE samples brought about by the electrochemical performance, SEI and CEI chemistries, Li plating morphologies, and CE cycling stabilities suggest that there are innate changes that occur in the bulk electrolyte structures between each LSE which warrants investigation.As such, 7Li NMR spectroscopy was conducted for LP57 and the D1–D7 electrolytes to further explore the bulk electrolyte properties shown in Fig. S9.† The LP57 7Li NMR is characterized by a single sharp and symmetric resonance peak. For a dynamically averaged cation environment, the resultant 7Li spectrum is a single exchanged narrowed Lorentzian line.39 The D1–D7 resonance peaks all present upfield shifts from the LP57 peak, which are attributed to the decreased distance between the Li+ and PF6 ions in the solvation structure due to the increased presence of CIPs and AGGs. We note that TTE was not added for the D1–D7 samples, as our previous study has shown that TTE influences the solvation structure and leads to line broadening and marked asymmetry of the peak. However, even with adding TTE, the broadened peaks still present upfield shifts to that of LP57, making the trend still valid with LSEs.21 Additionally, there is a further upfield shift in the resonance from the D7 to D1 samples, with D1 having the highest upfield shift among all electrolytes. This is due to the highest overall LiPF6 concentration in D1 with 3.85 M compared to that of D7 with 2.95 M as shown in Table 1. The higher the salt concentration, the more of an upfield shift is present. Gradually increasing line broadening and peak asymmetry are also seen from D7 to D1 due to high salt concentrations. Interestingly, the results seen from NMR do indicate the formation of CIP and AGG Li+ solvation structures for D1–D7, but do not distinguish the differences with Li+ coordination to FEC and EMC. Nonetheless, the results accumulated here from NMR well support the trends seen in our electrolyte data in Table 1 as well as the ionic conductivities in Table S1.†
Fourier-transform infrared spectroscopy (FTIR) characterization was performed on the LSEs as well as their saturated analogues in this study to better understand salt-solvent coordination changes. Fig. 6 presents the FTIR results of the various electrolyte samples in the range of 1600 to 2000 cm−1 to verify their solvation structures involving Li+ coordination with the solvent molecules. Fig. 6a shows the FTIR spectra of pure EMC and FEC solvents as well as saturated electrolyte (SE) versions of the LSEs, which do not include the TTE diluent like those analyzed with NMR. The spectra show different peaks pertaining to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond of the carbonate functional group, as this bond has a strong IR absorption in the carbonyl region (1650–1850 cm−1). Uncoordinated/coordinated CO groups for EMC and FEC are henceforth labeled as, respectively, COEMC and COFEC. Any shifts in the peak are indicative of coordination changes in the local solvation environment with Li+.40 The pure EMC spectrum at the top of Fig. 6a presents a clear peak around 1745 cm−1, which refers to its uncoordinated COEMC stretching frequency. Upon saturating the solvent with LiPF6 shown below the pure EMC spectra, a new peak at a lower wavenumber forms at around 1710 cm−1, which is the stretching frequency of the COEMC group coordinated to Li+. The uncoordinated COEMC peak almost entirely vanishes comparing the saturated and pure EMC solutions, indicating nearly all the EMC solvent molecules are effectively coordinated with Li+. Similarly, pure FEC and saturated FEC solutions have been analyzed and are shown at the bottom of Fig. 6a. The pure FEC spectrum presents two peaks in the carbonyl region around 1800 cm−1 and 1825 cm−1. The latter peak refers to the uncoordinated COFEC stretching band, while the former is the Fermi resonance band.41 The saturated FEC solvent spectrum above the pure FEC spectra has a new peak around 1790 cm−1, which is the coordinated COFEC species to Li+. However, this peak is broader than the other peaks seen in the spectra, which is most likely due to the overlap with the Fermi resonance of the uncoordinated COFEC peak, making it difficult for completely deconvoluting the peaks. Such overlap has been noted before for cyclic carbonate electrolytes.42,43 Additionally, a small remnant peak of the uncoordinated COFEC at 1825 cm−1 is still visible for the saturated FEC solvent, which may remain due to the lower solubility of LiPF6 in FEC compared to EMC.
O bond of the carbonate functional group, as this bond has a strong IR absorption in the carbonyl region (1650–1850 cm−1). Uncoordinated/coordinated CO groups for EMC and FEC are henceforth labeled as, respectively, COEMC and COFEC. Any shifts in the peak are indicative of coordination changes in the local solvation environment with Li+.40 The pure EMC spectrum at the top of Fig. 6a presents a clear peak around 1745 cm−1, which refers to its uncoordinated COEMC stretching frequency. Upon saturating the solvent with LiPF6 shown below the pure EMC spectra, a new peak at a lower wavenumber forms at around 1710 cm−1, which is the stretching frequency of the COEMC group coordinated to Li+. The uncoordinated COEMC peak almost entirely vanishes comparing the saturated and pure EMC solutions, indicating nearly all the EMC solvent molecules are effectively coordinated with Li+. Similarly, pure FEC and saturated FEC solutions have been analyzed and are shown at the bottom of Fig. 6a. The pure FEC spectrum presents two peaks in the carbonyl region around 1800 cm−1 and 1825 cm−1. The latter peak refers to the uncoordinated COFEC stretching band, while the former is the Fermi resonance band.41 The saturated FEC solvent spectrum above the pure FEC spectra has a new peak around 1790 cm−1, which is the coordinated COFEC species to Li+. However, this peak is broader than the other peaks seen in the spectra, which is most likely due to the overlap with the Fermi resonance of the uncoordinated COFEC peak, making it difficult for completely deconvoluting the peaks. Such overlap has been noted before for cyclic carbonate electrolytes.42,43 Additionally, a small remnant peak of the uncoordinated COFEC at 1825 cm−1 is still visible for the saturated FEC solvent, which may remain due to the lower solubility of LiPF6 in FEC compared to EMC.
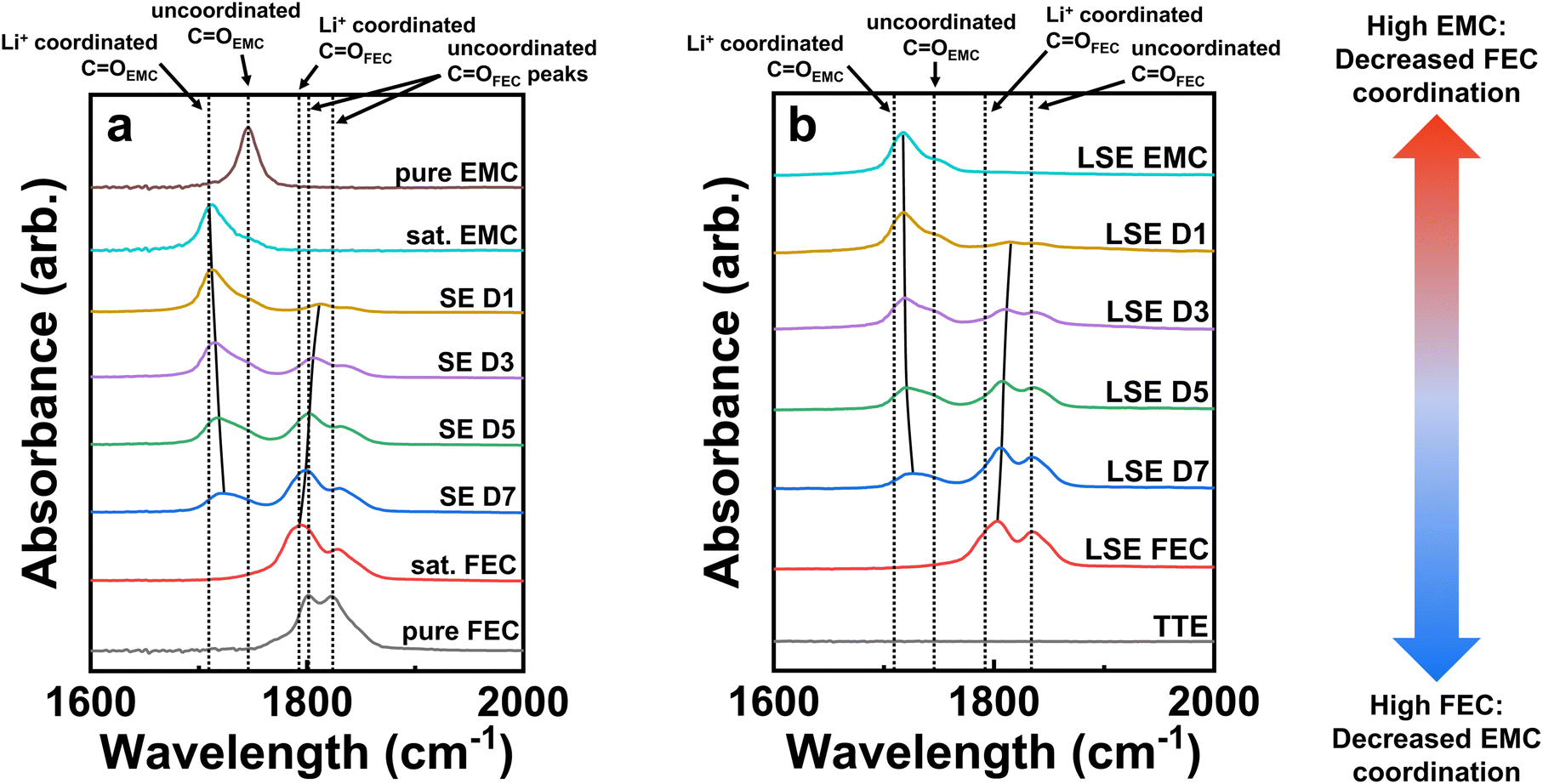 | ||
| Fig. 6 Characterization of the solvation structures of the various electrolytes. FTIR spectra of the (a) saturated electrolytes along with pure EMC and FEC solvents and (b) LSEs along with pure TTE from 1600 to 2000 cm−1 to study the Li+ coordination to solvent molecules. The solid black lines better show the shift in the peak positions with changing FEC to EMC ratios. | ||
The FTIR spectra for the LSE forms of the saturated EMC and FEC solvents, denoted as, respectively, LSE EMC and LSE FEC, are shown in Fig. 6b. The spectra indicate similar results to those seen with the SE spectra, in which the Li+-coordinated CO peaks for EMC and FEC develop and dominate over the uncoordinated peaks. Interestingly, however, both the LSE EMC and LSE FEC spectra still show noticeable peaks of, respectively, the uncoordinated EMC and FEC CO groups more so than in the SE spectra. Furthermore, there also is an apparent shift in the Li+-coordinated CO peaks seen with all the LSE sample spectra as they are shifted to slightly higher wavenumbers compared to the dashed lines of the coordinated peak positions, which are based on the SE spectra in Fig. 6a. This is likely because of the TTE diluent as it can affect the solvation structure of the localized CIPS and AGGs, but not to the extent that it disrupts the inner solvation sheath, as analyzed in our previous studies. The diluents still induce localized solvation cluster formations but can interact with the Li+ ions in very small quantities.21,44 The FTIR spectra for pure TTE are also shown in Fig. 6b and have no characteristic peaks within this wavenumber region, indicating that there are no significant interactions of TTE with the solvation structure among the LSEs. This makes the comparisons between the SEs and LSEs valid without worrying about the influence of TTE.
The SEs and LSEs are also characterized with increasing FEC to EMC ratios from D1 to D7 in Fig. 6b. Noticeable peak shifts of the Li+-coordinated COEMC to uncoordinated COEMC are seen with increasing FEC content from D1 to D7, as shown by the solid black line guiding the peak shift towards the right in both figures. This cannot just be from a lowered intensity of the coordinated COEMC peak because if it was, the peak intensity would simply diminish while maintaining the same wavenumber. The shift, therefore, must indicate the change in the solvation structure from coordinated to uncoordinated solvents. Likewise, the opposite trend is seen with increasing EMC content on going from D7 to D1 in both the SE and LSE spectra, where the Li+-coordinated COFEC peak in FEC gradually shifts rightwards towards the uncoordinated COFEC peak position. Therefore, at either extreme of high FEC or EMC content, it appears that the major saturated solvent will favor coordination with the Li+ ions and will outcompete the minor saturated solvent and induce a de-coordination of the solvent from the ions, resulting in unwanted free solvent molecules to exist in the solvation sheath. In the case with D1, which has the lowest FEC/EMC ratio, most of the coordination to Li+ is with the COEMC groups with very little COFEC coordination. As a result, the very slight amount of coordinated FEC molecules will reduce first and get permanently consumed, leaving mainly uncoordinated FEC molecules and Li+ complexes solvated only by EMC molecules.31 The uncoordinated FEC molecules are less likely to be reduced along with the Li+ and PF6− ions in CIPs and AGGs to form a well-passivated and inorganic-rich SEI. The main reduction species will instead be the EMC-solvated Li+ complexes. However, the decompositions of such Li+ complexes have been shown to be very detrimental to the Li-metal anode due to poor SEI formation and ensuing rapid capacity fade.45 In fact, we have also made LNO half cells in LSE only containing saturated EMC as the solvent in LSE EMC as part of the first electrolyte screening test shown in Fig. S10.† The resulting cell instantaneously drops to zero capacity even quicker than the D1 cell. This further indicates that LSEs with only saturated EMC as the solvent are poor electrolytes. The consequences of inadequate FEC coordination can, therefore, help explain the previous results of D1 seen with the inferior cycling performance, nonuniform and weakly formed SEI, along with the rapid fade in Li plating and stripping efficiencies.
In contrast, D7, which possesses the highest FEC/EMC ratio and thus the largest Li+-coordinated COFEC peak, also has the broadest COEMC peak that is shifting towards the uncoordinated position. Most of the Li+ ions in this case are solvated by FEC molecules, which correspondingly results in more uncoordinated EMC solvent molecules to be present. Interestingly, it appears that increasing the FEC content in the LSE spectra also increases the uncoordinated COFEC peak signal as both D7 and LSE FEC exhibit the largest peaks. This indicates that high saturated FEC content will still induce a notable amount of uncoordinated, free FEC solvent molecules to exist in the electrolyte, which is not favorable to forming CIPs and AGGs with the Li+ and PF6− ions. While the D7 LSE performance in LNO half cells in Fig. 2 displays far better cyclability compared to the D1 cell, it is still inferior to that of the D5 cell. This may be in part due to the larger presence of uncoordinated EMC and FEC molecules as free solvent reduction onto the Li-metal anode will generate unfavorable organic SEI species. This can help explain the XPS results seen back in Fig. 3c that display a larger P–F/C–F to LiF/LixPOyFz ratio for D7 than D5, which surprisingly indicates less inorganic F-based species formation for a higher saturated FEC electrolyte in D7. The fewer inorganic species formed from D7 may be due to the adverse passivation from the reduction of the greater free EMC solvent molecules. To test the limit of saturated FEC solvent in LSEs, LNO half cells were cycled with an LSE having an FEC/EMC ratio of 9/1 as well as an LSE FEC containing only saturated FEC in Fig. S10.† Both cells immediately fade close to zero capacity like that of the LSE EMC cell. The cycling performances reveal that either extreme of high EMC or FEC content in LSEs is detrimental to LMB stabilization.
The increased amount of FEC-solvated Li+ complexes may provide better SEI and CEI stabilization, but excessive contents evidently plague LMB performance now seen from the cycling data. Fig. S11† displays the images of all the formulated electrolytes used in this study. All electrolytes presented have clear, single-phase solutions except that of the LSE FEC, which has a distinct two-phase separation as shown. This indicates that with too high of a saturated FEC content, solvent separation from the TTE will occur, as FEC is not miscible with TTE. This is due to the low polarity of TTE coupled with the aprotic nature of FEC. As a result of this phase separation, the TTE may not entirely form good, localized solvation sheaths around the coordinated FEC molecules, which will cause unfavorable reductions with just FEC and just TTE yielding bad SEI products. Such phenomena help us understand the extremely poor fading shown by the LSEs with high FEC contents. Furthermore, this also shows why EMC is still needed in these electrolyte mixtures, as EMC is miscible in both FEC and TTE and can help attain a single-phase solution.46
Fig. S12a and b† compare the FTIR spectra of the electrolytes in their SE and LSE forms in the range of 800 to 900 cm−1 to study the effects of PF6− coordination to Li+ at high EMC and FEC contents. There are more noticeable peaks closer to the uncoordinated PF6− anions around 840 cm−1 for the LSEs compared to the SEs, but this is likely due to the influence of the TTE peak as shown with the TTE spectrum in Fig. S12b,† which makes it difficult to deconvolute the TTE to the uncoordinated PF6− peak contributions. Therefore, the SE spectra are analyzed to avert TTE convolution. From the SE spectra, the saturated FEC solvent has the largest uncoordinated PF6− peak compared to the saturated EMC solvent. In fact, it appears that increasing EMC seems to increase the PF6− coordination more while increasing FEC does the opposite, which may be correlated to the fact that too much FEC causes a stronger uncoordinated COFEC peak signal to form as previously discussed, indicating more free solvent formation. Such phenomena will induce less coordination to the corresponding PF6− anions as the Li+ cations will also be less coordinated to the solvent. This may be due to the lower solubility of the LiPF6 salt in FEC than in EMC. Therefore, with higher FEC content, not only does it seemingly induce higher amounts of uncoordinated FEC due to the lower solubility of the salt, but it also causes de-coordination of the EMC.
Conclusions
In summary, we have assessed the properties of LSEs with different FEC and EMC solvent ratios to uncover their ability to stabilize LMBs with the LNO cathode. By screening through four different FEC/EMC solvent ratios in LSEs, we found that the FEC/EMC (v/v) ratio of 1/1 in the D5 LSE brought about the best electrochemical performance with a remarkable 80.6% capacity retention after 600 cycles at a 1C rate compared to 53.5% retention after 400 cycles with commercial LP57 electrolyte in LNO half cells. Post-mortem XPS characterization studies reveal the greatest LiF composition and M–O lattice ratio on the cathode with D5 electrolyte, coupled with the largest LiF/LixPOyFz composition on the Li-metal anode, indicative of the most inorganic-rich and robust CEI and SEI passivation layers. The D5 LSE also presents the densest Li-plating morphology and the highest plating/stripping efficiency among all the electrolytes in Li|Cu cells.FTIR analyses uncover the differences in the solvation structures of the electrolytes and show that at either high saturated EMC or FEC contents, there is a competition between the majority and minority solvents. The majority solvent will dominate the Li+ coordination, which causes the minority solvent to de-coordinate and remain as free solvent molecules. Such presence of free solvents defeats the purpose of attaining LSEs and leads to inferior CEI and SEI formation and Li-plating morphologies as seen with the D1 and D7 LSEs. Specifically, low FEC/EMC ratios will promote free FEC and mainly form Li+-coordinated EMC complexes, the latter of which are also known to be detrimental for SEI development. At high FEC/EMC ratios, the opposite phenomenon is observed, where uncoordinated EMC and largely Li+-coordinated FEC clusters form. However, excessive FEC solvent content also can interestingly bring about more uncoordinated FEC molecules in addition to uncoordinated EMC, which are harmful for SEI formation. The dominant amounts of the coordinated FEC molecules will additionally lead to two-phase separation from the TTE diluent, ruining the solvation sheath. Overall, it is impossible to have both solvents be fully coordinated to Li+ and PF6− ions. Therefore, finding the optimal ratio where there are minimized free solvents that provide the best electrode–electrolyte interphase passivation is of paramount importance. This work provides new insights into rational design strategies for multi-solvent electrolyte systems to realize high-energy-density and long-life LMBs.
Conflicts of interest
The corresponding author (A. M.) is a co-founder of TexPower EV Technologies Inc., a start-up company focusing on cobalt-free cathode materials for lithium-based batteries.Acknowledgements
This work was supported by the Assistant Secretary for Energy Efficiency and Renewable Energy, Office of Vehicle Technologies of the U.S. Department of Energy through the Advanced Battery Materials Research (BMR) Program (Battery500 Consortium) award number DE-EE0007762 and Welch Foundation grant F-1254.References
- W. Li, E. M. Erickson and A. Manthiram, Nat. Energy, 2020, 5, 26–34 CrossRef CAS.
- R. Schmuch, R. Wagner, G. Hörpel, T. Placke and M. Winter, Nat. Energy, 2018, 3, 267–278 CrossRef CAS.
- A. Manthiram, ACS Cent. Sci., 2017, 3, 1063–1069 CrossRef CAS PubMed.
- S. Tan, W. Wang, Y. Tian, S. Xin and Y. Guo, Adv. Funct. Mater., 2021, 31, 2105253 CrossRef CAS.
- M. Winter, B. Barnett and K. Xu, Chem. Rev., 2018, 118, 11433–11456 CrossRef CAS PubMed.
- X.-B. Cheng, R. Zhang, C.-Z. Zhao and Q. Zhang, Chem. Rev., 2017, 117, 10403–10473 CrossRef CAS PubMed.
- K. Dokko, N. Tachikawa, K. Yamauchi, M. Tsuchiya, A. Yamazaki, E. Takashima, J.-W. Park, K. Ueno, S. Seki, N. Serizawa and M. Watanabe, J. Electrochem. Soc., 2013, 160, A1304–A1310 CrossRef CAS.
- X. Cao, H. Jia, W. Xu and J.-G. Zhang, J. Electrochem. Soc., 2021, 168, 010522 CrossRef CAS.
- J. Zheng, J. A. Lochala, A. Kwok, Z. D. Deng and J. Xiao, Adv. Sci., 2017, 4, 1700032 CrossRef PubMed.
- S. Chen, J. Zheng, D. Mei, K. S. Han, M. H. Engelhard, W. Zhao, W. Xu, J. Liu and J. Zhang, Adv. Mater., 2018, 30, 1706102 CrossRef PubMed.
- X. Ren, S. Chen, H. Lee, D. Mei, M. H. Engelhard, S. D. Burton, W. Zhao, J. Zheng, Q. Li, M. S. Ding, M. Schroeder, J. Alvarado, K. Xu, Y. S. Meng, J. Liu, J.-G. Zhang and W. Xu, Chem, 2018, 4, 1877–1892 CAS.
- J. Langdon, Z. Cui and A. Manthiram, ACS Energy Lett., 2021, 6, 3809–3816 CrossRef CAS.
- L. Yu, S. Chen, H. Lee, L. Zhang, M. H. Engelhard, Q. Li, S. Jiao, J. Liu, W. Xu and J.-G. Zhang, ACS Energy Lett., 2018, 3, 2059–2067 CrossRef CAS.
- S. Chen, J. Zheng, L. Yu, X. Ren, M. H. Engelhard, C. Niu, H. Lee, W. Xu, J. Xiao, J. Liu and J.-G. Zhang, Joule, 2018, 2, 1548–1558 CrossRef CAS.
- X. Zhang, L. Zou, Z. Cui, H. Jia, M. H. Engelhard, B. E. Matthews, X. Cao, Q. Xie, C. Wang, A. Manthiram, J.-G. Zhang and W. Xu, Mater. Today, 2021, 44, 15–24 CrossRef CAS.
- S. Jiao, X. Ren, R. Cao, M. H. Engelhard, Y. Liu, D. Hu, D. Mei, J. Zheng, W. Zhao, Q. Li, N. Liu, B. D. Adams, C. Ma, J. Liu, J.-G. Zhang and W. Xu, Nat. Energy, 2018, 3, 739–746 CrossRef CAS.
- L. Su, E. Jo and A. Manthiram, ACS Energy Lett., 2022, 7, 2165–2172 CrossRef CAS.
- X. Zhang, H. Jia, L. Zou, Y. Xu, L. Mu, Z. Yang, M. H. Engelhard, J.-M. Kim, J. Hu, B. E. Matthews, C. Niu, C. Wang, H. Xin, F. Lin and W. Xu, ACS Energy Lett., 2021, 6, 1324–1332 CrossRef CAS.
- J. Langdon and A. Manthiram, Adv. Mater., 2022, 51, 2205188 CrossRef PubMed.
- J.-G. Zhang, W. Xu, J. Xiao, X. Cao and J. Liu, Chem. Rev., 2020, 120, 13312–13348 CrossRef CAS PubMed.
- L. Su, X. Zhao, M. Yi, H. Charalambous, H. Celio, Y. Liu and A. Manthiram, Adv. Energy Mater., 2022, 12, 2201911 CrossRef CAS.
- M. Yi, A. Dolocan and A. Manthiram, Adv. Funct. Mater., 2023, 2213164 CrossRef CAS.
- Z. Cui, Z. Guo and A. Manthiram, Adv. Energy Mater., 2023, 2203853 CrossRef CAS.
- M. Bianchini, M. Roca-Ayats, P. Hartmann, T. Brezesinski and J. Janek, Angew. Chem., Int. Ed., 2019, 58, 10434–10458 CrossRef CAS PubMed.
- H. Li, N. Zhang, J. Li and J. R. Dahn, J. Electrochem. Soc., 2018, 165, A2985–A2993 CrossRef CAS.
- Q. Xie, W. Li, A. Dolocan and A. Manthiram, Chem. Mater., 2019, 31, 8886–8897 CrossRef CAS.
- M. Yi, W. Li and A. Manthiram, Chem. Mater., 2022, 34, 629–642 CrossRef CAS.
- L. Sharma, M. Yi, E. Jo, H. Celio and A. Manthiram, Chem. Mater., 2022, 34, 4514–4522 CrossRef CAS.
- K. Liu, Q. Zhang, S. Dai, W. Li, X. Liu, F. Ding and J. Zhang, ACS Appl. Mater. Interfaces, 2018, 10, 34153–34162 CrossRef CAS PubMed.
- Y. Zhang, D. Krishnamurthy and V. Viswanathan, J. Electrochem. Soc., 2020, 167, 070554 CrossRef.
- T. Hou, G. Yang, N. N. Rajput, J. Self, S.-W. Park, J. Nanda and K. A. Persson, Nano Energy, 2019, 64, 103881 CrossRef CAS.
- X. Zhang, X. Cheng, X. Chen, C. Yan and Q. Zhang, Adv. Funct. Mater., 2017, 27, 1605989 CrossRef.
- H. Li, A. Liu, N. Zhang, Y. Wang, S. Yin, H. Wu and J. R. Dahn, Chem. Mater., 2019, 31, 7574–7583 CrossRef CAS.
- R. Pan, E. Jo, Z. Cui and A. Manthiram, Adv. Funct. Mater., 2022, 2211461 Search PubMed.
- S. Klein, S. van Wickeren, S. Röser, P. Bärmann, K. Borzutzki, B. Heidrich, M. Börner, M. Winter, T. Placke and J. Kasnatscheew, Adv. Energy Mater., 2021, 11, 2003738 CrossRef CAS.
- L. Su, H. Charalambous, Z. Cui and A. Manthiram, Energy Environ. Sci., 2022, 15, 843–854 RSC.
- B. D. Adams, J. Zheng, X. Ren, W. Xu and J. Zhang, Adv. Energy Mater., 2018, 8, 1702097 CrossRef.
- R. Jung, M. Metzger, D. Haering, S. Solchenbach, C. Marino, N. Tsiouvaras, C. Stinner and H. A. Gasteiger, J. Electrochem. Soc., 2016, 163, A1705–A1716 CrossRef CAS.
- V. Kumar, R. R. Reddy, B. V. N. P. Kumar, C. V. Avadhani, S. Ganapathy, N. Chandrakumar and S. Sivaram, J. Phys. Chem. C, 2019, 123, 9661–9672 CrossRef CAS.
- D. J. Xiong, M. Bauer, L. D. Ellis, T. Hynes, S. Hyatt, D. S. Hall and J. R. Dahn, J. Electrochem. Soc., 2018, 165, A126–A131 CrossRef CAS.
- D. M. Seo, S. Reininger, M. Kutcher, K. Redmond, W. B. Euler and B. L. Lucht, J. Phys. Chem. C, 2015, 119, 14038–14046 CrossRef CAS.
- J. R. Durig, J. W. Clark and J. M. Casper, J. Mol. Struct., 1970, 5, 67–84 CrossRef CAS.
- B. Fortunato, P. Mirone and G. Fini, Spectrochim. Acta, Part A, 1971, 27, 1917–1927 CrossRef CAS.
- J. He, A. Bhargav, W. Shin and A. Manthiram, J. Am. Chem. Soc., 2021, 143, 20241–20248 CrossRef CAS PubMed.
- C. Su, M. He, J. Shi, R. Amine, J. Zhang and K. Amine, Angew. Chem., Int. Ed., 2020, 59, 18229–18233 CrossRef CAS PubMed.
- H. K. Bezabh, S.-F. Chiu, T. M. Hagos, M.-C. Tsai, Y. Nikodimos, H. G. Redda, W.-N. Su and B. J. Hwang, J. Power Sources, 2021, 494, 229760 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ta01061a |
| This journal is © The Royal Society of Chemistry 2023 |