
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhotosynthesis of hydrogen peroxide from dioxygen and water using aluminium-based metal–organic framework assembled with porphyrin- and pyrene-based linkers†
Yoshifumi
Kondo
a,
Kenta
Hino
a,
Yasutaka
Kuwahara
 abc,
Kohsuke
Mori
ab and
Hiromi
Yamashita
*ab
abc,
Kohsuke
Mori
ab and
Hiromi
Yamashita
*ab
aDivision of Materials and Manufacturing Science, Graduate School of Engineering, Osaka University, 2-1 Yamadaoka, Suita, Osaka 565-0871, Japan. E-mail: yamashita@mat.eng.osaka-u.ac.jp
bInnovative Catalysis Science Division, Institute for Open and Transdisciplinary Research Initiatives (OTRI), Osaka University, 2-1 Yamadaoka, Suita, Osaka 565-0871, Japan
cJST, PRESTO, 4-1-8 Honcho, Kawaguchi, Saitama 332-0012, Japan
First published on 12th April 2023
Abstract
Photocatalytic production of hydrogen peroxide (H2O2) from dioxygen (O2) and water (H2O) has shown promise for the artificial photosynthesis of liquid fuel. We previously demonstrated that an Al-based metal–organic framework (MOF) functions as a suitable platform for photocatalytic H2O2 production owing to the efficient suppression of H2O2 decomposition caused by the photocatalysts themselves, which increases the yield of H2O2. However, the photocatalytic efficiency of Al-based MOFs is often limited by their short-lived charge separation. The energy transfer process is a beneficial approach to promoting charge separation and thereby improving the photocatalytic activity; MOFs enable highly efficient energy transfer between organic linkers because they allow precise control of the arrangement of the building blocks. Herein, we demonstrate that an Al-based MOF composed of both porphyrin- and pyrene-based organic linkers (Al-TCPP(10-X)-TBAPyX) is a promising photocatalyst for producing H2O2 from O2 and H2O without additives under visible-light irradiation while simultaneously enabling efficient suppression of undesired H2O2 decomposition. Efficient energy transfer from 1,3,6,8-tetrakis(p-benzoic acid)pyrene (TBAPy) to tetrakis(4-carboxyphenyl)porphyrin (TCPP) was driven within Al-TCPP(10-X)-TBAPyX, resulting in dramatically enhanced photocatalytic H2O2 production through optimization of the linker mixture ratio in the MOF structure. The present work not only proposes a new reaction pathway for H2O2 generation via1O2 intermediates, which is quite different from well-accepted mechanisms involving O2˙−, but also provides a promising strategy for designing catalysts to realize efficient photosynthetic H2O2 production.
Introduction
Harvesting solar fuels by artificial photosynthesis has great value in the global mission to address climate change and energy issues.1,2 Solar fuels should have high energy density and be easy to store, transport and use. Hydrogen peroxide (H2O2) is attracting growing interest as a solar fuel because of its high volumetric energy density, easy storage and easy transportability.3–5 H2O2 can generate electricity via a single-compartment fuel cell.3,6 The single-compartment H2O2 fuel cell is an inexpensive and compact fuel cell because it does not require an expensive separation membrane, and its theoretical output potential of 1.09 V is comparable to that of a hydrogen fuel cell (1.23 V).3,6 Therefore, solar-driven H2O2 production from Earth-abundant dioxygen (O2) and water (H2O), in conjunction with H2O2 fuel cells, is expected to be a candidate technology for realizing a sustainable society.The photosynthesis of H2O2 from the coupling of O2 and H2O is an uphill reaction with a standard Gibbs free energy change (ΔG0) of 117 kJ mol−1 (eqn. (1)):7
| O2 + 2H2O → 2H2O2 (ΔG0 = 117 kJ mol−1) | (1) |
Over the past decade, substantial research has been focused on the development of efficient photocatalysts for H2O2 production.8–12 Following the first demonstration of ZnO photocatalysts for H2O2 production in 1927, inorganic and organic semiconductors for photocatalytic H2O2 production have emerged.8–12 However, current photocatalysts for H2O2 production have numerous associated challenges, such as poor light absorption properties and short photoexcited carrier lifetimes. Another challenging issue is suppressing H2O2 decomposition, which occurs simultaneously during the reaction and reduces the yield of H2O2. Thus, it is necessary to design photocatalysts that both improve activity and inhibit H2O2 decomposition.
To overcome the aforementioned problems, we introduced an energy transfer process into the photocatalytic system—specifically, energy transfer between chlorophyll (antenna molecule) and a carotenoid (pigment molecule) in natural photosynthesis (Scheme 1).13,14 An efficient energy transfer process has been reported to extend the fluorescence lifetime and improve the light-harvesting ability of acceptor moieties.15,16 The elongated charge separation enables interaction with more reactants and photocatalysts prior to charge recombination, leading to improved photocatalytic activity. The general requirements of photocatalysts for an efficient energy transfer process are (i) sufficient overlap between the emission spectrum of the donors and the absorption spectrum of the acceptors and (ii) appropriate spatial proximity of the donors and acceptors.15
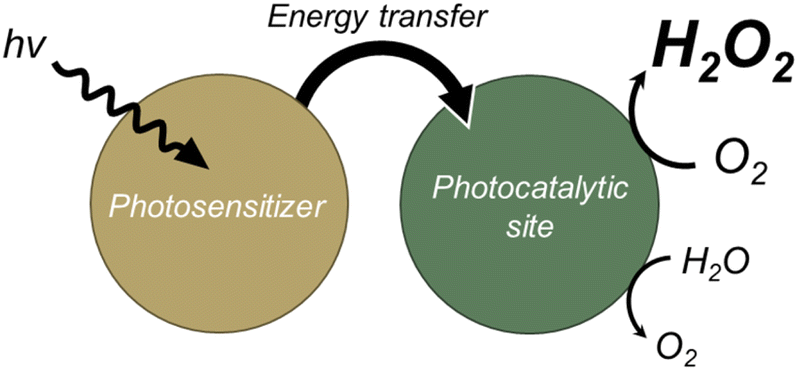 | ||
| Scheme 1 Strategy for development of photocatalysts for H2O2 production by driving an energy transfer process. | ||
Metal–organic frameworks (MOFs) are a new class of porous crystalline materials composed of metal-oxo clusters and organic linkers with well-defined structures.2,5,17–20 The highly ordered network of MOFs enables precise control of structural parameters such as the positions, mutual distances and relative orientations of organic linkers. Therefore, they are considered ideal platforms to hierarchically integrate photoactive ligands for synergistic catalysis, and can thus form antenna networks like those observed in natural photosynthesis.21–23 Interestingly, Morris and coworkers reported that the efficiency of energy transfer is sensitive to the geometric parameters of MOFs.13 The energy transfer process in MOFs deviates from the classical Förster model, and the structural arrangement of the linker affects the efficiency of the excitation energy transfer in MOFs. They demonstrated that energy transfer in a Zr-based MOF (ROD-7) should be highly anisotropic along the stacking direction, suggesting that MOFs provide a promising platform for efficient energy transfer.13
To that end, we selected a porphyrin-based Al-MOF (Al-TCPP) and a pyrene-based Al-MOF (Al-TBAPy) as supports because of their high water stability and good light-harvesting performance.24 Al-TCPP and Al-TBAPy consist of one-dimensional rod-like Al(OH) clusters linked by tetrakis(4-carboxyphenyl)porphyrin (TCPP) and 1,3,6,8-tetrakis(p-benzoic acid)pyrene (TBAPy), respectively (Fig. 1).16,24–26 The TCPP and TBAPy linkers act as the acceptor and donor moieties, respectively. TBAPy has a wide emission wavelength range (400–500 nm) that well matches the excitation wavelength for TCPP (410 nm).13–15 The TCPP and TBAPy linkers have similar connectivity because of their similar molecular size, which enables them to be introduced within the same MOF structure. Fortunately, Al-TCPP and Al-TBAPy possess a similar geometrical arrangement in ROD-7, which presumably favours efficient energy transfer.13 In addition, our group has previously demonstrated that Al-based MOFs exhibited low reactivity toward H2O2, leading to an increase in the yield of H2O2 in photocatalytic H2O2 production.18 Therefore, Al-MOFs assembled with TCPP (as an acceptor) and TBAPy (as a donor) are expected to function as a suitable photocatalyst for H2O2 production.
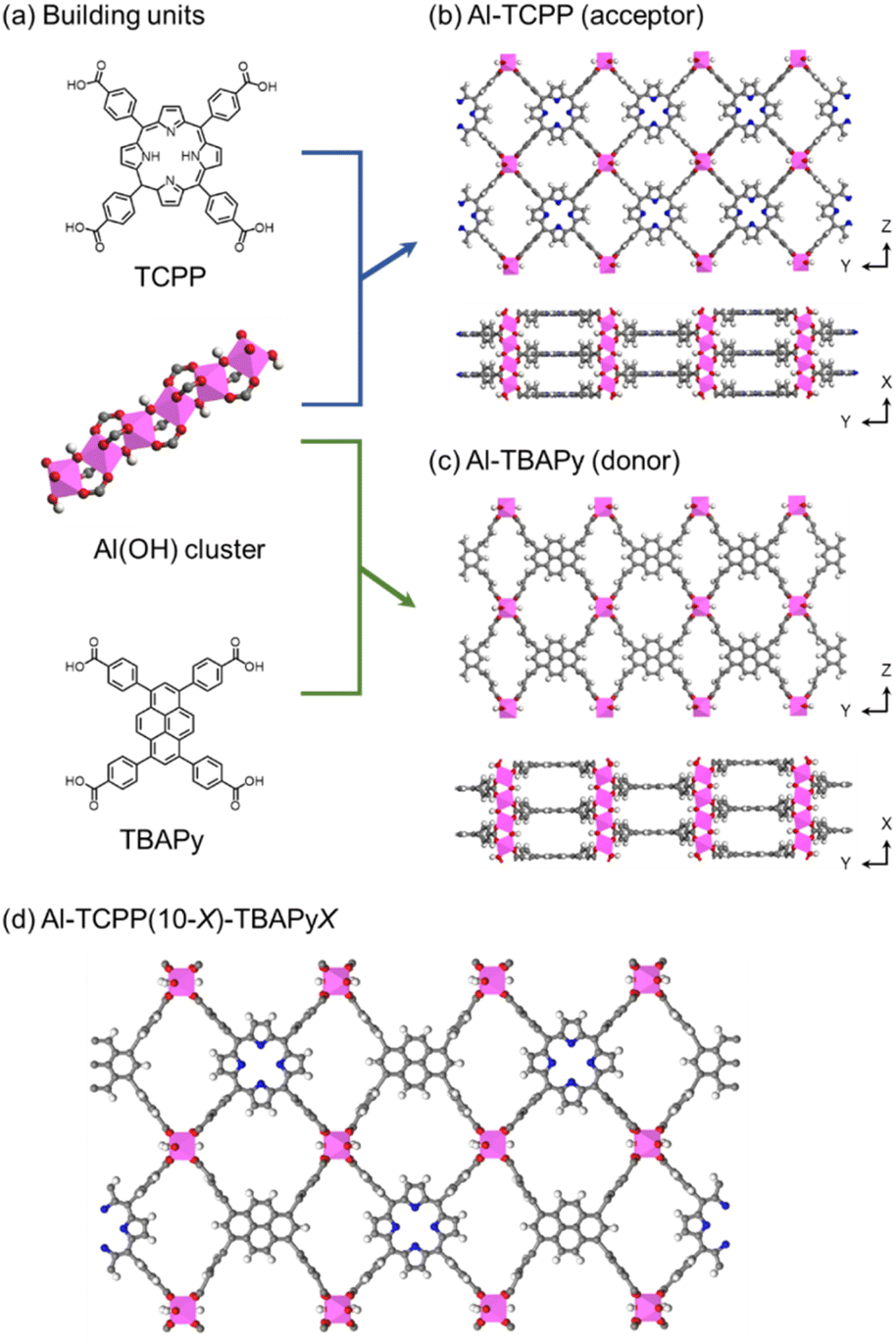 | ||
| Fig. 1 Schematics of (a) building units, (b) Al-TCPP, (c) Al-TBAPy and (d) Al-TCPP(10-X)-TBAPyX. | ||
Herein, we develop the porphyrin- and pyrene-based Al-MOF (Al-TCPP(10-X)-TBAPyX) as a photocatalyst for H2O2 production from O2 and H2O. Although a few recent investigations on MOFs containing porphyrin- and pyrene-based organic linkers have suggested preliminary influences on photocatalysis, to the best of our knowledge, there has never been a reported study that clarifies how mixing organic linkers in different proportions in MOFs systematically affects their photocatalytic performance. Photocatalytic H2O2 production from O2 and H2O without additives was significantly enhanced when both TCPP and TBAPy were incorporated within the framework of Al-TCPP(10-X)-TBAPyX. Photoluminescence measurements, electron-spin resonance (ESR) measurements and active-species trapping experiments were performed to elucidate the unique reaction mechanism. A comprehensive analysis of the results of the experiments revealed that the reaction pathway for H2O2 production using Al-TCPP(10-X)-TBAPyX differs substantially from that for previously reported MOF photocatalysts. The present study not only offers new insights into the rational design of MOF photocatalysts with an efficient energy transfer process, but could also drive a breakthrough in the field of photocatalytic H2O2 production.
Experimental
Materials
Aluminum chloride hexahydrate (AlCl3·6H2O), acetone, hydrogen peroxide (H2O2), N,N-dimethylformaldehyde (DMF),hydrochloric acid (HCl), perchloric acid (HClO4), barium sulfide (BaSO4), sodium azide (NaN3) and p-benzoquinone (p-BQ) were purchased from Nacalai Tesque. 1,3,6,8-tetrakis(p-benzoic acid)pyrene (TBAPy) was purchased by Ambeed Inc. Tetrakis(4-carboxyphenyl)porphyrin (TCPP)oxo[5,10,15,20-tetra(4-pyridyl)porphinato]titanium(IV) (TiO(tpypH4)4+), 5,5-dimethyl-1-pyrroline N-oxide (DMSO) and 2,2,6,6-tetramethylpiperidine (TEMP) were obtained from Tokyo Chemical Industry Co., Ltd. Sulfuric acid-d2 (D2SO4), dimethyl sulfoxide-d6 (DMSO-d6) and potassium bromate (KBrO3) were obtained from Aldrich Chemical Co. All the chemicals were used as received without further purification.Synthesis of Al-TCPP(10-X)-TBAPyX
Al-TCPP(10-X)-TBAPyX samples were prepared using a solvothermal method. 30 mg of AlCl3·6H2O was added in 15 mL of distilled water. TCPP and TBAPy were completely dissolved in 5.0 mL of DMF in the proportions shown in Table S1.† These solutions were mixed and sonicated for 10 min. The mixed solution was transferred to a 40 mL Teflon-liner in a stainless-steel autoclave and heated at 120 °C for 20 h. The obtained solid was collected by centrifugation and washed with DMF and acetone several times. The obtained product was dried and activated at 120 °C under vacuum overnight.Results and discussion
Characterizations of Al-TCPP(10-X)-TBAPyX
Al-MOFs containing porphyrin- and pyrene-based organic linkers (Al-TCPP(10-X)-TBAPyX) were prepared by a solvothermal method. Powder X-ray diffraction (PXRD) patterns for Al-TCPP10 and Al-TBAPy10 showed strong peaks and well matched the reported PXRD patterns for Al-TCPP and Al-TBAPy, respectively (Fig. 2a).24–26 The peaks at 7.6, 12.1, 13.5, and 15.4° for Al-TCPP10 correspond to the (201), (401), (110), and (402) crystal planes of Al-TCPP reported in the literature.24,27,28 Al-TBAPy10 exhibited diffraction peaks at 2θ = 8.1, 12.9, 13.9, and 16.2° which corresponded to (201), (401), (110), and (402) planes of Al-TBAPy reported in the literature.25,26,29 For the Al-TCPP(10-X)-TBAPyX samples, the Al-TCPP phase steadily changed to the Al-TBAPy phase with increasing TBAPy linker content in the MOF matrix. The diffraction intensity in the PXRD patterns decreased as the molar ratio of the organic linkers (TCPP and TBAPy) approached 1. This decrease in crystallinity might be attributable to homogenization of the crystalline phase. The 5° ≤ 2θ ≤ 10° range of the PXRD pattern for each sample is presented in Fig. 2b. The peak at ∼8°, which was assigned to (201) planes in Al-TCPP and Al-TBAPy, gradually shifted towards higher angles with increasing TBAPy linker content in the Al-TCPP(10-X)-TBAPyX samples. The lattice spacing for the (201) planes (d201) was calculated using the Bragg equation (Fig. 2c). The value of d201 decreased with increasing TBAPy linker content in the Al-TCPP(10-X)-TBAPyX samples. This trend is consistent with the larger value of d201 for Al-TCPP than for Al-TBAPy. These results indicate that the two different organic linkers were homogeneously incorporated into the matrix without separation into two phases. | ||
| Fig. 2 (a) PXRD patterns, (b) enlarged PXRD patterns and (c) lattice spacings for (201) planes (d201) for Al-TCPP(10-X)-TBAPyX samples. | ||
Nitrogen (N2) adsorption–desorption measurements were conducted to characterize the porous structures of the Al-TCPP(10-X)-TBAPyX samples. As shown in Fig. 3a, all the samples exhibit type-I isotherms with H4 hysteresis loops, indicating that they are microporous materials. The increase in N2 adsorption near a relative pressure of 0.9 is presumably due to inter-particle voids. No substantial differences are observed in their Brunauer–Emmett–Teller (BET) specific surface area (SBET) or total pore volume (Vtotal) because of the similarity in the crystal structures of Al-TCPP and Al-TBAPy (Table 1). These results are supported by the pore distributions estimated by the nonlocal density functional theory (NLDFT) method (Fig. 3b). The aforementioned results indicate that TCPP and TBAPy linkers are homogeneously incorporated into the Al-TCPP(10-X)-TBAPyX matrix while maintaining their well-defined microporous structures.
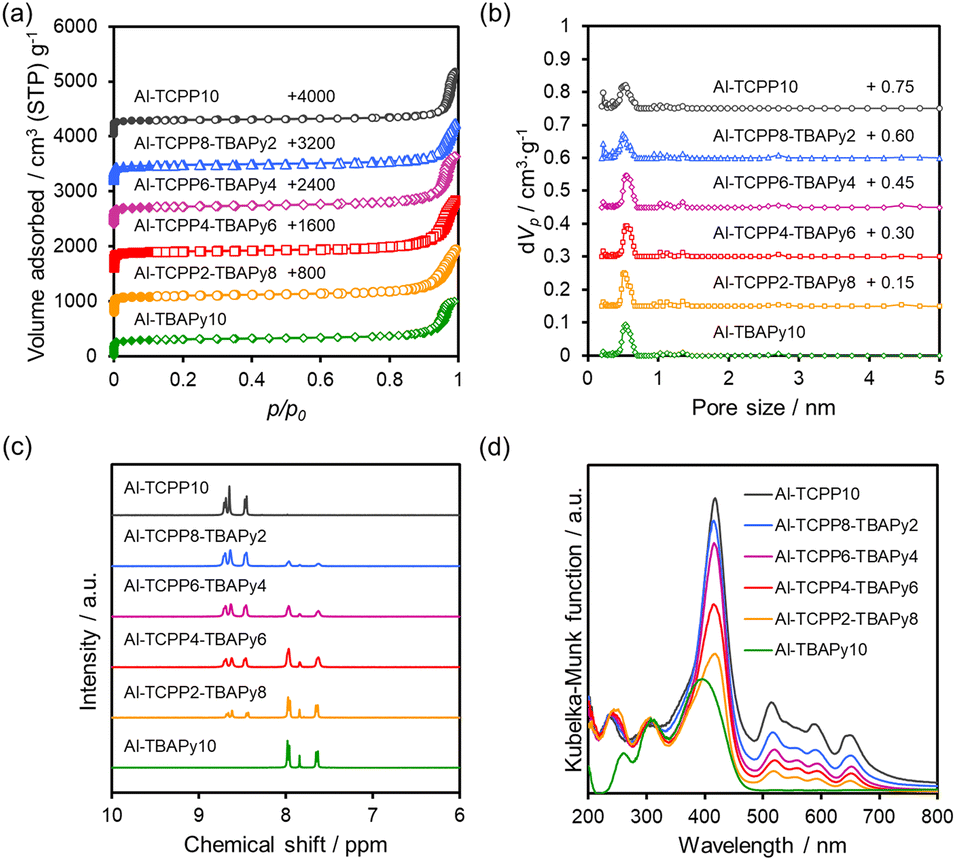 | ||
| Fig. 3 (a) N2 physisorption isotherms, (b) pore distributions calculated using NLDFT method, (c) dissolved/1H-NMR spectra and (d) diffuse-reflectance UV-vis spectra of Al-TCPP(10-X)-TBAPyX samples. | ||
| Sample | S BET (m2 g−1) | V p (cm3 g−1) | d (Å) |
|---|---|---|---|
| a Determined the BET method by N2 adsorption data using ISO 9227 standard. b Total pore volume reported at p/p0 = 0.99. c Peak pore size determined by the NLDFT method. | |||
| Al-TCPP10 | 1160 | 1.80 | 5.0 |
| Al-TCPP8-TBAPy2 | 1079 | 1.58 | 5.2 |
| Al-TCPP6-TBAPy4 | 1240 | 1.91 | 5.3 |
| Al-TCPP4-TBAPy6 | 1193 | 1.91 | 5.5 |
| Al-TCPP2-TBAPy8 | 1169 | 1.76 | 5.2 |
| Al-TBAPy10 | 1184 | 1.55 | 5.3 |
Proton nuclear magnetic resonance (1H-NMR) measurements were performed to quantify the molar ratio of TCPP linkers to TBAPy linkers. The dissolution/1H-NMR technique involves dissolving the MOFs in a deuterated dimethyl sulfoxide (DMSO-d6) medium containing deuterated sulfuric acid (D2SO4). As shown in Fig. 3c, the observed signals could be assigned to TCPP (8.4–8.8 ppm) or TBAPy (7.5–8.1 ppm).16 By integrating the peaks assigned to both the TCPP and TBAPy linkers, we estimated the ratio between them. The TCPP and TBAPy content in the Al-TCPP(10-X)-TBAPyX samples corresponded well to the molar ratio used during sample preparation (Table 2).
| Sample | TCPP (%) | TBAPy (%) |
|---|---|---|
| Al-TCPP10 | 100 | 0.0 |
| Al-TCPP8-TBAPy2 | 80.1 | 19.9 |
| Al-TCPP6-TBAPy4 | 59.5 | 40.5 |
| Al-TCPP4-TBAPy6 | 40.1 | 59.9 |
| Al-TCPP2-TBAPy8 | 23.5 | 76.5 |
| Al-TBAPy10 | 0.0 | 100 |
Photocatalytic H2O2 production from O2 and H2O over Al-TCPP(10-X)-TBAPyX
The photocatalytic H2O2 production performance of Al-TCPP(10-X)-TBAPyX samples was evaluated under visible-light (λ > 420 nm, 100 mW cm−2) irradiation in O2-saturated distilled water without any additive. All Al-TCPP(10-X)-TBAPyX samples produced H2O2 from O2 and H2O under irradiation by visible light (Fig. 4a). Compared with photocatalytic H2O2 production using bare Al-TCPP10 and Al-TBAPy10, photocatalytic H2O2 production was enhanced when the TCPP and TBAPy linkers were mixed in the Al-TCPP(10-X)-TBAPyX samples. In particular, Al-TCPP4-TBAPy6 exhibited the highest H2O2 production rate of 127 μmol L−1 h−1, which was 2.1-fold and 4.2-fold greater than the production rates for pristine Al-TCPP10 and Al-TBAPy10, respectively. This H2O2 production rate is, to our knowledge, among the best reported for a MOF photocatalyst (Table S2†). In addition, the activity was generally maintained even after a long–term reaction for 24 h and a 3-times recycle test (Fig. S1†). However, the crystallinity and BET surface area slightly declined, which may be due to the nanosheeting of Al-TCPP4-TBAPy6 by ultrasonic treatment before the reaction.27 These results suggest that the Al-TCPP4-TBAPy6 photocatalyst is both highly stable and reusable.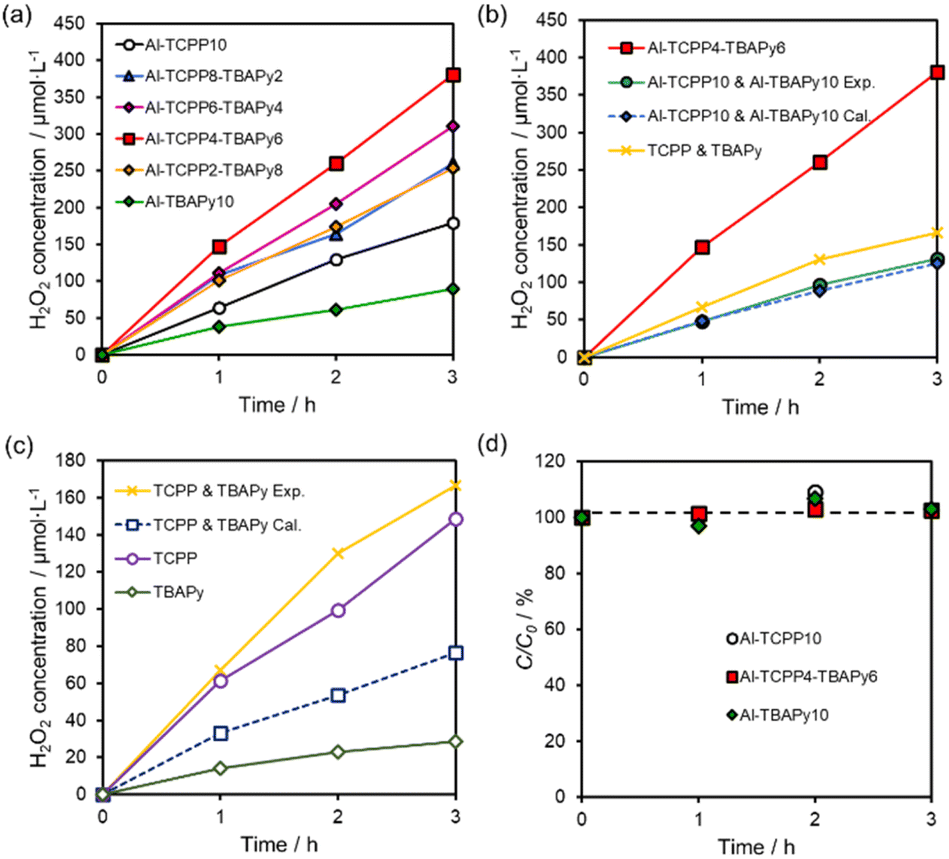 | ||
| Fig. 4 (a) Comparison of photocatalytic H2O2 production over Al-TCPP(10-X)-TBAPyX samples. (b) Photocatalytic H2O2 production using Al-TCPP4-TBAPy6, physical mixture of Al-TCPP10 and Al-TBAPy10 or physical mixture of TCPP and TBAPy. (c) Comparison of H2O2 production catalysed by TCPP, TBAPy and physical mixture of TCPP and TBAPy. (d) H2O2 decomposition in the presence of Al-TCPP10, Al-TCPP4-TBAPy6 and Al-TBAPy10 under dark conditions (C0,H2O2 = 0.5 mmol L−1). | ||
To investigate the effect of mixing two types of linkers in a single Al-MOF matrix, we compared the activity toward H2O2 production between Al-TCPP4-TBAPy6 and a physical mixture of Al-TCPP10 and Al-TBAPy10 MOFs or a mixture of the organic linker precursors. The mixing ratio for the Al-MOFs or linkers was determined to be equal to the molar ratio of the two linkers in Al-TCPP4-TBAPy6. Notably, Al-TCPP4-TBAPy6 exhibited 2.9-fold or 2.3-fold greater activity than the physical mixture of Al-TCPP10 and Al-TBAPy10 (Al-TCPP10 & Al-TBAPy10 Exp.) or TCPP and TBAPy molecules (Fig. 4b), respectively. The activity of the physical mixture of Al-TCPP10 and Al-TBAPy10 (Al-TCPP10 & Al-TBAPy10 Exp.) was found to be the same as the value calculated from the respective activities of Al-TCPP10 and Al-TBAPy10 (Al-TCPP10 & Al-TBAPy10 Cal., i.e., the value for Al-TCPP10 & Al-TBAPy10 Cal. is the sum of 0.4 times the activity of Al-TCPP10 and 0.6 times the activity of Al-TBAPy10). These results indicate that the internalization of TCPP and TBAPy linkers in a single MOF led to an increase in photocatalytic activity toward H2O2 production.
By contrast, the amount of H2O2 produced by the physical mixture of TCPP and TBAPy molecules (TCPP & TBAPy Exp.) was 2.2 times larger than the value calculated from the respective activity of TCPP and TBAPy (TCPP & TBAPy Cal.) (Fig. 4c). The activity improvement resulting from physical mixing of the linkers was presumably due to easier intermolecular energy transfer between TCPP and TBAPy, unlike the case of physical mixing of the Al-MOFs. Interestingly, Al-TCPP4-TBAPy6 exhibited superior catalytic performance compared with the physical mixture of TCPP and TBAPy molecules (TCPP & TBAPy Exp.). These results indicate that mixing the TCPP and TBAPy linkers within the MOF matrix provides a more efficient energy transfer process than a simple mixture of the two linker molecules. In addition, the formation of the MOF structures contributes to the suppression of activity loss originating from the aggregation of ligands.25,30,31 Therefore, we assumed that the accumulation of TCPP and TBAPy in the single Al-MOFs induced a strong interaction between TCPP and TBAPy and suppressed self-quenching of the ligands, resulting in a higher H2O2 production rate of Al-TCPP(10-X)-TBAPyX.
H2O2 decomposition experiments were conducted in a 0.5 mmol L−1 H2O2 aqueous solution using Al-MOFs (Al-TCPP10, Al-TCPP4-TBAPy6 and Al-TBAPy10) in the dark. The initial concentration of H2O2 was maintained for 3.0 h even in the presence of Al-MOFs (Fig. 4d). Thus, these results indicate that Al-TCPP(10-X)-TBAPyX exhibited poor decomposition ability toward H2O2, enhancing the H2O2 yield in photocatalytic H2O2 production.18 In other words, the Al clusters have two outstanding effects: stabilization of the MOF structure in the reaction solution and extremely low H2O2 degradability, resulting in a highly efficient H2O2 production by Al-TCPP(10-X)-TBAPyX.
Efficient energy transfer from TBAPy to TCPP in Al-TCPP(10-X)-TBAPyX
Steady-state photoluminescence (PL) measurements were performed to elucidate the energy transfer mechanism. Fig. 5a shows PL spectra of Al-TCPP(10-X)-TBAPyX in the wavelength range 430–600 nm, which originated from a TBAPy linker. Compared with the PL intensity for Al-TBAPy10, substantial quenching of the TBAPy-derived PL intensity was observed when the TBAPy and TCPP linkers were mixed. This result indicates the onset of energy transfer from the TBAPy donors to the TCPP acceptors.13,14 The peaks between 630 and 750 nm result from the fluorescence of TCPP linkers (Fig. 5b). Compared with the PL intensity for Al-TCPP10, the PL intensity was enhanced when the TBAPy linkers were mixed within an Al-MOF, with Al-TCPP4-TBAPy6 showing the greatest PL intensity. These results indicate that energy transfer from TBAPy to TCPP occurred within the MOFs, which is attributed to the overlap of the TBAPy-derived emission wavelength and the TCPP-derived absorption wavelength.14–16 In addition, the aforementioned trend in PL intensity is consistent with the activity in photocatalytic H2O2 production, indicating that the superior activity of Al-TCPP4-TBAPy6 is attributable to efficient energy transfer from TBAPy to TCPP, which increases the excitation efficiency of the reaction site, TCPP.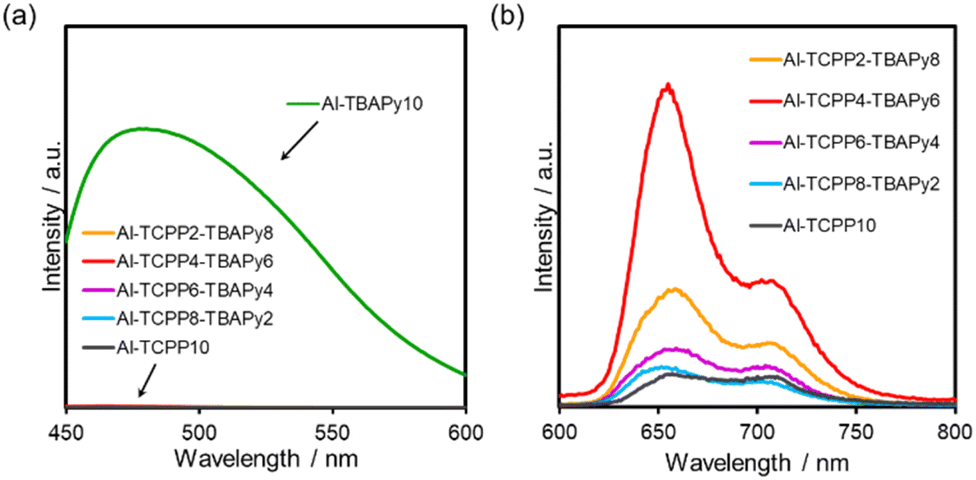 | ||
| Fig. 5 Photoluminescence spectra of Al-TCPP(10-X)-TBAPyX, where photoluminescence originated from (a) TBAPy (excitation wavelength = 420 nm) and (b) TCPP (excitation wavelength = 420 nm). | ||
Fig. 6a shows time-resolved PL spectra of TBAPy linkers in Al-TBAPy10 and Al-TCPP4-TBAPy6 excited at 405 nm and monitored at 480 nm. The average PL lifetime of the TBAPy linkers was dramatically decreased from 2.81 to 0.49 ns when TCPP was mixed into the Al-MOF matrix (Table S3†). This shortened PL lifetime suggests that energy transfer from TBAPy to TCPP occurred in Al-TCPP4-TBAPy6, consistent with the large quenching of the steady-state PL intensity observed upon the incorporation of TCPP linkers.16
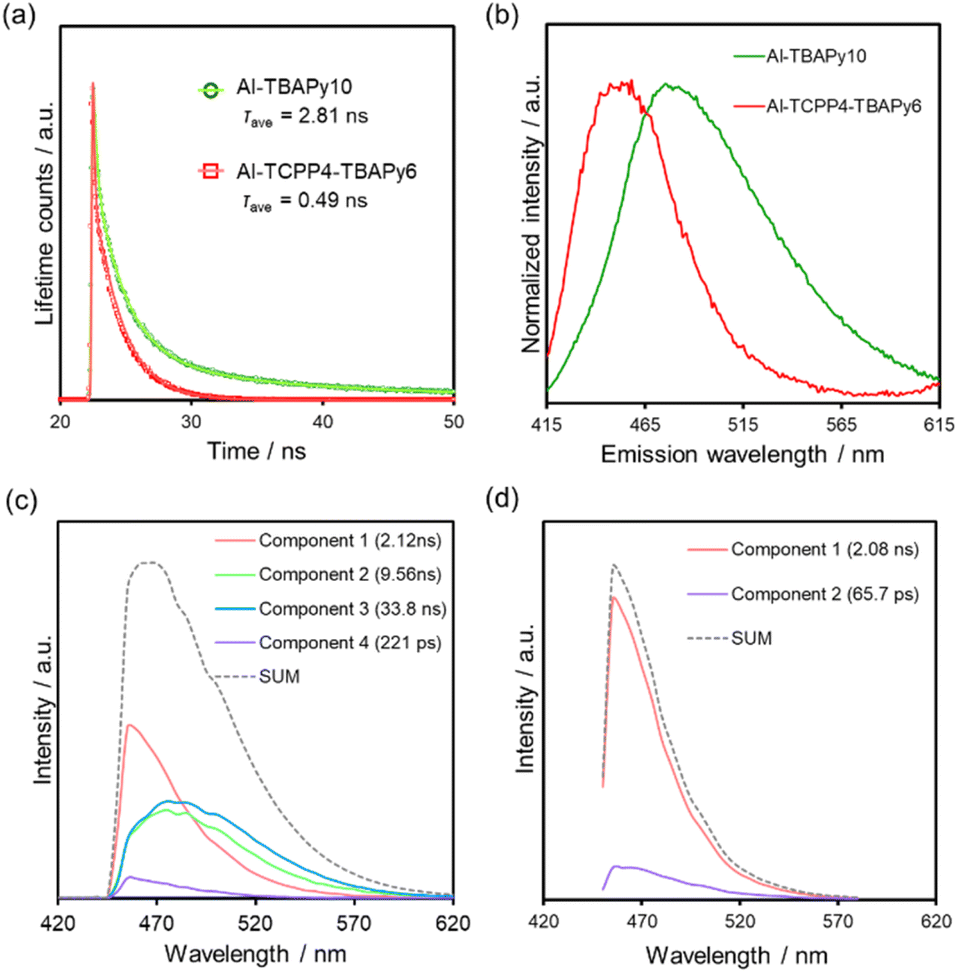 | ||
| Fig. 6 (a) PL lifetimes for Al-TBAPy10 and Al-TCPP4-TBAPy6 (excitation wavelength = 405 nm). (b) Static state of normalized PL spectra for Al-TBAPy10 and Al-TCPP4-TBApy6. TRES spectra of (c) Al-TBAPy10 and (d) Al-TCPP4-TBAPy6. The excitation wavelength for all spectra was 405 nm. | ||
Interestingly, the steady-state PL spectrum originating from the TBAPy of Al-TCPP4-TBAPy6 was blue-shifted compared with that originating from the TBAPy of Al-TBAPy10 (Fig. 6b). To elucidate the blue-shift in the PL spectrum, we conducted time-resolved emission spectroscopy (TRES) measurements. This is a technique used to obtain decay curves for PL lifetimes at multiple emission wavelengths and to determine the PL intensity of each PL lifetime component at each wavelength. As a result, the PL spectrum can be separated for each PL lifetime component. Fig. 6c and d present TRES spectra of Al-TBAPy10 and Al-TCPP4-TBAPy6, which were excited at 405 nm. A comparison of the spectra of Al-TBAPy10 and Al-TCPP4-TBAPy6 reveals that the components with longer PL lifetimes (blue and green) disappeared with the incorporation of the TCPP linkers. The disappearance of the long-lived and long-wavelength emission components is consistent with the shortening of the PL lifetime and the blue-shift of the emission spectrum derived from TBAPy due to the incorporation of TCPP. The longer-wavelength components originate from attenuation at the pyrene cores within the TBAPy linkers.25,32 The luminescence produced by the recombination of the pyrene cores is responsible for the energy transfer from the TBAPy linkers to the TCPP linkers.
Reaction pathway for H2O2 production over Al-TCPP(10-X)-TBAPyX
Fig. 7a shows H2O2 production over Al-TCCP4-TBAPy6. H2O2 was produced in the presence of O2, H2O and visible-light irradiation, demonstrating that H2O2 was produced from O2 and H2O by Al-TCCP4-TBAPy6 via a photocatalytic pathway. Activation of O2 is a key step in the photocatalytic production of H2O2 from O2 and H2O.11,18,33,34 ESR measurements were performed to confirm the active oxygen species involved in the reaction. The radical trapping agents 5,5-dimethyl-1-pyrroline N-oxide (DMPO) and 2,2,6,6-tetramethylpiperidine (TEMP) were used to identify O2˙− and 1O2, respectively.35 No DMPO–O2˙− adducts were detected after the light exposure time was extended (Fig. 7b). This result discounts the involvement of O2˙− as a key active species in this reaction system and suggests that the reaction mechanism differs from the reaction pathway involving O2˙− intermediates, which has been deemed as the main active species in previously reported MOF photocatalysts.5 Clear triplet signals were observed in the presence of TEMP under visible-light irradiation. These signals were identified as being associated with 2,2,6,6-tetramethylpiperidine 1-oxyl (TEMPO), indicating that Al-TCPP4-TBAPy6 produces 1O2via a light-induced energy transfer process (Fig. 7c). | ||
| Fig. 7 (a) Comparison of photocatalytic H2O2 production over Al-TCPP4-TBAPy6 under various conditions. (b and c) ESR spectra demonstrating active oxygen species of Al-TCPP4-TBAPy6 under visible-light irradiation. DMPO and TEMP were used as O2˙− and 1O2 trapping agents, respectively. (d) Comparison of H2O2 concentration over Al-TCPP4-TBAPy6 during 3 h of visible-light irradiation with p-BQ, NaN3 and KBrO3 as O2˙−, 1O2 and electron scavengers, respectively. | ||
To gain insight into the mechanism of photocatalytic H2O2 production over Al-TCCP4-TBAPy6, active-species trapping experiments were carried out using p-benzoquinone (p-BQ), sodium azide (NaN3) and potassium bromate (KBrO3) to capture superoxide radicals (O2˙−), singlet oxygen (1O2) and electrons, respectively.34–36 The addition of p-BQ did not substantially change the amount of H2O2 produced, whereas the addition of NaN3 dramatically suppressed the production of H2O2 (Fig. 7d). These results suggest that H2O2 was produced via a 1O2 intermediate, and not via general single-electron O2 reduction through O2˙− species. This suggests two possibilities for the H2O2 formation pathway: (i) direct two-electron reduction via1O2 species (eqn (2)–(4)) and (ii) H2O oxidation by 1O2 species (eqn (2) and (5)).37–40 The proposed reaction pathway to produce H2O2 over Al-TCPP(10-X)-TBAPyX is expressed as follows:
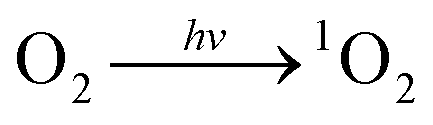 | (2) |
| 1O2 + 2H+ + 2e− → H2O2 | (3) |
| 2H2O + 4h+ → O2 + 4H+ | (4) |
| 1O2 + 2H2O → 2H2O2 | (5) |
The 1O2 intermediate is known to involve the selective two-electron reduction of O2.37,38 In this reaction system, electrons are supplied toward 1O2 from photoexcited electrons of MOFs or H2O. As shown in Fig. 7d, H2O2 production was inhibited in the presence of the electron-trapping agent. This indicates that photoexcited electrons of the MOF also contributed to H2O2 production via the direct two-electron reduction reaction (eqn (3)). Notably, however, energy transfer is also prevented by the electron-trapping agents. Electron transfer is more dominant than energy transfer because electron-trapping agents readily withdraw electrons from excited MOFs.41 In addition, 1O2 species can extract electrons from H2O to produce H2O2 (eqn (5)). A similar reaction has been reported elsewhere, where antibodies were found to produce H2O2 from 1O2via H2O oxidation.39,40 Wentworth et al. demonstrated that H2O3 is formed as an intermediate in the reaction that eventually leads to the formation of H2O2. Thus, 1O2 is generated through energy transfer between the ground state of O2 and triplet-state excited TCPP linkers, and acts as a main species in the formation of H2O2.
Furthermore, to determine whether holes are essential for H2O2 production, we performed a comparison experiment with methanol, a hole sacrificial agent. As shown in Fig. S2,† H2O2 production was dramatically enhanced by adding methanol in the reaction solution. This is because the oxidation reaction is easier in methanol than in water. This result indicates that photogenerated holes are necessary for photocatalytic H2O2 production using Al-TCPP(10-X)-TBAPyX. It is reported that the HOMO level of a TCPP linker in the reaction site of Al-TCPP(10-X)-TBAPyX is enough to undergo water oxidation.28 The above results and the comparison experiments in Fig. 7a present that Al-TCPP(10-X)-TBAPyX can utilize water as an electron source for photocatalytic H2O2 production.
On the basis of the aforementioned results, we propose mechanisms for H2O2 production over Al-TCPP(10-X)-TBAPyX, as follows (Scheme 2). The H2O2 production process is initiated by the photoexcitation of the TBAPy linkers under visible-light irradiation, followed by energy transfer from the TBAPy linkers to the TCPP linkers. The TCPP linkers transfer to the triplet state of the excited TCPP linkers. The triplet state of the TCPP linkers relaxes and provides energy transfer toward O2 to generate 1O2. The 1O2 species are reduced by photoexcited electrons of the MOFs, accompanied by a reaction with H+ in H2O to produce H2O2; by contrast, generated holes may produce O2via H2O oxidation. In addition, 1O2 species can directly oxidize H2O to generate H2O2. The mixing of TBAPy linkers and TCPP linkers in an Al-MOF matrix accelerates efficient energy transfer and inhibits relaxation.
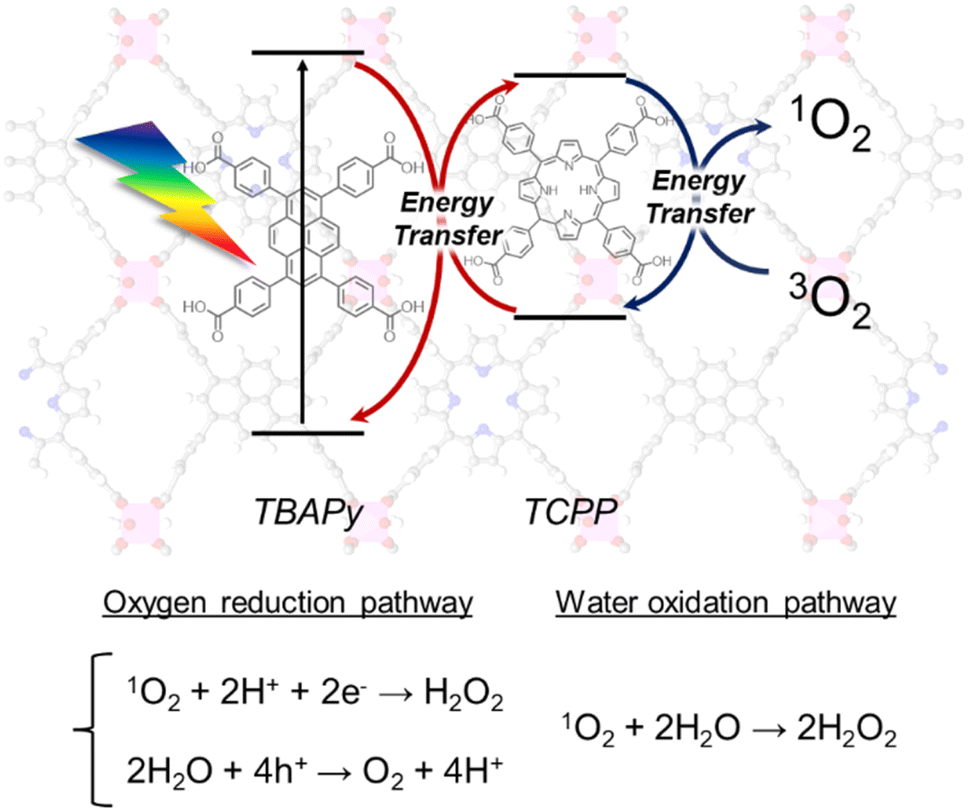 | ||
| Scheme 2 Proposed mechanism for photocatalytic H2O2 production from O2 and H2O over Al-MOFs containing both TBAPy and TCPP linkers via efficient energy transfer. | ||
Conclusions
In conclusion, we successfully synthesized a series of Al-based MOFs integrated with TBAPy and TCPP. Al-TCPP(10-X)-TBAPyX showed a phase transformation from Al-TCPP to Al-TBAPy without two-phase separation as the TBAPy-to-TCPP ratio was increased. These Al-MOF photocatalysts enabled the photocatalytic production of H2O2 from O2 and H2O without any additive under visible-light irradiation. In addition, the efficient energy transfer from the TCPP to the TBAPy enhanced the photocatalytic activity, which was maximal for Al-TCPP4-TBAPy6. The efficient energy transfer was maximized by the integration of both TCPP and TBAPy into a single Al-MOF. In addition, the poor H2O2 decomposition ability of Al-TCPP(10-X)-TBAPyX allowed for a high H2O2 yield. In this reaction system using Al-TCPP(10-X)-TBAPyX, 1O2 was found to be the main active species for H2O2 production, which is completely different from the H2O2 production pathway previously reported for MOF photocatalysts. This work provides new insights into MOF photochemistry and proposes a new reaction pathway for photocatalytic H2O2 production.Author contributions
The manuscript was written through the contributions of all authors. All authors approved the final version of the manuscript. Y. Kondo conceived the project, performed the catalyst preparation, characterization, catalytic reactions, and wrote the manuscript. K. H. performed the catalyst preparation, characterization, and catalytic reactions. Y. Kuwahara assisted in N2 sorption measurements and supported the project. K. M. helped PL measurements and supported the project. H. Y. supervised the project and the experiments. The manuscript was written through the discussion with all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by Grants-in-Aid for Scientific Research (KAKENHI) from the Japan Society for the Promotion of Science (JSPS) (No. 22H00275). Y. K. thanks JSPS for a Research Fellowship for Young Scientists (No. 21J10556). Y. K. thanks the cooperative research program of the “Network Joint Research Center for Materials and Devices” (No. 20211069 and 20221014).Notes and references
- X. Tao, Y. Zhao, S. Wang, C. Li and R. Li, Chem. Soc. Rev., 2022, 51, 3561–3608 RSC.
- P. Verma, Y. Kondo, Y. Kuwahara, T. Kamegawa, K. Mori, R. Raja and H. Yamashita, Catal. Rev., 2021, 63, 165–233 CrossRef CAS.
- Y. Yamada, M. Yoneda and S. Fukuzumi, Energy Environ. Sci., 2015, 8, 1698–1701 RSC.
- Y. Isaka, Y. Kondo, Y. Kawase, Y. Kuwahara, K. Mori and H. Yamashita, Chem. Commun., 2018, 54, 9270–9273 RSC.
- Y. Kondo, Y. Kuwahara, K. Mori and H. Yamashita, Chem, 2022, 8, 2924–2938 CAS.
- Y. Yamada, M. Yoneda and S. Fukuzumi, Inorg. Chem., 2014, 53, 1272–1274 CrossRef CAS PubMed.
- Y. Shiraishi, T. Takii, T. Hagi, S. Mori, Y. Kofuji, Y. Kitagawa, S. Tanaka, S. Ichikawa and T. Hirai, Nat. Mater., 2019, 18, 985–993 CrossRef CAS PubMed.
- H. Hou, X. Zeng and X. Zhang, Angew. Chem., Int. Ed., 2020, 59, 17356–17376 CrossRef CAS PubMed.
- X. Zeng, Y. Liu, X. Hu and X. Zhang, Green Chem., 2021, 23, 1466–1494 RSC.
- H. Cheng, J. Cheng, L. Wang and H. Xu, Chem. Mater., 2022, 34, 4259–4273 CrossRef CAS.
- S. Wu and X. Quan, ACS ES&T Eng., 2022, 2, 1068–1079 Search PubMed.
- Y. Kondo, K. Honda, Y. Kuwahara, K. Mori, H. Kobayashi and H. Yamashita, ACS Catal., 2022, 12, 14825–14835 CrossRef CAS.
- S. M. Shaikh, S. Ilic, B. J. Gibbons, X. Yang, E. Jakubikova and A. J. Morris, J. Phys. Chem. C, 2021, 125, 22998–23010 CrossRef CAS.
- K. C. Park, C. Seo, G. Gupta, J. Kim and C. Y. Lee, ACS Appl. Mater. Interfaces, 2017, 9, 38670–38677 CrossRef CAS.
- P. Cai, M. Xu, S. S. Meng, Z. Lin, T. Yan, H. F. Drake, P. Zhang, J. Pang, Z. Y. Gu and H. C. Zhou, Angew. Chem., Int. Ed., 2021, 60, 27258–27263 CrossRef CAS PubMed.
- M. Kim, J. S. Oh, B. H. Kim, A. Y. Kim, K. C. Park, J. Mun, G. Gupta and C. Y. Lee, Inorg. Chem., 2020, 59, 12947–12953 CrossRef CAS PubMed.
- X. Chen, Y. Kondo, Y. Kuwahara, K. Mori, C. Louis and H. Yamashita, Phys. Chem. Chem. Phys., 2020, 22, 14404–14414 RSC.
- Y. Kondo, K. Hino, Y. Kuwahara, K. Mori, H. Kobayashi and H. Yamashita, Chem. Commun., 2022, 58, 12345–12348 RSC.
- Y. Isaka, Y. Kondo, Y. Kuwahara, K. Mori and H. Yamashita, Catal. Sci. Technol., 2019, 9, 1511–1517 RSC.
- Y. Kondo, Y. Kuwahara, K. Mori and H. Yamashita, J. Phys. Chem. C, 2021, 125, 27909–27918 CrossRef CAS.
- P. Asselin and P. D. Harvey, ACS Appl. Nano Mater., 2022, 5, 6055–6082 CrossRef CAS.
- J. D. Xiao and H. L. Jiang, Acc. Chem. Res., 2019, 52, 356–366 CrossRef CAS.
- A. Dhakshinamoorthy, A. M. Asiri and H. García, Angew. Chem., Int. Ed., 2016, 55, 5414–5445 CrossRef CAS PubMed.
- A. Fateeva, P. A. Chater, C. P. Ireland, A. A. Tahir, Y. Z. Khimyak, P. V. Wiper, J. R. Darwent and M. J. Rosseinsky, Angew. Chem., Int. Ed., 2012, 124, 7558–7562 CrossRef.
- F. P. Kinik, A. Ortega-Guerrero, F. M. Ebrahim, C. P. Ireland, O. Kadioglu, A. Mace, M. Asgari and B. Smit, ACS Appl. Mater. Interfaces, 2021, 13, 57118–57131 CrossRef CAS PubMed.
- P. G. Boyd, A. Chidambaram, E. García-Díez, C. P. Ireland, T. D. Daff, R. Bounds, A. Gładysiak, P. Schouwink, S. M. Moosavi, M. M. Maroto-Valer, J. A. Reimer, J. A. R. Navarro, T. K. Woo, S. Garcia, K. C. Stylianou and B. Smit, Nature, 2019, 576, 253–256 CrossRef CAS PubMed.
- M. Jian, R. Qiu, Y. Xia, J. Lu, Y. Chen, Q. Gu, R. Liu, C. Hu, J. Qu, H. Wang and X. Zhang, Sci. Adv., 2020, 6, 1–10 Search PubMed.
- N. Sadeghi, S. Sharifnia and T. O. Do, J. Mater. Chem. A, 2018, 6, 18031–18035 RSC.
- K. C. Stylianou, R. Heck, S. Y. Chong, J. Bacsa, J. T. A. Jones, Y. Z. Khimyak, D. Bradshaw and M. J. Rosseinsky, J. Am. Chem. Soc., 2010, 132, 4119–4130 CrossRef CAS PubMed.
- T. Toyao, N. Ueno, K. Miyahara, Y. Matsui, T. H. Kim, Y. Horiuchi, H. Ikeda and M. Matsuoka, Chem. Commun., 2015, 51, 16103–16106 RSC.
- Z. Wei, Z. Y. Gu, R. K. Arvapally, Y. P. Chen, R. N. McDougald, J. F. Ivy, A. A. Yakovenko, D. Feng, M. A. Omary and H. C. Zhou, J. Am. Chem. Soc., 2014, 136, 8269–8276 CrossRef CAS.
- J. X. Wang, J. Yin, O. Shekhah, O. M. Bakr, M. Eddaoudi and O. F. Mohammed, ACS Appl. Mater. Interfaces, 2022, 14, 9970–9986 CrossRef CAS.
- L. Li, L. Xu, Z. Hu and J. C. Yu, Adv. Funct. Mater., 2021, 31, 2106120 CrossRef CAS.
- M. Sun, X. Wang, Y. Li, H. Pan, M. Murugananthan, Y. Han, J. Wu, M. Zhang, Y. Zhang and Z. Kang, ACS Catal., 2022, 12, 2138–2149 CrossRef CAS.
- Y. Nosaka and A. Y. Nosaka, Chem. Rev., 2017, 117, 11302–11336 CrossRef CAS PubMed.
- C. Liang, J. Xie, S. Luo, C. Huang, Q. Zhang, H. Huang and P. Zhang, Nat. Commun., 2021, 12, 2–10 CrossRef PubMed.
- S. Zhao and X. Zhao, Appl. Catal. B Environ., 2019, 250, 408–418 CrossRef CAS.
- J. Luo, Y. Liu, C. Fan, L. Tang, S. Yang, M. Liu, M. Wang, C. Feng, X. Ouyang, L. Wang, L. Xu, J. Wang and M. Yan, ACS Catal., 2021, 11, 11440–11450 CrossRef CAS.
- P. Wentworth, L. H. Jones, A. D. Wentworth, X. Zhu, N. A. Larsen, I. A. Wilson, X. Xu, W. A. Goddard, K. D. Janda, A. Eschenmoser and R. A. Lerner, Science, 2001, 293, 1806–1811 CrossRef CAS PubMed.
- X. Xu, R. P. Muller and W. A. Goddard, Proc. Natl. Acad. Sci., 2002, 99, 3376–3381 CrossRef CAS.
- X. Wang, K. Ma, T. Goh, M. R. Mian, H. Xie, H. Mao, J. Duan, K. O. Kirlikovali, A. E. B. S. Stone, D. Ray, M. R. Wasielewski, L. Gagliardi and O. K. Farha, J. Am. Chem. Soc., 2022, 144, 12192–12201 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ta01051a |
| This journal is © The Royal Society of Chemistry 2023 |