
Photothermal-coupled solar photocatalytic CO2 reduction with high efficiency and selectivity on a MoO3−x@ZnIn2S4 core–shell S-scheme heterojunction†
Renzhi
Xiong
,
Xiaoxue
Ke
,
Weifeng
Jia
,
Yanhe
Xiao
,
Baochang
Cheng
 and
Shuijin
Lei
*
and
Shuijin
Lei
*
School of Physics and Materials Science, Nanchang University, Nanchang, Jiangxi 330031, P. R. China. E-mail: shjlei@ncu.edu.cn
First published on 5th January 2023
Abstract
Photocatalytic technology to convert CO2 into a chemical fuel is one of the most promising ways to alleviate the greenhouse effect. However, due to the low photocatalytic efficiency and poor product selectivity, the application of photocatalytic CO2 reduction is seriously limited. In this study, a MoO3−x@ZnIn2S4 composite with a core–shell structure is designed for the first time, and the combination of an S-scheme heterojunction and photothermal synergistic catalysis is successfully applied to full-spectrum solar photocatalytic CO2 reduction. Thanks to the cooperative effects of its unique hierarchical architecture, close interface contact, special charge-transfer pathway and high photothermal efficiency, the MoO3−x@ZnIn2S4 composite photocatalyst exhibits average yields of CO and CH4 up to 4.65 and 28.3 μmol g−1 h−1 under UV-Vis-IR irradiation without a sacrificial agent and cocatalyst. The average yield of CH4 is 19.4 and 11.7 times that of pure MoO3−x and ZnIn2S4 samples, respectively. Moreover, it also shows a CH4 selectivity as high as 85.89%.
Introduction
With the rapid growth of population and the continuous expansion of industry, the consumption of fossil energy has increased sharply, which brings about a serious greenhouse effect and energy shortage. As a culprit of the greenhouse effect, CO2 can be converted into clean energy and additional chemical products by various methods, which is able to not only effectively alleviate environmental and climate problems, but also provide a new path for energy production.1,2 So far, the prevalent CO2 reduction methods include photocatalysis,3 electrochemical reduction,4 and biological immobilization.5 Among them, photocatalysis has attracted much attention as a sustainable, green and cost-effective technique. It has been widely applied in various catalytic reactions such as CO2 conversion,6 pollutant degradation,7 water splitting,8 and nitrogen fixation.9,10 However, the low photocatalytic efficiency and poor product selectivity have greatly limited the application of photocatalytic CO2 reduction.Since photocatalytic CO2 reduction is endothermic in thermodynamics, in order to improve its conversion efficiency and product selectivity, researchers have strived to introduce thermal effects in photocatalytic reactions to achieve photothermal synergistic catalysis.11 Higher temperature can be beneficial to improve the efficiency of photocatalytic CO2 reduction in several aspects: (1) accelerating the transfer of photogenerated charges to the catalyst surface and improving the separation efficiency of electrons and pores; (2) activating specific chemical bonds of reactants and increasing the selectivity of products; (3) providing some ability to cross the activation energy barrier and enhancing the reaction rate; (4) intensifying the thermal motion of molecules, promoting the adsorption and desorption of reactants and products, and enabling the rapid regeneration of active sites on the catalyst surface.12–16 Moreover, as a crucial factor affecting the reaction direction, temperature also has a critical influence on photocatalytic CO2 reduction. Generally, the increase of temperature can change the balance of the chemical reaction, thereby achieving higher reaction efficiency.17,18 The local surface plasmon resonance (LSPR) effect produced by noble metal nanoparticles can effectively convert light energy into thermal energy, thus significantly increasing the catalytic efficiency and product selectivity without an additional external heat source.19 In recent years, the LSPR effect has also been found in some non-stoichiometric compounds, such as WO3−x,20 MoO3−x,21 W18O49,22 Cu2−xS,23 and so on. Compared with noble metals, these compounds are not only low-cost and environmentally friendly, but also have semiconductor properties, allowing them to generate high-energy hot electrons under light irradiation. As a result, they themselves have the ability of photothermal synergistic catalytic reactions.
Due to the large number of oxygen vacancies in MoO3−x, Mo6+ ions are reduced to low-valent Mo5+, which leads to a great increase in the concentration of free electrons, and thus makes MoO3−x have a LSPR effect.24 In this case, the light absorption range of the material can be extended to the visible region and even to the near infrared region on account of the resonance absorption of free electrons. Under the excitation of light at a specific wavelength, electrons transition to form high-energy hot electrons and holes to participate in the corresponding reactions. At the same time, the conversion of these high-energy hot electrons into phonons converts chemical energy into thermal energy, which raises the surface temperature of the material, thus promoting certain catalytic reactions.25 Based on these advantages, the photothermal effect of MoO3−x has been widely used in photocatalytic reactions such as CO2 reduction,26 water splitting,27 and pollutant degradation.28 Unfortunately, because of the low efficiency of photogenerated charge separation, the photocatalytic performance of a single-phase MoO3−x material is greatly reduced, which limits its extensive application.
Combining two semiconductor materials with appropriate energy band structures to form a heterojunction has always been an effective strategy to improve the charge separation efficiency and catalyst performance. In addition, the reduction of CO2 to CH4 is an eight-electron reaction process, which requires more effective electrons to participate in the reaction to improve the CH4 selectivity.29 Therefore, the construction of a heterojunction can also increase the selectivity of products in photocatalytic CO2 reduction.30 Although the traditional type-I and type-II heterojunctions can improve the charge separation, the reducibility/oxidizability of photogenerated electrons/holes are greatly diminished. Furthermore, owing to the electrostatic effect, the electrons and holes in the heterojunction interact with each other, thereby inhibiting the charge transfer at interfaces.31 Recently, an S-scheme heterojunction has been demonstrated to provide a new way for the transfer of photogenerated charges under the action of an internal electric field (IEF) and electrostatic attraction, which not only essentially facilitates the separation and migration of photogenerated charges, but also maximizes the retention of the strong redox capacity of charges.32
ZnIn2S4 is a kind of multinary sulfides with excellent photocatalytic performance. Its suitable band gap and tunable morphology make it widely applicable in photocatalytic CO2 reduction.33 However, the poor light stability, narrow photoabsorption range and low charge separation efficiency are important bottlenecks restricting its practical application. Researchers have successfully combined numerous materials such as Bi4Ti3O12,34 g-C3N4,35 ZnWO4,36 and CeO2 (ref. 37) with ZnIn2S4 to construct S-scheme heterojunctions and applied them to various fields of photocatalysis. In this case, the photocorrosion phenomenon can be substantially improved, and the photocatalytic performance is greatly enhanced. Meanwhile, the effective contact area in the heterojunction also has great influence on the charge transfer. Generally, the construction of a core–shell architecture using one-dimensional (1D) and two-dimensional (2D) structures can form more effective interfacial contacts, which are more favorable for the transfer of photogenerated charges at the interface.38
Herein, 1D needle-like MoO3−x submicron rods are fabricated via a solvothermal reduction route, and the MoO3−x@ZnIn2S4 composites with a core–shell structure can be successfully prepared by in situ growth of ZnIn2S4 nanosheets on the surface of MoO3−x by a low-temperature reflux method. Although the ZnIn2S4@MoO3 composite has been previously reported for photocatalytic degradation of tetracycline hydrochloride,39 to the best of our knowledge, the construction of a MoO3−x@ZnIn2S4 composite has not been reported. In particular, the synergistic effect of photothermal conversion and the S-scheme heterojunction for photocatalytic CO2 reduction is rarely studied. The results demonstrate that the unique charge transfer mode of the S-scheme heterojunction in the prepared MoO3−x@ZnIn2S4 system is more beneficial to the photocatalytic reaction. Additionally, the LSPR effect due to the high concentration of oxygen vacancies in MoO3−x leads to more energetic hot electrons and higher catalyst surface temperature, which improves the reaction efficiency and product selectivity of CO2 reduction.
Experimental
Chemicals and materials
All chemicals and materials in this work were directly used without further purification. Anhydrous zinc chloride (ZnCl2) and thioacetamide (CH3CSNH2, TAA) were supplied by Macklin Biochemical Co., Ltd. (Shanghai, China). Nitric acid (HNO3) and indium chloride tetrahydrate (InCl3·4H2O) were purchased from Sinopharm Chemical Reagent Co., Ltd. (Shanghai, China). Ammonium molybdate tetrahydrate [(NH4)6Mo7O24·4H2O], glycerol (CH2OHCHOHCH2OH), and absolute ethanol (CH3CH2OH) were obtained from Xilong Science Co., Ltd. (Shantou, China). Deionized water was produced by using a Milli-Q pure water system (Merck, Germany).Synthesis of 1D MoO3
MoO3 rod-shaped crystals were prepared by a solvothermal method. (NH4)6Mo7O24·4H2O (1.5 mmol, 1.854 g) was dispersed in 40 mL of deionized water and stirred at room temperature for 5 min. Subsequently, 10 mL of concentrated nitric acid was added to the suspension and stirred for 30 min to obtain a transparent solution, which was then transferred to a 70 mL Teflon lined stainless steel autoclave (Anhui Kemi Machinery Technology Co., Ltd., Hefei, China) and heated at 180 °C for 17 h. After cooling down to room temperature naturally, the white precipitate was isolated by centrifugation, washed alternately with deionized water and absolute ethanol three times, and finally dried under vacuum at 60 °C for a day.Synthesis of 1D MoO3−x
MoO3 rod-shaped crystals were transformed into MoO3−x needle-like crystals by solvothermal reduction using absolute ethanol as the reducing agent. Typically, 0.5 g of 1D MoO3 precursor was added into 50 mL of absolute ethanol and stirred at room temperature for 30 min. The obtained suspension was transferred to a Teflon lined stainless steel autoclave and heated at 140 °C for 12 h. After cooling naturally to room temperature, a dark blue precipitate was collected by centrifugation, washed three times alternately with deionized water and absolute ethanol, and finally dried under vacuum at 60 °C for a day.Synthesis of MoO3−x@ZnIn2S4 composites
The MoO3−x@ZnIn2S4 core–shell composites were fabricated via a low-temperature reflux route. First, 100y mg (y = 1, 2, 3, 4, and 5) of the as-obtained 1D MoO3−x powder precursor was ultrasonically dispersed in 50 mL of glycerol aqueous solution (VH2O![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :VGly = 4:1) for 15 min. Afterwards, ZnCl2 (1 mmol, 0.1363 g), InCl3·4H2O (2 mmol, 0.5865 g) and TAA (6 mmol, 0.4508 g) were added to the above-mentioned MoO3−x suspension, and stirred at room temperature for 30 min. The resulting solution was transferred to a 100 mL round bottom flask, heated to 85 °C in a mantle, and refluxed under continuous stirring for 2 h. After reaction, it was cooled to room temperature naturally, and the powder could be separated by centrifugation. The product was then washed three times alternately with deionized water and absolute ethanol, and eventually dried under vacuum at 60 °C for 24 h. The prepared composite sample was labeled MO@ZIS-y. A ZnIn2S4 single phase sample was prepared under the same experimental conditions without the introduction of the MoO3−x precursor. The synthesis processes are illustrated in Scheme 1.
:VGly = 4:1) for 15 min. Afterwards, ZnCl2 (1 mmol, 0.1363 g), InCl3·4H2O (2 mmol, 0.5865 g) and TAA (6 mmol, 0.4508 g) were added to the above-mentioned MoO3−x suspension, and stirred at room temperature for 30 min. The resulting solution was transferred to a 100 mL round bottom flask, heated to 85 °C in a mantle, and refluxed under continuous stirring for 2 h. After reaction, it was cooled to room temperature naturally, and the powder could be separated by centrifugation. The product was then washed three times alternately with deionized water and absolute ethanol, and eventually dried under vacuum at 60 °C for 24 h. The prepared composite sample was labeled MO@ZIS-y. A ZnIn2S4 single phase sample was prepared under the same experimental conditions without the introduction of the MoO3−x precursor. The synthesis processes are illustrated in Scheme 1.
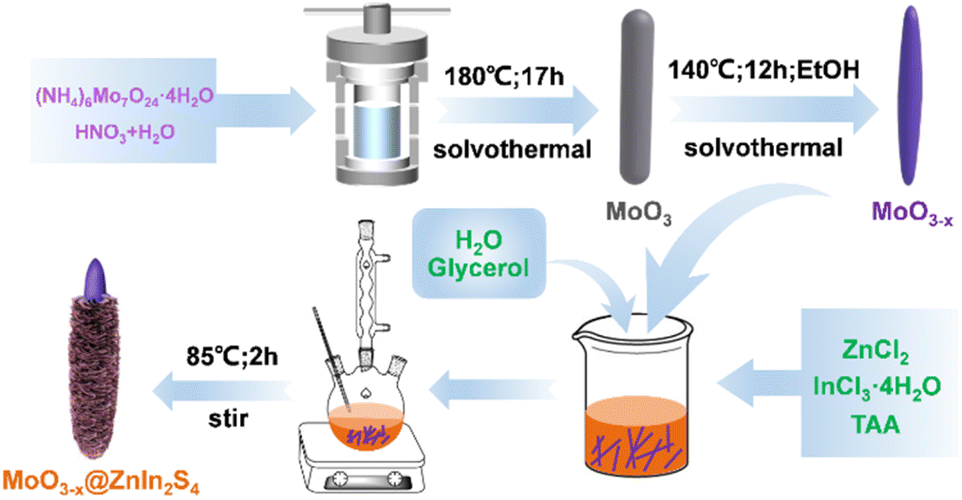 | ||
| Scheme 1 Schematic illustration for the preparation of the MoO3−x@ZnIn2S4 composites. | ||
Sample characterization and photoelectrochemical tests
The material characterization results including X-ray diffraction (XRD) patterns, scanning electron microscopy (SEM) images, energy dispersive spectrometer (EDS) spectra, transmission electron microscopy (TEM) images, high resolution transmission electron microscopy (HRTEM) images, selected area electron diffraction (SAED) patterns, X-ray photoelectron spectroscopy (XPS) spectra, valence band XPS (VB-XPS) spectra, nitrogen adsorption–desorption isotherms and pore size distribution curves, ultraviolet-visible-near infrared (UV-Vis-NIR) absorption spectra, photoluminescence (PL) emission spectra, and time-resolved photoluminescence (TR-PL) decay spectra, as well as the photoelectrochemical tests can refer to our previous work.40 The EDS elemental mapping images in this work were obtained by both SEM and TEM tests. In addition, to determine the presence of oxygen vacancies, 10 mg of product was put into the sample tube of a Bruker EMX-PLUS spectrometer for room temperature electron paramagnetic resonance (EPR) spectroscopy. Inductively coupled plasma optical emission spectroscopy (ICP-OES) tests were performed on an Agilent 730 device (Agilent Technologies, USA). In situ XPS data were obtained on an AXIS Supra X-ray photoelectron spectrometer (Kratos, Japan), during which a 300 W xenon lamp (PLS-SXE 300+, Beijing Perfect Light Technology Co., Ltd.) provided the simulated sunlight exposure. In the transient photocurrent tests under different light source conditions, the same 300 W xenon lamp as above was used as the simulated solar light source (UV-Vis-IR full-spectrum). An infrared cut-off filter (λ ≥ 780 nm) was used to provide ultraviolet-visible (UV-Vis) light irradiation, and an infrared cut-off filter (λ < 800 nm) was used to provide infrared (IR) light irradiation. The amperometric I–t curves of the transient photocurrent response were obtained by measuring the variation of photocurrent with time under continuous application of 1.5 V voltage and repeated intermittent illumination.Photothermal-assisted photocatalytic CO2 reduction tests
All the experiments of photothermal-assisted photocatalytic CO2 reduction were carried out in an enclosed reactor. First, 20 mg of catalyst was evenly coated on a round fiberglass membrane with a diameter of 30 mm, and fixed inside the reactor. Humid CO2 gas (VCO2:VH2O = 95:5) was continuously introduced into the reactor for 1 h to remove excess air from the reactor. Then, the reactor was closed and exposed to a light source for 3 h to conduct photothermal-coupled photocatalytic CO2 reduction experiments under UV-Vis-IR, UV-Vis and IR illumination, respectively. A gas chromatograph equipped with FID and TCD detectors regularly detected the output of CO and CH4, and calculated the average yield and selectivity of products. At the same time, a thermocouple was in contact with the surface of the catalyst to measure its surface temperature in real time. The experimental operations for the photothermal-assisted photocatalytic stability of the catalyst were the same as described above, except that the reactor was purged with moist CO2 gas for 2 h after each cycle of experiment to completely remove the gaseous products from the reactor for the next cycle of the catalytic experiment. The production rate of CH4 (RCH4) and CO (RCO) was evaluated by using the following equations:25,41,42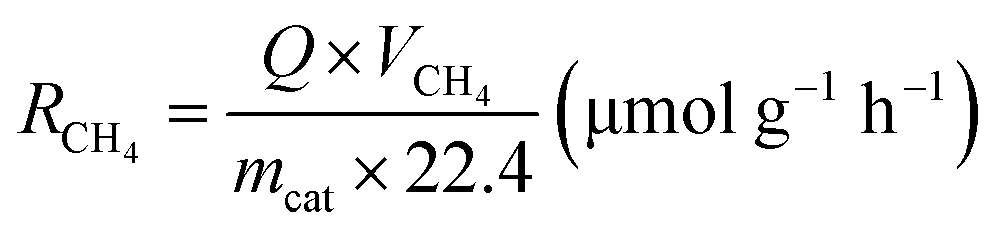 | (1) |
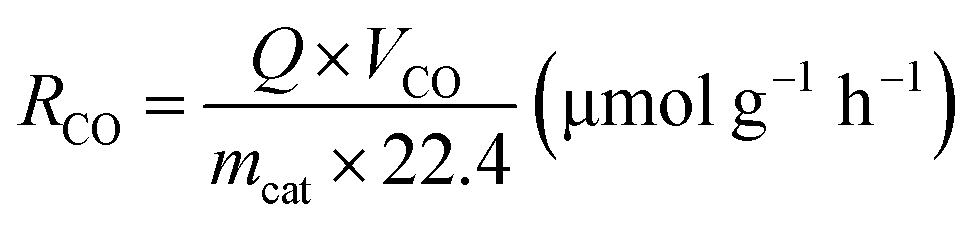 | (2) |
The product selectivity of CH4 and CO was estimated by using the equations as follows:25,41,42
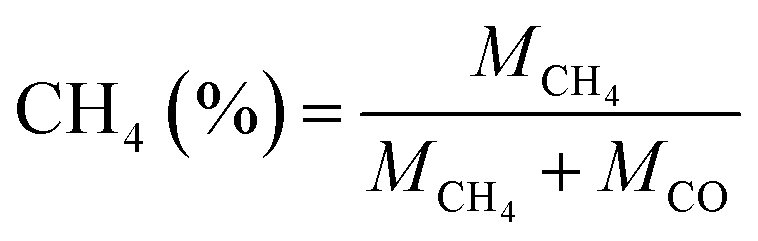 | (3) |
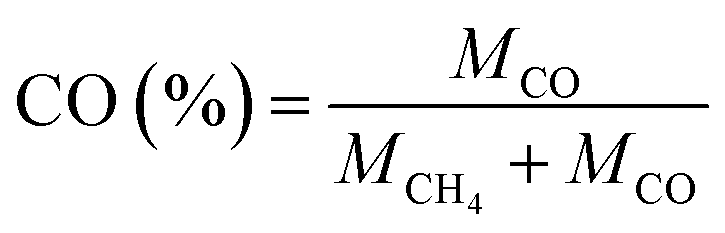 | (4) |
Results and discussion
Phase and composition analysis
In order to determine the phase structure of the prepared samples, they are characterized by XRD. The XRD patterns of the MoO3 sample as well as the corresponding MoO3−x sample obtained after solvothermal reduction are shown in Fig. S1.† For the MoO3 sample, it can be found that all the diffraction peaks can be readily indexed to the orthorhombic structure of MoO3 (JCPDS card file no. 76-1003).43 The diffraction patterns of the MoO3−x sample correspond perfectly to the standard data of the orthorhombic MoO3−x phase (JCPDS card file no. 70-0615). The diffraction peaks at 12.6°, 23.8°, 25.3°, 27.1°, 29.9°, 33.9° and 38.4° can be indexed to the (020), (110), (040), (020) and (130) planes of orthorhombic MoO3−x, respectively.44 It is clear that the relative diffraction intensity of (0k0) planes is extremely prominent for both MoO3 and MoO3−x samples, indicating that there should be a significant growth orientation along the b-axis in the prepared molybdenum oxides crystals. The XRD patterns of the ZnIn2S4 and MoO3−x single-phase samples together with the MoO3−x@ZnIn2S4 samples with different composition ratios are comparatively displayed in Fig. 1. Among them, all diffraction peaks of the ZnIn2S4 sample are in good agreement with those of the hexagonal ZnIn2S4 structure (JCPDS card file no. 65-2023). The characteristic diffraction peaks located at 21.6°, 27.2°, 30.4°, 39.8°, 47.2° and 52.4° correspond to the (006), (102), (104), (108), (110) and (116) reflections, respectively.45 As for the XRD spectra of those MO@ZIS-y composite samples, it can be observed that the diffraction peaks of the MoO3−x phase become more and more obvious with the increase of the y value (i.e., the increase of the MoO3−x content), while the diffraction intensity of the ZnIn2S4 phase gradually decreases. It should be noted that in the MO@ZIS-1 sample, the diffraction peaks of MoO3−x are difficult to identify due to its low content, and the whole diffraction patterns are almost dominated by the diffraction peaks of ZnIn2S4. Therefore, the XRD results demonstrate that MoO3−x@ZnIn2S4 composites have been successfully fabricated. No diffraction peaks of impurity phases are perceived. What's more, in order to prove that oxygen vacancies can be introduced into the MoO3 structure through the ethanol solvothermal reduction strategy, the prepared MoO3, MoO3−x and MO@ZIS-3 samples are tested with EPR, and the results are shown in Fig. S2.† As expected, no EPR signal is detected in MoO3, but a significant EPR signal peak appears at g = 2.003 in both MoO3−x and MO@ZIS-3 samples, which is a strong indication of the successful introduction of oxygen vacancies in MoO3 to produce the non-stoichiometric MoO3−x sample. Normally, the EPR peak intensity is positively related to the oxygen vacancy concentration. The signal intensity of oxygen vacancies in the MO@ZIS-3 composite is higher than that in the MoO3−x sample. This should be due to the strong reducibility of glycerol, which not only protects the oxygen vacancies in MoO3−x from oxidation, but also increases the number of oxygen vacancies in the sample. Oxygen vacancies can generally be used as a trap for photogenerated electrons in photocatalytic reactions, which inhibits the recombination of electrons and holes and makes more effective electrons participate in the CO2 reduction reaction, thus promoting its catalytic efficiency.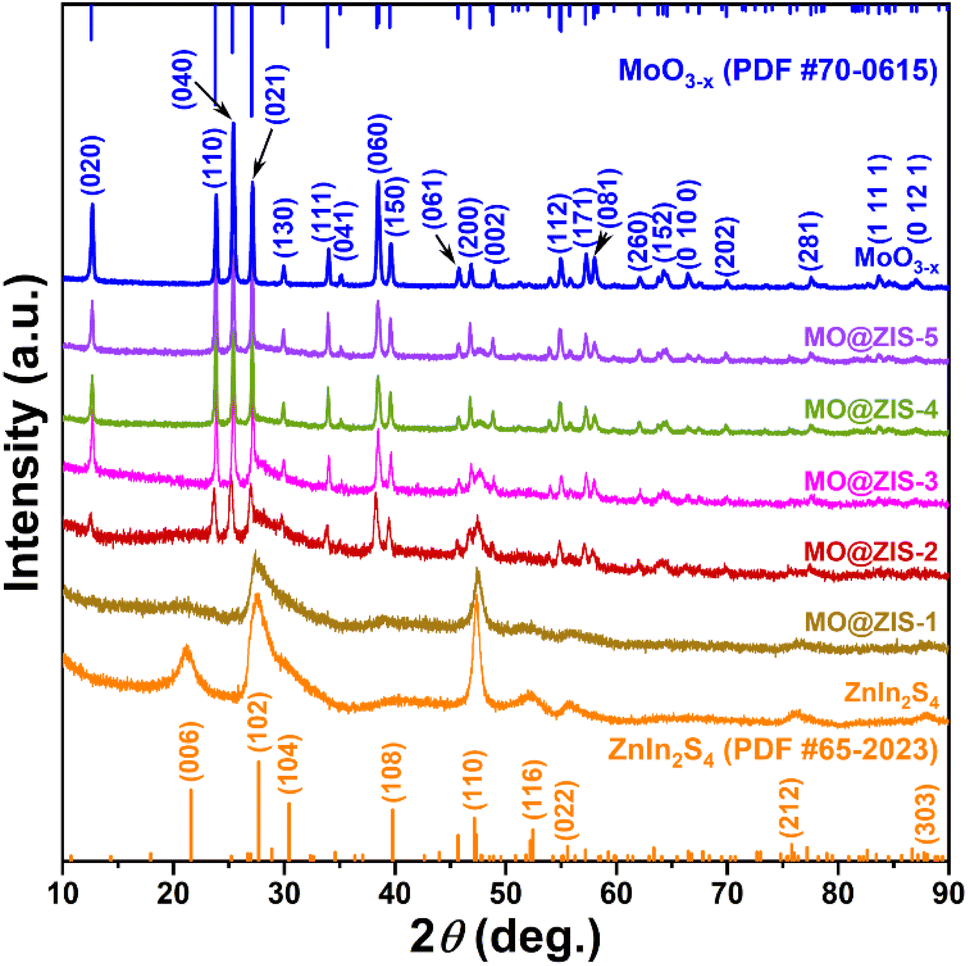 | ||
| Fig. 1 XRD patterns of the as-prepared MoO3−x, ZnIn2S4 and MO@ZIS-y samples. | ||
In order to understand the elemental composition and their valence states, the MoO3, MoO3−x, ZnIn2S4 and MO@ZIS-3 samples are characterized by XPS using C 1s binding energy (284.8 eV) as a reference. The XPS survey spectrum of the MoO3 sample is shown in Fig. S3a.† Except for the characteristic peaks of Mo and O elements, no signals of other elements are observed, suggesting the high purity of the prepared MoO3 sample. Meanwhile, there are only two characteristic peaks of hexavalent Mo6+ in the core-level spectrum of Mo 3d (Fig. S3b†), which can be attributed to Mo6+ 3d5/2 and Mo6+ 3d3/2, respectively.46 In the core-level spectrum of O 1s (Fig. S3c†), it can be divided into three peaks, which are ascribed to lattice oxygen (Olatt, i.e. O–Mo), adsorbed oxygen (Oads) and H2O.47 The survey spectra of the ZnIn2S4, MoO3−x and MO@ZIS-3 samples are presented in Fig. 2a. In the bare ZnIn2S4 and MoO3−x samples, only those peaks of the constituent elements can be detected. As expected, for the MO@ZIS-3 sample, the characteristic peaks of both MoO3−x and ZnIn2S4 phases are observed, confirming their successful compositing. The high-resolution XPS spectra of all elements in the ZnIn2S4, MoO3−x and MO@ZIS-3 samples are provided in Fig. 2b–f. In the ZnIn2S4 sample, the two strong peaks in the core-level spectrum of Zn 2p can be assigned to Zn 2p3/2 (1021.80 eV) and Zn 2p1/2 (1044.84 eV), which proves that Zn exists in the form of +2 valence.48 For the core spectrum of In 3d, the two prominent peaks are ascribed to In 3d5/2 (445.06 eV) and In 3d3/2 (452.64 eV), corresponding to the In3+ state.48 Meanwhile, the S 2p spectrum can be fitted into two component peaks, which are associated with S 2p3/2 (161.67 eV) and S 2p1/2 (162.76 eV).48 As for the MoO3−x sample, the core spectrum of the Mo 3d orbit has two more Mo5+ characteristic peaks (231.49 eV and 234.47 eV) in addition to two Mo6+ characteristic peaks (232.86 eV and 236.01 eV) compared with the MoO3 sample.49 This indicates that the use of ethanol as a solvent successfully reduces part of Mo6+ to Mo5+, resulting in a large number of oxygen vacancies.50 The core-level spectrum of O 1s for the MoO3−x sample is similar to that of MoO3 with three characteristic peaks corresponding to O–Mo (530.43 eV), Oads (531.86 eV), and H2O (533.20 eV). However, it should be noted that the relative intensity of the Oads peak is obviously enhanced in MoO3−x due to the formation of oxygen vacancies. By comparing the core spectrum of each element in ZnIn2S4, MoO3−x and MO@ZIS-3 samples, it can be found that there are two new characteristic peaks in the Mo 3d spectrum of MO@ZIS-3. Among them, the peak at 226.2 eV is related to S 2s, while the peak at 230.05 eV can be considered to be part of Mo4+ in the form of the Mo–S bond under the strong reduction of glycerol.51 Moreover, the peak intensity ratio of Oads to Olatt (Oads/Olatt) is greatly increased after compositing. The larger the Oads/Olatt value, the higher the oxygen vacancy concentration in the product,52 signifying a significant increase of oxygen vacancy concentration in the MO@ZIS-3 composite, which is consistent with the EPR results. The large number of oxygen vacancies allows Mo atoms to have more unpaired electrons, which greatly increases the chance of Mo atoms binding to S atoms to form Mo–S bonds. More importantly, after compositing, the characteristic peaks of Zn, In and S elements all shift to higher binding energy, while those of Mo and O elements all shift to lower binding energy, which means that the electron density in ZnIn2S4 decreases and becomes an electron donor, while the electron density in MoO3−x increases and becomes an electron acceptor. This result demonstrates that there is a strong interaction between ZnIn2S4 and MoO3−x, providing a suitable foundation of the IEF for the construction of an S-scheme heterojunction.53
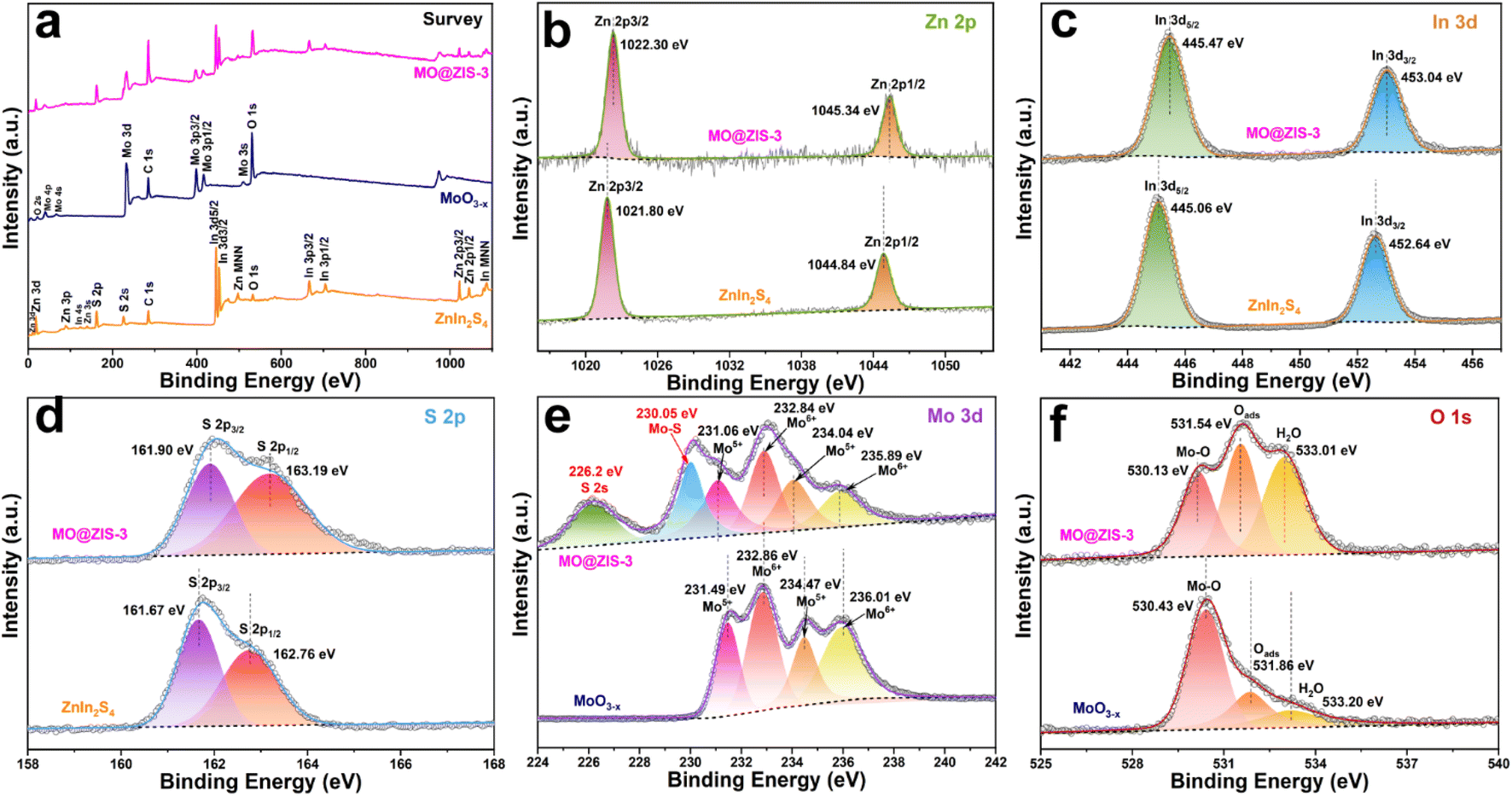 | ||
| Fig. 2 XPS spectra of the prepared MoO3−x, ZnIn2S4, and MO@ZIS-3 samples: (a) survey spectrum, and the corresponding core-level spectra of (b) Zn 2p, (c) In 3d, (d) S 2p, (e) Mo 3d, and (f) O 1s. | ||
Morphology and structure analysis
The interface and contact between the two components in composites usually have a huge impact on the transfer of photogenerated charges. The morphology and microstructure of the fabricated photocatalysts are observed by SEM and TEM measurements. The SEM images of the oxygen-vacancy-free MoO3 sample are shown in Fig. S4a and b,† which is composed of massive and uniform rod-shaped crystals with a length of about 10 μm and a diameter of about 200 nm. When the MoO3 sample is solvothermally reduced to MoO3−x with ethanol, its morphology changes from rod-shaped crystals to needle-like crystals, as shown in Fig. S4c and d.† Their length and diameter are similar to those of the MoO3 precursor, but the diameter at the tip is about 30 nm. The submicron needle-like structure of the MoO3−x sample is further confirmed by the TEM images (Fig. S4e and f†). The HRTEM image taken from a single MoO3−x crystal (Fig. S4g†) displays well-defined lattice fringes, demonstrating its good crystallinity. The lattice fringes with a d-spacing of 0.35 nm correspond to the (040) crystal plane of orthorhombic MoO3−x, revealing that these 1D crystals grow along the b-axis direction, which is totally consistent with the preferred orientation in the XRD results. The SAED patterns (Fig. S4h†) exhibit clear 2D diffraction spots, further confirming the good single crystal nature of MoO3−x 1D crystals. In the EDS spectra of MoO3 and MoO3−x (Fig. S5a and b†), except for Si, Au and C signals originating from the silicon substrate, gold spraying and carbon conductive tape during the SEM test, there are only the characteristic peaks of Mo and O elements. Additionally, from the elemental mapping images of the MoO3−x sample (Fig. S6a–d†), it is observed that Mo and O elements are uniformly distributed in the entire needle-shaped crystals. All results verify the successful synthesis of MoO3 and MoO3−x with high purity. The SEM images (Fig. S7a and b†) of the bare ZnIn2S4 sample show that it consists of nanosheets with a thickness of about 10–20 nm in flower-like clusters. The TEM images (Fig. S7c and d†) also confirm the hierarchical architecture composed of nanosheets. In the HRTEM image of ZnIn2S4 (Fig. S7e†), distinct 2D lattice fringes can also be observed, and the interplanar spacings can be measured to be 0.32 and 0.33 nm, corresponding to the (102) and (100) lattice planes of the hexagonal ZnIn2S4 phase, respectively. The polycrystalline diffraction rings in the SAED patterns (Fig. S7f†) can be readily indexed to the ZnIn2S4 hexagonal structure. The EDS spectrum (Fig. S8†) and the elemental mapping images (Fig. S9a–d†) of the ZnIn2S4 sample reveal the presence of Zn, In and S elements and their uniform distribution. Therefore, high purity ZnIn2S4 nanosheets have been successfully synthesized.The MO@ZIS-3 sample was taken as a representative to explore the composite and interface between ZnIn2S4 and MoO3−x. From the low-magnification SEM images in Fig. 3a and b, it can be observed that the original smooth surface of MoO3−x needle-like crystals is densely covered by a layer of interlaced nanosheets, thereby forming a unique core–shell structure. The morphology and thickness of these nanosheets are basically the same as those of the pure ZnIn2S4 sample. Therefore, it can be inferred that MoO3−x 1D crystals provide a good nucleation and growth substrate for the formation of ZnIn2S4 nanosheets, which makes ZnIn2S4 grow uniformly on the surface of MoO3−x instead of spontaneously aggregating into flower-like clusters. The high-magnification SEM image (Fig. 3c) evidently shows the close contact between ZnIn2S4 and MoO3−x at the interface, which is favourable for charge transfer in the photocatalytic reactions. The TEM images of the MO@ZIS-3 composite (Fig. 3d–f) further verify the core–shell structure with the tight growth of ZnIn2S4 nanosheets on 1D MoO3−x. This 1D/2D core–shell growth not only affords a large effective contact area and a convenient charge transfer channel, but also increases the specific surface area and the catalytically active sites. The microstructure of MO@ZIS-3 is studied by observing the HRTEM images at the interface between ZnIn2S4 and MoO3−x. As displayed in Fig. 3g, two groups of lattice fringes with a spacing of 0.32 nm and 0.35 nm can be ascribed to the (102) plane of ZnIn2S4 and the (040) plane of MoO3−x, respectively. Furthermore, the polycrystalline diffraction rings of ZnIn2S4 and the single crystal diffraction spots of MoO3−x can also be co-observed in the corresponding SAED patterns (Fig. 3h). These results clearly demonstrate that ZnIn2S4 2D nanosheets and MoO3−x 1D acicular crystals form a close contact composite material, rather than a simple mechanical mixture. The signal peaks of Mo, O, Zn, In and S are simultaneously observed in the EDS spectrum of the MO@ZIS-3 sample (Fig. S10†). Furthermore, except for Si, Au and C, there are no signal peaks of other impurity elements. As shown in Fig. 3i, the elemental mapping images of the MO@ZIS-3 sample reveal that Mo and O elements are distributed in the needle core, while Zn, In and S elements are distributed in the nanosheet shell. All these microscopic analyses once again prove that the MoO3−x@ZnIn2S4 core–shell composite with a direct intimate contact is successfully fabricated.
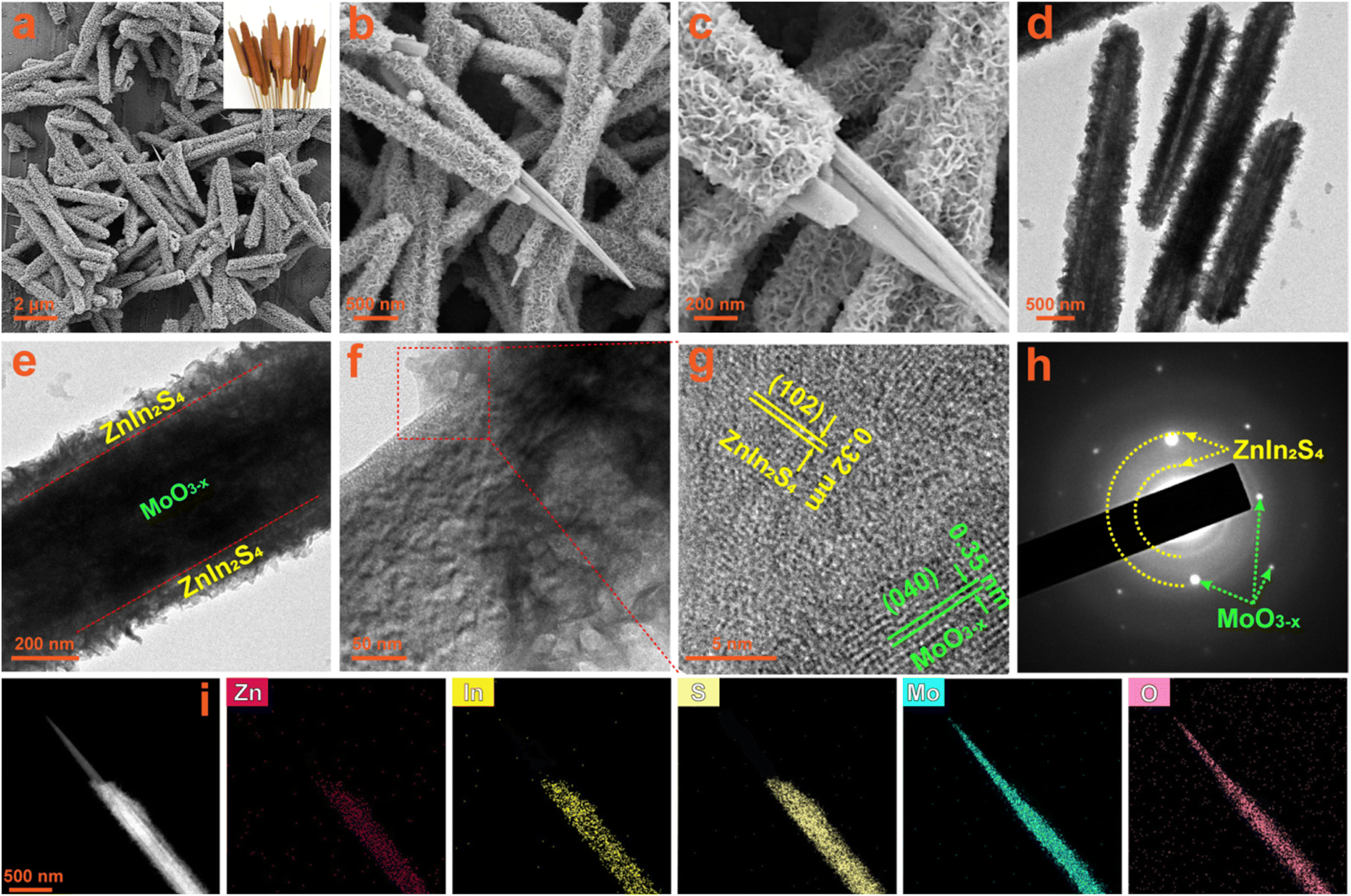 | ||
| Fig. 3 (a–c) SEM images, (d–f) TEM images, (g) HRTEM image, (h) SAED patterns, and (i) EDS elemental mapping images of the prepared MO@ZIS-3 composite. | ||
It is widely aware that a higher specific surface area can supply more adsorption sites for catalytic reactions, increase the contact probability between reactants and catalysts, and thus improve the catalytic activity. In order to explore the specific surface area and pore size distribution of samples, nitrogen (N2) adsorption–desorption isotherm tests are performed on the ZnIn2S4, MoO3−x and MO@ZIS-3 samples. Fig. 4a presents their adsorption–desorption isotherms, and the BET model is used for analysing and calculating the specific surface area. According to the IUPAC classification, ZnIn2S4, MoO3−x and MO@ZIS-3 all exhibit type IV adsorption–desorption isotherms with H3-type hysteresis loops, implying that these samples should be mesoporous materials. The specific surface area of MoO3−x and ZnIn2S4 is 166.8352 and 171.6381 m2 g−1, respectively, while that of the MO@ZIS-3 composite is as high as 237.3724 m2 g−1. The increase of the specific surface area can be attributed to the uniform growth of ZnIn2S4 nanosheets on the surface of MoO3−x 1D crystals, which effectively avoids the agglomeration of ZnIn2S4 nanosheets and then provides richer active sites for CO2 adsorption and photocatalytic reactions. The pore size distribution and average pore volume of the ZnIn2S4, MoO3−x and MO@ZIS-3 samples are determined using the BJH model, and the results are depicted in Fig. 4b. The porous structure of the MoO3−x sample is mainly provided by the surface pores, so its average pore diameter and average pore volume are relatively small. Hierarchical ZnIn2S4 flower clusters have porous structures formed by interlacing and stacking of nanosheets, and thus the average pore size is relatively large. When ZnIn2S4 nanosheets are in situ grown on the surface of MoO3−x crystals, the nanosheets are significantly closer to each other, and the average pore diameter become smaller, between ZnIn2S4 and MoO3−x. Meanwhile, the number of pores on the sample surface is greatly increased, resulting in an enhancement of the average pore volume.
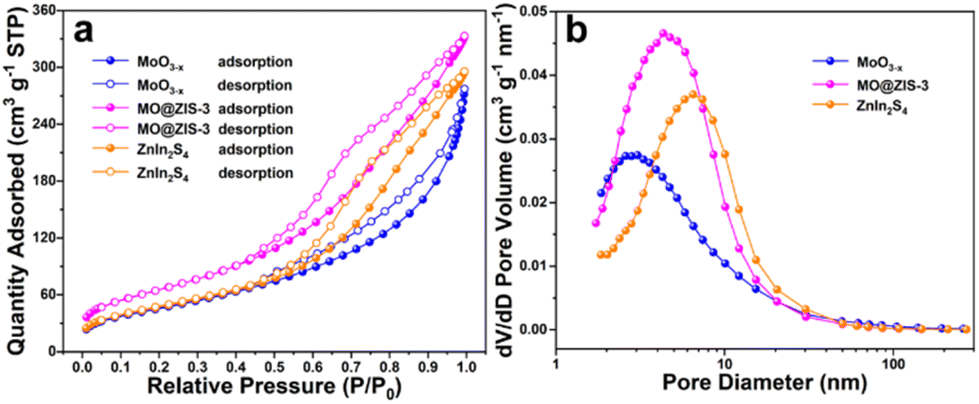 | ||
| Fig. 4 (a) N2 adsorption–desorption isotherms and (b) the corresponding pore size distribution curves for the prepared MoO3−x, MO@ZIS-3, and ZnIn2S4 samples. | ||
Optical absorption properties and energy band structure
As we all know, the low utilization rate of solar energy by photocatalysts has been one of the most important reasons restricting the development of photocatalysis technology. Since visible light accounts for only about 43% of sunlight, it is particularly vital to broaden the light absorption range of the catalysts. The UV-Vis-NIR absorption spectra of the MoO3 and MoO3−x samples are shown in Fig. S11.† Apparently, the light absorption of the MoO3 sample is mainly in the ultraviolet region, with low utilization of visible and near infrared light. However, as for the MoO3−x sample, the oxygen-vacancy-induced LSPR effect gives rise to strong absorption performance in both visible and near infrared regions. The absorption at 200–420 nm should be the intrinsic absorption of MoO3−x, while the broad absorption at 420–2500 nm can be attributed to the absorption band caused by the LSPR effect. The evident color change (from white to dark blue) of the powdery products also reflects a change in their light absorption. The optical absorption properties of the ZnIn2S4, MoO3−x and MoO3−x@ZnIn2S4 samples are contrastively analyzed in Fig. 5a. The absorption edge of the pure ZnIn2S4 sample is near 520 nm, while the optical absorption range can be extended to the near infrared region after ZnIn2S4 is composited with MoO3−x. With the increase of the MoO3−x content in the composites, the optical absorption intensity of the samples increases gradually, indicating that the compositing of MoO3−x and ZnIn2S4 can elevate the utilization efficiency of sunlight. The Tauc equation is employed to convert the optical absorption data, and the hν–(αhν)2 curves are plotted in Fig. 5b. By extending the linear part near the band edge to (αhν)2 = 0, the optical bandgap values of the synthesized ZnIn2S4 and MoO3−x samples are evaluated to be 2.53 eV and 2.42 eV, respectively. Moreover, the valence band (VB) potentials of ZnIn2S4 and MoO3−x samples are 1.61 V and 3.04 V (versus the normal hydrogen electrode, NHE) according to the VB-XPS results (Fig. 5c and d), and then the conduction band (CB) potentials of ZnIn2S4 and MoO3−x samples can be calculated to be −0.92 V and 0.66 V (vs. NHE), respectively. Based on the above results, the schematic diagram of the energy band structures of ZnIn2S4 and MoO3−x is illustrated in Fig. 5e. The staggered energy band structure between them is an essential condition for the formation of an S-scheme heterojunction.54 For the overpotential conditions required for CO2 reduction, although the conduction band electrons of MoO3−x are not negative enough to reduce CO2 to CO and CH4, the electrons in the ZnIn2S4 conduction band are fully satisfied.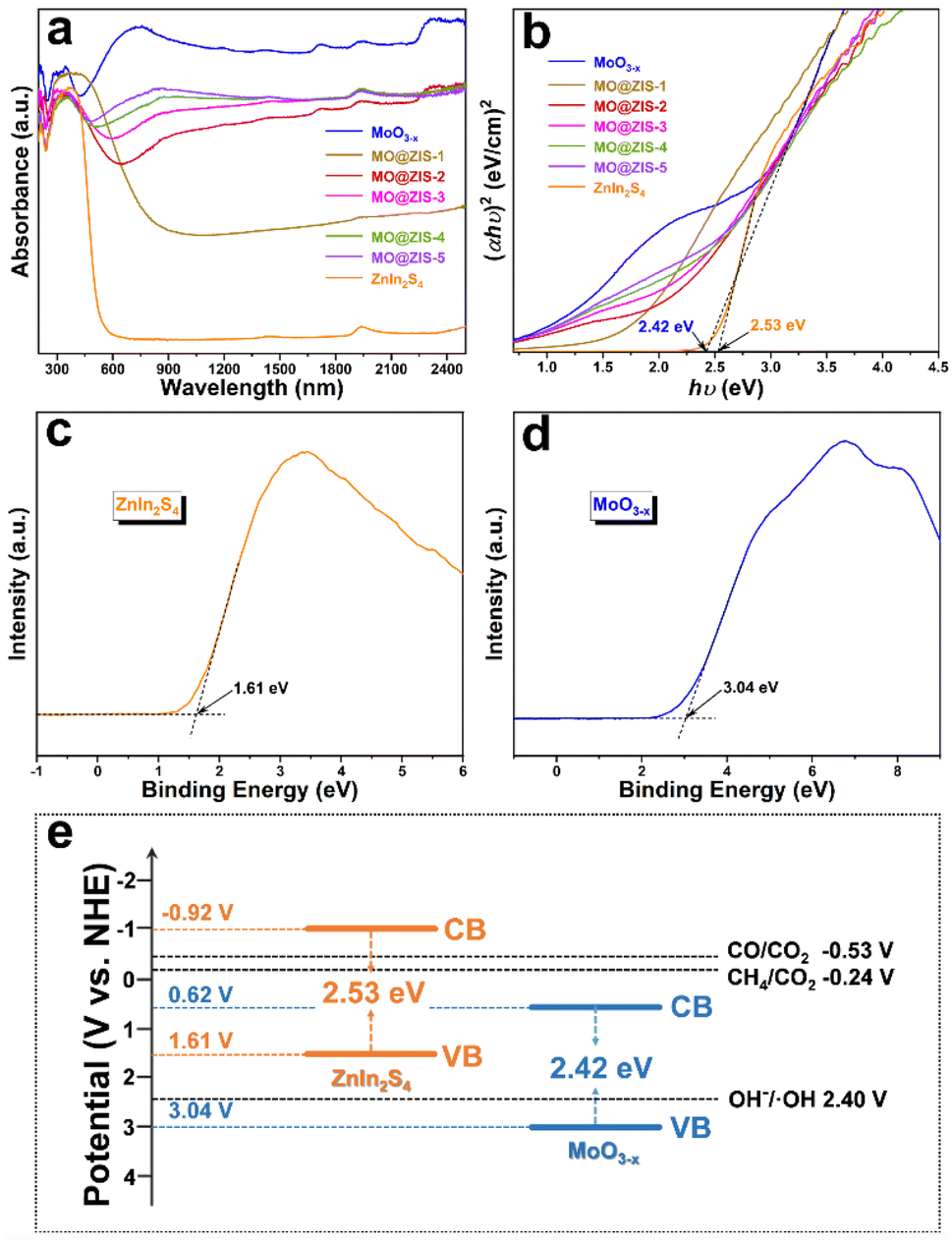 | ||
| Fig. 5 (a) UV-Vis-NIR absorption spectra and (b) the corresponding Tauc plots of all the as-prepared samples. VB-XPS spectra of (c) ZnIn2S4 and (d) MoO3−x. (e) Energy band structures of ZnIn2S4 and MoO3−x. | ||
Photothermal-coupled photocatalytic CO2 reduction performance
In order to evaluate the photothermal synergistic catalytic performance of the developed catalysts, photocatalytic CO2 reduction experiments are carried out in a gas–solid mode without sacrificial agents and co-catalysts. At the same time, the surface temperature of the catalysts is detected by using a thermocouple during the experiments, and the results are shown in Fig. 6. It can be evidently observed from Fig. 6a and b that the MoO3 sample fails to reduce CO2 and no gas products can be detected after 3 h of UV-Vis-IR full-spectrum irradiation. However, all other samples are subjected to photocatalytic CO2 reduction, and the presence of CH4 and CO is detected in the gaseous products. The production of CH4 and CO gases increases almost linearly with the irradiation time, indicating that these samples have stable photocatalytic CO2 reduction performance under continuous illumination. It is apparent that the yields of CH4 and CO for all composite samples are higher than those for pure MoO3−x and ZnIn2S4 samples. Fig. 6c shows the average yields of the CH4 and CO products, the selectivity of the CH4 product, and the surface temperature of the catalyst for all samples under UV-Vis-IR illumination for 3 h. The average CO production rate (RCO) and average CH4 production rate (RCH4) of the MoO3−x sample are 7.34 μmol g−1 h−1 and 1.46 μmol g−1 h−1, respectively, and the selectivity of CH4 is only 16.39%. The RCO and RCH4 of the ZnIn2S4 sample are 4.56 μmol g−1 h−1 and 2.43 μmol g−1 h−1, respectively, with a CH4 selectivity of 34.76%. However, after MoO3−x and ZnIn2S4 are successfully composited, the RCO of MO@ZIS-1, MO@ZIS-2, MO@ZIS-3, MO@ZIS-4 and MO@ZIS-5 is 9.23, 7.72, 4.65, 7.48 and 8.55 μmol g−1 h−1, respectively. Meanwhile, the RCH4 of each MO@ZIS-y composite is 10.81, 16.64, 28.3, 19.22 and 13.41 μmol g−1 h−1. Accordingly, in these MO@ZIS-y composite photocatalysts, the yield and selectivity of CH4 first rise and then fall with the increase of MoO3−x content. Among them, MO@ZIS-3 exhibits the best performance, and the average CH4 yield is 19.4 and 11.7 times that of MoO3−x and ZnIn2S4, respectively. Furthermore, for the MO@ZIS-3 catalyst, the selectivity to the CH4 product can reach 85.89%. Considering the high concentration of oxygen vacancies and their induced LSPR effect, the photothermal effect of MoO3−x as well as the composite samples is expected to effectively raise the surface temperature of the catalysts. During the photocatalytic CO2 reduction process, the surface temperatures of MoO3, MoO3−x, ZnIn2S4, MO@ZIS-1, MO@ZIS-2, MO@ZIS-3, MO@ZIS-4 and MO@ZIS-5 catalysts are measured, reaching 103 °C, 192 °C, 121 °C, 165 °C, 172 °C, 180 °C, 186 °C and 190 °C, respectively. It should be noted that although the oxygen vacancy concentration in MoO3−x is not as high as that in MO@ZIS-y, its surface temperature will be higher than that of the MO@ZIS-y composite, because it is a pure MoO3−x sample without the covering layer of ZnIn2S4. However, the surface temperature of all composite samples is much higher than that of the pure ZnIn2S4 sample, which indicates that the coating of ZnIn2S4 nanosheets affects the LSPR effect of MoO3−x, but does not cause its complete loss.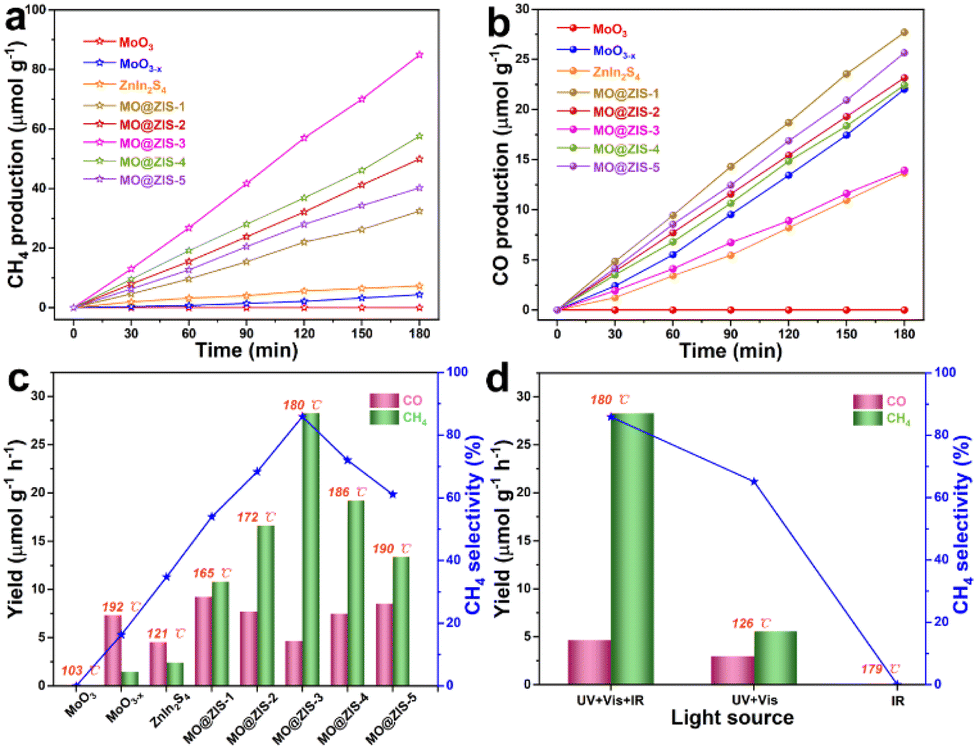 | ||
| Fig. 6 (a) CH4 production and (b) CO production as a function of irradiation time under full-spectrum illumination over the prepared samples. (c) Product yields, CH4 selectivity and surface temperature of the as-prepared samples under full-spectrum illumination. (d) Product yields, CH4 selectivity and surface temperature of the MO@ZIS-3 sample under different light irradiation. | ||
In order to verify that IR light itself cannot generate photoexcited charges, but only raises the catalyst surface temperature to promote the photocatalytic activity, the MO@ZIS-3 sample is used for photocatalytic CO2 reduction experiments with different light sources, and the results are presented in Fig. 6b. Firstly, under IR illumination, the sample does not cause the reduction of CO2, which indicates that IR light cannot excite the catalyst to produce photogenerated charges to participate in CO2 reduction. At the same time, the surface temperature of the sample reaches 179 °C under IR illumination, reflecting that thermocatalysis alone does not enable the reduction of CO2. Secondly, under UV-Vis illumination, the surface temperature of the MO@ZIS-3 sample only reaches 126 °C. The RCO and RCH4 are 2.98 and 5.56 μmol g−1 h−1, respectively, and the CH4 selectivity is only 65.11%. The above results reveal that the role of IR light is mainly to raise the surface temperature of the catalyst through the photothermal effect, rather than to stimulate the photocatalytic reactions. To investigate the effect of temperature on the photocatalytic CO2 reduction, the MO@ZIS-3 sample is taken as a representative catalyst, the surface temperature is controlled by using an external heater, and UV-Vis light is used as the illumination condition. Then thermal assisted photocatalytic CO2 reduction experiments are carried out at different temperatures. From Fig. S12,† it can be found that the higher the temperature, the higher the photocatalytic CO2 reduction efficiency and the better the CH4 selectivity, which unambiguously shows that the temperature is also a favorable factor affecting the photocatalytic CO2 reduction. In light of the above results, the main reasons for the substantially improved photocatalytic performance of the MoO3−x@ZnIn2S4 composites can be attributed to three aspects. First, the unique 3D hierarchical architecture together with the novel LSPR effect greatly enhances the light absorption efficiency of the catalyst. Second, the close contact growth and the matching band structures between MoO3−x and ZnIn2S4 allow them to build a good heterojunction, which can effectively promote the separation and transfer of photogenerated charges and enable more electrons to participate in the photocatalytic reaction. Finally, the photothermal effect effectively elevates the catalyst surface temperature and accelerates the catalytic reaction via photothermal synergistic catalysis.
The stability of the catalyst is always one of the decisive factors for its practical application. The best-performing MO@ZIS-3 sample is subjected to five rounds of photothermal-assisted photocatalytic CO2 reduction experiments, and the results are shown in Fig. S13a and b.† After five cycles, the yields of CH4 and CO do not decay significantly, indicating excellent catalytic stability. Fig. S13c† further presents their average yields during the five cycles of catalytic reactions. In the fifth round, RCO and RCH4 remain at 4.05 and 27.35 μmol g−1 h−1, respectively, with only a very slight decline in catalytic activity compared to the first round. Moreover, as revealed in Fig. S13d,† the surface temperature of the catalyst is still up to 172 °C in the fifth round of the catalytic experiment, which demonstrates that the as-prepared catalyst also possesses outstanding photothermal stability. The XRD patterns (Fig. S14†) of the MO@ZIS-3 sample before and after the cycling experiments do not change obviously. The characteristic peaks of both phases in the composite can be clearly observed, and there are no impurity peaks of newborn phases. Furthermore, the SEM images (Fig. S15a–d†) show that the morphology of MO@ZIS-3 is almost unchanged after five runs of catalysis, keeping the core–shell structure of ZnIn2S4 nanosheets tightly wrapped on the surface of MoO3−x needle crystals without perceptible dissociation and shedding. The ICP-OES results (Table S1†) reveal no significant decrease in the contents of Zn, In and S elements of the MO@ZIS-3 catalyst before and after the cycling catalytic tests, further proving the good stability of the ZnIn2S4 shell during the catalytic reaction. Therefore, the developed MoO3−x@ZnIn2S4 composite catalyst not only has superb performance stability, but also exhibits extremely high structural stability, which makes it promising for practical applications.
Charge separation and transfer analysis
The efficient separation and migration of photogenerated charges are prerequisites for remarkable photocatalytic performance. To this end, photoluminescence and photoelectrochemical tests are conducted on the prepared catalysts. As the catalyst is photoexcited, the photogenerated electrons transition and separate from the holes. After they are transferred to the surface of the photocatalyst, some of them will be recombined by emitting fluorescence. Therefore, the intensity of the steady-state PL emission spectrum can often reflect the separation efficiency of photogenerated carriers in the photocatalyst. The PL spectra of the MoO3−x and MO@ZIS-y samples are shown in Fig. 7a. Compared with the pure MoO3−x sample, the PL intensity of the MO@ZIS-y composite is obviously weaker, and the emission intensity of the best-performing MO@ZIS-3 sample is the lowest. This signifies that the compositing of MoO3−x and ZnIn2S4 provides a new pathway for the separation and transfer of photogenerated electrons and holes, thereby inhibiting their recombination and allowing more effective carriers to participate in the photocatalytic reactions on the catalyst surface. Fig. 7b exhibits the TR-PL spectra of the ZnIn2S4, MoO3−x and MO@ZIS-3 samples. The results reveal that the average fluorescence lifetime of the MO@ZIS-3 composite (τave = 2.26 ns) is much shorter than that of pure ZnIn2S4 (τave = 9.83 ns) and MoO3−x (τave = 3.60 ns), which implies that the photogenerated charges can be rapidly transferred at the interface to ensure more effective carriers.55 In addition, for the MoO3−x sample, the proportion of short-lived components (A1) due to non-radiative recombination is far greater than that of long-lived components (A2) due to free exciton interband recombination. Generally, the non-radiative recombination of photogenerated carriers is an important way to convert light energy into heat energy. That is, a higher proportion of non-radiative recombination will exert a more significant photothermal effect on catalysts.56 It can be found that the A1 value of MO@ZIS-3 is larger than that of pure MoO3−x, which may be due to the higher concentration of oxygen vacancies in MO@ZIS-3.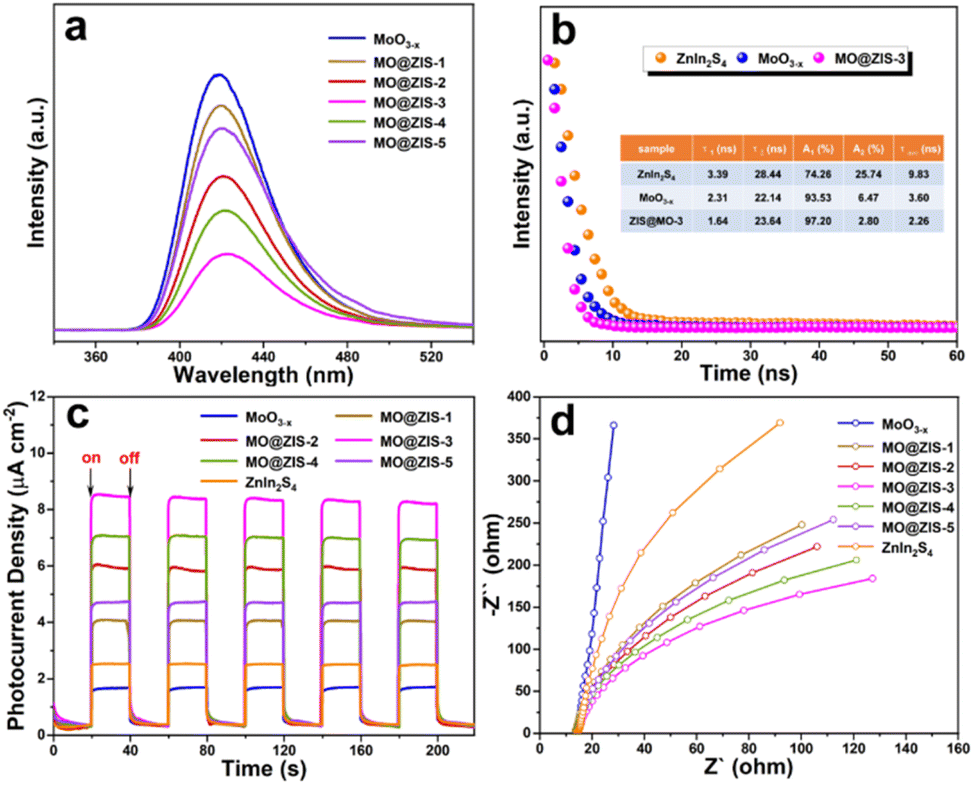 | ||
| Fig. 7 (a) Steady-state PL spectra of the MoO3−x and MO@ZIS-y samples, (b) TR-PL decay spectra of the ZnIn2S4, MoO3−x and MO@ZIS-3 samples, (c) transient photocurrent responses of the ZnIn2S4, MoO3−x and MO@ZIS-y samples, and (d) EIS Nyquist spectra of the ZnIn2S4, MoO3−x and MO@ZIS-y samples. | ||
As shown in Fig. 7c, the transient photocurrent response results of different catalysts are measured under UV-Vis-NIR full-spectrum illumination at a switching interval of 20 s. Obviously, the photocurrent response of the MO@ZIS-y composite is stronger than that of pure ZnIn2S4 and MoO3−x samples. The photocurrent intensity of all samples shows a positive correlation with their photocatalytic performance, and the best-performing MO@ZIS-3 composite has the largest photocurrent intensity as expected. The results further validate that the charge separation efficiency of the composites is much higher than that of the single-phase component. What's more, in order to ascertain that the photothermal effect accelerates the charge separation, the transient photocurrent responses of the ZnIn2S4, MoO3−x and MO@ZIS-3 samples are tested under different light sources, and the results are included in Fig. S16.† Compared with UV-Vis illumination, the photocurrent intensity of the ZnIn2S4 sample increases slightly under full-spectrum illumination, while that of MoO3−x and MO@ZIS-3 increases greatly, especially for the MO@ZIS-3 composite. This clearly demonstrates that the photothermal effect can significantly enhance the separation efficiency of photogenerated charges, thereby greatly boosting the photocatalytic performance of the catalyst. From the EIS Nyquist spectra of the samples (Fig. 7d), it can be observed that the arc radius of the MoO3−x@ZnIn2S4 composite is notably smaller than that of the others, suggesting less resistance to the transfer of photogenerated charges.57
S-scheme heterogeneous structure analysis
To probe the charge transfer mode in the MoO3−x@ZnIn2S4 heterojunction, an in situ XPS test is performed to investigate the transfer direction of photogenerated electrons, and the results are displayed in Fig. 8a–e. The XPS results in Fig. 2 have demonstrated that after the compositing of ZnIn2S4 and MoO3−x, an IEF is formed at their interface, with the ZnIn2S4 side becoming the electron depletion layer and the MoO3−x side being the electron accumulation layer. After the application of light, the in situ XPS results in Fig. 8 reveal that the characteristic peaks of Zn, In and S elements in ZnIn2S4 are shifted toward the low-energy region, while those of Mo and O elements in MoO3−x are shifted toward the high-energy region. These are exactly opposite to the XPS results of the sample without illumination, manifesting that the CB electrons of MoO3−x are transferred toward ZnIn2S4 and the VB holes of ZnIn2S4 move toward MoO3−x under the coulomb force of the IEF. This charge transfer mode is strictly in line with the S-scheme mechanism.58,59 In addition, the hydroxyl radicals (˙OH) in the photocatalytic process are detected by EPR radical capture experiments using DMPO as the trapping agent, and the results are provided in Fig. 8f. For the ZnIn2S4, MoO3−x and MO@ZIS-3 samples, ˙OH cannot be detected under dark conditions, proving that illumination is a prerequisite for triggering the photocatalytic reactions. After the application of light, ˙OH still fails to be detected in the ZnIn2S4 sample. Based on the band structure of ZnIn2S4, its VB potential (1.61 V vs. NHE) is far from the potential condition for ˙OH generation (2.40 V vs. NHE). However, the production of ˙OH is evidently observed in both MoO3−x and MO@ZIS-3 samples under light irradiation, and the signal intensity of MO@ZIS-3 is considerably higher than that of MoO3−x, suggesting that the VB holes of MoO3−x are more retained in the MO@ZIS-3 composite, inhibiting their transfer to the VB of ZnIn2S4. The EPR results once again provide strong evidence that the charge transfer between ZnIn2S4 and MoO3−x does not follow a conventional type-II heterojunction, but a unique S-scheme mechanism.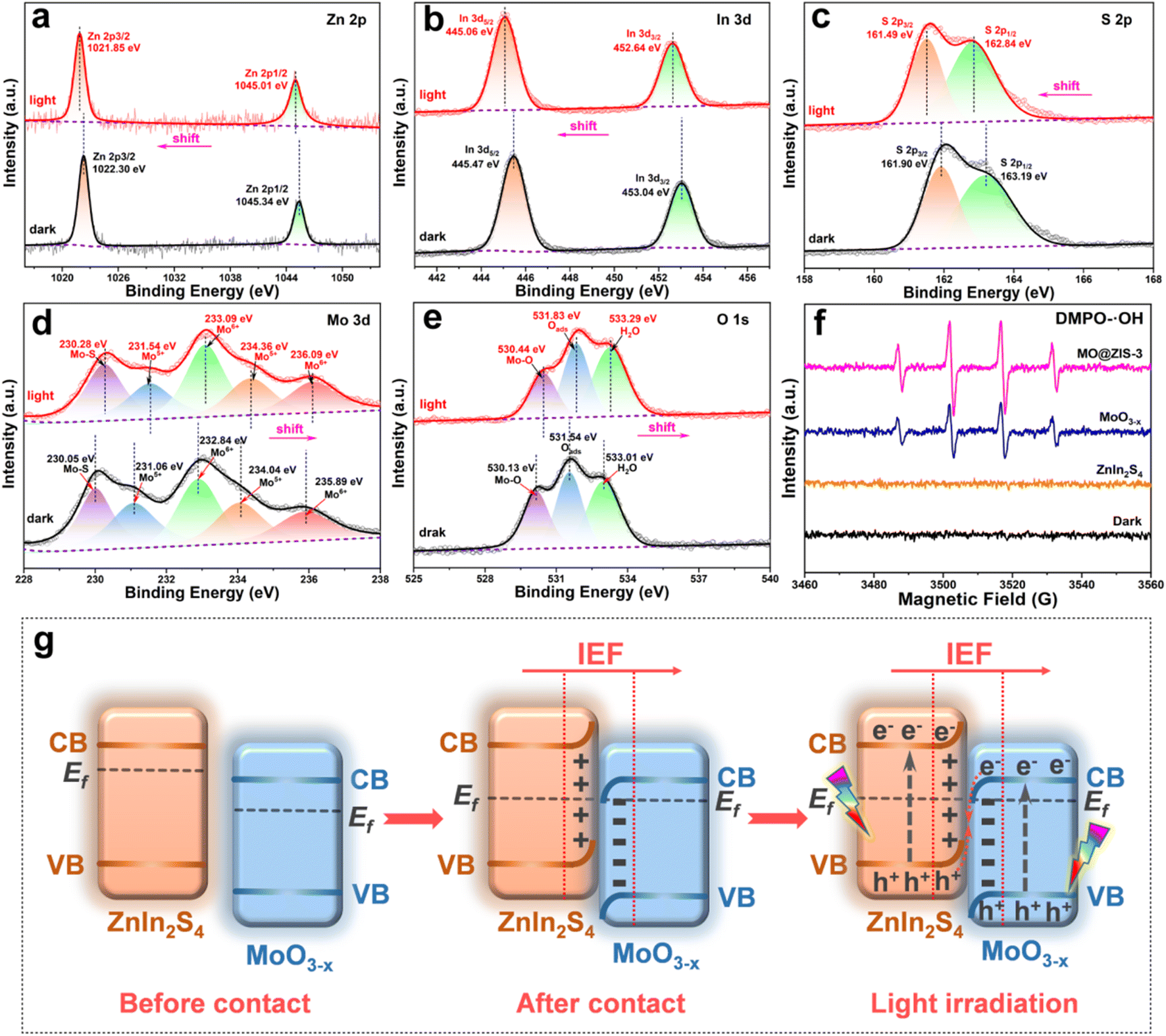 | ||
| Fig. 8 High-resolution in situ XPS spectra of the MO@ZIS-3 sample for (a) Zn 2p, (b) In 3d, (c) S 2p, (d) Mo 3d, and (e) O 1s measured in the dark and under full-spectrum illumination. (f) EPR spectra of DMPO-˙OH for the MoO3−x, ZnIn2S4 and MO@ZIS-3 samples. (g) Schematic illustration of the S-scheme transfer mechanism between MoO3−x and ZnIn2S4. | ||
Fig. 8g details the formation process and charge transfer path of the MoO3−x@ZnIn2S4 S-scheme heterojunction. In general, the Fermi level (Ef) of an n-type semiconductor is near the CB bottom. Both ZnIn2S4 and MoO3−x are n-type semiconductors, and the Ef of ZnIn2S4 is higher than that of MoO3−x.60,61 In this case, their compositing will generate an IEF at the interface, whereupon the energy bands of ZnIn2S4 and MO3−x are bent upward and downward, respectively, so that their Ef reaches equilibrium. Eventually, an electron depletion layer is formed on the ZnIn2S4 side and an electron accumulation layer is formed on the MoO3−x side.62 Under light excitation, the electrons in the VB of ZnIn2S4 and MoO3−x transition to the CB, leaving holes in their VB. Driven by the Coulomb attraction of the IEF and the Coulomb repulsion between the same charges, the photogenerated holes in the VB of ZnIn2S4 are transferred toward MoO3−x, while the photogenerated electrons in the CB of MoO3−x are transferred toward ZnIn2S4, which are thus recombined at the interface, leaving the VB holes of MoO3−x and the CB electrons of ZnIn2S4 to participate in the corresponding photocatalytic reactions. This charge transfer mechanism of the S-scheme heterojunction not only largely enhances the separation efficiency of photogenerated charges, but also preserves the strong reduction capacity of electrons and the strong oxidation capacity of holes to the greatest extent, bringing about a significant improvement in the performance of photocatalytic CO2 reduction.63
Photothermal synergistic photocatalysis mechanism
Scheme 2 elucidates the energy band structure and the charge transfer path in the MoO3−x@ZnIn2S4 heterojunction, as well as the positive effects of LSPR-induced high-energy hot electrons and high-efficiency photothermal conversion on the photocatalytic reactions, so as to discuss the mechanism of photothermal-coupled photocatalytic CO2 reduction. Based on band structure analysis, it is true that the CB potential of MoO3−x (0.62 V vs. NHE) is not negative enough to reduce CO2 to CO (−0.53 V vs. NHE) and CH4 (−0.24 V vs. NHE). The results of photocatalytic CO2 reduction experiments prove that a portion of high-energy hot electrons with plasma energy level potential higher than the CO2 reaction potential are involved in the photocatalytic reactions, which exactly confirms the generation of high-energy hot electrons in MoO3−x due to the LSPR effect. Based on the S-scheme mechanism as deliberated above, the VB holes of MoO3−x (3.04 V vs. NHE) are consumed by involvement in the OH−/˙OH reaction (2.40 V vs. NHE), while the CB electrons of ZnIn2S4 (−0.92 V vs. NHE) reduce CO2 to CO and CH4. Meanwhile, electrons near the Ef of MoO3−x reach a high-energy surface plasma state due to LSPR excitation, resulting in a large number of plasma hot electrons. They can radiate outward in the form of thermal energy, thereby drastically raising the catalyst surface temperature to boost the photocatalytic reduction efficiency and the CH4 product selectivity.64,65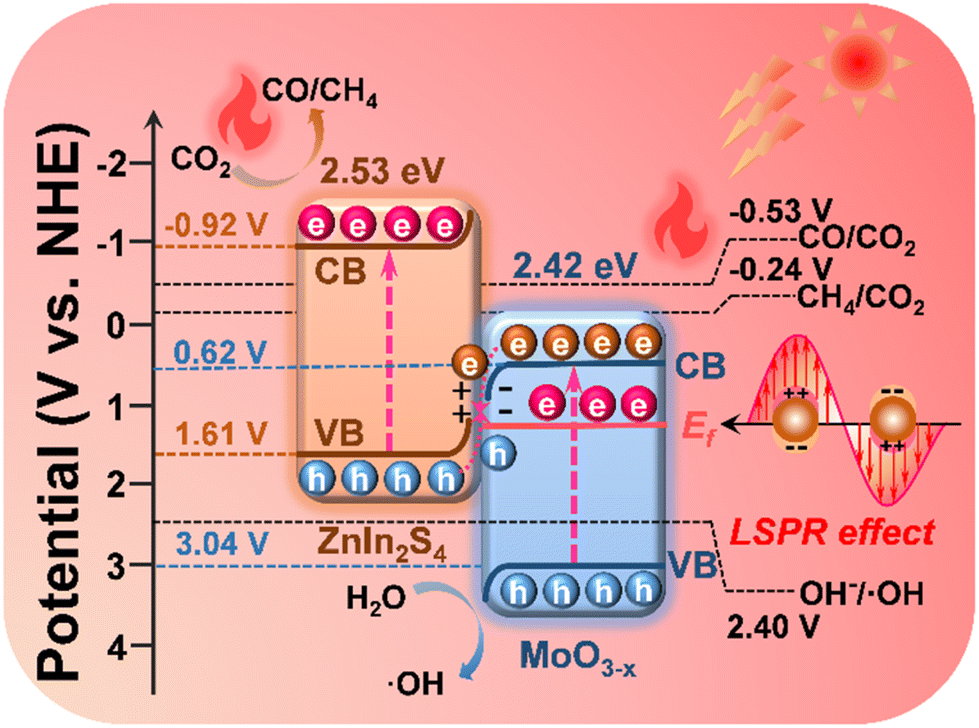 | ||
| Scheme 2 Diagram of the charge transfer and reaction paths of photothermal-coupled solar photocatalytic CO2 reduction at the MoO3−x@ZnIn2S4 core–shell S-scheme heterojunction. | ||
Conclusions
In summary, MoO3−x@ZnIn2S4 composites have been successfully constructed by in situ growth of ZnIn2S4 nanosheets on MoO3−x 1D needle-shaped crystals through low-temperature reflux. The unique 1D@2D core–shell architecture allows a tight interfacial contact and facilitates charge transfer. The results demonstrate that the transfer path of photogenerated charges in the MoO3−x@ZnIn2S4 composite follows a unique S-scheme mechanism, which improves the charge separation efficiency and enables the highest charge reactivity. Moreover, the LSPR effect triggered by abundant oxygen vacancies in MoO3−x produces a large number of high-energy hot electrons, which greatly increases the surface temperature of the catalyst and effectively accelerates the charge transfer and the CO2 adsorption–desorption. The MoO3−x@ZnIn2S4 composite is applied to photothermal-assisted photocatalytic CO2 reduction by combining the above multiple advantages. Under UV-Vis-IR full-spectrum illumination, the average yields of CO and CH4 over the optimal MO@ZIS-3 catalyst reach 4.65 and 28.3 μmol g−1 h−1, respectively, and the CH4 selectivity is as high as 85.89%. The average yield of CH4 is 19.4 and 11.7 times higher than those of bare MoO3−x and ZnIn2S4, respectively. This significantly enhanced photocatalytic CO2 reduction performance reveals that the combination of the S-scheme heterojunction and photothermal synergistic effect has very attractive application prospects in the field of photocatalysis.Author contributions
Renzhi Xiong: investigation, data curation, methodology, and writing – original draft. Xiaoxue Ke: investigation, data curation, and methodology. Weifeng Jia: investigation, data curation, and formal analysis. Yanhe Xiao: supervision and writing – review & editing. Baochang Cheng: supervision and writing – review & editing. Shuijin Lei: conceptualization, funding acquisition, investigation, methodology, project administration, resources, supervision, and writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support by the National Natural Science Foundation of China (Grant Number: 21961019 and 22265018) is gratefully acknowledged.Notes and references
- I. Sulivan, A. Goryachev, I. A. Digdaya, X. Li, H. A. Atwater, D. A. Vermaas and C. Xiang, Nat. Catal., 2021, 4, 952–958 CrossRef.
- M. Cai, Z. Wu, Z. Li, L. Wang, W. Sun, A. A. Tountas, C. Li, S. Wang, K. Feng, A. Xu, S. Tang, A. Tavasoli, M. Peng, W. Liu, A. S. Helmy, L. He, G. A. Ozin and X. Zhang, Nat. Energy, 2021, 6, 807–814 CrossRef CAS.
- L. Hurtadu, A. Mohan, U. Ulmer, R. Natividad, A. A. Tountas, W. Sun, L. Wang, B. Kim, M. M. Sain and G. A. Ozin, Chem. Eng. J., 2022, 435, 134864 CrossRef.
- Q. Fan, X. Zhang, X. Ge, L. Bai, D. He, Y. Qu, C. Kong, J. Bi, D. Ding, Y. Cao, X. Duan, J. Wang, J. Yang and Y. Wu, Adv. Energy Mater., 2021, 11, 2101424 CrossRef CAS.
- P. T. Fard, S. K. Albert, J. Ko, S. Lee, S. J. Park and J. Kim, ACS Catal., 2022, 12, 9698–9705 CrossRef CAS.
- T. Nakajima, Y. Tamaki, K. Ueno, E. Kato, T. Nishikawa, K. Ohkubo, Y. Yamazaki, T. Morimoto and O. Ishitani, J. Am. Chem. Soc., 2016, 138, 13818–13821 CrossRef CAS PubMed.
- Q. Hao, C. Xie, Y. Huang, D. Chen, Y. Liu, W. Wei and B. J. Ni, Chin. J. Catal., 2020, 41, 249–258 CrossRef CAS.
- X. Feng, W. Li, L. Yang, T. Song, Z. Xia, Q. Lai, X. Zhou, H. Xiao and C. Liu, Chem. Commun., 2022, 58, 13503–13506 RSC.
- D. Hao, J. Ren, Y. Wang, H. Arandiyan, M. Garbrecht, X. Bai, H. K. Shon, W. Wei and B. J. Ni, Energy Mater. Adv., 2021, 2021, 9761263 Search PubMed.
- D. Hao, Y. Liu, S. Gao, H. Arandiyan, X. Bai, Q. Kong, W. Wei, P. K. Shen and B. J. Ni, Mater. Today, 2021, 46, 212–233 CrossRef CAS.
- H. Han, K. Huang, Y. Yao, Z. Li and X. Meng, Chem. Eng. J., 2022, 450, 138419 CrossRef CAS.
- D. Mateo, J. L. Cerrillo, S. Durini and J. Gascon, Chem. Soc. Rev., 2021, 50, 2173–2210 RSC.
- C. Song, Z. Wang, Z. Yin, D. Xiao and D. Ma, Chem Catal., 2022, 2, 52–83 CrossRef.
- M. Sun, B. Zhao, F. Chen, C. Liu, S. Lu, Y. Yu and B. Zhang, Chem. Eng. J., 2021, 408, 127280 CrossRef CAS.
- L. Liu, C. Zhao, H. Zhao, D. Pitts and Y. Li, Chem. Commun., 2013, 49, 3664–3666 RSC.
- L. Wan, Q. Zhou, X. Wang, T. E. Wood, L. Wang, P. N. Duchesne, J. Guo, X. Yan, M. Xia, Y. F. Li, A. A. Jelle, U. Ulmer, J. Jia, T. Li, W. Sun and G. A. Ozin, Nat. Catal., 2019, 2, 889–898 CrossRef CAS.
- S. Cai, J. Chen, Q. Li and H. Jia, ACS Appl. Mater. Interfaces, 2021, 13, 14221–14229 CrossRef CAS PubMed.
- Y. Zhang, Y. Wu, L. Wan, H. Ding, H. Li, X. Wang and W. Zhang, Appl. Catal., B, 2022, 311, 121255 CrossRef CAS.
- M. Sun, B. Zhao, F. Chen, C. Liu, S. Lu, Y. Yu and B. Zhang, Chem. Eng. J., 2021, 408, 127280 CrossRef CAS.
- M. Guo, Z. Xing, T. Zhao, Z. Li, S. Yang and W. Zhou, Appl. Catal., B, 2019, 257, 117913 CrossRef CAS.
- Y. Guo, B. Chang, T. Wen, S. Zhang, M. Zeng, N. Hu, Y. Su, Z. Yang and B. Yang, J. Colloid Interface Sci., 2020, 567, 213–223 CrossRef CAS PubMed.
- G. Dong, X. Huang and Y. Bi, Angew. Chem., Int. Ed., 2022, 61, e202204271 CAS.
- Y. Wang, M. Liu, C. Wu, J. Gao, M. Li, Z. Xing, Z. Li and W. Zhou, Small, 2022, 18, 2202544 CrossRef CAS PubMed.
- J. Peng, J. Shen, X. Yu, H. Tang, Zulfiqar and Q. Liu, Chin. J. Catal., 2021, 42, 87–96 CrossRef CAS.
- Q. Li, Y. Gao, M. Zhang, H. Gao, J. Chen and H. Jia, Appl. Catal., B, 2022, 303, 120905 CrossRef CAS.
- J. Li, Y. Ye, L. Ye, F. Su, Z. Ma, J. Huang, H. Xie, D. E. Doronkin, A. Zimina, J. D. Grunwaldt and Y. Zhou, J. Mater. Chem. A, 2019, 7, 2821–2830 RSC.
- Z. Huang, J. Liu, S. Zong, X. Wang, K. Chen, L. Liu and Y. Fang, J. Colloid Interface Sci., 2022, 606, 848–859 CrossRef CAS PubMed.
- Y. Zhang, X. Ping, L. Hao, Y. He, Y. Guo, Q. Zhao, Z. Zheng and Y. Lu, J. Environ. Chem. Eng., 2021, 9, 1055565 Search PubMed.
- X. Jiang, J. Huang, Z. Bi, W. Ni, G. Gurzadyan, Y. Zhu and Z. Zhang, Adv. Mater., 2022, 34, 2109330 CrossRef CAS PubMed.
- M. Jiang, C. Li, K. Huang, Y. Wang, J. Liu, Z. Geng, X. Hou, J. Shi and S. Feng, ACS Appl. Mater. Interfaces, 2020, 12, 35113–35119 CrossRef CAS PubMed.
- X. Dai, S. Feng, W. Wu, Y. Zhou, Z. Ye, X. Cao, Y. Wang and C. Yang, Int. J. Hydrogen Energy, 2022, 47, 25104–25116 CrossRef CAS.
- L. Zhang, J. Zhang, H. Yu and J. Yu, Adv. Mater., 2022, 34, 2107668 CrossRef CAS PubMed.
- J. Wang, S. Sun, R. Zhou, Y. Li, Z. He, H. Ding, D. Chen and W. Ao, J. Mater. Sci. Technol., 2021, 78, 1–9 CrossRef CAS.
- Q. Zhou, L. Zhang, L. Zhang, B. Jiang and Y. Sun, J. Hazard. Mater., 2022, 438, 129438 CrossRef CAS PubMed.
- L. Li, D. Ma, Q. Xu and S. Huang, Chem. Eng. J., 2022, 437, 135153 CrossRef CAS.
- M. Dai, Z. He, P. Zhang, X. Li and S. Wang, J. Mater. Sci. Technol., 2022, 122, 231–242 CrossRef.
- X. Lu, L. Quan, H. Hou, J. Qian, Z. Liu and Q. Zhang, J. Alloys Compd., 2022, 925, 166552 CrossRef CAS.
- Y. Xiao, H. Wang, Y. Jiang, W. Zhang, J. Zhang, X. Wu, Z. Liu and W. Deng, J. Colloid Interface Sci., 2022, 623, 109–123 CrossRef CAS PubMed.
- C. Ouyang, X. Quan, C. Zhang, Y. Pan, X. Li, Z. Hong and M. Zhi, Chem. Eng. J., 2021, 424, 130510 CrossRef CAS.
- R. Xiong, C. Tang, S. Liu, Y. Xiao, B. Cheng and S. Lei, Sep. Purif. Technol., 2022, 295, 121267 CrossRef CAS.
- S. Saeidi, S. Najari, V. Hessel, K. Wilson, F. J. Keil, P. Concepción, S. L. Suib and A. E. Rodrigues, Prog. Energy Combust. Sci., 2021, 85, 100905 CrossRef.
- T. Mendiara, F. García-Labiano, A. Abad, P. Gayán, L. F. de Diego, M. T. Izquierdo and J. Adánez, Appl. Energy, 2018, 232, 657–684 CrossRef CAS.
- J. Li, Y. Ye, L. Ye, F. Su, Z. Ma, J. Huang, H. Xie, D. E. Doronkin, A. Zimina, J. D. Grunwaldt and Y. Zhou, J. Mater. Chem. A, 2019, 7, 2821–2830 RSC.
- Y. Wei, Q. Zhang, Y. Zhou, X. Ma, L. Wang, Y. Wang, R. Sa, J. Long, X. Fu and R. Yuan, Chin. J. Catal., 2022, 43, 2665–2677 CrossRef CAS.
- L. Sun, X. Liu, X. Jiang, Y. Feng, X. Ding, N. Jiang and J. Wang, J. Mater. Chem. A, 2022, 10, 25279–25294 RSC.
- X. Li, H. Yang, Q. Wu, C. Zhu, J. Tang, C. Ma, L. Tang, H. Su, X. Wang, C. Xie and D. Zeng, Appl. Surf. Sci., 2023, 612, 155845 CrossRef CAS.
- C. Zhao, C. Wang, H. Xin, H. Li, R. Li, B. Wang, W. Wei, Y. Cui and Q. Fu, ACS Appl. Mater. Interfaces, 2022, 14, 26194–26203 CrossRef CAS PubMed.
- M. Tan, C. Huang, C. Yu, C. Li, R. Yin, C. Liu, W. Dong, H. Meng, Y. Su, L. Qiao, L. Gao, Q. Lu and Y. Bai, Small, 2022, 18, 2205266 CrossRef CAS PubMed.
- W. Huang, K. Zhang, B. Yuan, L. Yang and M. Zhu, Energy Storage Mater., 2022, 50, 152–160 CrossRef.
- Z. Luo, R. Miao, T. D. Huan, I. M. Mosa, A. S. Poyraz, W. Zhong, J. E. Cloud, D. A. Kriz, S. Thanneeru, J. He, Y. Zhang, R. Ramprasad and S. L. Suib, Adv. Energy Mater., 2016, 6, 1600528 CrossRef.
- H. Su, H. Lou, Z. Zhao, L. Zhou, Y. Pang, H. Xie, C. Rao, D. Yang and X. Qiu, Chem. Eng. J., 2022, 430, 132770 CrossRef CAS.
- J. Hu, W. Zhao, R. Hu, G. Chang, C. Li and L. Wang, Mater. Res. Bull., 2014, 57, 268–273 CrossRef CAS.
- L. Wang, B. Cheng, L. Zhang and J. Yu, Small, 2021, 17, 2103447 CrossRef CAS PubMed.
- Q. Xu, L. Zhang, B. Cheng, J. Fan and J. Yu, Chem, 2020, 6, 1543–1559 CAS.
- K. Huang, C. Li and X. Meng, J. Colloid Interface Sci., 2020, 580, 669–680 CrossRef CAS PubMed.
- Y. Tang, W. Zhou, Q. Shang, Y. Guo, H. Hu, Z. Li, Y. Zhang, L. Liu, H. Wang, X. Tan, T. Yu and J. Ye, Appl. Catal., B, 2022, 310, 121295 CrossRef CAS.
- C. Chen, W. Hou and Y. Xu, Appl. Catal., B, 2022, 316, 121676 CrossRef CAS.
- F. Xu, K. Meng, B. Cheng, S. Wang, J. Xu and J. Yu, Nat. Commun., 2020, 11, 4613 CrossRef CAS PubMed.
- X. Han, B. Lu, X. Huang, C. Liu, S. Chen, J. Chen, Z. Zeng, S. Deng and J. Wang, Appl. Catal., B, 2022, 316, 121587 CrossRef CAS.
- Z. Yang, S. Li, X. Xia and Y. Liu, Sep. Purif. Technol., 2022, 300, 121819 CrossRef CAS.
- X. Liu, Y. Luo, C. Ling, Y. Shi, G. Zhan, H. Li, H. Gu, K. Wei, F. Guo, Z. Ai and L. Zhang, Appl. Catal., B, 2022, 301, 120766 CrossRef CAS.
- Y. Bao, S. Song, G. Yao and S. Jiang, Sol. RRL, 2021, 5, 2100118 CrossRef CAS.
- B. Su, H. Huang, Z. Ding, M. B. J. Roeffaers, S. Wang and J. Long, J. Mater. Sci. Technol., 2022, 124, 164–170 CrossRef.
- S. Cai, M. Zhang, J. Li, J. Chen and H. Jia, Sol. RRL, 2021, 5, 2000313 CrossRef CAS.
- X. Chen, Q. Li, M. Zhang, J. Li, S. Cai, J. Chen and H. Jia, ACS Appl. Mater. Interfaces, 2020, 12, 39304–39317 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: EPR spectra of the MoO3−x, MoO3 and MO@ZIS-3 samples; XRD, SEM, TEM, HRTEM, SAED, EDS, elemental mapping and UV-Vis-NIR absorption results of the MoO3 and MoO3−x samples; SEM, TEM, HRTEM, SAED, EDS and elemental mapping data of the ZnIn2S4 sample; EDS spectrum of the MO@ZIS-3 composite; product yields and CH4 selectivity of MO@ZIS-3 under different temperatures; cycling performance of photocatalytic CO2 reduction for MO@ZIS-3; XRD, SEM and elemental analysis of MO@ZIS-3 before and after cycling; transient photocurrent responses of MoO3−x, MO@ZIS-3 and ZnIn2S4 under UV-Vis and UV-Vis-IR illumination. See DOI: https://doi.org/10.1039/d2ta09255g |
| This journal is © The Royal Society of Chemistry 2023 |