
Heteroatomic interface engineering of an octahedron VSe2–ZrO2/C/MXene composite derived from a MXene-MOF hybrid as a superior-performance anode for lithium-ion batteries†
Hanbin
Li
 a,
Jinliang
Li
*b,
Liang
Ma
*bc,
Xinlu
Zhang
d,
Junfeng
Li
e,
Jiabao
Li
f,
Ting
Lu
a and
Likun
Pan
*a
a,
Jinliang
Li
*b,
Liang
Ma
*bc,
Xinlu
Zhang
d,
Junfeng
Li
e,
Jiabao
Li
f,
Ting
Lu
a and
Likun
Pan
*a
aShanghai Key Laboratory of Magnetic Resonance, School of Physics and Electronic Science, East China Normal University, Shanghai, 200241, China. E-mail: lkpan@phy.ecnu.edu.cn
bSiyuan Laboratory, Guangdong Provincial Key Laboratory of Nanophotonic Manipulation, Guangdong Provincial Engineering Technology Research Center of Vacuum Coating Technologies and New Materials, Department of Physics, Jinan University, Guangzhou, Guangdong 510632, China. E-mail: lijinliang@email.jnu.edu.cn; maliang2415@jnu.edu.cn
cSchool of Chemistry, Guangzhou Key Laboratory of Materials for Energy Conversion and Storage, South China Normal University, Guangzhou, 510006, China
dKey Laboratory for Liquid-Solid Structural Evolution & Processing of Materials (Ministry of Education), Research Center for Carbon Nanomaterials, School of Materials Science and Engineering, Shandong University, Jinan, 250061, China
eCollege of Logistics Engineering, Shanghai Maritime University, Shanghai, 201306, China
fSchool of Chemistry and Chemical Engineering, Yangzhou University, Yangzhou, Jiangsu 225002, China
First published on 5th January 2023
Abstract
As a promising electrode material with tremendous specific capacity, vanadium diselenide (VSe2) has recently attracted renewed attention. However, the application of VSe2 is still hindered by the difficulty in its synthesis and nature of volume expansion. In this work, we developed a practical solvothermal method and in situ selenization process to obtain the VSe2–ZrO2/C/MXene composite from the MXene-metal–organic framework (MOF) hybrid precursor. During the synthesis process, V2CTx is converted to VSe2/MXene, which firmly anchors on the porous carbon derived from UiO-66 with the assistance of another derivative ZrO2via chemical bonding. Remarkably, benefitting from the practical cooperation between VSe2/MXene, ZrO2 and porous carbon, VSe2–ZrO2/C/MXene displays an outstanding lithium storage performance with an enduring capacity rise from 461.2 to 1238.5 mA h g−1 at 100 mA g−1 after a short recession during cycling, which is investigated in detail as a “negative fading” phenomenon. Even at the high current density of 1.0 A g−1, the composite still presents a high reversible capacity of 430 mA h g−1 after 1000 cycles, highlighting its superior cycling stability. The application potential of the VSe2–ZrO2/C/MXene anode for LIBs has also been evaluated by assembling full cells. The strategy in this work inspires the construction design of novel selenide-based electrode materials for high-performance lithium-ion batteries.
Introduction
As an anticipant medium to realize energy conservation and emission reduction, rechargeable lithium-ion batteries (LIBs) have drawn wide attention in portable electronics, vehicle industry, and intelligent devices due to their ultra-high energy density, stability, and safety.1–4 Nevertheless, the intensifying global energy crisis as well as the eco-friendly ethic/policy demands urge a profound revolution of LIBs. Regarded as the primary commercial anode, graphite is restricted by a low theoretical capacity of 372 mA h g−1,5 which is inadequate for the ever-increasing electric-driving requirements.6–9 Because of this situation, numerous studies have been devoted to the exploration of battery materials pursuing higher performance.Among a series of anode species, transition-metal selenides (TMSes) have captured much research attention in recent years as an emerging material for lithium-ion storage due to their excellent charge transfer properties.10–12 Additionally, TMSes exhibit brilliant volumetric capacity and rapid electron migration rate, stemming from high conductivity (1 × 10−5 S m−1) and low electronegativity (2.4) of selenium during the full lithiation process.13,14 Zhang et al. used Cu-BTC as sacrificial templates to fabricate a Cu1.8Se@C decahedron through the co-precipitation process, which delivered a capacity of 824.4 mA h g−1 after 200 cycles at 100 mA g−1.15 Zhu et al. proposed ZnSe nanoparticle-embedded N-doped carbon nanocubes in a similar process, which displayed a high reversible capacity of 1166.6 mA h g−1 after 500 cycles at 0.5 A g−1.16 Among various TMSes, VSe2 with excellent electrochemical activity stands out for its high lithium-ion storage performance because of its light molecular mass and multi-electron transfer mechanism.17 Nevertheless, the preparation difficulty and insufficient capacity hinder the application of VSe2 in the energy storage field. For instance, among the traditional methods of VSe2 synthesis, chemical vapor deposition is capable of synthesizing high crystalline quality VSe2,18 but requires a high synthesis temperature (up to 900 °C), which makes the synthesis process complex and highly energy intensive.19 Therefore, it is necessary to develop methods with relatively simple synthesis processes, such as hydrothermal selenidation or surface annealing treatment selenidation. Wang et al. developed VSe2/graphene via a hydrothermal route using NH4VO3 and SeO2, which showed a reversible discharge capacity of 632 mA h g−1 at 100 mA g−1 during 60 cycles.20 Unfortunately, the reported lithium storage capacity is not enough for practical applications. Currently, MXenes, 2D layer transition metal carbonitrides/carbides with regulable physical and chemical properties have attracted much attention in energy storage fields21–23 because of their open structure, internal conductivity24,25 as well as terrific hydrophilicity endowing an abundance of active sits for ion storage and rapid charge transfer.26,27 Moreover, MXenes have been demonstrated as good sacrificial precursors for synthesizing numerous nanomaterials or their hybrids,3,28 where the incompletely converted MXenes can also act as a conductive support in these hybrids.29 The Sha group successfully constructed VSe2@V2CTx by surface selenization strategy. In this way, the surface metal atoms on MXene supplied a metal source for TMSe, and the layer structure preserved the nanoplates from restacking concurrently. The obtained VSe2@V2CTx delivered an excellent capacity of 158.1 mA h g−1 at 2.0 A g−1 after 600 cycles for aqueous zinc-ion batteries.30 Inspired by this encouraging work, the selenization of V2CTx can be a promising strategy for synthesizing stable VSe2.
Volume expansion and cycling have been widely reported to result in structural deterioration and even pulverization, remaining a critical challenge for TMSes.31,32 It is of great necessity to figure out advanced tactics to improve the cycling stability of selenide electrodes. It is well known that ZrO2 is a viable material not only for its high dielectric constant, but also for its structural protection role in mitigating capacity fading.33 Thus, introducing ZrO2 into TMSes is deemed to effectively solve the attenuation issue.34 Yao et al. found that the modified amorphous ZrO2 coating on LiCoO2 particles could improve the capacity retention to 82.5% after 100 cycles.35 Additionally, coupling with hollow/porous carbonaceous constructions,36–38 such as carbon aerogels, carbon nanotubes, carbon cages, and carbon nanofibers, as buffer materials is another rational strategy to relieve the volumetric strain.28,39–43 In particular, porous carbon derived from metal–organic frameworks (MOFs) can control the morphology and realize a high specific surface area, which provides sufficient active sites, shortens the pathways for lithium-ion transfer, improves the electrolyte infusion and enhances the electrical conductivity.44–47 Yang et al. synthesized a Ni–Co–Se@C bundle-like micro/nanostructure hybrid with bimetallic MOF as the precursor using a surfactant-assisted co-precipitation approach, which remarkably modified the rate performance with a capacity of 493 mA h g−1 at 8 A g−1 in LIBs.48 Jin et al. encapsulated ZnSe and CoSe in N-doped carbon polyhedra interconnecting with CNTs through in situ pyrolysis and selenization procedure, and the acquired LIBs anode established a capacity of 873 mA h g−1 after 500 cycles at 0.5 A g−1.49 Given the above, incorporating ZrO2 and porous carbon together into MXene-derived VSe2 (or VSe2/MXene) should be a feasible method to synergistically enhance its energy storage ability. Fortunately, UiO-66 can act as a sacrificial precursor to produce both ZrO2 and porous carbon with effective integration. However, up to now, the exploration of UiO-66 and its derivatives in LIBs is quite limited, much less for the design of the hybrid material containing VSe2, ZrO2 and porous carbon.
Herein, we propose a heteroatomic interface engineering of octahedron VSe2–ZrO2/C/MXene composite derived from the MXene-MOF hybrid via a two-step method. In the unique structure, V2CTx-derived VSe2/MXene contributed to a high reversible capacity, and the ZrO2-assisted VSe2 firmly anchoring on the octahedron carbon greatly improved the electrochemical stability, while the MOF-derived porous carbon suppressed the volumetric changes and enhanced the lithium-ion transport dynamics. Consequently, VSe2–ZrO2/C/MXene demonstrates a high reversible capacity reaching 1238.5 mA h g−1 at 100 mA g−1, and superior cycling stability with a capacity of 430 mA h g−1 even after 1000 cycles at 1.0 A g−1. Furthermore, we assembled a full cell by coupling LiFePO4 as the cathode and VSe2–ZrO2/C/MXene as the anode. This work could be regarded as a significant motivation for application exploration.
Experimental
For the etching process, 2.0 g V2AlC powder was slowly added into 40 mL HF (40%) solution for 1 h. The mixture was kept stirring for 5 days at a temperature of 35 °C to remove Al3+. After that, the black residue was washed with deionized water by centrifugation until the pH of the supernatant reached ∼7. Subsequently, the residue was transferred to a vacuum freeze dryer for drying to obtain V2CTx.The composite was synthesized via the typical hydrothermal method. First, 0.9 mmol H2BDC and 0.9 mmol ZrCl4 were dissolved in 60 mL DMF with 150 mg V2CTx under ultrasonic treatment. Then, 6 mL glacial acetic acid was dropwise injected into the above-mixed solution under ultrasound for 0.5 h. Subsequently, the mixed solution was transferred into a Teflon-lined stainless-steel autoclave for hydrothermal treatment at 120 °C for 16 h. The obtained black precipitate was washed by DMF and dried in a vacuum oven at 60 °C to attain the precursor of UiO-66/V2CTx. UiO-66 was prepared in the same way, except for adding V2CTx in the precursor. The as-prepared UiO-66/V2CTx precursor and Se powder were placed in two fused quartz boats with a dosage ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :20, and then calcinated at 600 °C for 2 h in nitrogen flow, which is denoted as VSe2–ZrO2/C/MXene. The corresponding synthesis schematic diagram is generalized in Fig. 1a. For comparison, Se–ZrO2/C and VSe2/MXene were obtained at the same selenization condition by directly heating UiO-66 and V2CTx MXene. Detailed characterizations and electrochemical tests are shown in ESI.†
:20, and then calcinated at 600 °C for 2 h in nitrogen flow, which is denoted as VSe2–ZrO2/C/MXene. The corresponding synthesis schematic diagram is generalized in Fig. 1a. For comparison, Se–ZrO2/C and VSe2/MXene were obtained at the same selenization condition by directly heating UiO-66 and V2CTx MXene. Detailed characterizations and electrochemical tests are shown in ESI.†
 | ||
| Fig. 1 (a) Schematic of the preparation of VSe2–ZrO2/C/MXene. FESEM images of (b) Se–ZrO2/C, (c) VSe2/MXene and (d and e) VSe2–ZrO2/C/MXene. (f) TEM (inset: HRTEM and corresponding IFFT image), and (g) EDS elemental mapping images of VSe2–ZrO2/C/MXene. | ||
Results and discussion
The morphologies of the samples were revealed in field-emission scanning electron microscopy (FESEM). As observed from Fig. S1a,† the original UiO-66 exhibits an octahedron configuration with an even size of 0.8 μm, while V2CTx shows a typical corrugated structure of MXene (Fig. S1b†).50Fig. 1b and S2a† show the FESEM and transmission electron microscopy (TEM) images of Se–ZrO2/C, which present the well-defined octahedral structure, except for slight size shrinkage owing to thermal contraction.51 The V2CTx layers turn into irregular stacking of VSe2/MXene with thicker flakes after selenization (Fig. 1c and S2b†). Fig. 1d and e demonstrates the retainability of the architecture after selenization of UiO-66/V2CTx to form VSe2–ZrO2/C/MXene. According to the morphology changes of different precursors (Fig. S1, S2† and 1), a relative non-uniform octahedral structure with a rough surface can be detected, which should be derived from the selenization process. A little flake also can be observed in Fig. 1d, caused by the collapse of octahedral structures during the selenization process. In this case, the auxetic specific surface area can supply more active sites and abundant Li-ion transfer paths to improve the reaction kinetics.52Fig. 1f shows the TEM image of the VSe2–ZrO2/C/MXene composite, demonstrating an interlink between flakes and octahedrons. The inset is the high-resolution transmission electron microscopy (HRTEM) and the corresponding inverse fast Fourier transform (IFFT) images of VSe2–ZrO2/C/MXene. Lattice fringes with spacings of 0.29 nm and 0.27 nm are observed, corresponding to the (101) planes of tetragonal ZrO2 (JCPDS 88-1007) and the (011) planes of hexagonal VSe2 (JCPDS 89-1641), respectively, implying the migration of V and in situ growth of VSe2. The MXene is too rare to be found in HRTEM. A uniform distribution of V, Zr, Se, C, and O elements is observed from energy dispersive spectroscopy (EDS) elemental mapping images in Fig. 1g. The V signal is found in the region of octahedrons, further verifying the transform of V2CTx to VSe2 on carbon. Therefore, the tiny particles on the rough surface of the octahedron are VSe2. The above tests confirm the existence of each component in VSe2–ZrO2/C/MXene. The contents of V, Zr, and Se are 15.7%, 18.2%, and 38.2%, respectively, determined by inductively coupled plasma-atomic emission spectrometry (ICP-AES). As a result, we can calculate the mass ratios of VSe2 and ZrO2 to be 50.53% and 24.55%, respectively. The remaining 24.92% should be porous carbon and a small number of MXene.Fig. 2a and b and S3† present the samples' X-ray diffraction (XRD) patterns. The XRD pattern of UiO-66 in Fig. S3a† is in line with the previous report.53 The sharp peak at 7.3° is characteristic of the typical UiO-66 diffraction peak. As seen from Fig. S3b,† the new peak that appears at 9.3° after etching treatment can be assigned to the (002) peak for V2CTx, indicating the removal of the Al layer. The complex pattern of UiO-66/V2CTx in Fig. S3c† incorporates the diffraction peaks of both UiO-66 and V2CTx, suggesting their successful integration. After selenization, the diffraction pattern of Se–ZrO2/C in Fig. 2a is in agreement with the standard ZrO2 peaks at 30.25°, 50.4°, and 60.2° that are ascribed to the (101), (112) and (211) planes, respectively, while the diffraction peaks of VSe2/MXene at 14.6°, 34.1° and 42.9° match well with the (001), (011) and (102) planes of Vse2, respectively. Notably, the peaks at 41.3° and 55.2° indicate the existence of residual V2CTx, which should be helpful in improving the conductivity of VSe2. Similar peaks detected in the pattern of VSe2–ZrO2/C/MXene in Fig. 2b with those for Se–ZrO2/C and VSe2/MXene can be rationalized as their co-existence in VSe2–ZrO2/C/MXene. In addition, the broad peak at around 22° belongs to amorphous carbon in the hybrid.54
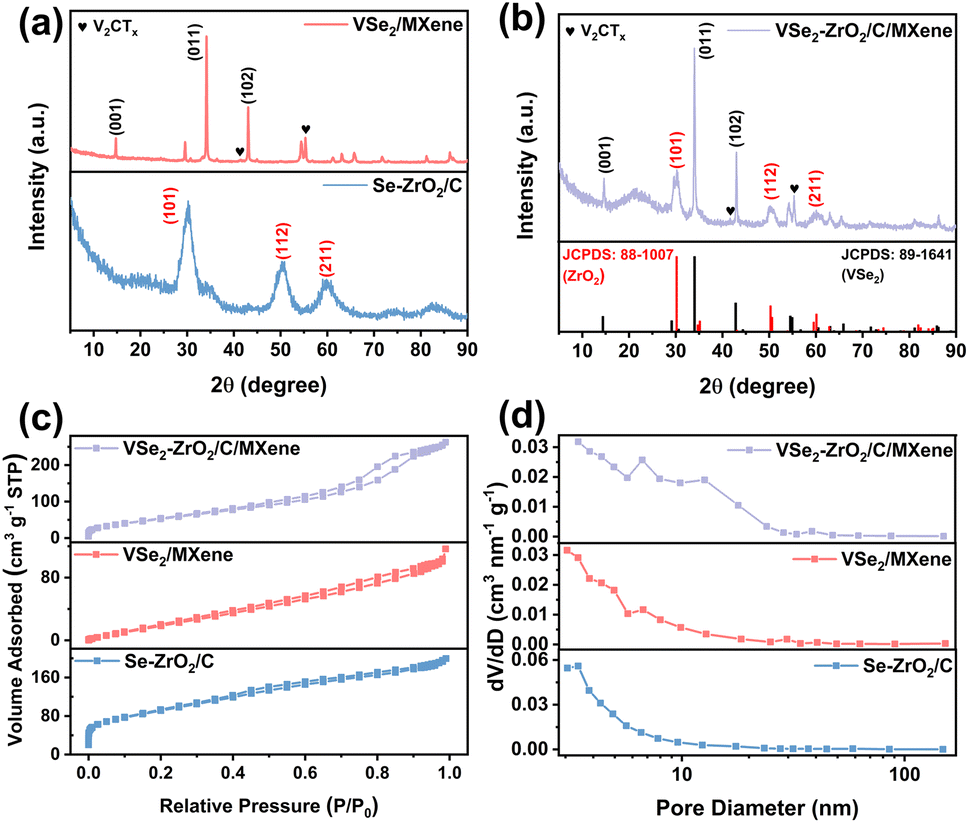 | ||
| Fig. 2 XRD patterns of (a) VSe2/MXene and Se–ZrO2/C, (b) VSe2–ZrO2/C/MXene. (c) Nitrogen adsorption–desorption isotherms and (d) pore size distributions of VSe2–ZrO2/C/MXene, VSe2/MXene and Se–ZrO2/C. | ||
N2 adsorption–desorption isotherms were conducted to investigate the specific surface area and pore nature based on the Brunauer–Emmett–Teller (BET) and Barrett–Joyner–Halenda (BJH) methods, respectively. All of the sample plots are in accordance with the type-IV hysteresis curve in Fig. 2c. The apparent hysteresis loop at a relative pressure range P/P0 of 0.2–0.8 and pore size distributions (Fig. 2d) indicate mesoporous constructions in these samples. The abundance of mesopores not only contributes to the specific surface area, but also provides more reaction sites. This type of pore structure will provide a site for the formation of the Li-cluster, which contributes to the high capacity of the composite, especially after activation. Furthermore, the hierarchical porous structure constructs abundant 3D channels to facilitate ion transport. The details of the specific surface areas, pore volumes, and mean pore diameters of V2CTx, Se–ZrO2/C, VSe2/MXene, and VSe2–ZrO2/C/MXene are summarized in Table 1. The specific surface area of VSe2–ZrO2/C/MXene is calculated to be 218.7 m2 g−1, which is higher than that of VSe2/MXene. Such an enhancement of the specific surface area relies on the carbon matrix from the MOF precursor. Furthermore, VSe2–ZrO2/C/MXene displays the largest pore volume among all of the samples due to its unique multi-component architecture. This structural improvement of the MOF-MXene hybrid intuitively explains the enhancement mechanism related to the ample ion storage, facile ion/charge diffusion, and void space for volume variation.55,56 Fig. S4† shows the nitrogen adsorption–desorption isotherms and pore size distributions of the pure V2CTx.
| Sample | Specific surface area (m2 g−1) | Pore volume (cm3 g−1) | Mean pore diameter (nm) |
|---|---|---|---|
| V2CTx | 88.2 | 0.173 | 3.063 |
| Se–ZrO2/C | 329.0 | 0.196 | 3.422 |
| VSe2/MXene | 132.7 | 0.176 | 3.059 |
| VSe2–ZrO2/C/MXene | 218.7 | 0.368 | 3.410 |
The chemical states of VSe2/MXene, Se–ZrO2/C, and VSe2–ZrO2/C/MXene were disclosed by X-ray photoelectron spectroscopy (XPS) analysis. Fig. 3a shows the XPS survey spectra and Table S1† demonstrates the atomic concentration (at%) of the elements for Se–ZrO2/C, VSe2/MXene and VSe2–ZrO2/C/MXene. The results confirm the existence of Zr, V, Se, C, N and O in VSe2–ZrO2/C/MXene, which is consistent with the elemental mapping results. As shown in the high-resolution Zr 3d spectrum (Fig. 3b), the peaks centered at 184.3 eV and 181.9 eV originate from the Zr 3d3/2 and Zr 3d5/2 orbitals of Zr4+.57 Compared to Se–ZrO2/C, a slight shift to higher binding energy for Zr 3d in VSe2–ZrO2/C/MXene can be detected, indicating the formation of strong chemical binding between VSe2 and ZrO2. Fig. 3c presents the V 2p spectrum of VSe2/MXene and VSe2–ZrO2/C/MXene. Three pairs of peaks at 524.9/517.0 eV, 523.4/516.6 eV, and 521.0/512.4 eV can be deconvoluted, representing the V 2p1/2 and V 2p3/2 orbitals of the V4+, V3+ and V2+ states, respectively.30 The XPS V 2p peak shape becomes less metallic after introducing ZrO2, which is mainly manifested as a higher binding energy shift and increased asymmetric shape at the high binding energy side.58 The Se 3d spectrum in Fig. 3d shows that the peaks observed at 55.9 eV and 53.1 eV are attributed to Se 3d3/2 and Se 3d5/2 orbitals, respectively. These two peaks appear at the relatively low binding energy position because of the weakened metal–Se bond in VSe2/MXene.59 Another peak at 58.9 eV is derived from the formation of SeOx on the surface of the composite.60 The increased atomic ratio of V4+ and V3+ at the high binding energy side and the change of the Se 3d orbital intensities reflect the partial electronic transfer upon the influence of ZrO2.61 Moreover, the presence of defects in the samples was investigated by electron paramagnetic resonance (EPR). From the spectra in Fig. S5,† the signal at g factor of 2.00 can be assigned to the amorphous MOF-derived carbon,62 which is detected in Se–ZrO2/C and VSe2–ZrO2/C/MXene. Accordingly, an additional signal at g factor of 1.97 originates from the paramagnetic V4+.63,64 The significant defect-related signal indicates the existence of abundant active sites in VSe2–ZrO2/C/MXene. Notably, more active nodes for lithium storage are favorable to coulombic efficiency because of the increased capacity and faster ion transport.65,66
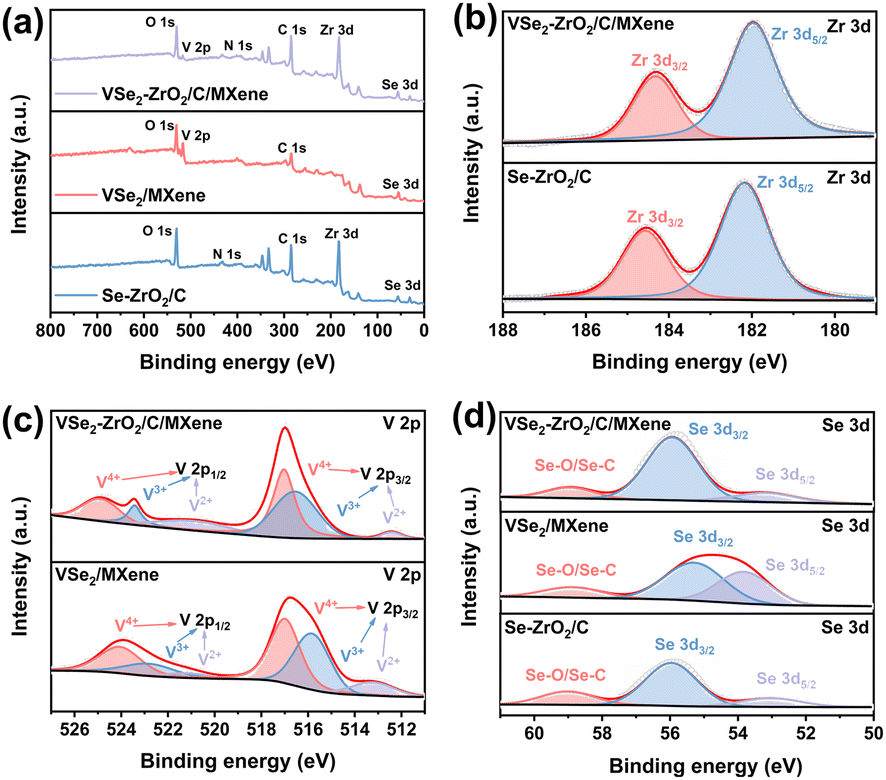 | ||
| Fig. 3 (a) Survey, (b) Zr 3d, (c) V 2p and (d) Se 3d XPS spectra of Se–ZrO2/C, VSe2/MXene and VSe2–ZrO2/C/MXene. | ||
Thermogravimetric analysis (TGA) from atmospheric temperature to 800 °C was tested in a nitrogen atmosphere to investigate the residue of free Se in these electrode materials. As previously reported, the Se is sublimated at around 400 °C in N2.67 It can be observed in Fig. S6† that except for the release of adsorbed or bonded water before 200 °C, the slight weight loss of 3.8% for Se–ZrO2/C results from the sublimation of the free Se, indicating that there is no residue of free Se in these samples. Notably, compared with Se–ZrO2/C, VSe2/MXene and VSe2–ZrO2/C/MXene exhibit much more significant weight loss after 400 °C, which should be ascribed to the fact that the unstable V4+ is prone to be converted into the V3+ or V5+ species.19
The electrochemical performances of the samples were evaluated in half cells. As depicted in Fig. 4a and S7a and b,† the cyclic voltammetry (CV) curves gradually overlap from cycle to cycle, which is typical for Se-based materials.68–70 In the initial cathodic scan of VSe2–ZrO2/C/MXene, two broad peaks from 0.9 to 0.01 V can be recorded, attributed to certain side reactions, including the electrolyte decomposition along with the generation of the solid-state electrolyte interface (SEI) layer.71 During the intercalation of lithium ions, the cathodic peaks centered at 1.9 V and 1.4 V can be assigned to the formation of LixVSe2 (eqn. (1))71 and intermediate product Li2Sen (n ≥ 4) (eqn. (2)),67 respectively. Another reduction peak at 0.5 V can be attributed to the conversion reaction to V and Li2Se (eqn. (3)).71 In the subsequent curves, two intercalation peaks shift slightly to lower voltage due to the polarization and merge into a wide-ranging peak at ∼1.3 V, which could be certified by the decreased capacity in the first few cycles in Fig. 4b. This phenomenon for the deviation of CV curves is common for TMDs.72,73 During the initial anodic scan, the peaks at 1.9 V and 2.2 V are associated with delithiation to gain the intermediate product LixVSe2 (eqn. (4)) and fully charged product VSe2 (eqn. (5))19 and Se (eqn. (6)).74 It is noticed that from the subsequent cycles, two anodic peaks remain at the original position with weakened intensity, resulting from the irreversible formation of the SEI layer. The CV curves of Se–ZrO2/C and VSe2/MXene show the corresponding redox peaks. Notably, the reduction peaks for VSe2/MXene are not obvious at 1.8 V, demonstrating weaker insertion of lithium ions. The phenomenon should be ascribed to its low specific surface area and small pore volume, which leads to stiff lithium-ion insertion and poor storage site. According to the above electrochemical reaction analysis, we suggest the reaction process of the VSe2–ZrO2/C/MXene electrode for LIBs as below:
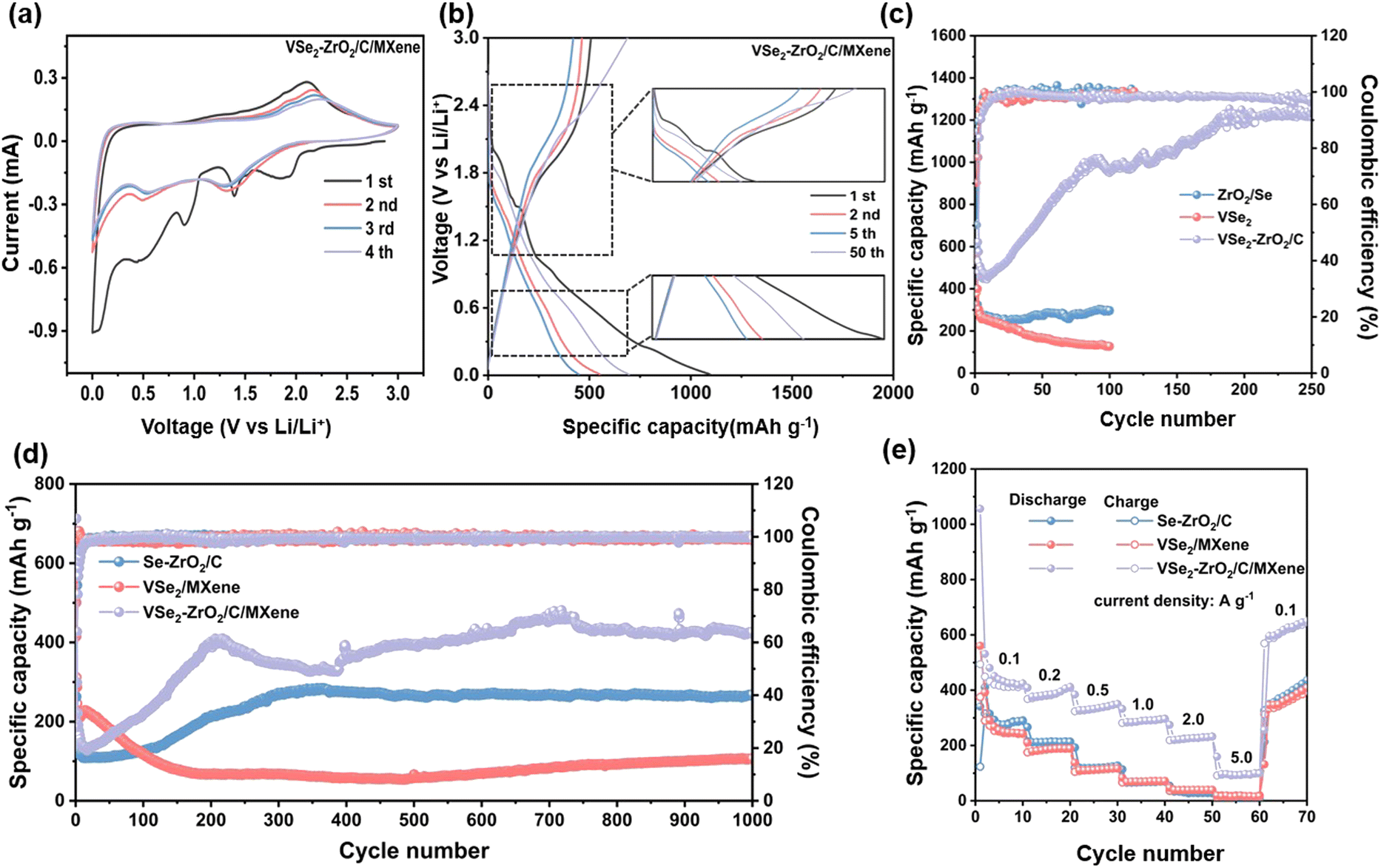 | ||
| Fig. 4 (a) CV curves at 0.2 mV s−1, and (b) galvanostatic charge/discharge profiles of VSe2–ZrO2/C/MXene (inset shows the magnified images for charge/discharge curves at the range of 0.2–0.8 V and 1.0–2.5 V). (c) Cycling performances and corresponding coulombic efficiencies of Se–ZrO2/C, VSe2/MXene, and VSe2–ZrO2/C/MXene at 100 mA g−1. (d) Long-term cyclabilities at 1.0 A g−1 and (e) rate performances of Se–ZrO2/C, VSe2/MXene and VSe2–ZrO2/C/MXene. | ||
The discharge reactions:
| xLi+ + VSe2 + xe− → LixVSe2 | (1) |
| 2Li+ + Sen + 2e− → Li2Sen | (2) |
| LixVSe2 + (4 − x)Li+ + (4 − x)e− → 2Li2Se + V | (3) |
The charge reactions:
| 2Li2Se + V → LixVSe2 + (4 − x)Li+ + (4 − x)e− | (4) |
| LixVSe2 → xLi+ + VSe2 + xe− | (5) |
| Li2Sen → 2Li+ + Sen + 2e− | (6) |
Fig. 4b and S7c and d† show the galvanostatic charge/discharge (GCD) profiles of the electrodes. Several distinct voltage plateaus at ∼2.0 V, 1.4 V, and 0.9 V can be detected in the initial discharge curve, which is in good agreement with the CV curves. The current measurement of the state of charge (SOC) of the battery is currently still a difficult point. Thus, for the linear discharge curve of the anode, we suggest that this behavior may provide an opportunity to accurately measure the SOC of the battery. During the charging process, the plateau is of negligible change. It can be observed that the initial charge/discharge capacities of VSe2–ZrO2/C/MXene, Se–ZrO2/C and VSe2/MXene are 508/1094.4 mA h g−1, 315.4/598.3 mA h g−1 and 381/562.6 mA h g−1, respectively, giving the coulombic efficiencies of 46.42%, 52.71% and 67.72%, respectively. The initial fading irreversible capacity is mainly related to the formation of the SEI layer. For the porous material, the high pore volume and a larger surface area offer abundant contact sites between the electrolyte and electrode, which leads to the polarization and lower coulombic efficiency.75,76 On the other side, the reversible capacity of VSe2–ZrO2/C/MXene surpasses those of Se–ZrO2/C and VSe2/MXene to a large extent, illustrating that the architecture of VSe2–ZrO2/C/MXene is more conducive to the storage of lithium ions. With the cycling proceeding, the reversible capacity of VSe2–ZrO2/C/MXene manifests a continuous elevation derived from the activation of the electrode after a transitory deterioration in the first ten cycles. The details are elaborated in Fig. 4c. Surprisingly, the specific capacity of VSe2–ZrO2/C/MXene becomes stable up to 1238.5 mA h g−1 after 200 cycles. Compared to VSe2–ZrO2/C/MXene, Se–ZrO2/C achieves stable cycling performance with a specific capacity of 298 mA h g−1, while VSe2/MXene only presents a specific capacity of 128.2 mA h g−1 after 100 cycles. The visual insight of the morphology change after cycling by FESEM is presented in Fig. S8.† As depicted, the obtained VSe2/MXene flakes become one giant bulk, and Se–ZrO2/C is subjected to severe structural damage on account of the mechanical strains in repeated cycling. Fortunately, the VSe2–ZrO2/C/MXene composite retains the octahedral shape, ensuring its outstanding cyclic and structural stability. All of the samples retain stable coulombic efficiencies close to 100% after ten cycles, as shown in Fig. 4c. The continuous activation is also reflected by the long-term cycling performance in Fig. 4d. The capacity of VSe2–ZrO2/C/MXene continues to increase until 200 cycles. VSe2–ZrO2/C/MXene delivers excellent cycling performance even over 1000 cycles at 1.0 A g−1, achieving a high capacity retention of nearly 100%. The reversible capacity can be maintained at 430 mA h g−1 after 1000 cycles, which is superior to Se–ZrO2/C (265 mA h g−1) and VSe2/MXene (105 mA h g−1). It can be detected that the electrode underwent complex activation during cycling, which is common in anodes.48,77–79 Furthermore, the activation behavior depends on many factors, such as the mechanical stress and volume change rate at different current densities.80 This fading-reactivation phenomenon is probably driven by the delayed soaking of electrolytes into porous carbon and the gradual establishment of the SEI layer.81–83 Small inorganic species in the composite can also induce the enhancement.84 The Li-cluster in porous carbon material can also provide extra ultra-high capacity.85,86 The increasing capacity of the anode does not cause a capacity decay of the battery. However, we suggest that the increased capacity of the composite can couple with some capacity attenuation anode materials, including Si, Sn, Sb, and Bi, to achieve capacity complementarity.87–90 This behavior should provide an opportunity to settle the capacity-fading situation.
Fig. 4e shows the rate performances of VSe2–ZrO2/C/MXene, Se–ZrO2/C and VSe2/MXene. The reversible specific capacities of VSe2–ZrO2/C/MXene are 427.8, 385.1, 335.3, 288.9 and 225.9 mA h g−1 at the current density of 0.1, 0.2, 0.5, 1.0 and 2.0 A g−1, respectively. Even at a high current density of 5.0 A g−1, VSe2–ZrO2/C/MXene still exhibits excellent cyclability. The gratifying stability demonstrates that the multi-hole carbon matrix derived from the MOF structure can act as a flexible buffering to relieve volume variety and suppress electrode pulverization at high current density. With the recovery of the current density to 0.1 A g−1, a reversible capacity of 612.2 mA h g−1 can be obtained, indicating the excellent reversibility of the VSe2–ZrO2/C/MXene electrode. The versatile rate performance is to a great extent attributed to the inimitable structural construction. Expressly, the multi-channel carbon derived from the MOF precursor not only guarantees the architectural integrity during the charge/discharge process, but also provides ample reaction sites for lithium ions. Furthermore, ZrO2 chemically bonds with VSe2 to assist it in anchoring firmly on carbon. By contrast, Se–ZrO2/C and VSe2/MXene establish much lower reversible capacities than VSe2–ZrO2/C/MXene. Thereinto, Se–ZrO2/C presents higher capacities of 243.5, 213.4, 120.9, 68.0, 30.3 and 9.3 mA h g−1 than VSe2/MXene at 0.1, 0.2, 0.5, 1.0, 2.0 and 5.0 A g−1, respectively, which further proves that the introduction of ZrO2 is beneficial to better stability for lithium-ion storage. The relative growth of reversible capacity for these selenide samples indicates the different extent of the activation process.71,75,91 To the best of our knowledge, our prepared VSe2–ZrO2/C/MXene exhibits the best electrochemical properties among the similar type of LIBs anode materials reported in the literature in terms of the reversible capacity, cycling stability, and rate capability, as shown in Table S2†, highlighting the promising application of VSe2–ZrO2/C/MXene in LIBs.
The evaluation of the electrochemical performance of anode materials is not only the capacity, but also its voltage plateau. Therefore, we adopted a concept of relative energy density (ER).92,93 We compared our anode with meso-carbon microbeads to evaluate the electrochemical performance of anode materials more accurately and conveniently. The corresponding equations are as follows:
| ER = ΔUQ | (7) |
| ΔU = −PK − VA | (8) |
| VA = ∫UdQ/Q | (9) |
In the equations, Q, PK and VA represent the specific capacity, electrode potential of Li (−3.04 V vs. Li/Li+) and average voltage, respectively. Fig. 5a and b present the determination of these parameters. According to the calculation from the above equations, Fig. 5c shows that the electrode presents an initial discharge ER of 2715.616 W h kg−1, and maintains the discharge ER of 2171.12 W h kg−1 after 100 cycles at 100 mA g−1. Compared with Fig. S9b and c,† it was found that the ER of VSe2–ZrO2/C/MXene is relatively higher than the conventional carbon material. Fig. S9a† is the cycling performance of meso-carbon microbeads.
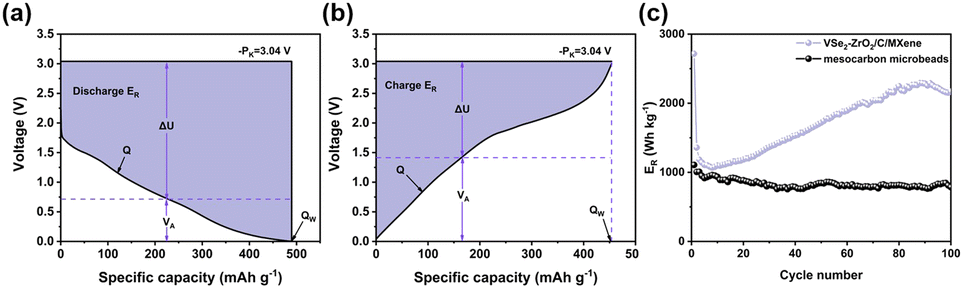 | ||
| Fig. 5 Typical legends for the determination of the relative energy density (ER) of (a) discharge ER and (b) charge ER. (c) Discharge ER of VSe2–ZrO2/C/MXene and meso-carbon microbeads. | ||
To better understand the electrochemical reaction along with cycling and prove the above-mentioned phase transformation, the ex situ XRD measurement of the VSe2–ZrO2/C/MXene electrode was carried out. Fig. 6a records the charge/discharge curve of the VSe2–ZrO2/C/MXene electrode after 250 cycles, and Fig. 6b shows the corresponding XRD patterns during the different insertion/extraction states. It was found that the (101) peak of ZrO2 presents the left shift, suggesting a lattice expansion in the Li-ion insertion process to form LixVSe2, whereas the lattice regresses after delithiation. In the discharge process, the peak of the (011) lattice plane becomes broader and the intensity weakens. After discharging to 0.05 V, the new characteristic peaks of metal V and Li2Se appear. These changes indicate the occurrence of a partial conversion reaction wherein LixVSe2 is converted to Li2Se and metal V. Li2Se and metal V reversibly disappear in the charge process. A similar result was obtained from ex situ Raman spectra, as shown in Fig. S10.† The peaks at ∼1351 and 1590 cm−1 can be characterized as the D and G bands of amorphous carbon, respectively.94 The vague peak at around 640 cm−1 confirms the existence of ZrO2 with low crystallinity.95 Specifically, two broad peaks at ∼280 and 406 cm−1 are assigned to the E2g in-plane vibration mode and A1g out-of-plane vibration mode of VSe2, respectively.71 These two characteristic peaks broadened, and the intensities tended to vanish during the discharging process, which suggests the intercalation of lithium ions and conversion reaction of VSe2. In the charging process, the intensities of the VSe2 peaks gradually recovered. The appearance of a peak located at around 1087 cm−1 is due to the symmetric stretching vibrations of Li2CO3,96,97 which indicates certain side reactions. Additionally, the electrochemical impedance spectroscopy (EIS) plots during the 40th cycle in the activation process were measured at different charge/discharge states, namely, open-circuit voltage (OCV), 0.8 V and 0.05 V (discharging), 1.8 V and 3.0 V (charging). As presented in Fig. 6c, the large Rct (charge transfer resistance) value of 155 Ω in the OCV state decreases to 143 Ω after discharging to 0.8 V. It further decreases to 55.5 Ω when discharging to 0.05 V, which can be attributed to the activation process as a result of enhanced infiltration of the electrolyte into the electrode.98 The semicircle at high frequency nearly vanishes at the delithiation state of 3.0 V, indicating the partial decomposition of the SEI layer at this stage. Fig. S11† records the TEM images of VSe2–ZrO2/C/MXene after discharging down to 0.05 V and fully charging to 3.0 V. The thickness of the SEI layer is approximately 880 nm at 0.05 V in Fig. S11a.† Hereafter, the electrode undergoing reconversion only preserves a thin SEI layer in Fig. S11b,† which further proves the decomposition of the SEI layer. We also compared the EIS plots of VSe2–ZrO2/C/MXene, Se–ZrO2/C and VSe2/MXene cells after 100 cycles, as shown in Fig. 6d. The corresponding equivalent circuit model is shown in the inset. The fitted Rct values of Se–ZrO2/C, VSe2/MXene and VSe2–ZrO2/C/MXene are 9.67, 38.6, and 13.2 Ω, respectively. The decreased impedance of VSe2–ZrO2/C/MXene indicates that the conductive carbon plays an essential role in kinetic behavior. Given that the value of the second semicircle is caused by the charge transfer resistance, the small Rct unveils the surface layer's low resistance, which greatly facilitates the electron transport.99 To further confirm the increase of ionic conductivity in the battery, we calculated the lithium-ion diffusion coefficients (DLi) according to the EIS results. The corresponding equation is as follows:
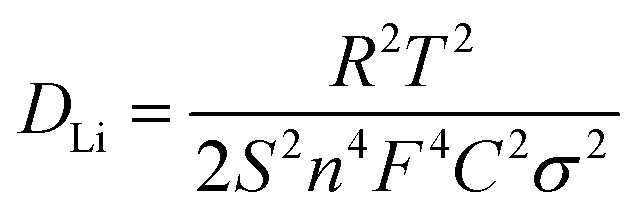 | (10) |
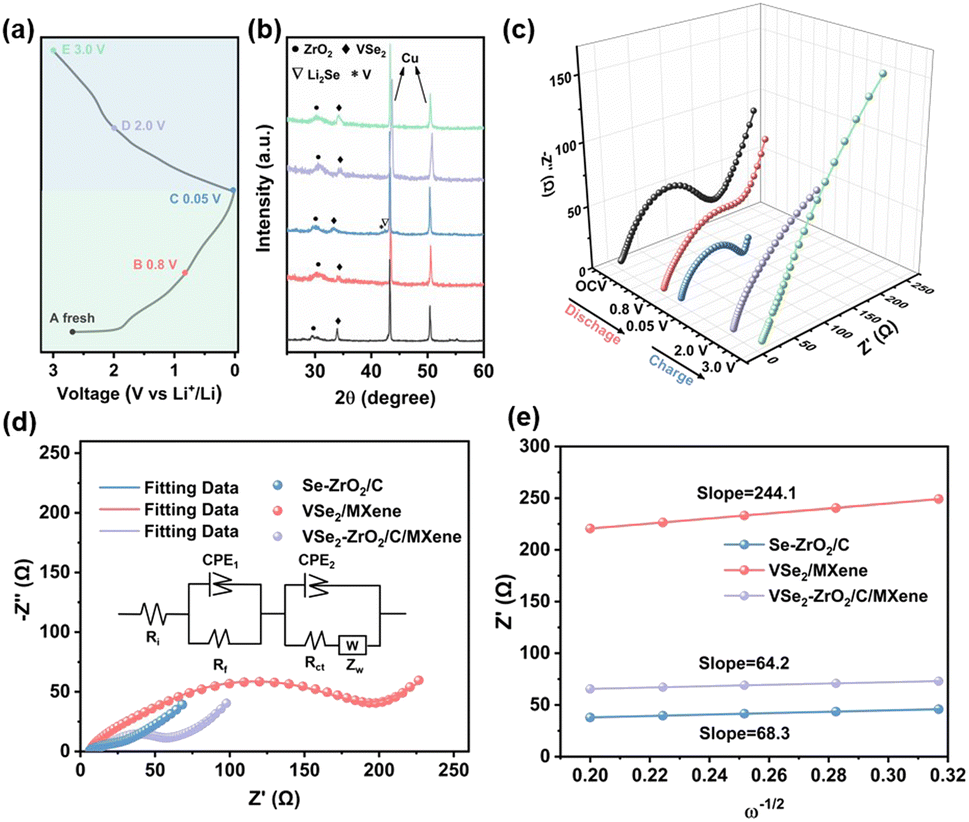 | ||
| Fig. 6 (a) Discharge/charge curve and corresponding voltage positions of VSe2–ZrO2/C/MXene. (b) Ex situ XRD characterizations of the corresponding potential states. (c) EIS plots of VSe2–ZrO2/C/MXene at various states in the 40th cycle at 100 mA g−1. (d) Nyquist plots of Se–ZrO2/C, VSe2/MXene and VSe2–ZrO2/C/MXene after 100 cycles at 100 mA g−1 (inset: corresponding equivalent circuit model). (e) Fitted lines of Z′ versus ω−1/2 at low frequency for VSe2–ZrO2/C/MXene. | ||
Based on the above results, we find a very interesting phenomenon wherein the abnormal charge storage behavior observed in the cycling performance is in contrast with capacity fading in VSe2–ZrO2/C/MXene. The behavior has been referred to as “negative fading” in previous works.100,101 Despite the extensive observations on this subject, its origin has not been identified by far for the reason of complex reactions, involving bulk materials, interface and electrolyte constituents.100,102 To study the internal mechanism of the capacity increase, a series of tests for cells after cycles were thoroughly explored for comparison. Fig. S12b† shows the capacity difference between the 100th cycle (C100) and the 10th cycle (C10), and its calculated differential vs. voltage d(C100–C10)/dV. We suggest that the “negative fading” results from the capacity enhancement in the low potential region.103 The sharp differential peaks at around 0.7 V and 2.0 V can be attributed to the discharge plateaus in Fig. S12a.† When fully discharging to ∼0.01 V, the capacity difference increases to 463.1 mA h g−1. The exceeding reversible capacity for the 100th cycle reveals prominent “negative fading”.
The capacity of the composite should be attributed to the synergistic effect, including the insertion reaction in carbon and MXene, conversion reaction in VSe2, adsorption of Li on the surface of the composite, and the formation of Li-cluster in the space of the composite. To further understand the kinetic behavior, CV curves at different scan rates from 0.2 to 2.0 mV s−1 were measured (Fig. 7a). Especially, the CV curves with the same shapes illustrate the excellent reversibility for Li-ion storage of VSe2–ZrO2/C/MXene. As the two vital mechanisms, the capacitive contribution mainly comprises the adsorption of Li on the surface and the formation of a Li-cluster in the space of the composite, and the diffusion-controlled contributions from the insertion reaction in carbon and MXene, and conversion reaction in VSe2.104 The relationship between the scan rate (v) and current (i) can be summarized as eqn (8):
| i = avb | (11) |
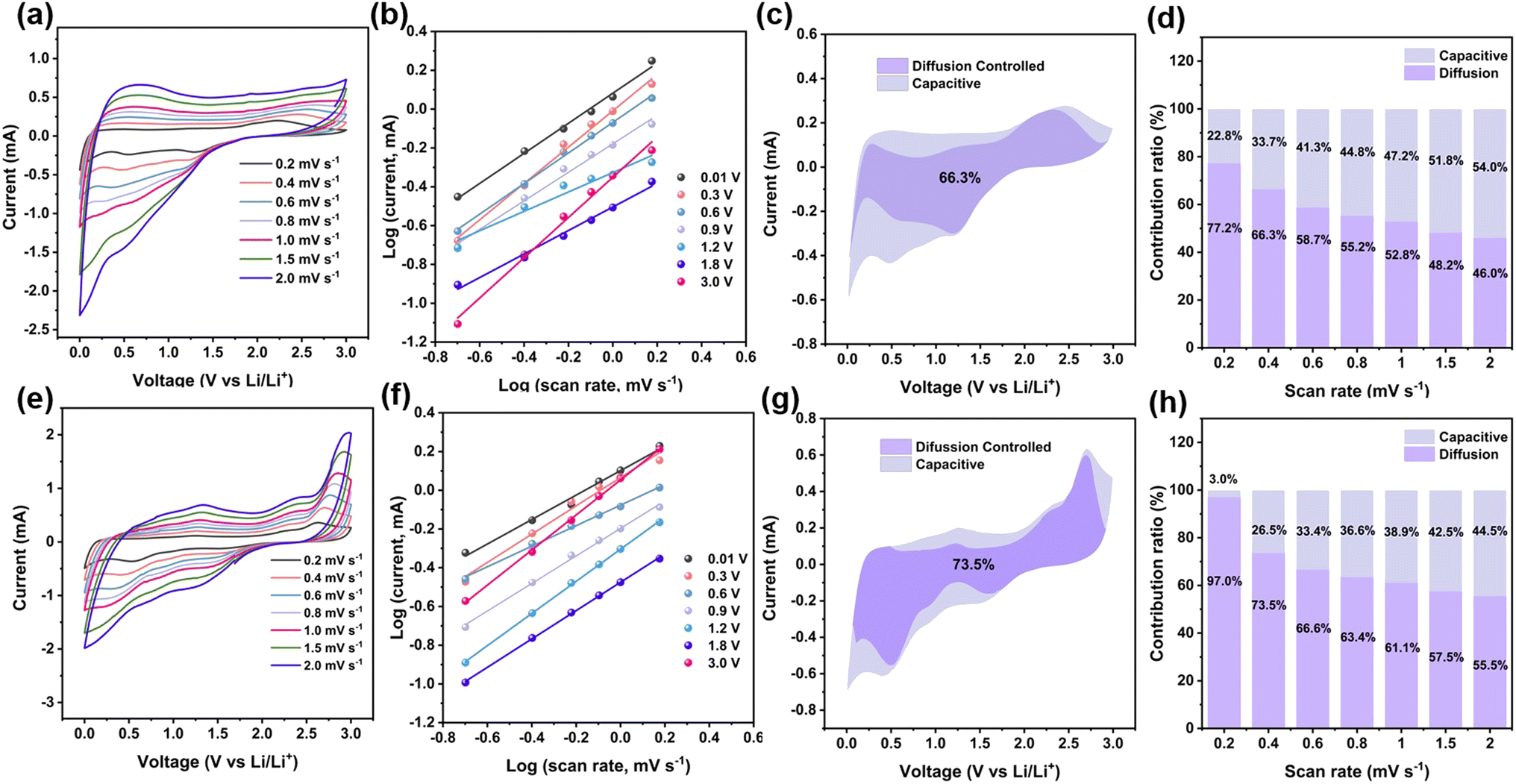 | ||
| Fig. 7 (a and e) CV curves of the as-prepared VSe2–ZrO2/C/MXene and after cycling for 250 cycles at different scan rates. (b and f) Linear relationship between log(i) and log(v) at different voltages for the as-prepared VSe2–ZrO2/C/MXene and after cycling for 250 cycles. (c and g) Separation of capacitive and diffusion-controlled contribution areas at 0.4 mV s−1 for the as-prepared VSe2–ZrO2/C/MXene and after cycling for 250 cycles. (d and h) Ratios of capacitive and diffusion-controlled contributions at different scan rates for the as-prepared VSe2–ZrO2/C/MXene and after cycling for 250 cycles. | ||
Eqn (8) can be converted to eqn (9):
| log(i) = log(a) + blog(v) | (12) |
The specific contribution ratios for capacitive and diffusion-controlled behaviors can be quantified by eqn (10):
| i(V) = k1v + k2v1/2 | (13) |
To fully explain the evolution of the lithium-ion storage performance, the XPS analysis, CV curves and EIS plots for the cells after 250 cycles were performed. As shown in Fig. S13,† the changes in XPS can be found on the V and Se elements, indicating that the fading-reactivation phenomenon is mainly caused by VSe2 in the composite. Fig. 7e displays the CV curves at different sweep rates of 0.2–2.0 mV s−1. The linear relationship between log(i) and log(v) in Fig. 7f reflects the fitted b values of 0.63, 0.73, 0.53, 0.71, 0.83, 0.73, and 0.91. The variations of the b values indicate the change of dominating mechanism. We also calculated the diffusion-controlled contribution of VSe2–ZrO2/C/MXene, as shown in Fig. 7g and h. An enhanced diffusion behavior was observed. The diffusion mechanism of the batteries mainly contributes to the Li-ion insertion or conversion reaction. These continuous behaviors will lead to the continuous change of the material structure, resulting in the occurrence of the fading-reactivation phenomenon. The Nyquist plots for the VSe2–ZrO2/C/MXene anode are also displayed after different cycles in Fig. S14.† The resistance continuously decreases during the activation process, demonstrating the facile reaction kinetics. Even after 200 cycles, the Rct value of 105 Ω is still smaller than the original value of 155 Ω, further supporting the improved capacity activation during cycling. The above evidence explains the enhancement phenomenon in negative fading.
To confirm the applicability of the VSe2–ZrO2/C/MXene electrode, the LiFePO4‖VSe2–ZrO2/C/MXene (LFP‖VSe2–ZrO2/C/MXene) full cells were assembled. It should be noted that the anodes were pre-lithiated at 2 A g−1 for five cycles in half cells to achieve rapid activation. Based on the cycle performance of commercial LiFePO4 and VSe2–ZrO2/C/MXene in Fig. 8a, the mass loading of LiFePO4 should be 2.5 times of that of VSe2–ZrO2/C/MXene, achieving an anode/cathode capacity ratio of ∼1.1. Fig. 8b depicts the galvanostatic charge/discharge curves of the LFP‖VSe2–ZrO2/C/MXene full cell in the 1st, 2nd, 15th and 20th cycles. It can be seen that the full cell exhibits a clearly extended platform. Benefitting from the pre-lithiation before assembly, the initial coulombic efficiency reaches 90.2%, indicating that the effective replenishment of lithium to anode can offset the irreversible lithium loss during the formation of the SEI layer in the initial cycle.105,106 From Fig. 8c and d, the reversible capacity for the LFP‖VSe2–ZrO2/C/MXene full cell retains 113.2 mA h g−1 after 100 cycles at 100 mA g−1 and good rate capability is manifested with a capacity of 71 mA h g−1 at 500 mA g−1, demonstrating a successful operation of the full cell. In addition, the EIS test of the full cell is provided to evaluate the reaction dynamics in Fig. 8e. A small Rct of 113 Ω can be obtained, indicating that our electrode in the full cell also presents an excellent electron transfer characteristic.
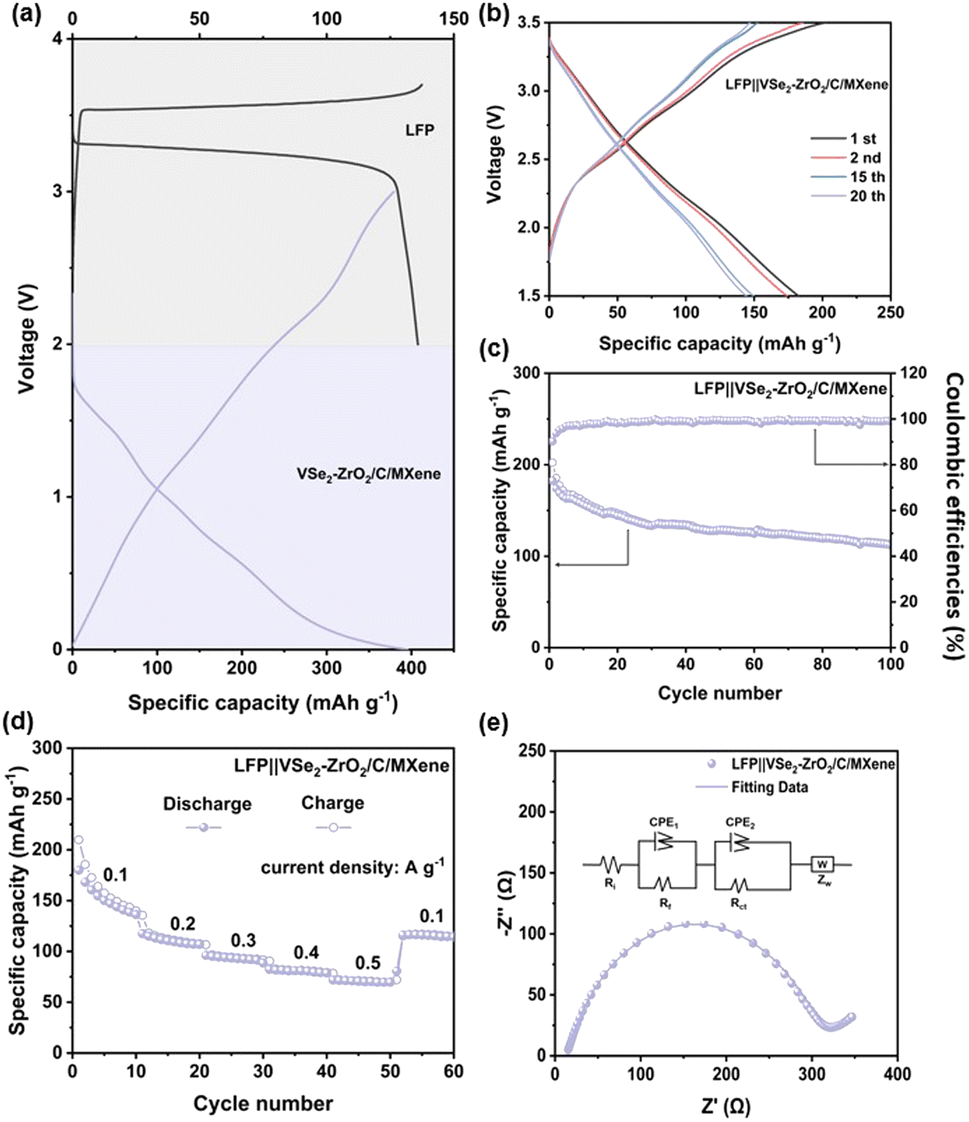 | ||
| Fig. 8 (a) Half-cell charge–discharge profiles of LFP and VSe2–ZrO2/C/MXene electrodes at 100 mA g−1. (b) Galvanostatic charge/discharge profiles and (c) cycling performances for LFP‖VSe2–ZrO2/C/MXene full cell at 100 mA g−1. (d) Rate performances and (e) Nyquist plot for LFP‖VSe2–ZrO2/C/MXene full cell (inset: equivalent electrical circuit model). | ||
The mechanism schematic is summarized in Fig. 9. VSe2 provides the capacity, while the remaining MXene can enhance the conductivity of the hybrid. The derived carbon and ZrO2 play a significant role in Li-ion storage. The ZrO2 in the abundant channel can act like steel to reinforce the carbon structure during repeated lithiation/delithiation. Moreover, it assists the anchoring of VSe2. It may also help the accommodation of Li cluster for storage to provide extra ultra-high capacity.85,86 As shown in Fig. S15,† without the participation of ZrO2 and porous carbon, VSe2/MXene suffers during the repeated cycles, which leads to degenerative performance.
 | ||
| Fig. 9 Schematic of VSe2–ZrO2/C/MXene for the Li-ion storage mechanism. | ||
Conclusions
To summarize, after a simple solvothermal method and in situ selenization, V2CTx-derived VSe2/MXene was incorporated with UiO-66-derived ZrO2 and porous carbon. Due to the synergistic effect of electrochemically active VSe2/MXene, stable ZrO2 and conductive porous carbon matrix in lithium-ion kinetics, the constructed VSe2–ZrO2/C/MXene composite shows an excellent reversible specific capacity of 1238.5 mA h g−1 at 100 mA g−1 with a “negative fading” behavior and a superior rate capability with a capacity retention of 143%. In addition, VSe2–ZrO2/C/MXene manifests a long-term cycling stability with a capacity of 430 mA h g−1 after 1000 cycles at 1.0 A g−1. The LFP‖VSe2–ZrO2/C/MXene full cell retains 113.2 mA h g−1 after 100 cycles at 100 mA g−1, and remains stable even at 500 mA g−1. This new attempt should guide future investigations on MOF/MXene-derived selenide electrode materials in energy storage exploration.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
Financial support from the Shanghai Domestic Science and Technology Cooperation Project (21015801000), the Fundamental Research Funds for the Central Universities (21621406), the Science and Technology Program of Guangzhou, China (202102020737), the Shenzhen Science and Technology Program (JCYJ20200109113606007) is gratefully acknowledged.Notes and references
- Y. Li, R. Wang, Z. Guo, Z. Xiao, H. Wang, X. Luo and H. Zhang, J. Mater. Chem. A, 2019, 7, 25227–25246 RSC.
- J. Li, J. Yu, I. S. Amiinu, J. Zhang, J. Sheng, Z. Kou, Z. Wang, Q. Yu, L. Mai and S. Mu, J. Mater. Chem. A, 2017, 5, 18509–18517 RSC.
- X. Zhang, J. Li, J. Li, L. Han, T. Lu, X. Zhang, G. Zhu and L. Pan, Chem. Eng. J., 2020, 385, 123394 CrossRef CAS.
- Y. Zhang, J. Li, H. Li, H. Shi, Z. Gong, T. Lu and L. Pan, J. Colloid Interface Sci., 2022, 607, 145–152 CrossRef CAS PubMed.
- Y. Zhao, L. P. Wang, M. T. Sougrati, Z. Feng, Y. Leconte, A. Fisher, M. Srinivasan and Z. Xu, Adv. Energy Mater., 2017, 7, 1601424 CrossRef.
- X. Cai, W. Yi, J. Chen, L. Lu, B. Sun, Y. Ni, S. A. T. Redfern, H. Wang, Z. Chen and Y. Chen, J. Mater. Chem. A, 2022, 10, 6551–6559 RSC.
- H. Hou, L. Shao, Y. Zhang, G. Zou, J. Chen and X. Ji, Adv. Sci., 2017, 4, 1600243 CrossRef PubMed.
- F. Wang, B. Wang, J. Li, B. Wang, Y. Zhou, D. Wang, H. Liu and S. Dou, ACS Nano, 2021, 15, 2197–2218 CrossRef CAS PubMed.
- Y. Zhang, X. Yuan, T. Lu, Z. Gong, L. Pan and S. Guo, J. Colloid Interface Sci., 2021, 585, 347–354 CrossRef CAS PubMed.
- Z. Wei, L. Wang, M. Zhuo, W. Ni, H. Wang and J. Ma, J. Mater. Chem. A, 2018, 6, 12185–12214 RSC.
- J. Li, W. Liu, C. Chen, X. Zhao, Z. Qiu, H. Xu, F. Sheng, Q. Hu, Y. Zheng, M. Lin, S. J. Pennycook, C. Su and J. Lu, J. Mater. Chem. A, 2019, 7, 23958–23963 RSC.
- X. Hu, X. Liu, K. Chen, G. Wang and H. Wang, J. Mater. Chem. A, 2019, 7, 11016–11037 RSC.
- S.-K. Park, J. K. Kim and Y. C. Kang, Chem. Eng. J., 2017, 328, 546–555 CrossRef CAS.
- P. Ge, C. Zhang, H. Hou, B. Wu, L. Zhou, S. Li, T. Wu, J. Hu, L. Mai and X. Ji, Nano Energy, 2018, 48, 617–629 CrossRef CAS.
- J. Xiao, H. Liu, J. Huang, Y. Lu and L. Zhang, Appl. Surf. Sci., 2020, 526, 146746 CrossRef CAS.
- L. Zhu, Z. Wang, L. Wang, L. Xie, J. Li and X. Cao, Chem. Eng. J., 2019, 364, 503–513 CrossRef CAS.
- C. Yang, J. Feng, F. Lv, J. Zhou, C. Lin, K. Wang, Y. Zhang, Y. Yang, W. Wang, J. Li and S. Guo, Adv. Mater., 2018, 30, 1800036 CrossRef PubMed.
- Z. Zhang, J. Niu, P. Yang, Y. Gong, Q. Ji, J. Shi, Q. Fang, S. Jiang, H. Li, X. Zhou, L. Gu, X. Wu and Y. Zhang, Adv. Mater., 2017, 29, 1702359 CrossRef PubMed.
- F. Ming, H. Liang, Y. Lei, W. Zhang and H. N. Alshareef, Nano Energy, 2018, 53, 11–16 CrossRef CAS.
- Y. Wang, B. Qian, H. Li, L. Liu, L. Chen and H. Jiang, Mater. Lett., 2015, 141, 35–38 CrossRef CAS.
- J. Li, D. Yan, S. Hou, Y. Li, T. Lu, Y. Yao and L. Pan, J. Mater. Chem. A, 2018, 6, 1234–1243 RSC.
- L. Wan, Y. Tang, L. Chen, K. Wang, J. Zhang, Y. Gao, J. Y. Lee, T. Lu, X. Xu, J. Li, Y. Zheng and L. Pan, Chem. Eng. J., 2021, 410, 128349 CrossRef CAS.
- C. Cui, R. Dai, C. Zhang, B. Fan and X. Wang, J. Mater. Chem. A, 2022, 10, 15474–15484 RSC.
- A. Lipatov, M. Alhabeb, M. R. Lukatskaya, A. Boson, Y. Gogotsi and A. Sinitskii, Adv. Electron. Mater., 2016, 2, 1600255 CrossRef.
- Y. Yu, Z. Guo, Q. Peng, J. Zhou and Z. Sun, J. Mater. Chem. A, 2019, 7, 12145–12153 RSC.
- M. K. Aslam, Y. Niu and M. Xu, Adv. Energy Mater., 2021, 11, 2000681 CrossRef CAS.
- Y. Zhang, J. Li, Z. Gong, J. Xie, T. Lu and L. Pan, J. Colloid Interface Sci., 2021, 587, 489–498 CrossRef CAS PubMed.
- L. Wan, D. H. C. Chua, H. Sun, L. Chen, K. Wang, T. Lu and L. Pan, J. Colloid Interface Sci., 2021, 588, 147–156 CrossRef CAS PubMed.
- Z. Chen, X. Xu, Z. Ding, K. Wang, X. Sun, T. Lu, M. Konarova, M. Eguchi, J. G. Shapter, L. Pan and Y. Yamauchi, Chem. Eng. J., 2021, 407, 127148 CrossRef CAS.
- D. Sha, C. Lu, W. He, J. Ding, H. Zhang, Z. Bao, X. Cao, J. Fan, Y. Dou, L. Pan and Z. Sun, ACS Nano, 2022, 16, 2711–2720 CrossRef CAS PubMed.
- Z. Ali, T. Zhang, M. Asif, L. Zhao, Y. Yu and Y. Hou, Mater. Today, 2020, 35, 131–167 CrossRef CAS.
- Z. Ye, Y. Jiang, L. Li, F. Wu and R. Chen, Adv. Mater., 2021, 33, 2101204 CrossRef CAS PubMed.
- J. Ahn, E. K. Jang, S. Yoon, S.-J. Lee, S.-J. Sung, D.-H. Kim and K. Y. Cho, Appl. Surf. Sci., 2019, 484, 701–709 CrossRef CAS.
- J. Cho, Y. J. Kim, T.-J. Kim and B. Park, Angew. Chem., Int. Ed., 2001, 40, 3367–3369 CrossRef CAS PubMed.
- L. Yao, F. Liang, J. Jin, B. V. R. Chowdari, J. Yang and Z. Wen, Chem. Eng. J., 2020, 389, 124403 CrossRef CAS.
- C. Yang, D. Liu, S. Huang and W. Lei, Energy Storage Mater., 2020, 28, 393–400 CrossRef.
- J. H. Park, H. J. Lee, J. Y. Cho, S. Jeong, H. Y. Kim, J. H. Kim, S. H. Seo, H. J. Jeong, S. Y. Jeong, G.-W. Lee and J. T. Han, ACS Appl. Mater. Interfaces, 2020, 12, 1322–1329 CrossRef CAS PubMed.
- X.-h. Zhou, K.-m. Su, W.-m. Kang, B.-w. Cheng, Z.-h. Li and Z. Jiang, Chem. Eng. J., 2020, 397, 125271 CrossRef CAS.
- J. Li, L. Han, X. Zhang, H. Sun, X. Liu, T. Lu, Y. Yao and L. Pan, Chem. Eng. J., 2021, 406, 126873 CrossRef CAS.
- J. Li, J. Li, Z. Ding, X. Zhang, Y. Li, T. Lu, Y. Yao, W. Mai and L. Pan, Chem. Eng. J., 2019, 378, 122108 CrossRef CAS.
- Z. Ding, X. Xu, J. Li, Y. Li, K. Wang, T. Lu, M. S. A. Hossain, M. A. Amin, S. Zhang, L. Pan and Y. Yamauchi, Chem. Eng. J., 2022, 430, 133161 CrossRef CAS.
- J. Wang, H. Wang, F. Li, S. Xie, G. Xu, Y. She, M. K. H. Leung and T. Liu, J. Mater. Chem. A, 2019, 7, 3024–3030 RSC.
- Y. Zhang, S. Li, L. Cheng, Y. Li, X. Ren, P. Zhang, L. Sun and H. Y. Yang, J. Mater. Chem. A, 2021, 9, 3388–3397 RSC.
- J. Li, L. Han, X. Zhang, G. Zhu, T. Chen, T. Lu and L. Pan, Chem. Eng. J., 2019, 370, 800–809 CrossRef CAS.
- J. Li, D. Yan, T. Lu, Y. Yao and L. Pan, Chem. Eng. J., 2017, 325, 14–24 CrossRef CAS.
- Q. Cheng and X. Yu, J. Mater. Chem. A, 2021, 9, 11381–11396 RSC.
- K. Liu, C. Li, L. Yan, M. Fan, Y. Wu, X. Meng and T. Ma, J. Mater. Chem. A, 2021, 9, 27234–27251 RSC.
- T. Yang, Y. Liu, D. Yang, B. Deng, Z. Huang, C. D. Ling, H. Liu, G. Wang, Z. Guo and R. Zheng, Energy Storage Mater., 2019, 17, 374–384 CrossRef.
- J. Jin, Y. Zheng, L. B. Kong, N. Srikanth, Q. Yan and K. Zhou, J. Mater. Chem. A, 2018, 6, 15710–15717 RSC.
- C. Liu, Y. Bai, W. Li, F. Yang, G. Zhang and H. Pang, Angew. Chem., Int. Ed., 2022, 61, e202116282 CAS.
- F. Chen, S. Zhang, B. Ma, Y. Xiong, H. Luo, Y. Cheng, X. Li, X. Wang and R. Gong, Chem. Eng. J., 2022, 431, 134007 CrossRef CAS.
- H. Zou, B. He, P. Kuang, J. Yu and K. Fan, ACS Appl. Mater. Interfaces, 2018, 10, 22311–22319 CrossRef CAS PubMed.
- Y. Jiang, G. Cheng, Y. Li, Z. He, J. Zhu, W. Meng, L. Dai and L. Wang, Chem. Eng. J., 2021, 415, 129014 CrossRef CAS.
- Z. Wang, S. Wang, A. Wang, X. Liu, J. Chen, Q. Zeng, L. Zhang, W. Liu and L. Zhang, J. Mater. Chem. A, 2018, 6, 17227–17234 RSC.
- T. Li, Y. Bai, Y. Wang, H. Xu and H. Jin, Coord. Chem. Rev., 2020, 410, 213221 CrossRef CAS.
- Y. Zhang, Z. Mu, J. Lai, Y. Chao, Y. Yang, P. Zhou, Y. Li, W. Yang, Z. Xia and S. Guo, ACS Nano, 2019, 13, 2167–2175 CAS.
- R. J. Toh, Z. Sofer and M. Pumera, J. Mater. Chem. A, 2016, 4, 18322–18334 RSC.
- W. Zhang, L. Zhang, P. K. J. Wong, J. Yuan, G. Vinai, P. Torelli, G. van der Laan, Y. P. Feng and A. T. S. Wee, ACS Nano, 2019, 13, 8997–9004 CrossRef CAS PubMed.
- Y. Bai, H. Zhang, B. Xiang, X. Liang, J. Hao, C. Zhu and L. Yan, ACS Appl. Mater. Interfaces, 2021, 13, 23230–23238 CrossRef CAS PubMed.
- Z. Liu, C. Zhang, H. Liu and L. Feng, Appl. Catal., B, 2020, 276, 119165 CrossRef CAS.
- L. Kang, C. Huang, J. Zhang, M. Zhang, N. Zhang, S. Liu, Y. Ye, C. Luo, Z. Gong, C. Wang, X. Zhou, X. Wu and S. C. Jun, Chem. Eng. J., 2020, 390, 124643 CrossRef CAS.
- R. Daiyan, X. Tan, R. Chen, W. H. Saputera, H. A. Tahini, E. Lovell, Y. H. Ng, S. C. Smith, L. Dai, X. Lu and R. Amal, ACS Energy Lett., 2018, 3, 2292–2298 CrossRef CAS.
- G. Che, D. Wang, C. Wang, F. Yu, D. Li, N. Suzuki, C. Terashima, A. Fujishima, Y. Liu and X. Zhang, Chem. Eng. J., 2020, 397, 125381 CrossRef CAS.
- Z. Chen, N. Mi, L. Huang, W. Wang, C. Li, Y. Teng and C. Gu, Sci. Total Environ., 2022, 808, 152083 CrossRef CAS PubMed.
- R. Xue, N. Liu, L. Bao, L. Chen, Y. Su, Y. Lu, J. Dong, S. Chen and F. Wu, J. Energy Chem., 2020, 50, 378–386 CrossRef.
- Y. Tian, Y. An and J. Feng, ACS Appl. Mater. Interfaces, 2019, 11, 10004–10011 CrossRef CAS PubMed.
- P. Xue, Y. Zhai, N. Wang, Y. Zhang, Z. Lu, Y. Liu, Z. Bai, B. Han, G. Zou and S. Dou, Chem. Eng. J., 2020, 392, 123676 CrossRef CAS.
- X. Yang, S. Wang, D. Y. W. Yu and A. L. Rogach, Nano Energy, 2019, 58, 392–398 CrossRef CAS.
- J. Li, J. Zhao, R. Tang, Q. Chen, Z. Niu, M. Li, C. Guo, J. Su and L. Zhang, J. Power Sources, 2020, 449, 227517 CrossRef CAS.
- J. Jin, Y. Zheng, L. B. Kong, N. Srikanth, Q. Yan and K. Zhou, J. Mater. Chem. A, 2018, 6, 15710–15717 RSC.
- Q. Jiang, J. Wang, Y. Jiang, L. Li, X. Cao and M. Cao, Nanoscale, 2020, 12, 8858–8866 RSC.
- Y. Liu, Y. Fang, Z. Zhao, C. Yuan and X. W. Lou, Adv. Energy Mater., 2019, 9, 1803052 CrossRef.
- M. Wan, R. Zeng, K. Chen, G. Liu, W. Chen, L. Wang, N. Zhang, L. Xue, W. Zhang and Y. Huang, Energy Storage Mater., 2018, 10, 114–121 CrossRef.
- H. Ye, Y.-X. Yin, S.-F. Zhang and Y.-G. Guo, J. Mater. Chem. A, 2014, 2, 13293–13298 RSC.
- H. Wang, X. Wang, Q. Li, H. Li, J. Xu, X. Li, H. Zhao, Y. Tang, G. Zhao, H. Li, H. Zhao and S. Li, ACS Appl. Mater. Interfaces, 2018, 10, 38862–38871 CrossRef CAS PubMed.
- D. Zhao, J. Qin, L. Zheng and M. Cao, Chem. Mater., 2016, 28, 4180–4190 CrossRef CAS.
- Q. Yi, M. Du, B. Shen, J. Ji, C. Dong, M. Xing and J. Zhang, Sci. Bull., 2020, 65, 233–242 CrossRef CAS.
- J. Yan, C. Gao, S. Qi, Z. Jiang, L. R. Jensen, H. Zhan, Y. Zhang and Y. Yue, Nano Energy, 2022, 103, 107779 CrossRef CAS.
- T. Yang, M. Fang, J. Liu, D. Yang, Y. Liang, J. Zhong, Y.-J. Yuan, Y. Zhang, X. Liu, R. Zheng, K. Davey, J. Zhang and Z. Guo, Adv. Funct. Mater., 2022, 32, 2205880 CrossRef CAS.
- H. Sun, G. Xin, T. Hu, M. Yu, D. Shao, X. Sun and J. Lian, Nat. Commun., 2014, 5, 4526 CrossRef CAS PubMed.
- H. Wu, G. Yu, L. Pan, N. Liu, M. T. McDowell, Z. Bao and Y. Cui, Nat. Commun., 2013, 4, 1943 CrossRef PubMed.
- X. Huang, H. Yu, J. Chen, Z. Lu, R. Yazami and H. H. Hng, Adv. Mater. Technol., 2014, 26, 1296–1303 CAS.
- C. Huan, X. Zhao, X. Xiao, Y. Lu, S. Qi, Y. Zhan, L. Zhang and G. Xu, J. Alloys Compd., 2019, 776, 568–574 CrossRef CAS.
- H. Song, L. Shen, J. Wang and C. Wang, Nano Energy, 2017, 34, 47–57 CrossRef CAS.
- F. Wang, L. Mao, X. Wei and J. Mao, Chem. Eng. J., 2022, 450, 138235 CrossRef CAS.
- S. Huang, J. Yang, L. Ma, J. Ding, X. Wang, C. Peng, B. Zhao, M. Cao, J. Zheng, X.-X. Zhang and J. Chen, Adv. Sci., 2021, 8, 2101584 CrossRef CAS PubMed.
- S. Liang, Y.-J. Cheng, J. Zhu, Y. Xia and P. Müller-Buschbaum, Small Methods, 2020, 4, 2000218 CrossRef CAS.
- A. N. Preman, H. Lee, J. Yoo, I. T. Kim, T. Saito and S.-k. Ahn, J. Mater. Chem. A, 2020, 8, 25548–25570 RSC.
- W.-J. Zhang, J. Power Sources, 2011, 196, 13–24 CrossRef CAS.
- C.-M. Park, J.-H. Kim, H. Kim and H.-J. Sohn, Chem. Soc. Rev., 2010, 39, 3115–3141 RSC.
- R. Wu, D. P. Wang, X. Rui, B. Liu, K. Zhou, A. W. K. Law, Q. Yan, J. Wei and Z. Chen, Adv. Mater., 2015, 27, 3038–3044 CrossRef CAS PubMed.
- X. Li, J. Li, L. Ma, C. Yu, Z. Ji, L. Pan and W. Mai, Energy Environ. Mater., 2022, 5, 458–469 CrossRef CAS.
- M. Kim, J. F. S. Fernando, Z. Li, A. Alowasheeir, A. Ashok, R. Xin, D. Martin, A. Kumar Nanjundan, D. V. Golberg, Y. Yamauchi, N. Amiralian and J. Li, Chem. Eng. J., 2022, 445, 136344 CrossRef CAS.
- D. Wu, Z. Yao, X. Sun, X. Liu, L. Liu, R. Zhang and C. Wang, Chem. Eng. J., 2022, 429, 132449 CrossRef CAS.
- A. Cabañas, J. A. Darr, E. Lester and M. Poliakoff, J. Mater. Chem., 2001, 11, 561–568 RSC.
- P. Pasierb, S. Komornicki, M. Rokita and M. Rekas, J. Mol. Struct., 2001, 596, 151–156 CrossRef CAS.
- A. Halder, H.-H. Wang, K. C. Lau, R. S. Assary, J. Lu, S. Vajda, K. Amine and L. A. Curtiss, ACS Energy Lett., 2018, 3, 1105–1109 CrossRef CAS.
- Y. Yi, X. Du, Z. Zhao, Y. Liu, H. Guan, X. Liu, X. Pei, S. Zhang and D. Li, ACS Nano, 2022, 16, 7772–7782 CrossRef CAS PubMed.
- X. Li, L. Qiao, D. Li, X. Wang, W. Xie and D. He, J. Mater. Chem. A, 2013, 1, 6400–6406 RSC.
- H. Kim, W. Choi, J. Yoon, J. H. Um, W. Lee, J. Kim, J. Cabana and W.-S. Yoon, Chem. Rev., 2020, 120, 6934–6976 CrossRef CAS PubMed.
- Y. S. Choi, W. Choi, W.-S. Yoon and J. M. Kim, ACS Nano, 2022, 16, 631–642 CrossRef CAS PubMed.
- T. Kim, K. Ho Kim, W. Lee, W. Choi, H. Park, H. Kim, J. Yoon, Y. S. Choi, J. Lee, J. M. Kim and W.-S. Yoon, Appl. Surf. Sci., 2022, 575, 151744 CrossRef CAS.
- A. Ponrouch and M. R. Palacín, J. Power Sources, 2012, 212, 233–246 CrossRef CAS.
- J. Zhu, D. Wang, D. Chen, B. Song, Y. Li, F. Liu and J. Chen, J. Alloys Compd., 2020, 848, 156593 CrossRef CAS.
- D. Sun, L. Zhao, Z. Xiao, K. Zhao, R. Lin, H. Song, X. Zhang, X. Ma, C. Peng, X. Huang, X. Li, J. Gao and C. Xu, J. Energy Chem., 2022, 74, 100–110 CrossRef CAS.
- L. Ma, Z. Li, J. Li, Y. Dai, C. Qian, Y. Zhu, H. Wang, K. N. Hui, L. Pan, M. A. Amin, Y. Yamauchi and W. Mai, J. Mater. Chem. A, 2021, 9, 25445–25452 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ta09043k |
| This journal is © The Royal Society of Chemistry 2023 |