
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Investigation of the impact of zeolite shaping and salt deposition on the characteristics and performance of composite thermochemical heat storage systems†
Quentin
Touloumet
,
Georgeta
Postole
 *,
Laurence
Massin
,
Chantal
Lorentz
and
Aline
Auroux
*
*,
Laurence
Massin
,
Chantal
Lorentz
and
Aline
Auroux
*
Univ Lyon, Université Claude Bernard Lyon 1, CNRS, IRCELYON, F-69626 Villeurbanne, France. E-mail: georgeta.postole@ircelyon.univ-lyon1.fr; aline.auroux@ircelyon.univ-lyon1.fr
First published on 23rd December 2022
Abstract
Zeolites are promising candidate materials for water-based thermochemical heat storage applications. In this study, 13X and LiX commercial zeolites of different shapes were investigated due to their high thermal stability and storage capacities. Composite materials were prepared by impregnating matrices of 13X zeolite beads (13X(b)), 13X zeolite powder (13X(p)), or LiX beads (LiX(b)) with CaCl2, MgSO4 and LiCl salts (5 wt%). The different zeolites and zeolite@salt composites were characterized using N2 and H2O sorption isotherms, X-ray diffraction and NMR. The heat and water storage capacities were studied by TG–DSC analysis, as well as the hydration kinetics and the stability over time of the storage capacity. The Dubinin–Astakhov model was applied to the water sorption isotherms in order to better understand the sorption process. It turns out that zeolite shaping has a strong impact on hydration kinetics; moreover, the presence of an inert binder reduces the sorption capacity of 13X(b) compared to 13X(p). The pure zeolites and composites present a very high reversibility of water and heat storage capacities, with a good preservation of storage capacities after 50 simulated cycles of hydration in a water saturated environment. Salt incorporation impacts the porosity (especially for beads) and the environment of the zeolite component, as well as the water sorption behavior, kinetics and process, with a greater influence for LiCl based composites. The impregnation of 13X(b) and LiX(b) zeolites negatively affects the sorption capacities of the composites. This is due to blockage of the zeolite pores, which limits the accessibility of the zeolite structure for water vapor and decreases the contribution of the host matrix to the storage capacity to an extent that is not offset by the relatively small amount of salt added to the system.
1. Introduction
Increasing energy demand in various fields and decreasing reserves of fossil fuels have motivated the world to shift attention to sustainable development.1,2 Improved energy efficiency, valorization of waste heat, and a shift towards renewable energy sources represent the main approaches leading to energy conservation and emission reductions.3,4 The mismatch between energy supply and demand, however, represents a major roadblock to these efforts.5,6 Thermal energy storage (TES) has recently become the focus of renewed attention, due to its great potential to capture surplus energy from waste heat and/or renewable sources and release it at times of high demand. Thermal energy storage systems can exploit sensible,7 latent8 or thermochemical heat storage.9,10 Thermochemical heat storage (TCHS) is a promising technology for the loss-free storage of heat which has amply been reviewed in the recent literature.11–15 Thermochemical heat storage systems have the highest energy storage density compared to sensible and latent heat storage systems, and are generally based on the following equation:| AB + heat = A + B | (1) |
Two components A and B can be separated by adding heat, via an endothermic reaction. This is the charging process (from left to right in eqn (1)). When the two components (sorbent and sorptive) are brought together, heat is released via an exothermic reaction. This process represents the discharging process (from right to left in eqn (1)). Thermochemical heat storage systems thus operate via reversible reaction and store heat by breaking physical or chemical bonds.16 Depending on the thermochemical heat storage medium, the energy storage density can be as high as 2000 MJ m−3.13 Adsorbent materials such as alumina, silico-aluminophosphates, ordered silica (SBA-15, MCM-41), MOF and zeolites have been used to store thermochemical heat by adsorption.17–21 The main desirable properties for adsorbent materials to be used in thermochemical heat storage applications are thermal stability, high heat and water storage capacities, stability over successive sorption/desorption cycles, high affinity for the sorptive, existence of an abundant supply, and sustainability. Zeolites, an important class of crystalline aluminosilicate microporous materials, are widely used in various industrial applications as catalysts, absorbents, gas storage materials, ion-exchangers and heat exchangers.22,23 They are characterized by framework structures with uniform open pore and channel systems capable of adsorbing large quantities of water molecules at room temperature and at relatively low pressures.24,25 Synthetic zeolites present the advantage of being industrially commercialized in a wide range of shapes such as granulates, extrudates, pellets or powder. The hydrophilicity of zeolites, an important factor for their application in solar or waste heat driven systems, mainly depends on the framework Si/Al molar ratio, the framework structure type and the related exchangeable extra-framework cation sites.26 Aluminum-rich zeolites (A, Y and X-type) are commonly studied for thermochemical heat storage system applications.27–30 In particular, the low Si/Al ratio of faujasite-type zeolite X gives it a strong hydrophilic character, leading to water sorption capacities ranging from 0.28 to 0.33 kg H2O kg−1.31–33
A common strategy to improve the storage capacities of zeolites is the impregnation by a hygroscopic salt. Different hygroscopic salts have been investigated, including CaCl2, SrBr2, MgSO4, MgCl2 and LiCl.34–38 Deliquescence, associated with salt agglomeration and washing out of active materials, can be avoided by salt deposition into porous media. Numerous studies on zeolite-salt composites can be found in the literature and contradictory effect of the salt incorporation were reported. Hongois et al.39 first studied by microcalorimetry the energy released by 13X zeolite–MgSO4 (15 wt%) composite during hydration (over 3 cycles of dehydration/hydration of 100 mg of samples, with dehydration at 150 °C and hydration at 30 °C). The authors reported that the composite achieved an energy density 27% higher than of pure zeolite tested in similar conditions. Xu et al.40 reported an improved water storage capacity (0.188 vs. 0.197 kg H2O kg−1) and higher dehydration enthalpy (598.25 vs. 635.34 kJ kg−1) for 13X pellets after impregnation by 15 wt% of MgSO4 (determined after water saturation at 60% RH at 30 °C). Despite a drastic reduction of specific surface area and pore volume after impregnation of 13X beads by [3.22–10.8 wt%] of MgSO4, Wang and coworkers41 reported increased water and heat storage capacities in comparison with parent zeolite (hydration at 25 °C for 24 h, with RH between 50 and 80% RH). The improvement in water sorption capacity reached 40% for the zeolite-salt composite with 10.8% of MgSO4 when hydration was performed at 80% RH. An improvement of energy storage density of 13X zeolite in beads after incorporation of 15 wt% of MgCl2/CaCl2, for hydration between 15 and 20 °C with 90% RH during 120 min, has been reported by Ji et al.42 Moreover composites with 10% salt hydrates, ZnSO4/13X and MgSO4/13X, absorbed 42 and 64% more water respectively than pure salts upon hydration at 75% RH and 25 °C.43
Mahon et al.44 compared the storage capacities of a 13X zeolite before and after impregnation with MgSO4 (12.9 wt% maximum) after hydration at 56% RH, 20 °C for more than 18 h. Contrary to the previous cited results in ref. 39–43, they observed a small decrease of the hydration enthalpies after salt deposition on a 10 mg sample. The water sorption isobars at 12.3 mbar and temperatures from 50 to 170 °C have been determined by Cortés et al.45 for 13X zeolite, both pure and impregnated by 17 wt% of CaCl2. In this temperature range, a lower water uptake capacity was measured for the zeolite-salt composite than for pure zeolite. These results agree with a previously reported study by our group on 13X beads impregnated by around 17 wt% of MgSO4.46 The reduction in storage performances of the microporous zeolites upon impregnation was explained by a pore blocking effect. The porosity of the zeolite is indeed blocked by the impregnated salt, which limits the access of water molecules to the sorption sites and induces a decrease of the overall storage performance at low RH.46 It has to be noticed that the heat and water storage capacities depend strongly on the experimental conditions for dehydration (temperature, flow, heating rate…) and hydration (temperature, heating rate, relative humidity, flow rate…).24
Recent publications have reported on studies of 13X composites after ion exchange (with Ca2+ and Mg2+) followed by impregnation by CaCl2 or MgSO4.29,47–49 Nonnen et al.47 reported on Na+ exchange of 13X zeolite followed by impregnation by 9 to 18 wt% of CaCl2 and MgSO4. Both water loading and heat storage density decreased with increasing salt content at humidity levels below the deliquescence relative humidity (DRH) of the salt. At higher humidity, high salt loadings in combination with high humidity lead to higher heat and water storage capacities compared to pure zeolites (up to 73 and 54% increase in water and heat storage, respectively). Similar observations have been made by Chan et al.48 concerning the water sorption capacity of 13X (Na+ exchange by Ca2+) impregnated by 10 to 46 wt% of CaCl2 solution at 8.7 mbar for temperatures ranging from 25 to 200 °C. It is worth noting that at humidity levels above the DRH of the salt, leaking of the salt solution from the porosity may occur, which increases the corrosive character of the material and decreases the stability of the storage capacities after numerous hydration/dehydration cycles, due to salt agglomeration and leaching.
With the aim to reduce the extent of micropore blockage by the salt and therefore to increase the storage capacities of zeolites at relatively low RH (30%), three commercial zeolites (13X in powder and bead forms and LiX in bead form) have been impregnated by 5 wt% of salt. Three different salts have been used: CaCl2, MgSO4 and LiCl. In this work, we report on the physico-chemical characteristics, storage capacities and stability, as well as hydration behaviour and kinetics, in order to analyze the role of the zeolite shaping and the impact of relatively low amounts of salt incorporation on these parameters. In addition, the Dubinin–Astakhov adsorption model was applied in order to study the water vapor sorption process.
2. Experimental part
2.1 Preparation of materials
2.2 Characterization of materials
The water sorption capacity was determined using eqn (2) during the second dehydration and hydration:
 | (2) |
3. Results and discussion
3.1 Structural and textural properties
Nitrogen sorption/desorption isotherms of 13X(b), 13X(b) crushed and LiX(b) are a combination of type I and IV according to IUPAC classification indicating that the porous structure is composed of both micropores and mesopores (Fig. 1a and b). This is confirmed by data presented in Table 1. The pore size distributions of the 13X based samples are presented in Fig. S1† and those of the LiX(b) based samples in Fig. S2.† The presence of narrow mesopores in LiX(b) and 13X(b) is highlighted, characteristic of type IV isotherm. 13X(b) exhibits mesopores with size distribution between 2 and 50 nm, with a main contribution coming from narrow mesopores (2-3 nm), without any significant main pore size. LiX(b) shows only narrow mesopores between 2 and 2.7 nm without any noticeable main pore size. The further increase in adsorption at P/P0 > 0.95 is attributed to inter-particle condensation, i.e. the space formed between the particles is filled with condensed nitrogen. The slight hysteresis loop at P/P0 > 0.5 is related to the presence of narrow mesopores. The high microporous volume of pure zeolites (92 and 91% for LiX(b) and 13X(p), respectively) and consequently the high surface areas account for an important amount of available adsorption sites.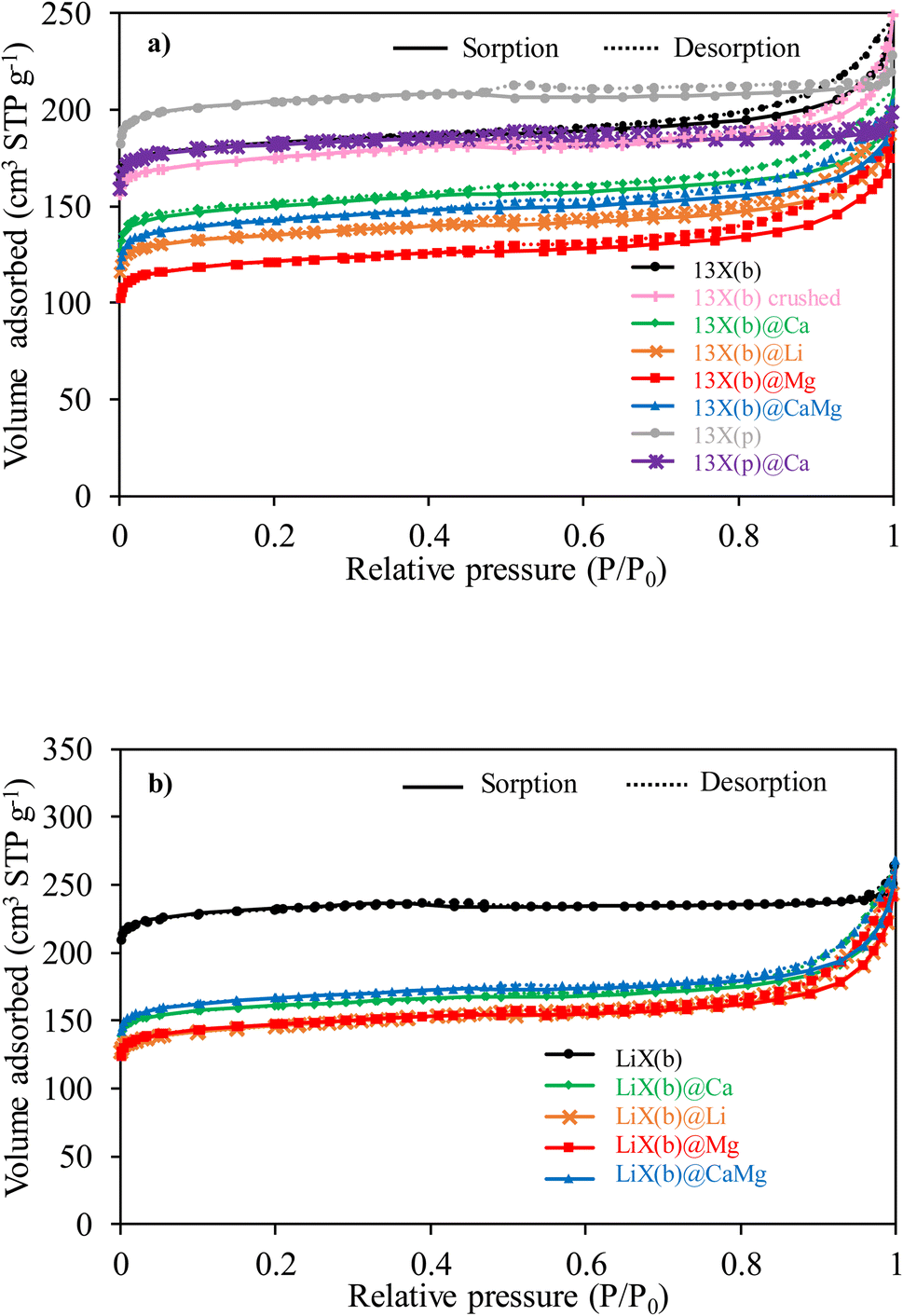 | ||
| Fig. 1 N2 adsorption/desorption isotherms of 13X- (a) and LiX- (b) based materials. | ||
| Name | Amount of salta (wt%) | Apparent BET surface area (m2 g−1) | Total pore volumeb (cm3 g−1) | Micropores volumec (cm3 g−1) | Theoretical pore volumed (cm3 g−1) |
|---|---|---|---|---|---|
| a Determined by ICP-OES (±2%). b Determined for P/P0 = 0.98. c Determined using the t-plot N2. d Determined with eqn (3). | |||||
| 13X(b) | — | 733 | 0.37 | 0.26 | — |
| 13X(b) crushed | — | 672 | 0.36 | 0.25 | — |
| 13X(b)@Ca | 4.8 | 547 | 0.29 | 0.21 | 0.33 |
| 13X(b)@Mg | 4.8 | 463 | 0.26 | 0.17 | 0.33 |
| 13X(b)@Li | 5.1 | 518 | 0.28 | 0.19 | 0.33 |
| 13X(b)@CaMg | 2.6/2.5 | 521 | 0.28 | 0.20 | 0.33 |
| 13X(p) | — | 787 | 0.33 | 0.30 | — |
| 13X(p)@Ca | 4.6 | 699 | 0.30 | 0.27 | 0.32 |
| LiX(b) | — | 894 | 0.38 | 0.35 | — |
| LiX(b)@Ca | 5.2 | 614 | 0.36 | 0.22 | 0.34 |
| LiX(b)@Mg | 5.4 | 536 | 0.35 | 0.20 | 0.34 |
| LiX(b)@Li | 4.7 | 556 | 0.33 | 0.20 | 0.34 |
| LiX(b)@CaMg | 2.7/2.5 | 607 | 0.35 | 0.23 | 0.34 |
For the 13X(b) zeolite, after the first steep increase at P/P0 < 0.05, the sorption/desorption isotherm curve shows a continuous increase up to P/P0 < 0.8 followed by a sharp adsorption at higher pressures. The mild slope of the curve in the middle range and the wide hysteresis loop indicate the presence of pores >2 nm, generated by the binder used to form beads, according to the pore size distribution. Similar behaviour was reported by Q. Wang et al.41 for 13X zeolite beads with particle diameter between 3 and 5 mm and attributed to the mesoporosity generated by the presence of the binder in the material structure. The influence of zeolite shaping and the textural changes induced by the binder can be also pointed out. Without any binder the proportion of microporous volume upon the total pore volume (91% for the powder form versus 70% for zeolite in beads) as well as the apparent specific surface area are higher (Table 1). Contrary to 13X(b), 13X(p) exhibits narrow mesopores between 2 and 3.1 nm with a main pore size at 2.9 nm, showing that some mesoporosity (at pore diameters higher than 3 nm) is created by the binder (Fig S1†), thus remaining as a type I isotherm.50 The presence of non-porous binder for around 20% of the total weight reduces the microporous volume and the apparent specific surface area when these are expressed per gram of sample. Although the binder-free zeolite possesses higher microporous volume, the new porosity generated by the binder results in a higher total pore volume for 13X(b) in comparison with 13X(p). The mechanical grinding of 13X(b) led to a lower apparent specific surface area measured for 13X(b) crushed than 13X(b).
Pore volume retention as well as apparent specific surface area are important factors in water sorption behaviour. Since it is well known that salt incorporation in porous sorbents can influence the porosity of the host material, in order to avoid a decrease in pore volume and consequently the loss of storage properties44,45 of zeolite matrices, relatively low amounts of salt have been used in this work. Indeed, the salt contents (wt%, ±2% error) in the zeolite@salt composites, as presented in Table 1, range from 4.6 to 5.2 wt%. The volume of N2 adsorbed by 13X(b) and 13X(p) based composites are lower in the entire range of relative pressure compared to the corresponding host matrices. In comparison with the pure zeolite, 13X(b) based composites present lower apparent specific surface areas and pore volumes, with a pore distribution shifted towards larger mesopores. This could be caused by (i) penetration of salt particles into the host zeolite micropores (ii) pore access blockage by an excess of salt (iii) a breakdown/enlargement of part of the micropores induced by the impregnation of the salt.
Pore blocking can be evidenced by determining the theoretical pore volume after salt incorporation, eqn (3), considering the salt amount (wt%) determined by chemical analysis and assuming the salt occupies the difference of pore volume between zeolite and composites:51
 | (3) |
The shapes of the isotherms of the LiX(b)-salt composites do not match those of LiX(b) host matrix. Indeed, after the first increase at P/P0 < 0.05, the isotherm curves show a continuous slight increase followed by a sharp increase in adsorption at P/P0 > 0.8, eventually surpassing the host matrix at P/P0 > 0.98. A larger hysteresis loop is also observed for LiX(b) based composites than for pure LiX(b). After salt deposition, LiX(b) shows drastic decreases in apparent specific surface area and microporous volume, while the total pore volume remains stable. Enhanced capillary condensation due to the deposited salt may be responsible for the increase in the total adsorbed volume.
The proportion of microporous volume in the total pore volume drastically decreases from 92% for LiX(b) to [57–66%] for the composites, which may suggest pore filling and/or blocking by the salt. The pore size distribution is shifted toward higher pore diameters with a range between 2 and 50 nm. Nonetheless, the LiX(b) materials exhibit less pore access blockage by the salt than NaX based composites. Indeed, the total pore volumes of the composites remain similar to that of host LiX(b). However, a breakdown and/or enlargement of part of the micropores upon salt impregnation, together with an enhancement of the capillary condensation due to the presence of salt on the zeolite surface, cannot be excluded.39
The impact of salt deposition on the structure of the host zeolites was further determined by XRD. The obtained diffractograms are given in Fig. 2a–c. 13X(b), 13X(p) and LiX(b) host matrices exhibit typical FAU reflection peaks at 2θ = 6.1, 10.0, 11.8, 15.5, 23.3, 26.8 and 30.9° assigned to (111), (220), (311), (331), (642) and (555) facets respectively.53 For zeolite–salt composite materials, all reflections match with those of parent zeolites. As reported in ref. 54 for 13X impregnated by <10% of salt, crystalline salt hydrates can be detected by XRD on the zeolite surface even at low loadings. No visible XRD reflections characteristic of salt hydrates can be assigned to salt incorporated in the zeolite porosity and/or salt deposited in amorphous hydrate phase. Indeed, the materials obtained by impregnation of the zeolites were calcined at 250 °C overnight in order to eliminate residual water. Pore blockage of the zeolites could hinder the complete removal of absorbed water molecules from the hydrated salt, leading to an amorphous phase. Highly dispersed salt located at the surface and in the host matrix porosity would also be consistent with the lack of salt hydrate diffraction peaks. Meanwhile, a decrease in intensity of the peaks corresponding to the porous matrix is also observed after impregnation by a salt.55,56 This decrease can be explained by (i) a decrease in the zeolite amount for the same quantity of material and/or by (ii) a reduction of the contrast due to host matrix pore filling by the salt and its hydrated forms.
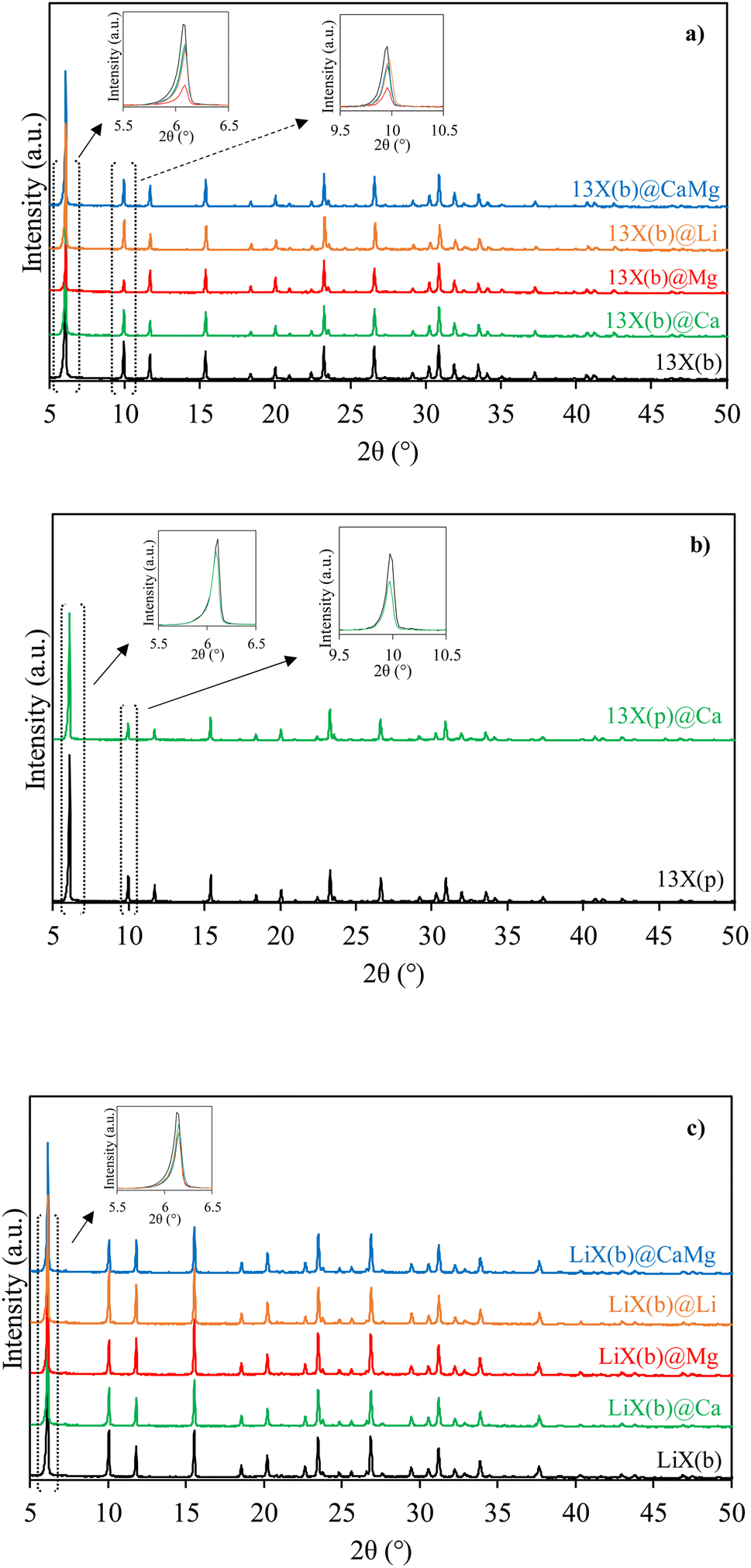 | ||
| Fig. 2 XRD patterns of 13X(b) (a), 13X(p) (b), LiX(b) (c) based materials. | ||
27Al NMR profiles of the 13X(b), 13X(p) and LiX(b) series of materials are shown in Fig. 3a–c, respectively. 13X(b), 13X(p) and LiX(p) exhibit one single signal at δ = 62, 61.5 and 64 ppm, respectively, characteristic of tetracoordinated aluminum (AlIV), confirming the regular systems of intercrystallite cavities and channels of the zeolite frameworks and their corner-sharing AlO45− tetrahedra. After impregnation of 13X(b) and LiX(b), no diffraction peak characteristic of the presence of hexacoordinated Al (AlVI) has been observed, thus showing the preservation of the AlIV coordination. Interestingly, among the different studied composites, 13X(b)@Mg, 13X(b)@Li, 13X(p)@Ca and LiX(b)@Li show a wider main resonance signal than the corresponding host zeolites. In the quadripolar MAS NMR, a modification of the symmetry around the element causes an increase of the quadripolar constant and therefore the resulting NMR signals become wider.57 A modification of the electronic environment around Al due to new interactions and/or distortions caused by salt impregnation explains the widening of the 27Al NMR signals. Other composites such as 13X(b)@Ca, 13X(b)@CaMg, LiX(b)@Ca, LiX(b)@Mg and LiX(b)@CaMg exbibit small shifts of the signal due to slight modification of the AlIV environment. In order to study in greater depth these electronic and/or symmetry modifications of the zeolite constituents, 23Na and 7Li MAS NMR analyses of the materials were also performed; the profiles obtained are presented in Fig. 4 and 5.
 | ||
| Fig. 3 27Al-MAS NMR profiles of 13X(b) (a), 13X(p) (b) and LiX(b) (c) based samples. | ||
 | ||
| Fig. 4 23Na-MAS NMR profiles of 13X(b) (a) and 13X(p) (b) based samples. | ||
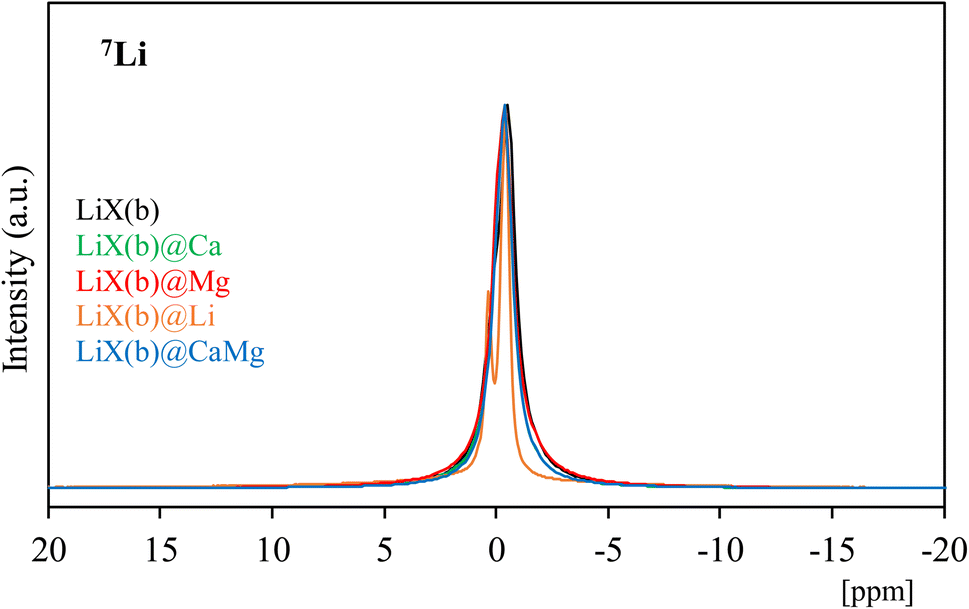 | ||
| Fig. 5 7Li-MAS NMR profiles of LiX(b) and the corresponding composites. | ||
Na+ cations, acting as counter-ions to the negatively charged zeolite structure, are usually found in 6 different sites of the faujasite framework: (I) at the center of the double six-rings (D6R) which tetrahedrally connect the sodalite cavities by bridging oxygens (I′) in the sodalite supercage, at the opposite of site I, (II) at the center of the single six-rings (S6R) shared by the supercages and sodalite (II′) inside the sodalite cavity near a S6R, (III) in the supercage on the opposite axis of a four-ring between two 12-rings and (III′) near the inner walls of the supercages or the edges of 12-rings. The position and distribution of the Na+ sites can vary with the hydration state of the zeolite and the Si/Al ratio. Olson58 reported Na+ ions at sites I, I′, II and III′ with a total of seven Na+ equipoints in 13X with Si/Al = 1. The presence of Na+ in four distinct sites: I′, II and two different III′ positions, in 13X containing Si/Al = 1.08 has been reported by Seff et al.59 Feuerstein et al.60 performed 23Na MAS NMR simulation and experimental analysis of 13X (Si/Al = 1). Five signals were identified by simulation for Na+ cations at sites I, I′, II and two different III′ sites, and the experimental signal represents a large overlapped peak of five different signals corresponding to the simulation ones. In this study, one single Lorentzian-type signal is observed in 23Na MAS NMR spectra of 13X(b) and 13X(p) with a maximum of δ = 1 and −4.8 ppm, respectively, which can be attributed to Na+ positioned in crystallographic site I. Quadrupole interactions of the 23Na nuclei (I = 3/2) at distinct crystallographic cationic sites can lead to overlapped signals in the 23Na NMR spectra, making the separation of the resonance peaks and their assignment difficult. Therefore, the presence of additional resonance signals in the 23Na MAS NMR profiles, corresponding to another Na+ position in the framework structure of 13X, cannot be excluded. Moreover, not all the 23Na nuclei in X-type zeolites are observable by NMR.61
The 23Na NMR profiles of 13X(b)@Ca and 13X(b)@CaMg are very similar to that of the parent zeolite (slightly shifted towards lower δ), suggesting that in these composites most of the Na+ cations occupy the same crystallographic sites and present very similar symmetries to pure 13X(b). In contrast, the NMR signals obtained for 13X(b)@Li, 13X(b)@Mg, and 13X(p)@Ca do not match those of the host zeolites. 13X(b)@Li exhibits one resonance peak at δ = 7 ppm followed by a broader one with a maximum at δ = −7 ppm, showing the non-equivalence of Na+ positions in 13X(b) before and after impregnation by LiCl. The presence of two signals in the NMR spectrum of 13X(b)@Li indicates different symmetries around Na+ cations in this composite, although the exact positions of the Na+ sites remain difficult to assess. It must be noticed that the analysed materials were not dehydrated before the MAS NMR measurements, in order to be as close as possible to a thermochemical heat storage application system. The deliquescence relative humidity (DRH) of LiCl·H2O, defined as the RH at which deliquescence takes place, is relatively low (11%). Therefore, salt liquefaction, which leads to greater mobility of the salt through the zeolite, may take place, with direct impact on the Na+ cation environment. The presence of at least two types of non-equivalent Na+ cations can also be deduced from the spectrum of 13X(p)@Ca. The 23Na NMR profile exhibits a thin resonance peak centred at δ = 7 ppm, characteristic of a quasi-symmetric environment around Na+, and a second wider peak with broad shoulders which overlap the first one and extend all the way to −80 ppm with quadrupolar pattern. In the high-field region, broad shoulder peaks appear due to several overlapped signals which indicate the presence of Na+ in several crystallographic sites. An easier diffusion of CaCl2 particles through 13X in powder form compared to bead shaped zeolite can explain the different structural modifications observed between 13X(p)@Ca and 13X(b)@Ca composites. The only broad signal measured for 13X(b)@Mg, with a maximum at δ = −5 ppm, suggests a deformation around Na+ cations compared to the parent zeolite and thus a symmetry loss. The presence of overlapped peaks under the broad signal and so the presence of Na+ in different site positions cannot be completely ruled out. A breakdown/enlargement of part of the zeolite porosity induced by the salt incorporation, in addition to the pore access blockage previously highlighted for 13X(b)@Mg and 13X(b)@Li, could facilitate the diffusion of salt through the host matrix and thus influence the environment around the zeolite components. The creation of new interactions and/or distortion around the zeolite components is highlighted for 13X(b)@Mg, 13X(b)@Li and 13X(p)@Ca, which agrees with the 27Al MAS NMR results.
Li+ cations are found in three different crystallographic sites, in SI′ inside the β-cages, in SII inside the faujasite supercages and in the position SIII, near the four-ring windows inside the faujasite supercages.62,63 The resonance peak assignments in the 7Li NMR spectrum of LiX (Si/Al = 1) have been studied by Feuerstein et al.60 in correlation with neutron diffraction data analysis. The authors60 concluded that the low-field component is attributed to Li+ at SI’ (δ = 0.4 ppm), the peak at δ = −0.3 ppm to SII sites and the signal at higher field (δ = −0.9 ppm) to SIII. It has to be noticed that the SI′ and SII components are much broader in the 7Li NMR spectra than the SIII signal, due to smaller quadrupole interaction on the Li cations at SIII. The 7Li NMR spectrum of LiX(b) presents a single signal between approximatively δ = −5 and 5 ppm with a maximum at −0.52 ppm, indicating that quadrupolar interactions are not important. LiX(b)@Ca, LiX(b)@Mg and LiX(b)@CaMg present very similar 7Li NMR profiles with a single signal with a maximum at δ = −0.40 ppm showing that salt impregnation proceeds without significant modification of the position in crystallographic sites and the symmetry around Li+ cations in LiX zeolite. In contrast, LiX(b)@Li presents two thin peaks with maxima at δ = +0.36 and −0.36 ppm, indicating that Li+ cations are mainly in two different environments. Similarly to LiCl incorporation in 13X(b), the easier formation of a salt solution facilitates the diffusion of salt through the zeolite and therefore greatly influences the symmetry around the cation. Also, the lower ionic radius of Li+ in comparison with Ca2+ and Mg2+ can lead to an easier incorporation of LiCl in the faujasite structure of zeolites, involving a greater modification of environment and/or symmetry around zeolite components.
3.2 Water vapor sorption isotherms
The hydration/dehydration behaviour of the 13X and LiX based samples has been studied by performing water sorption/desorption experiments in closed environment at 20 °C. The water sorption/desorption isotherms over the entire range of relative water pressure are presented in Fig. 6a and b, respectively. For the sake of clarity, the water uptake at P/P0 < 0.1 is presented in Fig. S3a (13X(b), 13X(p) and the corresponding composites) and b (pure LiX(b) and LiX(b) – based composites).† | ||
| Fig. 6 Water vapor sorption/desorption isotherms at 25 °C of 13X (a) and LiX(b) (b) and corresponding composites. | ||
We discuss first the influence of the zeolite shaping. The type I isotherm exhibited by 13X(p) is consistent with its predominantly microporous character, and agrees with previous reported water sorption isotherm curves of 13X zeolites.64 13X(b), 13X(b) crushed and LiX(b) also exhibit type I adsorption isotherms, but the curves show a slight convex increase from P/P0 = 0.1 to 0.9 probably due to water sorption in the porosity generated by the binder. Pure zeolites present minor irreversibility of the water adsorption/desorption behaviour (the sorption and desorption branches do not match perfectly) due to a strong affinity of zeolites for water. The water sorption capacity follows the order 13X(p) > 13X(b) crushed > 13X(b) in the entire relative pressure range, to reach 0.300 to 0.313 kg H2O kg−1 at P/P0 = 0.9, showing the effect of both binder and shaping on the water vapor sorption capacity. The presence of around 20 wt% of inert binder contributes to the reduction in water uptake per mass unit for the shaped zeolites. The higher water uptake of LiX(b) compared to 13X(b), in the entire range of water pressure, and the more pronounced hysteresis loop indicate a stronger hydrophilicity of LiX(b) zeolite.
The impregnation of 13X(b) by CaCl2 salt (i.e. 13X(b)@Ca and 13X(b)@CaMg composites) resulted in a decrease in the water uptake capacity of the matrix without changing the shape of the isotherms. Based on the N2 sorption isotherms data the partial pore blockage observed after salt deposition, can be correlated with the reduction of the number of available sorption sites (i.e. reduction of both surface area and pore volume). The similarity between the behaviors of 13X(b)@Ca and 13X(b)@CaMg upon water vapor hydration may be explained by the similar structures around the aluminum and Na+ counterions, as determined by solid MAS NMR for these two composites. Lower water uptakes compared to 13X(b) can be also observed for 13X(b)@Mg and 13X(b)@Li, at P/P0 < 0.73 and P/P0 < 0.89, respectively. At higher relative pressures, the water uptakes increase to reach 0.35 kg H2O kg−1 for 13X(b)@Mg at P/P0 = 0.94 and 0.65 kg H2O kg−1 for 13X(b)@Li at P/P0 = 0.91. This increase could be attributed to the easier hydration of the incorporated salts in these composites, due to (i) a breakdown/enlargement of part of the micropores upon salt impregnation, facilitating the diffusion of water vapor and (ii) the presence of salt partly on the zeolite surface. 13X(p)@Ca presents a similar water sorption equilibrium to the host zeolite in the range of relative pressure lower than 0.76, followed by a sharp increase at high pressure to reach 0.82 kg H2O kg−1 at P/P0 = 0.99. The different behaviour of 13X(p)@Ca in comparison with 13X(b)@Ca at high relative pressures evidences the effect of the host matrix shaping on the water sorption performance of the composites. Easier water vapor diffusion through the powder compared to the beads facilitates the formation of a salt solution, and thus greatly increases the water uptake at high relative pressures.
The isotherms of LiX(b)@Ca, LiX(b)@Mg and LiX(b)@CaMg present sharp water adsorption uptakes in the low relative pressure region, mainly attributed to micropore filling, followed by a gentle rise between 0.01 < P/P0 < 0.7, assigned to the filling of narrow mesopores, and a slight increase at P/P0 > 0.7 attributed mainly to hydration of the incorporated salt. At P/P0 < 0.3, LiX(b)@Ca and LiX(b)@Mg show similar water uptakes. At higher relative pressures, the CaCl2 based composite shows better hydration properties than LiX(b)@Mg in the whole studied range. Interestingly, the water uptake of LiX(b)@Ca surpasses that of the host zeolite for P/P0 > 0.73, indicating an easier hydration of the incorporated CaCl2 compared to MgSO4. The water uptake and the shape of the isotherm for LiX(b)@Li are comparable to those of LiX(b)@Ca and LiX(b)@Mg at P/P0 < 0.3, followed by a sharp increase with relative pressure, as previously observed for 13X(b) impregnated by LiCl. This sharp increase in water uptake can be explained by the easier formation of a salt solution in LiCl-based composites, and to a slightly lesser extent in CaCl2-based composites, compared to MgSO4-based composites, due to the lower deliquescence relative humidity (DRH) of LiCl·H2O (11%) than CaCl2·6H2O (28–29%) and MgSO4·7H2O (87–89%) and/or by a higher amount of available salt on the material surface.
No synergetic effect is observed between the deposited salt and the host zeolite matrix in the water sorption performances of the composites. The zeolite undergoes a partial loss of sorption properties due to the partial blocking of its microporosity by the salt incorporation. The low amount of salt confined inside the microporous zeolite due to diffusional issues is not sufficient to offset the reduction in hydration ability of the host matrices at low water pressure. The sharp rise in water uptake capacity of the composites for water vapor relative pressures starting at P/P0 = 0.3 is mainly caused by the salt deliquescence, which can appear at a water vapor partial pressure above the DRH of the hydrated salt, and therefore lead to a leakage of the salt solution.
3.3 Water and heat storage capacities: TG-DSC hydration/dehydration cycles
 | ||
| Fig. 7 Weight evolution during the second dehydration of 13X (a) and LiX (b) and corresponding composites. | ||
All the samples exhibit first a small loss of weight which occurs during the first 20 min at 25 °C, followed by a sharp weight decrease when the temperature is increased from 25 to 300 °C (2 °C min−1). The effect of the binder on the dehydration behaviour can be studied by comparing 13X(p) and 13X(b) crushed. Binder-free 13X(p) presents a higher weight loss and higher heat storage capacity (in kJ kg−1). Even though shaping could hinder water desorption, these two samples display very similar weight loss curves. After 30 min at 300 °C, only 1.7 and 1.2% of the total amount of water sorbed is still present in the 13X(b) and 13X(b) crushed, respectively, showing the slight hindrance effect of the shaping on water desorption. In addition, 3% of the total amount of water adsorbed by LiX(b) is still present in the material at the end of the dehydration step, suggesting a stronger interaction between LiX(b) and water than between 13X zeolites and water.
The water sorption (amount of water sorbed per dehydrated mass, determined using eqn (2)) and heat storage (in kJ per dehydrated mass or per amount of water sorbed) capacities were determined from TG-DSC analysis and are summarized in Table 2. For the sake of comparison, the first dehydration–hydration cycle was used as conditioning treatment. The second dehydration–hydration cycle was further considered to gain insight into the performances of different zeolites and composites as thermochemical heat storage materials.
| Name | Water sorption capacitya (kg H2O kg−1) | Heat storage capacityb (kJ kg−1) | Heat storage capacityc (kJ mol−1 H2O) |
|---|---|---|---|
| a Water sorption capacity determined with eqn (2) on dehydrated sample at 150 °C (±0.02). b Dehydration heat determined by 2nd dehydration heat flow integration (±20 kJ kg−1). c Dehydration heat expressed in kJ mol−1 H2O (±0.5 kJ mol−1). | |||
| 13X(b) | 0.28 | 1007 | 64.9 |
| 13X(b) crushed | 0.28 | 980 | 65.5 |
| 13X(b)@Ca | 0.21 | 800 | 63.1 |
| 13X(b)@Mg | 0.23 | 813 | 64.6 |
| 13X(b)@Li | 0.25 | 860 | 62.8 |
| 13X(b)@CaMg | 0.25 | 892 | 62.4 |
| 13X(p) | 0.31 | 1080 | 63.1 |
| 13X(p)@Ca | 0.29 | 972 | 59.5 |
| LiX(b) | 0.28 | 1058 | 69.6 |
| LiX(b)@Ca | 0.28 | 900 | 58.6 |
| LiX(b)@Mg | 0.28 | 890 | 60.4 |
| LiX(b)@Li | 0.26 | 892 | 62.2 |
| LiX(b)@CaMg | 0.26 | 897 | 62.8 |
With the highest apparent specific surface area among the studied zeolites, LiX shows a water sorption capacity similar to those of 13X(b) and 13X(b) crushed (0.28 kg H2O kg−1), but a higher heat storage capacity (1058 kJ kg−1vs. 1007 and 980, respectively). This is attributed to a stronger interaction between LiX and water. Indeed the amount of heat stored per mol of sorbed water (69.6 kJ mol−1 H2O), which reflects the strength involved in the sorbent–sorptive reaction, surpasses those of 13X(b) (64.6 kJ mol−1 H2O) and 13X(b) crushed (65.5 kJ mol−1 H2O). As reported by Yang et al.65 smaller cations with higher ionic potential are more apt to coordinate water molecules. In addition, the smaller amount of binder used to shape smaller beads of LiX can have an impact on the heat and water storage capacities. The heat and water storage capacities of studied zeolites are higher than those of other porous host matrices reported in the literature (see Table S1†). It has to be noticed that the heat storage capacities of low the Si/Al ratio zeolites provided by ARKEMA outperform those of other commercial zeolites (Table S1†).
In order to study the reversibility of the water and heat storage/release capacities, the heat and water storage capacities during first hydration (H1), second dehydration (D2) and second hydration (H2) are presented in Fig. S5–S17.† Pure zeolites exhibit very good reversibility between water uptake (H1, H2) and water release capacities (D2) with a difference of 1.5% at most. Zeolites exhibit similar water uptakes between the first (H1) and second cycles (H2). Pure zeolites also present good reversibility of the released capacities (H1 and H2), with difference of only 3.4%. The difference between hydration and dehydration enthalpies is however greater (with a maximum of 6.7%), which can be attributed to the integration terminals and/or to the blank subtraction (the blank was not subtracted from the hydration heat flow). LiX(b) represents the most promising host zeolite in terms of heat and water storage capacities, and the bead shape allows its use in many large-scale reactors.
Surprisingly, while a strong chemical interaction between hygroscopic salt and water is expected, the amount of water still presents in the composites after 30 min at 300 °C is lower than in the corresponding host zeolites. The relative amount of water not desorbed in the experimental conditions used here is less than 0.5% for 13X based composites and between 1.2 and 1.6% for LiX based composites (Fig. 7).
Impregnation of 13X(b) results in a decrease in the water sorption capacities (between 0.21 and 0.25 kg H2O kg−1 for the composites) as well as the heat storage capacities (between 800 and 892 kJ kg−1). The heat storage capacities of LiX(b) composites are also lower (between 892 and 900 kJ kg−1) than that of pure LiX(b), although pore blockage by the salt is limited in comparison with NaX based composites. In the case of non-shaped composites, 13X(p)@Ca also presents lower water and heat storage capacities than 13X(p). The decrease in the storage capacities of zeolites after impregnation by hygroscopic salt(s) can be explained by the physico-chemical properties of the composite materials. Indeed, these results are in accordance with the previous conclusions on water vapor sorption isotherms, showing a pore blockage by salt which reduces the sorption capacity of the zeolite and a reduction of salt hydration ability due to diffusional issues. The latter is correlated with a lower diffusion coefficient of the confined salt solution in the smallest pores.67 Ostrovskii et al.68 studied the limiting steps of water sorption in composites and reported the possible formation of a salt solution layer on the channel surface, limiting the amount of water sorbed and thus the heat storage capacities of the composite materials. The water and heat storage capacities of some previously studied composites are summarized in Table S1.† Our resulting composites present higher storage performances compared to other composites with relatively low amount of incorporated salt (4–15 wt%) cited in the literature (despite the decrease of heat and water storage capacities of host zeolites after salt incorporation). On the other hand, due to the large amount of added salt, reported composites with amount of salt between 46 and 62 wt% outperform the storage capacities of studied composites. The composites present very high reversibility of the water uptake capacities (maximum difference of 2.2% between H1 and H2) and between the amount of water sorbed and desorbed (maximum difference of 1.1%), as shown in Fig. S5–S17.† Regarding the heat storage/release, the reversibility between hydration enthalpies is lower for the composite materials (maximum difference of 6.2%) than for the pure host matrices. Also, the gap between the hydration and dehydration enthalpies reaches a maximum of 5.1%. Addition of the fusion heat of the incorporated salts as well as agglomeration and/or leaching of the salt deposited on the surface of the materials can explain this difference. It has to be mentioned that no deliquescence of the salt solution outside zeolite porosity and thus no leaching of the salt were observed after hydration/dehydration cycles.
Regarding the heat storage capacity in kJ mol−1 H2O, the interaction between composites and water results in a less energetic process than between pure zeolites and water. This behaviour was already observed after impregnation of 5A zeolite by 5 and 10 wt% of NaOH.46 The high hydrophilicity (i.e. the high-water uptake at low water pressure) of zeolites has been shown in Fig. 6. Pore blocking limits the water sorption ability of the zeolites, reducing the impact of the highly hydrophilic zeolite on the interaction of composites with water. Pure zeolites exhibit higher storage capacities than rehydrated pure CaCl2 (with 2.7 water molecules under the same hydration conditions), as reported in our previous study.56 CaCl2 based composites, 13X(b)@Ca, 13X(p)@Ca and LiX(b)@Ca, present lower storage capacities than bulk rehydrated salt. The dehydration conditions used in this study allow the full dehydration of bulk salts,56,69,70 and the stabilization of the weight of the studied composites at the end of the isothermal plateau suggests that salt is dehydrated. But the reduction of the water–composite interaction strength could be explained by the presence of a small amount of amorphous phase of partially hydrated salt at the end of the dehydration. The presence of an amorphous phase can be suggested by the lack of salt hydrate crystals in XRD analysis (Fig. 2). Pore blockage could also hinder the release of water from the composite's porosity.
A dish filled with water was used in order to ensure 100% relative humidity in the environment. The studied samples were analysed by TG-DSC using the procedure described in section 2.2.5., after 800 h in the closed environment, in order to mimic 50 cycles of 16 h of hydration at 100% RH. The water sorption and heat storage capacities are summarized in Table 3. For easier comprehension “-ws” is added to the name of samples subjected to 800 h in water-saturated closed environment.
| Name | Water sorption capacitya (kg H2O kg−1) | Heat storage capacityb (kJ kg−1) |
|---|---|---|
| a Water sorption capacity determined with eqn (2) on dehydrated sample at 150 °C (±0.02). b Dehydration heat determined by 2nd dehydration heat flow integration (±20 kJ kg−1). | ||
| 13X(b)-ws | 0.28 ± 0.02 | 960 ± 20 |
| 13X(b)@Ca-ws | 0.25 ± 0.02 | 910 ± 20 |
| LiX(b)-ws | 0.28 ± 0.02 | 1015 ± 20 |
| LiX(b)@Ca-ws | 0.26 ± 0.02 | 900 ± 20 |
The heat storage capacities of 13X(b)-ws and LiX(b)-ws are 4.6 and 4.1% less than the “fresh” materials, respectively. It is difficult to reach a definitive conclusion about a decrease in the amount of heat stored, because the differences in heat amounts are close to the experimental uncertainty. However, the stability of 13X(b) upon cycling is confirmed by the results obtained by nitrogen sorption isotherm measurements after ageing the sample for 800 h (see 13X(b)-ws in Table S2†), with no evident degradation of the structural properties. In the case of zeolite LiX(b), an enlargement of the porosity during ageing process could explain the observed lower apparent BET surface area and microporous volume and thus a loss of the initial performance. The heat and water storage capacities of 13X(b)@Ca-ws are higher than those of the corresponding fresh 13X(b)@Ca (by 14 and 19% for the heat and water storage capacities, respectively). The increase in the surface area revealed after cycling could assign for a better distribution of the salt throughout the zeolite which can allow the clear passage of adsorbent molecules such as N2 or H2O. However, both salt hydration and water sorption by the zeolites can be facilitated by salt deliquescence and leakage of salt solution, improving the storage capacities. Although no deliquescence of salt solution outside matrix porosity is observed, such phenomenon cannot be completely ruled out under the high-water pressure used for ageing, and can impact the stability of the storage system over numerous hydration/dehydration cycles as well as the corrosivity of the material.
Stable thermochemical properties are observed for LiX(b)@Ca after 800 h under humid atmosphere in closed environment, in spite of about 9% loss of its surface area after ageing. This could be explained by a synergetic contribution of the incorporated salt and zeolite matrix to the final storage properties of the composite. The possible redistribution of the salt not only in the pores, but also on the external surface may allow the rapid diffusion of adsorbent molecules throughout the pores, and hence does not have a detrimental impact on the heat released.
The water loading coefficients of the 13X and LiX based samples were determined using eqn (4),
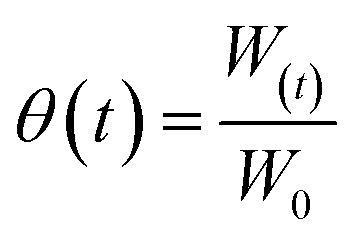 | (4) |
 | ||
| Fig. 8 Evolution of the water loading coefficient during the second hydration of 13X (a) and LiX (b) based samples. | ||
Water vapor sorption is faster on 13X(p) than on 13X(b) crushed and 13X(b) zeolites, showing the effect of both the binder and the material shaping on the kinetics. The slowdown of water sorption for shaped 13X is likely due to slower diffusion through condensed beads than crushed or pure powder. 13X(b)@Mg and 13X(b)@Li exhibit faster sorption kinetics than corresponding pure 13X(b), contrary to 13X(b)@CaMg. Faster water sorption on the composites can be explained by the large amount of hygroscopic salt on the composite surface, which enhances the water sorption kinetics. Although 13X(b)@Ca reaches θ = 0.95 faster than pure 13X(b), θ = 0.99 is attained after a longer time. 13X(b)@CaMg presents slower water vapor sorption kinetics than the corresponding host zeolite during the entire hydration process. These observations can be correlated with a lower water uptake than 13X(b) in water sorption isotherms and TG-DSC experiments, and could be related to better incorporation of the salts in the zeolite structure, which may slow down the water vapor sorption kinetics. It was concluded in section 3.1 that a better diffusion of salt through the material limits the amount of pore blocking for 13X(p) based composites. This leads to a higher amount of salt inside the zeolite porosity, and thus slows down the water sorption kinetics of 13X(p)@Ca compared to 13X(p).
Although LiX based composites exhibit less pore blockage than 13X(b) ones, the preferential deposition of salt in the micropores and/or blockage of access to the micropores of LiX(b) increases the water vapor sorption kinetics of the composites compared to pure host LiX(b). On the other hand, the longer time needed to reach θ = 0.99 for LiX(b)@Ca and LiX(b)@Li compared to LiX(b) can be attributed to a larger amount of salt on the zeolite surface, limiting water vapor diffusion through the composites. This observation is correlated to the easier hydration of CaCl2 and LiCl deposited in LiX(b), as shown by the water sorption isotherms. Thus, the presence of salt on the surface of the microporous materials can have a dual influence on water vapor sorption kinetics: (i) enhancing the kinetics due to the high affinity of the salt for water and (ii) induce water vapor diffusion limitations.
Microporous materials possess by definition a large amount of micropores (<2 nm), but they also contain a large variety of pores such as narrow mesopores and transitional pores. Generally, micropores are the key factor for adsorption of water vapor. Dubinin72 developed the “theory of volume filling in micropores”, which considers the mechanism of filling a limited amount of volume during micropore adsorption. In the theory, two functions are associated with the parameters of the adsorption equilibrium, the amount adsorbed, the temperature and the equilibrium pressure.
The first of these functions is expressed by:
 | (5) |
The second function is the filled volume of the adsorption space W:
| W = av′ | (6) |
Thanks to the Polanyi adsorption potential theory, the volume sorbed (W) can be plotted as a function of the adsorption potential ε. The curves ε = f(W) with W representing the volume adsorbed in the water sorption isotherm and ε determined by eqn (5) are presented in Fig. S19a and b† for 13X and LiX based materials, respectively.
For all the studied materials, the adsorption potential of water molecules decreases as the adsorbed volume increases, and eventually vanishes when the maximum sorption capacity of the material is reached. At low sorbed volumes (∼0.01 cm3 g−1dehydrated sample) the adsorption potential is high for the pure zeolites due to the highly energetic interactions between the microporous structure of zeolites and water. The adsorption potentials of zeolite-based composites do not match those of the parent host matrices, showing the effect of deposited salt.
The decrease in water sorption potential after salt deposition, as seen for 13X(b)@Ca, 13X(b)@Mg and 13X(b)@CaMg, could be caused by zeolite pore blockages, reducing the ability of the host matrices to adsorb water. The confinement of salt in the porosity can also reduce the contribution of the salt to the sorption potential. The higher water sorption potentials observed for LiX(b)@Ca, LiX(b)@CaMg, LiX(b)@Mg and LiX(b)@Li compared to LiX(b) correlate with the higher water uptakes of these materials at low relative pressures (see Fig. S3b†). 13X(b)@Li, 13X(b)@Mg 13X(p)@Ca, LiX(b)@Li and LiX(b)@Ca reach ε = 0 at higher adsorbed volumes due to the higher hydration ability of the deposited salt. As shown by water vapor sorption isotherms, the increase of water uptake at high relative pressure is mainly due to the salt hydration.
To better understand the sorption process as well as the nature of the interactions between sorbent and sorptive, Dubinin and Astakhov73 proposed a model of the sorption isotherm which considers the heterogeneity of the adsorption phenomenon, described in the ESI.†
The linear transforms of the characteristic adsorption curves of the water vapor sorption isotherms are plotted in Fig. S20–S32.† The parameters of the Dubinin–Astakhov (D–A) model (n and θ) and the correlation coefficient R2 of the linear regression are summarized in Table S3.†
The D–A transform of pure zeolite is represented by two straight lines, which confirms the hypothesis of energy heterogeneity. Indeed, the sorption process for the zeolite–water pair is characterized by heterogeneous interactions, with two types of interaction forces: a dispersion force (water–water interaction) and an attraction force due to the adsorption of the first molecules on the active center. Indeed, inside the zeolite cavity, compensating cations (Na+ or Li+) create active sorption centers. Each straight line corresponds to a type of interaction, either attraction or dispersion. The values of n vary between 3.28 and 3.74 for low pore filling ratios (θ) and between 0.57 and 1.19 for high pore filling ratios. Dubinin and co-workers74 reported that the pore filling process can be expressed by two term equations, with each term having the mathematical form of eqn (S1) (ESI†). The attractive interaction is predominant at low filling ratios, while dispersion interactions become more important at high filling ratios.
The D–A transform of pure composites still presents two straight lines corresponding mainly to water sorption by the zeolite structure, but also a third convex curve at high filling ratios. This last contribution shows the predominant effect of the salt hydration at high relative pressure (as observed in Fig. 6) which corresponds to high filling ratios. Thus, the application of the Dubinin–Astakhov model brings light on the effect of the salt in the water vapor sorption process of the zeolite-based composites.
The effect of salt addition on the water sorption by the zeolite structure is also highlighted. Indeed, the modification of D–A model parameters in comparison with the respective parent zeolites is particularly significant for 13X(b)@Mg, 13X(b)@Li, 13X(p)@Ca and LiX(b)@Ca. The filling ratios (θ) at which the transition occurs between the dispersive and attractive portions of the curves are considerably reduced. Also, the reduction of the n values for the high and low filling ratio regions suggests increased structural heterogeneity, caused by the salt addition. These results are in agreement with the NMR analysis of these materials, which showed a modification of the electronic environment around zeolite components due to new interactions and/or distortions caused by salt impregnation. These composites also present wider convex curves in the D–A transform curve compared to the other composites, likely due to the strong hydration ability of the deposited salt (previously shown in Fig. 6).
4. Conclusions
In this study, the physicochemical characteristics, storage capacities, stability, as well as hydration process and kinetics of pure zeolites and zeolite@salt composites were studied. The impact of host zeolite shaping and of the choice of deposited salt was investigated. It has been confirmed that pure zeolites exhibit high heat and water storage capacities and high stability of the storage performances. Pure zeolites are also saturated by water at low relative humidity, which is beneficial for applications given their total potential storage capacities. The non-shaped 13X(p) zeolite presents faster hydration kinetics, and the lack of binder to form beads makes it possible to achieve higher heat and water sorption capacities.The storage capacities in the open system are totally reversible for all the zeolites and corresponding composites. Although all salt incorporation impacts the characteristics and properties of zeolites, LiCl deposition has a stronger influence, greatly modifying the environment and/or symmetry around zeolite components and influencing the water sorption behaviour in closed system as well as the hydration process and kinetics.
The presence of salt dramatically decreases the heat and water storage capacities due to blocking of the zeolite pores, which limits the access of water molecules. The reduction of porosity is more limited for powder 13X, which leads to a smaller loss of storage capacities.
In conclusion, the key influence of the available porosity on the storage performances of zeolite composites was highlighted. It was also shown that the use of very low amounts of salt does not allow the preservation of the porosity of the zeolite while maintaining the hydration ability of the salt, reducing the storage capacities of zeolite@salt composites.
Therefore, this study adds fundamental knowledge on the impact of (i) zeolite shaping, (ii) deposition of a low amount of salt, and (iii) the choice of the hygroscopic salt, on the storage capacities of zeolite-based composites for potential thermochemical heat storage applications.
Author contributions
Q. T., G. P., and A. A. contributed to conceptualization of research in the manuscript. Q. T., and G. P. were involved in data curation, analysis, and methodology. Q. T., L. M., and C. L. performed experimental investigations. G. P. and A. A. supervised the research activities and handled project administration, data validation and visualization. Q. T. writing of the original draft. G. P., and A. A. writing-review & editing. All authors contributed to review and editing of the final manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the French National Research Agency (Agence Nationale de la Recherche, Projet ANR-18-CE05-0044). The authors are thankful for the scientific services of IRCELYON (P. Mascunan, N. Bonnet, Y. Aizac) for the material characterizations. Authors gratefully acknowledge C. Lutz from ARKEMA for zeolites supply and discussion.References
- M. van der Hoeven, World Energy Outlook Special Report 2015: Energy and Climate Change – Executive Summary, International Energy Agency (IEA), 2015 Search PubMed.
- S.-Y. Liu, Y.-H. Perng and Y.-F. Ho, The effect of renewable energy application on Taiwan buildings : What are the challenges and strategies for solar energy exploitation?, Renewable Sustainable Energy Rev., 2013, 28, 92–106, DOI:10.1016/j.rser.2013.07.018.
- I. Dincer and C. Acar, A review on clean energy solutions for better sustainability, Int. J. Energy Res., 2015, 39(5), 585–606, DOI:10.1002/er.3329.
- J. Fitó, J. Ramousse, S. Hodencq and F. Wurtz, Energy, exergy, economic and exergoeconomic (4E) multicriteria analysis of an industrial waste heat valorization system through district heating, Sustain. Energy Technol. Assess., 2020, 42, 100894, DOI:10.1016/j.seta.2020.100894.
- I. Gur, K. Sawyer and R. Prasher, Searching for a better thermal battery, Science, 2012, 335, 1454, DOI:10.1126/science.1218761.
- H. Nazir, M. Batool, F. J. B. Bolivar, M. Isaza-Ruiz, X. Xu, K. Vignarooban, P. Phelan, Inamuddin and A. M. Kannan, Recent developments in phase change materials for energy storage applications: a review, Int. J. Heat Mass Transfer, 2019, 129, 491–523, DOI:10.1016/j.ijheatmasstransfer.2018.09.126.
- A. Elouali, T. Kousksou, T. El Rhafiki, S. Hamdaoui, M. Mahdaoui, A. Allouhi and Y. Zeraouli, Physical models for packed bed: Sensible heat storage systems, J. Energy Storage, 20199, 23, 69–78, DOI:10.1016/j.est.2019.03.004.
- L. Qiu, Y. Ouyang, Y. Feng and X. Zhang, Review on micro/nano phase change materials for solar thermal applications, Renewable Energy, 2019, 140, 513–538, DOI:10.1016/j.renene.2019.03.088.
- Z. Ma, H. Bao and A. P. Roskilly, Seasonal solar thermal energy storage using thermochemical sorption in domestic dwellings in the UK, Energy, 2019, 166, 213–222, DOI:10.1016/j.energy.2018.10.066.
- G. Krese, R. Koželj, V. Butala and U. Stritih, Thermochemical seasonal solar energy storage for heating and cooling of buildings, Energy Build., 2018, 164, 239–253, DOI:10.1016/j.enbuild.2017.12.057.
- A. J. Carrillo, J. Gonzalez-Aguilar, M. Romero and J. M. Coronado, Solar Energy on Demand: A Review on High Temperature Thermochemical Heat Storage Systems and Materials, Chem. Rev., 2019, 119, 4777–4816, DOI:10.1021/acs.chemrev.8b00315.
- M. Felderhoff, R. Urbanczyk and S. Peil, Thermochemical heat storage for high temperature applications, Green, 2013, 3, 113–123, DOI:10.1515/green-2013-0011.
- P. Tatsidjodoung, N. Le Pierres and L. Luo, A review of potential materials for thermal energy storage in buildings applications, Renewable Sustainable Energy Rev., 2013, 18, 327–349, DOI:10.1016/j.rser.2012.10.025.
- J. Cot-Gores, A. Castell and L. F. Cabeza, Thermochemical heat storage and coversion: a-state-of-art review of the experimental research under practical conditions, Renewable Sustainable Energy Rev., 2012, 16, 5207–5224, DOI:10.1016/j.rser.2012.04.007.
- W. Hue, H. Yan, X. Zang, X. Xu, L. Zhang and Y. Shi, Review of salt hydrates-based thermochemical adsorption thermal storage technologies, J. Energy Storage, 2022, 56, 106158, DOI:10.1016/j.est.2022.106158.
- A. H. Abedin and M. A. Rosen, A critical review of thermochemical energy storage systems, Open Renewable Energy J., 2011, 4, 42–46, DOI:10.2174/1876387101004010042.
- H. Wu, F. Salles and J. Zajac, A critical review of solid materials for low-temperature thermochemical storage of solar based on solid-vapour adsorption in view of space heating uses, Molecules, 2019, 45, 945, DOI:10.3390/molecules24050945.
- M. de Lange, K. Verouden, T. Vlugt, J. Gascon and F. Kapteijn, Adsorption-driven heat pumps: the potential of metal-organic frameworks, Chem. Rev., 2015, 115(22), 12205–12250, DOI:10.1021/acs.chemrev.5b00059.
- S. Henninger, S.-J. Ernst, L. Gordeeva, P. Bendix, D. Fröhlich, A. Grekova, L. Bonaccorsi, Y. Aristov and J. Jaenchen, New materials for adsorption heat transformation and storage, Renewable Energy, 2017, 110, 59–68, DOI:10.1016/j.renene.2016.08.041.
- G. Basina, D. AlShami, K. Polychronopoulou, V. Tzitzios, V. Balasubramanian, F. Dawaymeh, G. Karanikolos and Y. Al Wahedi, Hierarchical AlPO4-5 and SAPO-5 microporous molecular sieves with mesoporous connectivity for water sorption applications, Surf. Coat., 2018, 3523, 378–386, DOI:10.1016/j.surfcoat.2018.08.083.
- P. L. Llewellyn, F. Schüth, Y. Grille, F. Rouquerol, J. Rouquerol and K. K. Unger, Water sorption on mesoporous aluminosilicate MCM-41, Langmuir, 1995, 11, 574–577, DOI:10.1021/la00002a036.
- Zeolites in Industrial Separation and Catalysis, ed. S. Kulprathipanja, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2010, pp. 1–593 Search PubMed.
- B. Yilmaz, N. Trukhan and U. Müller, Industrial outlook on zeolites and metal organic frameworks, Chin. J. Catal., 2012, 33, 3–10, DOI:10.1016/S1872-2067(10)60302-6.
- S. Yamanaka, P. B. Malla and S. Komarneni, Water sorption and desorption isotherms of some naturally occurring zeolites, Zeolites, 1989, 9, 18–22, DOI:10.1016/0144-2449(89)90004-3.
- H. S. Sherry, Handbook of Zeolite Science and Technology, Marcel Dekker, New York, 2003, pp. 1007–1061 Search PubMed.
- J. J. D. Ackermann, H. Stach and W. Brösicke, Studies of the water adsorption on zeolites and modified mesoporous materials for seasonal storage of solar heat, Solar Energy, 2004, 76, 339–344, DOI:10.1016/j.solener.2003.07.036.
- R. Shigeishi, C. Langford and B. Hollebone, Solar energy storage using chemical potential changes associated with dying of zeolites, Solar Energy, 1979, 23, 489–495, DOI:10.1016/0038-092X(79)90072-0.
- MODESTORE – Deliverable D14, Final Report on Austrian Field Test Period (Performance and Evaluation), AEE – Institute for Sustainable Technologies, 2005 Search PubMed.
- T. S. Yan, T. X. Li, J. X. Xu and R. Z. Wang, Water sorption properties, diffusion and kinetics of zeolite NaX modified by ion-exchange and salt impregnated, Int. J. Heat Mass Transfer, 2019, 139, 990–999, DOI:10.1016/j.ijheatmasstransfer.2019.05.080.
- H. Stach, J. Mugele, J. Jänchen and E. Weiler, Influence of cycle temperatures on the thermochemical heat storage densities in the systems water/microporous and water/mesoporous adsorbents, Adsorption, 2005, 11, 393–404, DOI:10.1007/s10450-005-5405-x.
- D. Chen, X. Hu, L. Shi, Q. Cui, H. Wang and H. Yao, Synthesis and characterization of zeolite X from lithium slag, Appl. Clay Sci., 2012, 59–60, 148–151, DOI:10.1016/j.clay.2012.02.017.
- T. Yamamoto, Y. H. Kim, B. C. Kim, A. Endo, N. Thongprachan and T. Ohmori, Adsorption characteristics of zeolites for dehydration of ethanol: Evaluation of diffusivity of water in porous structure, Chem. Eng. J., 2012, 181–182, 443–448, DOI:10.1016/j.cej.2011.11.110.
- J. Jänchen and H. Stach, Shaping adsorption properties of nano-porous molecular sieves for solar thermal energy storage and heat pump applications, Solar Energy, 2014, 104, 16–18, DOI:10.1016/j.solener.2013.07.018.
- H. A. Zondag, V. M. van Essen, L. P. J. Bleijendaal, B. Kikkert and M. Bakker, Application of MgCl2·6H2O for thermochemical seasonal solar heat storage, 5th Int Renew Energy Storage Conf. IRES 2010, Berlin, Germany, 2010 Search PubMed.
- H. U. Rammerlberger, T. Schmidt and W. Ruck, Hydration and dehydration of salt hydrates and hydroxides for thermal energy storage – kinetics and energy release, Energy Procedia, 2012, 30, 362–369, DOI:10.1016/j.egypro.2012.11.043.
- K. E. NTsoukpoe, T. Schmidt, H. U. Rammerlberg, B. A. Watts and W. K. L. Ruck, A systematic multi-step screening of numerous salt hydrates for low temperature thermochemical energy storage, Appl. Energy, 2014, 124, 1–16, DOI:10.1016/j.apenergy.2014.02.053.
- L. Scapino, H. A. Zondag, J. V. Bael, J. Diriken and C. C. M. Rindt, Sorption heat storage for long-term low-temperature applications: A review on the advancements at material and prototype scale, Appl. Energy, 2017, 190, 920–948, DOI:10.1016/j.apenergy.2016.12.148.
- K. 1 Posern and A. Osburg, Determination of the heat storage performance of thermochemical heat storage materials based on SrCl2 and MgSO4, J. Therm. Anal. Calorim., 2018, 131, 2769–2773, DOI:10.1007/s10973-017-6861-8.
- S. Hongois, F. Kuznik, P. Stevens and J.-J. Roux, Development and characterisation of a new MgSO4 zeolite composite for long-term thermal energy storage, Sol. Energy Mater. Sol. Cells, 2011, 95, 1831–1837, DOI:10.1016/j.solmat.2011.01.050.
- S. Z. Xu, Lemington, R. Z. Wang, L. W. Wanga and J. Zhu, A zeolite 13X/magnesium sulfate–water sorption thermal energy storage device for domestic heating, Energy Convers. Manage., 2018, 171, 98–109, DOI:10.1016/j.enconman.2018.05.077.
- Q. Wang, Y. Xie, B. Ding, G. Yu, F. Ye and C. Xu, Structure and hydration state characterizations of MgSO4-zeolite 13x composite materials for long-term thermochemical heat storage, Sol. Energy Mater. Sol. Cells, 2019, 200, 110047, DOI:10.1016/j.solmat.2019.110047.
- W. Ji, H. Zhang, S. Liu, S. Wang and S. Deng, An experimental study on the binary hydrated salt composite zeolite for improving thermochemical energy storage performance, Renewable Energy, 2022, 194, 1163–1173, DOI:10.1016/j.renene.2022.06.024.
- A. Ur Rehman, M. Zahir Shah, S. Rasheed, W. Afzal, M. Arsala, H. Ur Rahman, M. Ullah, T. Zhao, I. Ullah, A. Ud Din, S. Ullah, R. Shah, M. Iqbal, Z. Maosheng and Z.-H. Guan, Inorganic salt hydrates and zeolites composites studies for thermochemical heat storage, Z. Phys. Chem., 235(11), 1481–1497, DOI:10.1515/zpch-2021-3012.
- D. Mahon, G. Claudio and P. Eames, An experimental investigation to assess the potential of using MgSO4 impregnation and Mg2+ ion exchange to enhance the performance of 13X molecular sieves for interseasonal domestic thermochemical energy storage, Energy Convers. Manage., 2017, 150, 870–877, DOI:10.1016/j.enconman.2017.03.080.
- F. B. Cortés, F. Chejne, F. Carrasco-Marín, A. F. Pérez-Cadenas and C. Moreno-Castilla, Water sorption on silica- and zeolite-supported hygroscopic salts for cooling system applications, Energy Convers. Manage., 2012, 53, 219–223, DOI:10.1016/j.enconman.2011.09.001.
- G. Whiting, D. Grondin, S. Bennici and A. Auroux, Heats of water sorption studies on zeolite-MgSO4 composites as potential thermochemical heat storage materials, Sol. Energy Mater. Sol. Cells, 2013, 112, 112–119, DOI:10.1016/j.solmat.2013.01.020.
- (a) A. Jabbari-Hichri, A. Auroux and S. Bennici, Effect of NaOH addition on the thermochemical heat storage capacity of nanoporous molecular sieves, Int. J. Energy Res., 2017, 41, 1134–1149, DOI:10.1002/er.3697; (b) T. Nonnen, H. Preißl, S. Kött, S. Beckert and R. Gläser, Salt inclusion and deliquescence in salt/zeolite X composites for thermochemical heat storage, Microporous Mesoporous Mater., 2020, 303, 110239, DOI:10.1016/j.micromeso.2020.110239.
- K. C. Chan, C. Y. H. Chao, G. N. Sze-To and K. S. Hui, Performance predictions for a new zeolite 13X/CaCl2 composite adsorbent for adsorption cooling systems, Int. J. Heat Mass Transfer, 2012, 55, 3214–3224, DOI:10.1016/j.ijheatmasstransfer.2012.02.054.
- T. Nonnen, S. Beckert, K. Gleichmann, A. Brandt, B. Unger, H. Kerskes, B. Mette, S. Bonk, T. Badenhop, F. Salg and R. Gläser, A thermochemical long-term storage system based on a salt/zeolite composite, Chem. Ing. Tech., 2016, 88, 363–371, DOI:10.1002/ceat.201600301.
- H. Mousavi, J. T. Darian and B. Mokhtarani, Enhanced nitrogen adsorption capacity on Ca2+ ion-exchanged hierarchical X zeolite, Sep. Purif. Technol., 2021, 264, 118442, DOI:10.1016/j.seppur.2021.118442.
- P. D’ans, E. Courbon, A. Permyakova, F. Nouar, C. Simmonet-J'egat, F. Bourdreux, L. Malet, C. Serre, M. Frère and N. Steunou, A new strontium bromide MOF composite with improved performance for solar energy storage application, J. Energy Storage, 2019, 25, 100881, DOI:10.1016/j.est.2019.100881.
- Yu. I. Aristov, in Nanocomposite Sorbents for Multiple Applications, Jenny Standford Publishing, 1 edn, 2020 Search PubMed.
- M. M. J. Treacy, J. B. Higgins and R. von Ballmos, Collection of Simulated XRD Powder Patterns for Zeolite, Elsevier, UK, 1996 Search PubMed.
- G. Whiting, D. Grondin, D. Stosic, S. Bennici and A. Auroux, Zeolite-MgCl2 composites as potential heat storage materials: influence of zeolite properties on heats of water sorption, Sol. Energy Mater. Sol. Cells, 2014, 128, 289–295, DOI:10.1016/j.solmat.2014.05.016.
- A. Permyakova, S. Wang, E. Courbon, F. Nouar, N. Heymans, P. D'Ans, N. Barrier, P. Billemont, G. De Weireld, N. Steunou, M. Frère and C. Serre, Design of salt-metal organic framework composites for seasonal heat storage applications, J. Mater. Chem. A, 2017, 5, 12889–12898, 10.1039/C7TA03069J.
- Q. Touloumet, G. Postole, L. Silvester, L. Bois and A. Auroux, Hierarchical aluminium fumarate metal-organic framework – alumina host matrix: Design and application to CaCl2 composites for thermochemical heat storage, J. Energy Storage, 2022, 50, 104702, DOI:10.1016/j.est.2022.104702.
- J. Klinowski, Nuclear magnetic resonance studies of zeolites, Prog. Nucl. Magn. Reson. Spectrosc., 1984, 16, 237–309, DOI:10.1016/0079-6565(84)80007-2.
- D. H. Olson, The crystal structure of dehydrated NaX, Zeolites, 1995, 15, 439–443, DOI:10.1016/0144-2449(95)00029-6.
- L. Zhu and K. Seff, Reinvestigation of the Crystal Structure of Dehydrated Sodium Zeolite X, J. Phys. Chem. B, 1999, 103, 9512–9518, DOI:10.1021/jp991817l.
- M. Feuerstein, G. Engelhardt, P. L. McDaniel, J. E. MacDougall and T. R. Gaffney, Solid-state nuclear magnetic resonance investigation of cation sitint in LiNaLSX zeolites, Microporous Mesoporous Mater., 1998, 26, 27–35, DOI:10.1016/S1387-1811(98)00211-X.
- W. D. Basler, Sodium ions in hydrated faujasites as studied by 23Na and 1H NMR, Colloids Surf., 1984, 12, 59–67, DOI:10.1016/0166-6622(84)80089-X.
- C. Forano, R. C. T. Slade, E. K. Andersen, I. G. K. Andersen and E. Prince, Neutron diffraction determination of full structure of anhydrous LiX and LiY zeolite, J. Solid State Chem., 1989, 82, 95–102, DOI:10.1016/0022-4596(89)90227-2.
- M. Feuerstein, M. Hunger, G. Engerlhardt and J. P. Amoureux, Characterisation of sodium cations in dehydrated zeolite NaX by 23Na NMR spectroscopy, Solid State Nucl. Magn. Reson., 1996, 7, 95–103, DOI:10.1016/S0926-2040(96)01246-5.
- D. Chen, X. Hu, L. Shi, Q. Cui, H. Wang and H. Yao, Synthesis and characterization of zeolite X from lithium slag, Appl. Clay Sci., 2012, 59–60, 148–151, DOI:10.1016/j.clay.2012.02.017.
- S. Yang and A. Navrotsky, Energetics of formation and hydration of ion-exchanged zeolite Y, Microporous Mesoporous Mater., 2000, 37, 175–186, DOI:10.1016/S1387-1811(99)00264-4.
- H. U. Rammelberg, T. Osterland, B. Priehs, O. Opel and W. K. L. Ruck, Thermochemical heat storage materials – Performance of mixed salt hydrates, Solar Energy, 2016, 136, 571–589, DOI:10.1016/j.solener.2016.07.016.
- B. Dawoud and Y. I. Aristov, Experimental study on the kinetics of water vapor sorption on selective water sorbents, silica gel and alumina under typical operating conditions of sorption heat pumps, Int. J. Heat Mass Transfer, 2003, 46, 273–281, DOI:10.1016/S0017-9310(02)00288-0.
- N. M. Ostrovskii, N. A. Chumakova, N. M. Bukhavtsova, N. V. Vernikovskaya and Y. I. Aristov, Modeling of the Limiting Step of Water Sorption by Composite Sorbents of the “Calcium Chloride in Porous Matrix” Type, Theor. Found. Chem. Eng., 2007, 41, 83–90, DOI:10.1134/S0040579507010095.
- Q. Touloumet, L. Silvester, L. Bois, G. Postole and A. Auroux, Water sorption and heat storage in CaCl2 impregnated aluminium fumarate MOFs, Sol. Energy Mater. Sol. Cells, 2021, 231, 111332, DOI:10.1016/j.solmat.2021.111332.
- P. J. Masset, Thermogravimetric study of the dehydration reaction LiCl·H2O, J. Therm. Anal. Calorim., 2009, 96, 439–441, DOI:10.1007/s10973-008-9399-y.
- M. Fasano, L. Bergamasco, A. Lombardo, M. Zanini, E. Chiavazzo and P. Asinari, Water/ethanol and 13X zeolite pairs for long-term thermal storage at ambient pressure, Front. Energy Res., 2019, 7, 148, DOI:10.3389/fenrg.2019.00148.
- M. M. Dubinin, Adsorption in micropores, J. Colloid Interface Sci., 1967, 23, 487–499, DOI:10.1016/0021-9797(67)90195-6.
- M. M. Dubinin and V. A. Astakhov, Development of the concepts of volume filling of micropores in the adsorption of gases and vapors by microporous adsorbents: communication 1. Carbone adsorbents, Bull. Acad. Sci. USSR, Div. Chem. Sci., 1971, 20, 3–7, DOI:10.1007/BF00849307.
- M. M. Dubinin and V. A. Astakhov in Molecular Sieve Zeolites-II ed. E. M. Flanigen and L. B. Sand, American Chemical Society, 1971, vol. 101, pp. 69–85 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ta07615b |
| This journal is © The Royal Society of Chemistry 2023 |