
Photocatalytic hydrogen evolution and simultaneously converting high-concentration of thiols into disulfides with excellent yield under visible-light†
Shaosen
Shi
a,
Huajing
Li
a,
Yagang
Zhang
a,
Yonghong
Shi
b,
Nana
Zhang
a,
Tian
Li
a,
Yating
Zhang
 a,
Qing
Li
a,
Pengfei
Duan
*b and
Yuangang
Li
*a
a,
Qing
Li
a,
Pengfei
Duan
*b and
Yuangang
Li
*a
aCollege of Chemistry and Chemical Engineering, Xi'an University of Science and Technology, Xi'an 710054, China. E-mail: liyuangang@xust.edu.cn
bCAS Center for Excellence in Nanoscience, CAS Key Laboratory of Nanosystem and Hierarchical Fabrication, National Center for Nanoscience and Technology (NCNST), Beijing 100049, China. E-mail: duanpf@nanoctr.cn
First published on 12th January 2023
Abstract
The simultaneous efficient utilization of photon-generated carriers to realize the selectively photocatalytic oxidation of organics coupled with hydrogen evolution has gained ever-increasing attention in the energy sector. A series of CdS/P25/Ni2P (SxOyP) was prepared, in which, S1O1P showed the highest activity. The conversion, selectivity, and yield are 97.47, 100, and 97.47%, respectively. Moreover, in consideration of the practical application of photocatalysis, the substrate concentration was expanded to 300 mM, which significantly exceeded that of the usually reported benchmark for photocatalysis, and the rate of hydrogen evolution could reach up to 16![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 697.86 μmol gcat−1 h−1 under light irradiation for 3 h. Based on the experimental data and analysis, a possible mechanism for this reaction was proposed. This paper provides a strategy for making full use of photocarriers and explores the field of dual-function photocatalytic systems, which will be inspirational for the development of photocatalysis technology.
697.86 μmol gcat−1 h−1 under light irradiation for 3 h. Based on the experimental data and analysis, a possible mechanism for this reaction was proposed. This paper provides a strategy for making full use of photocarriers and explores the field of dual-function photocatalytic systems, which will be inspirational for the development of photocatalysis technology.
Introduction
Solar-to-energy conversion is an alluring and sustainable technique to solve the energy crisis owing to its greenness and sustainability.1–3 Hydrogen (H2) is known as the future energy carrier on account of its high calorific value, extensive element distribution, and pollution-free characteristics.4–6 Notably, photocatalytic hydrogen production technology has recently attracted extensive attention for its non-toxicity, solar-powering, and inexhaustible availability, which is identified as a promising approach to harvesting and converting solar energy into chemical compounds.7–10 After decades of development, although the photocatalytic technology has become increasingly mature, to date most of the reports on photocatalytic hydrogen evolution have been achieved with the help of sacrificial agents.11–13 From theoretical considerations, only half of the photo-generated charge carriers are used to produce useful energy-intensive hydrogen, which suffers from the waste of energy and produces a myriad of by-products (CO2, etc.), limiting the increase in the economic value. Therefore, it is desirable to explore an approach for simultaneously combining the oxidation and reduction capabilities of photocatalysts to facilitate energy conversion.Fortunately, the dual-function photocatalytic system has provided an ideal way to solve this problem, wherein photo-generated electrons and holes are simultaneously used to reduce and oxidize reactants, respectively.14–16 Theoretically, the dual-function photocatalytic systems can maximize the usage of solar energy and be extremely efficient.17,18 Nevertheless, there is still a big challenge in the dual-function photocatalytic system, that is, mostly the concentration of substrates is still at the level of a few millimoles under ambient temperature and pressure and the yield is not satisfactory, which is far from the requirement of the large-scale practical applications.19,20 Thus, it is very urgent to develop a photocatalysis system with high efficiency in a high concentration of the substrate to meet the demand from industrial production.
In recent years, coupling reactions, such as the formation of the S–S bond,21,22 C–N bond,23,24 C–S bond,25 and C–C bond,26,27 have gained tremendous momentum owing to their great significance for the synthesis of pharmaceuticals and new materials. Among them, the selective formation of disulfide (S–S) bonds by thiols is very important. Such reactions play an important role in the formation of tertiary structures of proteins, the vulcanization of rubber materials, and the development of nano-bio materials.28,29 For example, L-cystine can be achieved by the selective coupling of L-cysteine, which assists in skin formation and has been employed as compound pharmaceuticals for its inhibition of hepatitis, alopecia, and leukopenia.30,31 A seemingly facile S–S coupling reaction is, in reality, not facile at all. Current methods of synthesizing disulfides require a large number of energy and oxidants, resulting in a waste of energy and environmental pollution.32,33 Thus, it is meaningful to discover a green and efficient approach for carrying out the S–S coupling reactions.
It is widely believed that the interaction between nanostructured semiconductor photocatalysts and substrates has an important effect on photocatalytic performance. Recently, CdS, as a general semiconductor photocatalyst, has attracted extensive attention on account of its simple preparation, narrow band gap (∼2.4 eV), and wide light absorption range.34,35 There is a strong interaction between CdS and thiols, for instance, L-cysteine,36 mercaptoproic acid,37 and glutathione can modify CdS,38 and the thiol ligand exchange can improve the surface coordination of CdS, reduce the degree of catalyst agglomeration and improve its photocatalytic performance.39 In solutions at pH values near the pKa of the thiol groups, thiolates, which can coordinate easily to cadmium(II) ions at the CdS surfaces, are present.40 In addition, heterojunction structures have expanded new avenues for both technological applications and scientific research of photocatalysts, owing to their ability to accelerate the transformation of photogenerated carriers and broaden the light response range.41,42 Therefore, CdS was selected as a major composite for this experiment to construct heterojunction-structured photocatalysts to catalyze hydrogen-producing and thiols-coupling simultaneously.
Based on the above ideas, in this study, CdS/P25/Ni2P (SxOyP) photocatalysts with a heterojunction structure were successfully prepared and characterized using a suite of techniques. Wherein, Ni2P acts as a hydrogen production cocatalyst, and P25 is a good semiconductor for heterojunction formation to achieve high photocatalytic activity. The obtained photocatalysts are capable of efficient utilization of photogenerated holes (h+) and electrons (e−) for the photocatalytic coupling of thiols into disulfides paired with hydrogen production under visible light. Under optimal conditions, the photocatalyst has the ability to promote the couple reactions of a wide variety of thiols with excellent yield. In addition, considering the practical application of photocatalysis in the future, we carried out cycle experiments and scale-up experiments. The results showed that the photocatalyst has ultra-stability and achieved high conversion after the substrate concentration was amplified to as high as 300 mM, which is one order of magnitude higher than the usually reported benchmark. A possible mechanism for the reaction was proposed. This article not only provides an efficient strategy for the development of photocatalytic technology but also contributes to the green synthesis of pharmaceuticals and materials.
Experimental
Preparation of photocatalysts
The preparation of the photocatalyst process is shown in Fig. S1.†Physicochemical characterization
X-ray diffraction (XRD) patterns were collected on a Bruker D8 Advance diffractometer with Cu Kα radiation at room temperature and the scanning angle ranged from 10° to 90° of 2θ at a scan rate of 1.5° min−1. The Raman spectra of the samples were recorded on a confocal Raman spectrometer (inVia Reflex) using an excitation wavelength of 532 nm. The morphology and structure of the materials were observed using a Hitachi S-4800 scanning electron microscope (SEM) under an accelerating voltage of 15 kV. An X-ray energy spectrometer (EDS) was used to characterize the photocatalysts and study the distribution of elements on the photocatalyst. The morphology and microstructure of the photocatalyst were analyzed using a Tecnai G2 F20 transmission electron microscope (TEM) and a high-resolution transmission electron microscope (HRTEM) with an acceleration voltage of 200 kV. The X-ray photon energy spectrum (XPS) was measured using the Al Kα X-ray beam of the Kratos Axis Ultra DLD system. The XPS data were fitted with CasaXPS software, which was used to characterize the surface chemical composition and valence of the samples. The properties of the photocatalysts were evaluated on a Shimadzu UV-2600 with BaSO4 as a reflectance standard to obtain the UV-vis diffuse reflectance spectroscopy (DRS). 1H NMR spectroscopy was used to characterize the products; it was performed at 298 K using a Bruker Avance III instrument operating at 400 MHz. The pore size distribution and specific surface area of the photocatalysts were calculated by the Barrett–Joyner–Halenda (BJH) and the Brunauer–Emmett–Teller (BET) methods, respectively. The Mott–Schottky (MS), chronoamperometric curve (I–t), and electrochemical impedance spectroscopy (EIS) were investigated in a three-electrode system by the electrochemical workstation (CHI 660E).Evaluation of photocatalytic activity
Photocatalytic hydrogen evolution and conversion of thiols into disulfides was carried out using photocatalytic reaction equipment (Merry Change, MC-SPH20-A) and the temperature of this reaction was controlled at 8 °C by using a cooling system. To ensure that the reaction occurred in a closed vacuum environment, the solution was degassed using a vacuum pump for 30 minutes. A 300 W Xe lamp (CEL-HXF300, Beijing China Education Au-light Co., Ltd) equipped with a 400 nm cutoff filter (λ ≥ 400 nm) was used as a light source in this system. Generally, 50 mg of the samples were dispersed in a 50 mL solution with 1.5 mmol, 3.0 mmol, 15 mmol, and 30 mmol thiols (c = 30, 60, 300, and 600 mM). The generated H2 during the experimental process was quantified and analyzed using the online gas chromatograph (GC 7920, Beijing China Education Au-light Co., Ltd) using N2 as the carrier gas. Besides, the products were characterized by 1H NMR spectroscopy. The conversion, selectivity, and yield of thiols were calculated with the following eqn (1)–(3): | (1) |
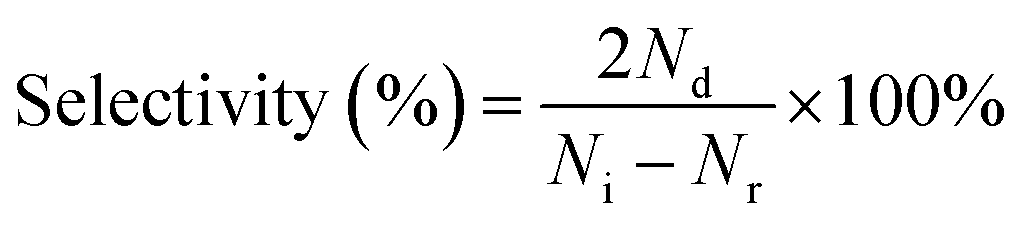 | (2) |
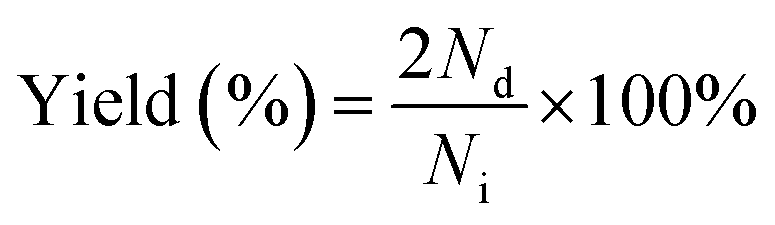 | (3) |
Results and discussion
Catalysts characterization
The crystal phase and crystallographic structure of S1O1P and corresponding precursors were characterized using X-ray diffraction (XRD) patterns. As shown in Fig. 1a, the primordial CdS has characteristic diffraction peaks at 2θ = 25.8°, 26.9°, 28.9°, 37.3°, 44.1°, 45.5°, and 52.4° that could be indexed as (100), (002), (101), (102), (110), (103) and (112) facets, respectively, suggesting that the XRD peaks of CdS can be assigned to a hexagonal phase (JCPDS No. 41-1049).44 In addition, as for P25, the peaks at 2θ = 25.8°, 38.4°, 48.4°, 54.3°, 55.6°, and 63.1° can be ascribed to the (101), (103), (200), (105), (211) and (204) planes (JCPDS No. 21-1272),45 respectively, while for Ni2P, the peaks at 2θ = 40.8°, 44.9°, 47.8°, 54.4°, and 55.2° are consistent with the (111), (201), (210), (300) and (211) planes (JCPDS No. 03-0953),46 respectively. The positions of the diffraction peaks of S1O1P are consistent with the characteristic diffraction peaks of CdS and P25, indicating that the composition process of S1O1P did not change the crystal structure of the precursors. It is notable that no characteristic diffraction peaks of Ni2P were found in S1O1P, which can be ascribed to the high dispersion or the low loading amount of Ni2P. Moreover, the S1O1P exhibited relatively good crystallinity, which can be conducive to promoting its photocatalytic ability.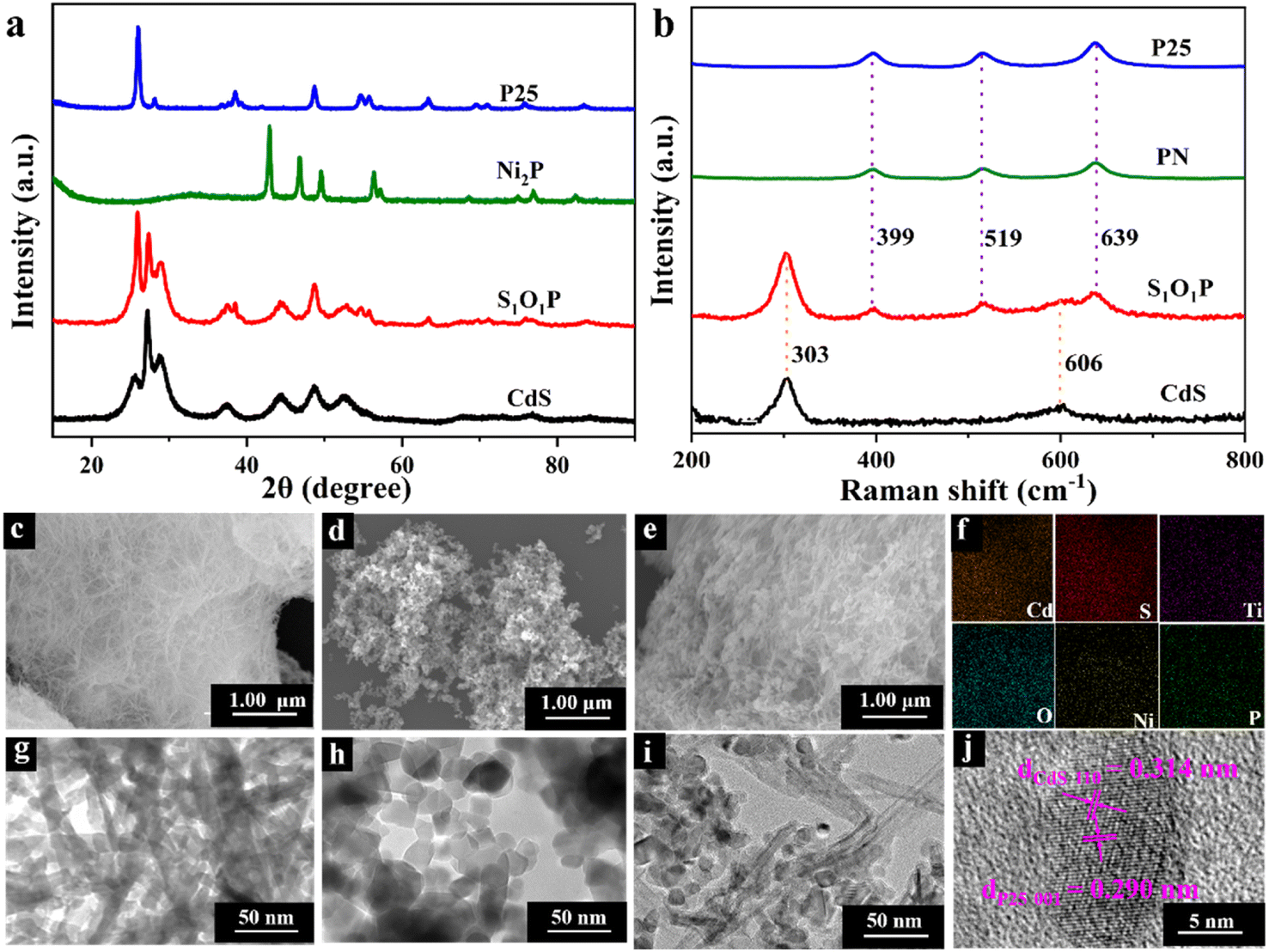 | ||
| Fig. 1 (a) XRD patterns of P25, Ni2P, S1O1P and CdS; (b) Raman spectra of P25, PN, S1O1P and CdS; SEM images of (c) CdS, (d) P25, (e) S1O1P and (f) energy dispersive spectrometer elemental mapping of S1O1P; TEM images of (g) CdS, (h) P25, (i) S1O1P and (j) HRTEM image of S1O1P. | ||
Raman spectroscopy was used to further investigate the structures of S1O1P and its precursors. As shown in Fig. 1b, the Raman spectrum of P25 showed peaks at 399, 519, and 639 cm−1, which can be assigned to Eg, B1g, and A1g modes,47 respectively. Ni2P decorated P25 sample showed a scattering pattern identical to that of the original P25, indicating that the loading of Ni2P was low and the deposition of Ni2P had no impact on the P25 structure. It was observed that for the prepared CdS, Raman data indicated two dominant peaks, indicating the longitudinal optical (LO) phonon mode at approximately 303 cm−1 (1LO) and its overtone (2LO) at about 606 cm−1, which indicated the typical CdS hexagonal phase and is consistent with the XRD results.48 It can be observed that S1O1P contained five peaks, corresponding to the characteristic peaks of PN and CdS, respectively, and the positions were basically the same, which can be attributed to the successful combination of PN and CdS.
The microstructures and morphology of the samples were observed using the scanning electron microscope (SEM) and transmission electron microscope (TEM) (Fig. 1c–j). As shown in Fig. 1c, the SEM image of the original CdS showed a porous network structure consisting of crisscrossed nanowires and its surface was smooth. The structure of CdS nanowires can be further confirmed from the TEM image (Fig. 1g), and a width of ca. 10 nm was determined. The SEM and TEM images of P25 nanospheres are shown in Fig. 1d and h, respectively, showing the average size to be about 25 nm. Fig. 1e presents the microscopic structure of S1O1P prepared by a simple calcination method with CdS and PN. It can be noted that S1O1P still had a porous network structure of CdS nanowires and PN nanospheres were uniformly dispersed on the surface, facilitating the migration of reactants and products. The TEM image of the as-prepared S1O1P shown in Fig. 1i demonstrates that the photocatalyst is composed of CdS nanowires and PN nanospheres, in accordance with the SEM results (Fig. 1e). In addition, to further clarify the element distribution of S1O1P, the energy dispersive spectroscopy (EDS) mapping was carried out. Fig. 1f shows that the sulfur (S), cadmium (Cd), oxygen (O), titanium (O), phosphorus (P), and nickel (Ni) elements are uniformly located in the photocatalyst. Furthermore, two lattice fringes can be observed from the HRTEM image of S1O1P (Fig. 1j) and the distance between the two adjacent fringes are about 0.314 and 0.290 nm, respectively, corresponding to the (110) crystal plane of CdS (JCPDS No. 41-1049) and the (001) crystal plane of P25 (JCPDS No. 21-1272). These above results show that S1O1P was successfully prepared with a porous network structure, which is conducive to further improving the photocatalytic performance.
To further investigate the surface chemical composition and element valence, S1O1P and the precursors were examined by XPS. The results are shown in Fig. S2.† Fig. S2a† is the full XPS spectra of samples and it can be seen from the figure that the prepared S1O1P showed characteristic peaks of Ni 2p, O 1s, Ti 2p, Cd 3d, S 2p, and P 2p. The spectrum of S1O1P is the superimposition of CdS and PN spectra, reflecting the successful combination of PN nanoparticles and CdS nanowires, and further proving the above conclusion. The high-resolution spectra of Cd, S, Ti, O, Ni, and P are shown in Fig. S2b–g.†
The high-resolution XPS peaks of Cd 3d (Fig. S2b†) are located at 411.9 and 405.2 eV, corresponding to Cd 3d5/2 and Cd 3d3/2 of Cd2+, respectively. The binding energy of Cd 3d in the prepared S1O1P is 0.7 eV lower than that in pristine CdS. The spectrum of S 2p in CdS (Fig. S2c†) can be deconvolved into two peaks centered at 162.4 and 161.1 eV, which belong to S 2p1/2 and S 2p3/2 of S2−,49 respectively. Compared with the original CdS, the binding energy of the S 2p peak in S1O1P is also reduced by 0.7 eV, in accordance with the results of Cd 3d. Both the shifts of Cd 3d and S 2p to the lower binding energy in S1O1P suggest there is a strong interaction between CdS and PN. Meanwhile, the XPS spectra of Ti 2p peaks (Fig. S2d†) at 458 and 463.5 eV and O 1s peaks (Fig. S2e†) at 532.6, 531.3, and 530.3 eV, respectively, can be attributed to P25. The Ni 2p peaks (Fig. S2f†) at 873 and 855.6 eV and P 2p peak (Fig. S2g†) at 133.5 eV can be assigned to Ni 2p3/2, Ni 2p1/2 and P 2p1/2, respectively, corresponding to Ni2P.50 To sum up, the XPS analysis corroborates the presence of P25 and Ni2P in S1O1P, showing that the chemical character of each component has not changed significantly after the combination.
The specific surface area is one of the most important factors to determine photocatalytic activity. The adsorption–desorption curves and pore size distribution of the samples are shown in Fig. 2a. It can be observed that all the photocatalysts correspond to type III adsorption–desorption isotherms, meaning that they all have adsorption interactions and porous structures. The specific surface area and pore volume of the four samples were calculated, as shown in Table S2.† The specific surface area of S1O1P is 113.55 m2 g−1, which is between that of the CdS and PN, indicating that PN nanoparticles are uniformly distributed on the surface of CdS. Furthermore, the specific surface areas of P25 and PN are 50.44 and 55.09 m2 g−1, respectively, which indicates that the loadings of Ni2P cannot change the structure of P25. These characteristics are conducive to the uniform distribution of the photocatalyst nanoparticles, which can improve the photocatalytic performance.
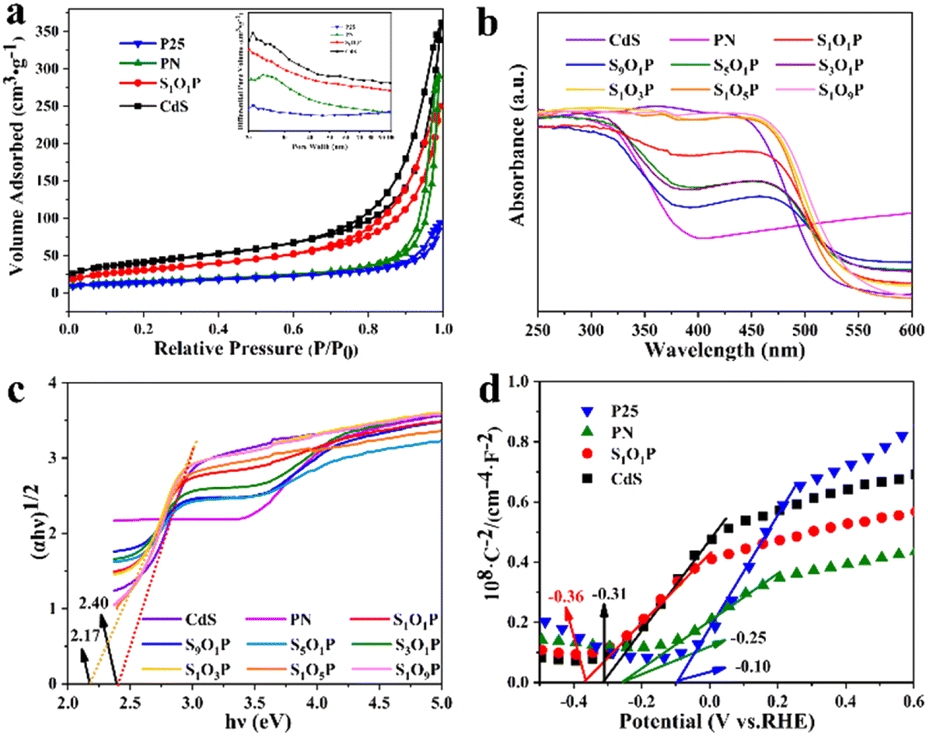 | ||
| Fig. 2 (a) N2 adsorption–desorption and pore size distribution plots (inset) of S1O1P and its precursors; (b) UV-vis DRS spectra, (c) (αhυ)1/2versus hυ curves and (d) Mott–Schottky of the photocatalysts. | ||
The optical properties of the prepared SxOyP samples and precursors were studied using a UV-vis diffuse reflectance spectrometer (DRS). Fig. 2b clearly shows that the strong absorption of PN is in the range of 320–385 nm, reflecting that it mainly absorbs UV light.
The light absorption range gradually expands from the UV-light to the visible-light region with the increase of the CdS in SxOyP, which can be attributed to the narrow band gap of CdS. The specific band gaps (Eg) of the photocatalysts were calculated using the Kubelka–Munk formula. The specific calculation process is shown in ESI Note 1† and the results are shown in Fig. 2c. The band gap of the hydrothermally prepared CdS is 2.40 eV by calculation, which is consistent with the reported value.39 In addition, the Eg of S1O1P is 2.17 eV, which is smaller than that of CdS, indicating that more visible light can be absorbed. Meanwhile, the Mott–Schottky experiments were conducted to explore the flat-band potential (Efb) of S1O1P and its precursors. As shown in Fig. 2d, it can be found that the slopes of the four sample curves are all positive, indicating that all the photocatalysts are n-type semiconductors. The Efb of the photocatalysts was calculated by eqn (4).
| RHE = 0.059 × pH + 0.197 + Ag/AgCl | (4) |
The Efb of S1O1P, CdS, PN, and P25 were −0.36, −0.31, −0.25, and −0.10 eV vs. RHE, respectively. As the bottom of the CB is more negative by 0.1 V than Efb for many n-type semiconductors,51 the CB values are −0.46, −0.41, −0.35 and −0.20 eV vs. RHE for S1O1P, CdS, PN, and P25, respectively. The reduction potential of H+ to H2 is ca. 0 V (vs. RHE) and the CB potential of S1O1P is more negative than the reduction potential of H+. The above results display that the photocatalytic H2 evolution over S1O1P can be achieved thermodynamically under light irradiation, which is consistent with the experimental results.
Catalytic performance
The photocatalytic performance of S1O1P and other samples was tested using a dual-function photocatalytic system under the irradiation of a 300 W Xe lamp (AM 1.5G), in which, the released protons were reduced to hydrogen by photogenerated electrons and thiols were selectively oxidized to disulfides by photogenerated holes. Herein, mercaptopropionic acid (MPA) as a representative of thiols was used to explore the optimal pH and photocatalyst formula for the reaction.The photocatalytic performance of the S1O3P under different pH is shown in Fig. 3a. An increase in the H2 amount can be observed with irradiation time at different pH. As can be seen, under light illumination for 4.5 h, the rate of hydrogen evolution is slow when pH = 3 (no NaOH added), and the total amount of hydrogen production is only 2.5 mL, which is far less than the theoretical yield (16.8 mL). The above result may be caused by the dissolution of the photocatalyst in the acidic solution. In order to verify the above speculation, the hydrogen evolution performance at various pH values was observed. It is clear that the rate of hydrogen evolution can be substantially improved by the increase in pH. As shown in Fig. S3,† under light illumination for 4.5 h, the evolution amounts of H2 are 10.2, 15.1, 15.3, 15.4, and 15.9 mL for pH = 5, 6, 7, 8, and 9, respectively. However, as the pH value increases continuously, the total amount of hydrogen production displays a downward trend when pH = 10, indicating that the –SH of MPA (pKaSH = 10.84, 25 °C) was ionized at high pH.52 To prove the assumption, the pH of the system was turned up to 12. The rate of H2 evolution dropped dramatically and the released hydrogen was only 6.9 mL, which can be attributed to the inhibition of hydrogen evolution under strongly basic conditions. Additionally, the conversions of MPA at different pH are shown in Table S3.† It can be observed that the conversions at pH = 3, 5, 6, 7, 8, 9, 10, and 12 are 15.13, 60.85, 89.96, 91.27, 91.55, 94.49, 87.06, and 41.50%, respectively. Therefore, the optimal pH value is 9 for this experimental system and other samples were explored for the optimal photocatalyst as described in the following experiments.
 | ||
| Fig. 3 The effects of (a) pH and (b) the formula of photocatalysts on H2 evolution during the conversion of MPA to 3,3′-dithiodipropanoic acid; (c) 1H NMR spectra of liquid-phase products at different reaction periods; (d) comparison of hydrogen production under the irradiation of a 300 W Xe lamp with and without a cutoff filter (λ ≥ 400 nm); (e) photocatalytic hydrogen evolution coupled with different thiols to corresponding disulfides; (f) H2 evolution coupled with the conversion of high-concentration MPA to 3,3′-dithiodipropanoic acid; (g) five-consecutive photocatalytic tests. Photocatalyst: 50 mg S1O1P, pH = 9, 8 °C. | ||
Fig. 3b displays the dependence of the photocatalytic performance on the photocatalyst formula under pH = 9. A linear increase of H2 amount can be seen with irradiation time over the prepared CdS and P25. The experiments with the prepared CdS and PN show that the H2 evolution amounts in 4.5 h were only 5.39 and 0.34 mL, respectively, which are far below the theoretical value. The low efficiency of photocatalysis is due to the serious recombination of the photogenerated carriers. As far as the photocatalysis performance of a series of CdS/P25/Ni2P (SxOyP) photocatalysts with a heterojunction structure was evaluated. The rate of hydrogen evolution is fast over all the SxOyP. Under the light illumination for 4.5 h, the total amounts of H2 evolution were 14.02, 15.49, 15.59, 16.29, 15.87, 15.22, and 12.47 mL (Fig. S4†) and the conversions of MPA were 83.43, 92.24, 92.82, 97.01, 94.49, 90.62 and 74.27% (Table S4†) for S9O1P, S5O1P, S3O1P, S1O1P, S1O3P, S1O5P, and S1O9P, respectively. All the results demonstrate that S1O1P is the best photocatalyst for the photocatalytic transformation of MPA to 3,3′-dithiodipropanoic acid coupled with the H2 production among the prepared samples. The excellent photocatalytic activity of S1O1P can be attributed to the formation of the heterojunction with CdS and PN, which can expand the absorption of light and accelerate the transport of carriers. Furthermore, there was hardly any H2 produced in the absence of light and photocatalyst, which confirmed that light and photocatalyst are indispensable for this system. Based on the above experimental results and analysis, it is demonstrated that the optimal condition of this system is pH = 9 and the optimal formula of photocatalyst is S1O1P. Under these optimal conditions, the substrates are almost completely transformed within 3 h, and the rate of H2 evolution could reach 4999.98 μmol gcat−1 h−1.
The experimental supernatants were collected after centrifugation and examined by liquid UV-vis. The results are shown in Fig. S5.† Only one peak can be observed in the substrate (MPA), but two peaks are seen in supernatants after photocatalytic reactions. This phenomenon can be explained by the appearance of new chemicals. In order to figure out the specific structure and concentration of products, 1H NMR technique was adopted. The idiographic calculation process and results are shown in ESI Note 2.† Compared with the 1H NMR spectra of MPA and product, two characteristic peaks at δ = 2.51 and 2.73 ppm of MPA disappeared and two new peaks at δ = 2.94 and 2.60 ppm appeared, corresponding to the generation of 3,3′-dithiodipropanoic acid.22 The concentration of 3,3′-dithiodipropanoic acid was 14.6 mmol L−1 from the calculation, which is close to the theoretical value (15.0 mmol L−1). The conversion of MPA was 97.47% determined by the 1H NMR measurement, which is consistent with 97.01% calculated by the evaluated hydrogen as the standard. As shown in Fig. 3c, it is clear that the content of MPA decreased continuously as the reaction time increased and only two peaks of 3,3′-dithiodipropanoic acid were produced during the whole reaction process. After performing calculations using eqn (1)–(3) according to the 1H NMR spectra, the conversion, selectivity, and yield were 97.47, 100 and 97.47%, respectively.
To evaluate the activity of S1O1P under visible light, a 400 nm cut-off filter (λ ≥ 400 nm) was employed to filter out UV light. A comparison of hydrogen production under the irradiation of a 300 W Xe lamp with and without the cutoff filter is shown in Fig. 3d. It can be seen that the hydrogen production increases rapidly at first and remains unchanged after reaching the maximum at 3 h without the cutoff filter. The reason for this phenomenon is that the substrate concentration is high at the beginning and the reaction speed is fast, however, with the consumption of reactants and the accumulation of products, the reaction gradually tends to cease. The total amount of H2 evolved with a 400 nm cut-off filter is consistent with that from the original experiment under the light illumination after 3.5 h. Nevertheless, the rate of hydrogen production before 3.5 h was slower with the filter than that without the filter, which can be attributed to the absence of UV light in the latter case. The S1O1P still has excellent photocatalytic performance under visible light based on the above experiment and analysis.
In order to learn the universality of S1O1P, the photocatalytic hydrogen evolution coupled with selective transformations of various thiols to corresponding disulfides was examined. As shown in Fig. 3e, the total amounts of H2 produced were 16.30, 15.56, 15.46, and 15.90 mL for MPA, 3-mercapto-1-propanol (MP), L-cysteine, and glutathione, respectively. The plots of hydrogen evolution with different substrates are shown in Fig. S6† and an increase in the H2 amount can be observed with the irradiation time. The plots of hydrogen evolution with MPA and MP are parallel as their structures are similar. However, it is worth noting that the rates of hydrogen evolution with L-cysteine and glutathione are relatively slow, probably because of their large molecular mass and complex structures, increasing steric hindrance and hampering the reaction progress. In addition, the products were tested by 1H NMR and the results are shown in ESI Note 2.† It can be observed that all the structures of the products are consistent with corresponding disulfides. As shown in Table 1, the corresponding product yields and conversions of MPA, MP, L-cysteine, and glutathione were 14.62, 14.01, 14.28 and 13.95 mmol L−1 and 97.47, 92.62, 95.20, and 92.03%, respectively, from the 1H NMR spectra. These results indicate that the prepared S1O1P sample possesses excellent universality for photocatalytic H2-evolution and converting thiols into disulfides.
|
|
|||
|---|---|---|---|
| Entry | Substrate | Yield (mmol L−1) | Conversions (%) |
| a Conditions: substrate (15 mmol) and S1O1P (50 mg) in 50 mL reaction solution under a 300 W Xe lamp, pH = 9, 8 °C. Calculate with 1H NMR spectra: conversions (%) = (product produced)/15. | |||
| 1 | MPA | 14.62 | 97.47 |
| 2 | MP | 14.01 | 92.62 |
| 3 | L-Cysteine | 14.28 | 95.20 |
| 4 | Glutathione | 13.95 | 92.03 |
Although this reaction system showed excellent yield at the current concentration (30 mM), it was far from the requirements for the industrial application and unfavorable for further large-scale practice. Therefore, the concentration of MPA was increased to explore the photocatalytic hydrogen evolution and thiol coupling at high concentrations. In this part of the experiments, a 300 W Xe lamp with an AM 1.5G filter (simulated sunlight) was adopted as a light source. Fig. S7† shows the plot of hydrogen production versus the reaction time when the initial concentration was expanded to 60 mM. As the reaction time increases, the total amount of hydrogen production increased, and the rate of hydrogen evolution slows down. The conversions are 99.28% under light illumination for 9 h (Table S5,† entry 3). This result shows that it is possible to achieve 100% yield at a high concentration. Hence, the initial concentration of MPA was increased by a factor of 10 to 300 mM. As shown in Fig. 3f, an increase in H2 amount can be observed with irradiation time. Under the light illumination for 8.5 and 18 h, the total amount of H2 evolved were 106.86 and 121.02 mL and the conversion reached 65.54% and 82.36%, respectively (Table S5,† entry 4 and 5). Satisfyingly, the conversion of MPA was 83.81% under the light illumination for 22 h (Table S5,† entry 6), which can be confirmed from the 1H NMR spectrum of high-concentration liquid phase products. As shown in Fig. S8,† the characteristic peaks of the product at δ = 2.94 and 2.60 ppm and the characteristic peak of the interior label (CH3OH, 0.12 M) can be observed, resulting in the concentration and conversion of the product to be 12.59 mM and 83.93%, respectively. The above results have shown that the selectivity of this photocatalytic reaction is 100% at high concentrations and the stoichiometry is the same as that at the low concentration. The above results could be promising for the feasibility of large-scale photocatalytic synthesis under solar irradiation. However, when the initial concentration was expanded to 600 mM, the plot of hydrogen production versus reaction time was the same as that with 300 mM (Fig. S9†), which could be due to the inadequate photocatalysts. The rate of hydrogen evolution under different concentrations of MPA is shown in Fig. S10.† Under the light illumination for 3 h, the rates of H2 evolution were 4999.98, 9007.32, 16697.86, and 13950.18 μmol gcat−1 h−1 when the concentrations of MPA were 30, 60, 300, and 600 mM, respectively. Among them, the maximum hydrogen production rate was obtained when the concentration of MPA was 300 mM, which is higher than that from most of the previously reported literature (Table S6†). Meanwhile, the apparent quantum yields (AQY) were calculated and detailed procedures are shown in ESI Note 3.† As shown in Table 2, a significant decrease in AQY can be observed by increasing the concentration of MPA from 30 to 300 mM, the tendency being similar to the rate of hydrogen evolution. It should be noted that the maximum AQY value was 3.92% when the concentration of MPA was 300 mM, indicating that S1O1P exhibited high photon utilization. Furthermore, an analogical experiment of 300 mM MP was held, and the result is shown in Fig. S11.† The plot of the hydrogen production versus reaction time is similar to Fig. 3f and the total amount of H2 evolution reaches 124.4 mL under the light illumination for 20 h (Table S5,† entry 14). After calculation, the rate of H2 evolution realizes to 16282.83 μmol gcat−1 h−1 and AQY is 3.82% under light irradiation for 3 h. The above result shows the feasibility of photocatalysis to achieve high-concentration of thiol conversion coupled with hydrogen production, making a contribution to the practical application of photocatalytic technology.
| Entry | Substrate | Concentration (mM) | Volume of H2 (mL) | AQY (%) |
|---|---|---|---|---|
| a Conditions: S1O1P (50 mg) in 50 mL reaction solution under a 300 W Xe lamp, pH = 9, 8 °C, under light irradiation for 3 h. | ||||
| 1 | MPA | 30 | 15.93 | 1.11 |
| 2 | MPA | 60 | 30.26 | 2.11 |
| 3 | MPA | 300 | 56.11 | 3.92 |
| 4 | MPA | 600 | 46.87 | 3.28 |
| 5 | MP | 300 | 54.71 | 3.82 |
Besides the photocatalytic activity, the stability of a photocatalyst also is a vital property. To evaluate the stability of S1O1P, the cycle experiments were carried out 5 times with 50 mg S1O1P. As shown in Fig. 3g, the conversion of MPA was 82.39% after 5 cycles, indicating that the photocatalyst still has excellent photocatalytic ability. In order to further explore the reason for the decrease in photocatalytic activity, XRD was employed to detect the used S1O1P. As shown in Fig. S12,† the characteristic peaks of S1O1P are disappeared, indicating that the crystal structure has been destroyed. Therefore, it is reasonably considered that the reason for the decreased activity is due to the damaged catalyst structure. There are two main factors affecting the stability of CdS. On the one hand, CdS is not stable at low pH, which can react with acids to form cadmium salts and release hydrogen sulfide. On the other hand, as a semiconductor photocatalyst, CdS can produce holes (h+) to undergo oxidation reactions under light irradiation. However, S2− will be oxidized if the holes are not consumed in time and accumulate continuously, affecting the stability of CdS. Considering the experimental conditions, it is believed that the decreased activity of S1O1P is mainly due to the photocorrosion of CdS. Despite the photocorrosion of CdS existing in the experiment, S1O1P revealed good stability in 5 cycles of photocatalytic thiol conversion coupled with hydrogen production. In the next step, we will adopt some methods to improve the photocatalytic stability of S1O1P.
Reaction mechanism
To explain why the S1O1P photocatalyst exhibits excellent photocatalytic performance, the photoelectric chemistry experiments including chronoamperometric curve (I–t) and electrochemical impedance spectroscopy (EIS) were performed. The detailed steps of electrochemical testing are shown in ESI Note 4† and the results are shown in Fig. 4a and b. The quick separation of photon-generated carriers produced by S1O1P can be studied from the I–t curve. As shown in Fig. 4a, S1O1P exhibits the best photoelectric response capability and the largest photocurrent density among the samples. It is generally accepted that the photo-generated carriers are produced by the light excitation of the photocatalyst. Then, the photogenerated holes are captured by MPA and the production of H2 is achieved by the photogenerated electrons. Moreover, S1O1P also showed an extremely sensitive photocurrent response (Fig. S13†). Thus, the S1O1P can generate abundant photogenerated hole–electron pairs and accelerate their transfer process. The charge transfer resistance on the photocatalyst could be represented by the radius on the EIS Nyquist plots, thus, electrochemical impedance spectroscopy (EIS) was implemented to study the charge transfer and separation efficiency of photogenerated carriers. As shown in Fig. 4b, S1O1P exhibits the smallest semicircle among the samples in the Nyquist plots and the promotion effect can be partly attributed to the reduction in the interface charge transfer resistance between CdS and PN. Besides, the charge transfer resistances (Rct) of S1O1P and other samples were calculated. The results are shown in Table S7,† and the Rct of S1O1P is 5205 Ω, which is far less than those of other photocatalysts. Based on the above discussion, we can draw a conclusion that the separation and transfer of light-generated carriers on S1O1P are the most efficient, which is consistent with the experimental results.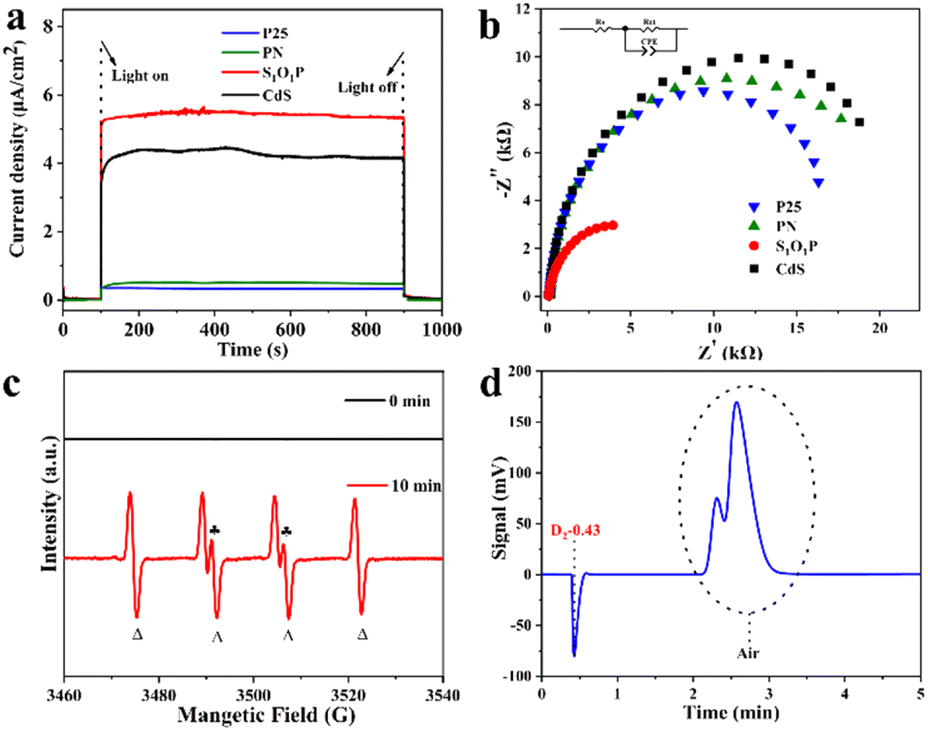 | ||
| Fig. 4 The electrochemical tests of S1O1P and precursors for (a) chronoamperometric curves (I–t) and (b) EIS Nyquist plots; (c) EPR signals from a solution containing MPA (30 mM), S1O1P (1 mg mL−1) and DMPO (20 mM) at pH = 9.0 before and after the irradiation of 300 W Xe lamp for 10 min; (d) GC characterization for produced hydrogen: employing D2O as the solvent. | ||
In order to further investigate the reaction mechanism of the photocatalytic process, the electron paramagnetic resonance (EPR) spin trapping technique was utilized to monitor the reaction intermediates.53,54 The tested EPR signals of the photocatalytic system containing MPA (30 mM), S1O1P (1 mg mL−1) and DMPO (20 mM) at pH = 9.0 are shown in Fig. 4c. After irradiation with a 300 W Xe lamp for 10 min, the spin adducts of sulfenyl radicals with DMPO were successfully detected and the characteristic peaks (labeled with  and Δ) stand for the adducts of sulfenyl radicals (RS˙) and hydrogen radicals (H˙) with DMPO, respectively.55,56 However, no signal can be observed in the dark, confirming that this reaction indeed goes through the photocatalytic transformation of MPA to 3,3′-dithiodipropanoic acid through a radical pathway including both RS˙ and H˙ radicals under light illumination.
and Δ) stand for the adducts of sulfenyl radicals (RS˙) and hydrogen radicals (H˙) with DMPO, respectively.55,56 However, no signal can be observed in the dark, confirming that this reaction indeed goes through the photocatalytic transformation of MPA to 3,3′-dithiodipropanoic acid through a radical pathway including both RS˙ and H˙ radicals under light illumination.
Isotope labeling is an advanced method for tracking the movement and change of substances.57 The chemical and biological properties of the nuclides and their compounds used by isotope labeling are identical to the chemical and biological properties between the corresponding ordinary elements and their compounds present in nature, only with different nuclear physical properties. Therefore, isotope labeling can be used as a marker to make labeled compounds to explore the source of an element in the target product.58 When the reaction was conducted in H2O, there was a positive peak signal of H2 presented at 0.48 minutes on a gas chromatograph (GC) (Fig. S14†) retention profile. In the controlled experiment, D2O was used instead of H2O as the reaction solvent. The result of the GC retention curve is shown in Fig. 4d, from which a negative peak signal at a similar retention time is observed. It is reported that the negative signal is attributed to D2.59 It becomes obvious from the figure that D2 is the gaseous product instead of H2 with D2O as the solvent, indicating that the source of the H element in the released H2 is from the solvent H2O. Similar phenomena were observed by others.22,59
Based on the above experimental analysis, a feasible mechanism for the photocatalytic transformation of MPA into 3,3-dithiodipropanoic acid coupled with H2 evolution by S1O1P under light irradiation is proposed in Scheme 1. Firstly, S1O1P can be excited by light irradiation, and generate electron–holes pairs simultaneously. The h+ is produced on the VB of CdS and the e− is transferred to the CB of CdS. The photoinduced e− is injected from the CB of CdS to that of P25, then, the electrons are transferred from P25 to Ni2P nanoparticles due to the lower reduction potential of Ni2P. Meanwhile, the photogenerated holes migrate from the VB of P25 to CdS, which are the active sites for the oxidation reaction. Ultimately, the photogenerated electrons accumulated on the outer surface of Ni2P, and the photogenerated holes gathered at the surface of CdS, which is conducive to the efficient separation of the photo-generated charge carriers. In the presence of MPA, there is an ionization balance between MPA and MPA−, as shown in Scheme 1. Besides, due to the interaction between CdS and thiols, the obtained MPA− are easily adsorbed and bind to the surface of S1O1P through Cd–S bonds to form S1O1P/thiolate conjugates. The excited state of the S1O1P is subsequently reductively quenched by the bound Cd–S species, thereby, producing MPA˙ radicals, confirmed by EPR, which couple to form the 3,3-dithiodipropanoic acid on the surface of CdS. Finally, the products are released into water, then, additional MPA− will bind to the vacant active sites and repeat the reaction cycle. On the other hand, the H+ from the water ionization is reduced into H˙ radicals (also proved by the EPR experiment) by e− on the surface of Ni2P, which is finally coupled to generate H2 and be released to surroundings. Thus, the presence of heterogeneous structures can improve the utilization of photogenerated electron–hole pairs and promote the efficiency of the photocatalytic synergistic reaction.
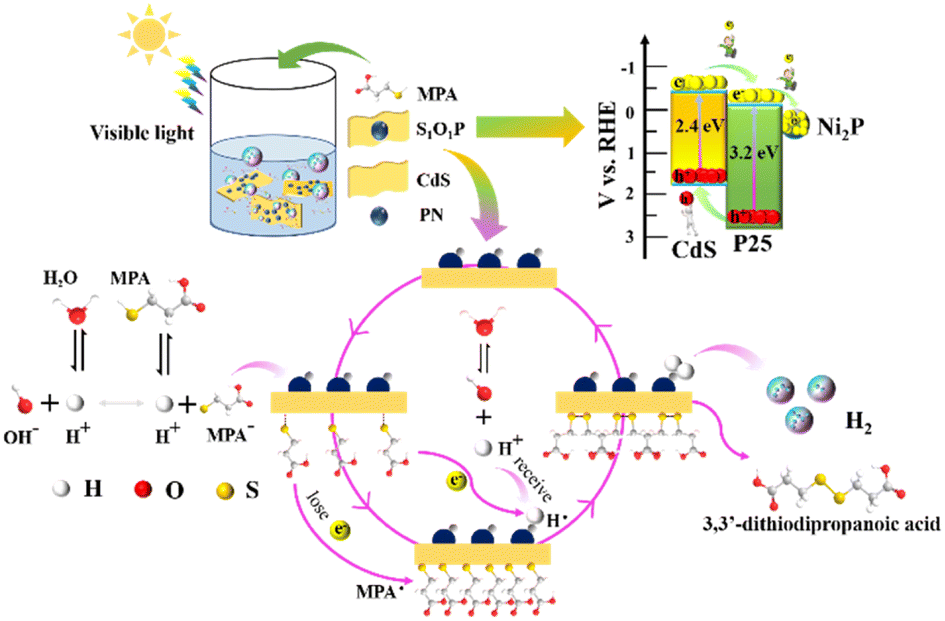 | ||
| Scheme 1 Reaction mechanism of photocatalytic transformation of thiols into disulfides coupled with H2 evolution over S1O1P under visible light irradiation. | ||
Conclusion
In summary, SxOyP photocatalysts were synthesized by a simple hydrothermal and calcination method and showed excellent photocatalytic activity and chemical stability. It is remarkable that the photogenerated carriers are effectively utilized for the photocatalytic transformation of thiols to corresponding disulfides coupled with hydrogen production. Under optimal conditions, S1O1P has the ability to promote the couple reactions with excellent yield, and the reasons are considered from the formation of the heterojunction, the interaction of the substrate with a photocatalyst, and so on. Moreover, for realizing industrialization and practical applications, the concentration of thiol was increased tenfold and the conversion was still satisfactory, indicating the feasibility of photocatalytic synthesis. This work achieved important breakthroughs in photocatalytic synthesis, providing significant theoretical and experimental support by performing the synthesis of pharmaceuticals and preparation of new materials in a way.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this study.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (22072113); and the CAS Key Lab of Colloids, Interfaces, and Thermal Dynamics. We also thank Prof. Kaiqiang Liu and Mr Xiangyang Yan (School of Chemistry and Chemical Engineering, Shaanxi Normal University) for their technical support in TEM and XPS measurements.Notes and references
- C. S. Ponseca, P. Chabera, J. Uhlig, P. Persson and V. Sundstrom, Chem. Rev., 2017, 117, 10940–11024 CrossRef CAS PubMed.
- J. Gong, C. Li and M. R. Wasielewski, Chem. Soc. Rev., 2019, 48, 1862–1864 RSC.
- M. Kokkonen, P. Talebi, J. Zhou, S. Asgari, S. A. Soomro, F. Elsehrawy, J. Halme, S. Ahmad, A. Hagfeldt and S. G. Hashmi, J. Mater. Chem. A, 2021, 9, 10527–10545 RSC.
- T. He, P. Pachfule, H. Wu, Q. Xu and P. Chen, Nat. Rev. Mater., 2016, 1, 16059 CrossRef CAS.
- M. Chatenet, B. G. Pollet, D. R. Dekel, F. Dionigi, J. Deseure, P. Millet, R. D. Braatz, M. Z. Bazant, M. Eikerling, I. Staffell, P. Balcombe, Y. Shao-Horn and H. Schafer, Chem. Soc. Rev., 2022, 51, 4583–4762 RSC.
- Y. Wang, P. Hu, J. Yang, Y. Zhu and D. Chen, Chem. Soc. Rev., 2021, 50, 4299–4358 RSC.
- X. Tao, Y. Zhao, S. Wang, C. Li and R. Li, Chem. Soc. Rev., 2022, 51, 3561–3608 RSC.
- Q. Wang and K. Domen, Chem. Rev., 2020, 120, 919–985 CrossRef CAS PubMed.
- M. Schröder, K. Kailasam, J. Borgmeyer, M. Neumann, A. Thomas, R. Schomäcker and M. Schwarze, Energy Technol., 2015, 3, 1014–1017 CrossRef.
- H. Nishiyama, T. Yamada, M. Nakabayashi, Y. Maehara, M. Yamaguchi, Y. Kuromiya, Y. Nagatsuma, H. Tokudome, S. Akiyama, T. Watanabe, R. Narushima, S. Okunaka, N. Shibata, T. Takata, T. Hisatomi and K. Domen, Nature, 2021, 598, 304–307 CrossRef CAS PubMed.
- C. Marchal, T. Cottineau, M. G. Méndez-Medrano, C. Colbeau-Justin, V. Caps and V. Keller, Adv. Energy Mater., 2018, 8, 1702142 CrossRef.
- D. Seo, G. Park and H. Song, J. Am. Chem. Soc., 2011, 134, 1221–1227 CrossRef PubMed.
- H. Ahmad, S. K. Kamarudin, L. J. Minggu and M. Kassim, Renewable Sustainable Energy Rev., 2015, 43, 599–610 CrossRef CAS.
- W. Shang, Y. Li, H. Huang, F. Lai, M. B. J. Roeffaers and B. Weng, ACS Catal., 2021, 11, 4613–4632 CrossRef CAS.
- M. Y. Qi, M. Conte, M. Anpo, Z. R. Tang and Y. J. Xu, Chem. Rev., 2021, 121, 13051–13085 CrossRef CAS PubMed.
- S. L. Meng, C. Ye, X. B. Li, C. H. Tung and L. Z. Wu, J. Am. Chem. Soc., 2022, 144, 16219–16231 CrossRef CAS PubMed.
- D. K. Chauhan, M. Sarkar, A. Patra and K. Kailasam, J. Mater. Chem. A, 2022, 10, 22289–22300 RSC.
- V. R. Battula, A. Jaryal and K. Kailasam, J. Mater. Chem. A, 2019, 7, 5643–5649 RSC.
- H. Kasap, C. A. Caputo, B. C. Martindale, R. Godin, V. W. Lau, B. V. Lotsch, J. R. Durrant and E. Reisner, J. Am. Chem. Soc., 2016, 138, 9183–9192 CrossRef CAS PubMed.
- Y. Wu, M. Qi, C. Tan, Z. Tang and Y. Xu, Chin. J. Catal., 2022, 43, 1851–1859 CrossRef CAS.
- M. Arisawa, K. Fukumoto and M. Yamaguchi, ACS Catal., 2020, 10, 15060–15064 CrossRef CAS.
- X. Li, Z. Li, Y. Gao, Q. Meng, S. Yu, R. G. Weiss, C. H. Tung and L. Wu, Angew. Chem., Int. Ed., 2014, 53, 2085–2089 CrossRef CAS PubMed.
- X. Li, S. Yang, F. Zhang, L. Zheng and X. Lang, Appl. Catal., B, 2022, 303, 120846 CrossRef CAS.
- W. Sheng, J. Shi, H. Hao, X. Li and X. Lang, Chem. Eng. J., 2020, 379, 122399 CrossRef CAS.
- C. Huang, R. Ci, J. Qiao, X. Wang, K. Feng, B. Chen, C. Tung and L. Wu, Angew. Chem., Int. Ed., 2021, 60, 11779–11783 CrossRef CAS PubMed.
- S. Xie, Z. Shen, J. Deng, P. Guo, Q. Zhang, H. Zhang, C. Ma, Z. Jiang, J. Cheng, D. Deng and Y. Wang, Nat. Commun., 2018, 9, 1181 CrossRef PubMed.
- A. D. Samue, M. L. Colleen, A. G. Laurel, A. R. Magaly, T. B. Gunnoe, L. P. Jeffrey and D. B. Paul, Inorg. Chem., 2007, 46, 2365–2367 CrossRef PubMed.
- M. H. Moreira, F. C. L. Almeida, T. Domitrovic and F. L. Palhano, Comput. Struct. Biotechnol. J., 2021, 19, 6255–6262 CrossRef CAS PubMed.
- H. Huang, R. Jiang, H. Ma, Y. Li, Y. Zeng, N. Zhou, L. Liu, X. Zhang and Y. Wei, Mater. Sci. Eng., C, 2021, 118, 111437 CrossRef CAS PubMed.
- F. D'Agostini, P. Fiallo, M. Ghio and S. De Flora, Arch. Dermatol. Res., 2013, 305, 25–34 CrossRef PubMed.
- J. Duperray, R. Sergheraert, K. Chalothorn, P. Tachalerdmanee and F. Perin, J. Cosmet., Dermatol., 2022, 21, 802–813 CrossRef PubMed.
- I. V. Koval, Chem. Rev., 1994, 63, 735–752 Search PubMed.
- X. Wang, M. Lu, J. Zeng, Y. Weng and Y. Li, Green Chem., 2021, 23, 307–313 RSC.
- Z. Zhang, M. Wang, H. Zhou and F. Wang, J. Am. Chem. Soc., 2021, 143, 6533–6541 CrossRef CAS PubMed.
- Y. Yuan, D. Chen, Z. Yu and Z. Zou, J. Mater. Chem. A, 2018, 6, 11606–11630 RSC.
- Q. Bi, J. Wang, J. Lv, J. Wang, W. Zhang and T. Lu, ACS Catal., 2018, 8, 11815–11821 CrossRef CAS.
- S. Iqbal, Appl. Catal., B, 2020, 274, 119097 CrossRef CAS.
- Y. Ben-Shahar, F. Scotognella, N. Waiskopf, I. Kriegel, S. Dal Conte, G. Cerullo and U. Banin, Small, 2015, 11, 462–471 CrossRef CAS PubMed.
- P. Wang, J. Zhang, H. He, X. Xu and Y. Jin, Nanoscale, 2015, 7, 5767–5775 RSC.
- J. Aldana, N. Lavelle, Y. Wang and X. Peng, J. Am. Chem. Soc., 2005, 127, 2496–2504 CrossRef CAS PubMed.
- H. Wang, L. Zhang, Z. Chen, J. Hu, S. Li, Z. Wang, J. Liu and X. Wang, Chem. Soc. Rev., 2014, 43, 5234–5244 RSC.
- C. Xu, P. Ravi Anusuyadevi, C. Aymonier, R. Luque and S. Marre, Chem. Soc. Rev., 2019, 48, 3868–3902 RSC.
- S. Iqbal, Z. Pan and K. Zhou, Nanoscale, 2017, 9, 6638–6642 RSC.
- Y. K. Kim and H. Park, Energy Environ. Sci., 2011, 4, 685–694 RSC.
- W. Wei, D. Liu, Z. Wei and Y. Zhu, ACS Catal., 2016, 7, 652–663 CrossRef.
- K. Mi, Y. Ni and J. Hong, J. Phys. Chem. Solids, 2011, 72, 1452–1456 CrossRef CAS.
- M. L. de Souza, D. C. Tristão and P. Corio, RSC Adv., 2014, 4, 23351–23358 RSC.
- N. Venkatesh, K. Sabarish, G. Murugadoss, R. Thangamuthu and P. Sakthivel, Environ. Sci. Pollut. Res. Int., 2020, 27, 43212–43222 CrossRef CAS PubMed.
- S. Hong, D. P. Kumar, E. H. Kim, H. Park, M. Gopannagari, D. A. Reddy and T. K. Kim, J. Mater. Chem. A, 2017, 5, 20851–20859 RSC.
- D. Dai, L. Wang, N. Xiao, S. Li, H. Xu, S. Liu, B. Xu, D. Lv, Y. Gao, W. Song, L. Ge and J. Liu, Appl. Catal., B, 2018, 233, 194–201 CrossRef CAS.
- X. Ye, Y. Chen, Y. Wu, X. Zhang, X. Wang and S. Chen, Appl. Catal., B, 2019, 242, 302–311 CrossRef CAS.
- J. J. Gooding, P. S. Hale, L. M. Maddox and J. G. Shapter, J. Chem. Educ., 2005, 82, 775–777 CrossRef.
- Y. Chen, Y. Li, N. Luo, W. Shang, S. Shi, H. Li, Y. Liang and A. Zhou, Chem. Eng. J., 2022, 429, 132577 CrossRef CAS.
- J. Li, W. Zhu, Y. Gao, P. Lin, J. Liu, J. Zhang and T. Huang, Sep. Purif. Technol., 2022, 285, 120362 CrossRef CAS.
- K. Huvaere, M. L. Andersen, M. Storme, J. Van Bocxlaer, L. H. Skibsted and D. De Keukeleire, Photochem. Photobiol. Sci., 2006, 5, 961–969 CrossRef CAS PubMed.
- C. A. Walenta, C. Courtois, S. L. Kollmannsberger, M. Eder, M. Tschurl and U. Heiz, ACS Catal., 2020, 10, 4080–4091 CrossRef CAS.
- O. Al-Madanat, Y. AlSalka, M. Curti, R. Dillert and D. W. Bahnemann, ACS Catal., 2020, 10, 7398–7412 CrossRef CAS.
- J. Chen, J. Yi, W. Zhu, W. Zhang and T. An, Environ. Sci. Technol., 2021, 55, 16617–16626 CrossRef CAS PubMed.
- L. L. Liao, G. M. Cao, Y. X. Jiang, X. H. Jin, X. L. Hu, J. J. Chruma, G. Q. Sun, Y. Y. Gui and D. G. Yu, J. Am. Chem. Soc., 2021, 143, 2812–2821 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ta08783a |
| This journal is © The Royal Society of Chemistry 2023 |