
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePyrolyzed cobalt hexacyanocobaltate dispersed on reduced-graphene-oxide as an electrocatalyst of the oxygen reduction reaction in an alkaline medium†
B.
Zakrzewska
a,
A.
Jabłońska
a,
L.
Adamczyk
b,
B.
Dembińska
a,
A.
Kostuch
 a,
M.
Strawski
a,
I. A.
Rutkowska
a,
P. J.
Kulesza
a,
M.
Marcinek
c,
J. A.
Cox
d and
K.
Miecznikowski
*a
a,
M.
Strawski
a,
I. A.
Rutkowska
a,
P. J.
Kulesza
a,
M.
Marcinek
c,
J. A.
Cox
d and
K.
Miecznikowski
*a
aUniversity of Warsaw, Faculty of Chemistry, L. Pasteura 1, 02-093 Warsaw, Poland. E-mail: kmiecz@chem.uw.edu.pl; Fax: +49-22-8225996; Tel: +48-22-5526340
bCzestochowa University of Technology, Faculty of Production Engineering and Materials Technology, Al. Armii Krajowej 19, 42-200 Czestochowa, Poland
cDepartment of Chemistry, Warsaw University of Technology, Noakowskiego 3, 00-664 Warsaw, Poland
dDepartment of Chemistry and Biochemistry, Miami University, Oxford, OH 45056, USA
First published on 17th March 2023
Abstract
Described is the development of a unique electrocatalytic material prepared by heat-treating a composite of a Prussian blue analogue, cobalt hexacyanocobaltate, and immobilizing it on reduced graphene oxide (rGO). The pyrolysis process, heating at 500 °C, forms a material with catalytic cobalt centers that are active toward the electrochemical oxygen reduction reaction in alkaline media. Physicochemical properties of this material containing cobalt in different oxidation states have been elucidated using transmission electron microscopy, Raman spectroscopy, X-ray photoelectron spectroscopy, and various electrochemical diagnostic techniques. Experimental results have shown that this material displayed comparable electrocatalytic activity (e.g., onset potential) toward the oxygen reduction reaction in alkaline media to that of Vulcan-supported platinum nanoparticles. Formation of undesirable peroxide species, as demonstrated by monitoring ring currents during rotating ring-disk electrode measurements, was somewhat higher at the pyrolyzed cobalt hexacyanocobaltate on rGO than with platinum nanoparticles on a carbon black support; however, even in the worst case, these values were below 15%, which demonstrates a high activity of cyanide-bridged cobalt sites. This thermally prepared electrocatalytic material exhibits high stability and tolerance to alcohols such as methanol.
Introduction
The oxygen reduction reaction (ORR) is a bottlenecking process in the development of various electrochemical devices, especially fuel cells and air batteries. The preparation of non-precious-metal materials to serve as cost-effective catalysts of the ORR was an objective of numerous studies. These materials include conductive-polymer-based complexes (pyrolyzed and non-pyrolyzed), non-pyrolyzed transition metal macrocycles, metal oxide/carbide/nitride, and metal/nitrogen/carbon.1–13Ideally the ORR is a four-electron transfer accompanied by four protons to form water; however, even when using highly efficient catalytic systems such as Vulcan-supported Pt nanoparticles, intermediate species are produced, especially hydrogen peroxide in acidic media or peroxide ions in alkaline media.14,15 Alkaline media have gained attention for the ORR because of a lower overpotential and a less corrosive environment relative to acidic media. They also result in a less pronounced crossover effect in alcohol fuel cells.14,16,17 Moreover, alkaline media are compatible with non-precious-metal candidates for ORR catalyst such as nitrogen-coordinated compounds of metals (e.g. Fe, Co, Ag, Au, Mn), transition metal oxides, and metal-free carbon based systems.18–35
It is commonly accepted that three active-site structures are primarily responsible for the activity of non-precious metal catalysts, namely nitrogen-coordinated transition metal sites, metal-free carbon structures containing CNx, and iron species encapsulated in a carbon structure.36 Among the compounds that have such structures are metal-hexacyanometallates; they are the series of compounds comprised of Prussian blue, PB (iron(II) hexacyanoferrate) and its transition metal analogues, M1(M2CN) where M1 and M2 can be the same or different metals.37–40 A network that is common to most of the PB series is a three-dimensional, face-centered cubic lattice. Here, one metal ion is octahedrally coordinated by the nitrogen of the cyano group, and the second metal ion is octahedrally coordinated by the carbon. The positively charged metals at the cube center are charge-balanced by counter-ions (e.g. potassium ions). The open-framework structure and electrical conductivity of hexacyanometallates contributes to their use as precursors in the design of electrocatalysts that are free of platinum-group metals.41–45
Hexacyanometallates films on electrodes were initially investigated as electron-transfer mediators, but their efficacy as electrocatalysts was enhanced by electrochemical conversion to mixed-valent compounds with centers for both electron and oxygen transfer.46,47 The resulting films have been applied widely, especially for the electrocatalytic oxidation of organic compounds such as insulin.48,49 These analytical applications used a RuCl3, K4Ru(CN)6 mixture for electrode modification, the cost of which was not a factor for electrode areas used in analysis; however, for applications to batteries and fuel cells, a more cost-efficient means of synthesizing these films is desired.
Recently, pyrolysis of compounds in the PB series on conducting supports was tested as a method to produce materials with properties amenable to application as electrochemical catalysts.38–40,44,50 The hypothesis of the present study is that the combination of adsorption of a PB analogue on a high surface area support followed by pyrolysis will yield a material that can be applied as an electrocatalyst of the oxygen reduction reaction. Cognizant of the potential importance of both the selection of the support and the identity of the metal centers, the hypothesis was tested with cobalt hexacyanocobaltate in combination with a reduced graphene oxide (rGO) support. Among important issues is feasibility of the coexistence of both carbon- and nitrogen-coordinated cobalt (rather than iron) active sites at the electrocatalytic interface. Indeed, recent studies with use of N–C core–shell nanocages and Co–N–C metal–organic frameworks showed that cobalt coordinated by nitrogen or carbon resulted in a material rich in exposed catalytic sites with a high surface area and a high electrical conductivity.18,51 A cobalt-substituted PB analogue, cobalt hexacyanoferrate, immobilized on carbon black (Vulcan XC-72) was previously used after heat treatment by Sawai et al. to enhance the ORR.52,53 To our best knowledge, there was no attempt so far to utilize well-defined cyanocobaltate polynuclear network as a source of unique and distinct cobalt centers for ORR. In the present study, reduced graphene oxide (rGO) has been selected as the support because of its potential to improve efficiency of the electron transport as well as the recognized utility as a carrier for immobilization and distribution of catalytic nanocenters.54–56
Experimental
The chemicals were reagents of analytical grade; they were used as received. Potassium sulfate, 2-propanol, cobalt sulfate, potassium hexacyanocobaltate(III), 5 wt% Nafion™ solution, and reduced graphene oxide (rGO) were from Sigma-Aldrich. The reference catalytic material, 20% Pt/Vulcan nanoparticles, was from Premetek, and the gases, nitrogen and oxygen (purity 99.999%), were purchased from Air Products (Poland). All solutions were prepared using triply distilled and deionized water.The rGO modified with Co(II) hexacyanocobaltate (CoHCNCo@rGO) was produced by first mixing 50 mg of rGO with 5 cm3 of 0.01 mol per dm CoSO4 and placing it in an ultrasonic bath for 30 min to get an aqueous dispersion. Next, 5 cm3 of aqueous K3[Co(CN)6] (0.01 mol dm−3) was introduced, and the slurry was stirred continuously for 12 h. The resulting suspension was centrifuged and washed several times with water. Finally, the separated solid was dried at approximately 50 °C to yield CoHCNCo@rGO. Considering the amount of rGO and CoHCNCo utilized during synthesis as well as accounting for the structure of the precipitate, the content of Co in the sample, CoHCNCo@rGO, was estimated as 20% by weight. The powder was placed in a quartz tube and pyrolyzed under an argon atmosphere at 500 °C for 2 h. The choice of 500 °C was based on the current–voltage curves for the reduction of O2 obtained at rotating disk electrode where the CoHCNCo@rGO component was treated at 300 °C, 500 °C and 900 °C (Fig. S1†); the blank was not pyrolyzed. The diagnostic parameters were the current density and the half-wave potential. The data in the Results and discussion section suggested the abbreviation of the product of pyrolysis at 500 °C as NCoC@rGO.
To produce catalytic inks, a measured amount, typically 10 mg, of NCoC@rGO was suspended in a mixture of 1 cm−3 of 2-propanol and 0.015 cm−3 of the 20% by weight Nafion solution, which served as the solvent and binder, respectively. The ensuing ink was mixed in an ultrasonic bath for 30 minutes, after which it was magnetically stirred for 24 h to obtain a homogeneous suspension. Subsequently, a 0.002 cm−3 aliquot was placed on the surface of glassy carbon disk electrode (GC) and was air-dried at room temperature. The typical catalyst load on the surface was 200 μg cm−2. Finally, the resulting films were activated by applying voltammetric potential cycles from 0 to 1 V at 50 mV s−1 in 0.1 mol per dm KOH under nitrogen until stable current responses were obtained (typically about 30 scans).
The electrochemical experiments were carried out with a CH Instruments (Austin, TX, USA) model 760E workstation. Rotating ring-disk electrode (RRDE) experiments were with an assembly (RRDE-3A ver. 2.0) from ALS Co., Ltd, Japan. The disk was glassy carbon (geometric area 0.197 cm−2), and the ring was Pt. In all experiments, a mercury/mercuric oxide electrode (Hg/HgO) with a potential that was 98 mV more negative than the reversible hydrogen electrode (RHE) was used as a reference electrode. Before voltammetric experiments the potential of the reference electrode was calibrated relative to a RHE in the same supporting electrolyte. Herein, all potentials are reported vs. the RHE. A carbon rod was used as counter electrode to avoid any contamination from platinum. The glassy carbon bases were polished with successively finer grade aqueous alumina slurries (grain size, 5–0.05 μm) on a Buehler polishing cloth.
The RRDE was characterized by the collection efficiency (N), which is determined as the ratio of the ring-to-disk currents at various rotation rates.57 Based on five independent measurements with the Fe(CN)63−/Fe(CN)64− couple as the electroactive system and with rotation rates up to 2500 rpm, N was 0.43. During the RRDE experiments in oxygen saturated solutions, the potential of the ring electrode was kept at 1.2 V (vs. RHE). At this potential, peroxide ions that are formed at the disk are oxidized and quantified at the Pt ring. Each linear sweep voltammetry (LSV) curve at the RRDE was recorded at a scan rate of 10 mV s−1.
All current densities presented herein were calculated with respect to the geometric surface area of the working electrode. The electrochemical measurement were performed either after deaeration with nitrogen or in an electrolyte saturated with oxygen using a 30 min sparge. Following this step, nitrogen (or oxygen) flow over the electrolyte was maintained. All experiments were conducted at room temperature (22 ± 1 °C).
Thermogravimetric analyses (TGA) were done on a TA instruments Q50 thermal gravimetric analyzer under an inert atmosphere (high purity nitrogen). The profiles were obtained by depositing on a TGA pan a sample of ca. 5 mg of CoHCNCo powder that was dried at 25 °C; a scans over the range, 25 °C to 900 °C at 5 K min−1 were recorded. The X-ray diffraction experiments used a Bruker D8 discover diffractometer with a Cu anode (1.5406 Å) source, a Goebel focusing mirror, and a LynxEye detector. The basic phase analyses were performed in Match software using the Crystallography Open Database (COD) with calculated diagrams based on single crystal structures corresponding to the following phases: KCo[Co(CN)6]·H2O,58 Co2C and Co2N,59–61 K3[Co(CN)6].62
Samples for TEM measurements were prepared by placing drops of the diluted CoNC@rGO suspension onto the TEM grid, which had a Formvar film (Agar Scientific). The TEM images were taken on a Talos F200X HRTEM microscope equipped with a four-detector, super-EDS systems (FEI). Several locations on each sample were targeted to ensure that the outcomes were representative. The nitrogen adsorption/desorption isotherm was measured with a Micromeritics ASAP2060 instrument. The surface areas of the of NCoC@rGO was determined using the Brunauer–Emmett–Teller (BET) method. Pore sizes and volumes were evaluated using the Barrett–Joyner–Halenda (BJH) method. The average grain sizes of NCoC@rGO were estimated based on an assumed powder density of 2.0 g cm−3. Raman spectra were obtained with a DXR Raman spectrometer (Thermo Scientific) equipped with a 50×/NA 0.75 objective and a laser set at 532 nm. Sample spectra were obtained from various surface locations to evaluate sample uniformity. X-ray photoelectron spectroscopy (XPS) images were collected with a Keratos Axis Supra (Keratos, UK) spectrometer equipped with a nonmonochromatic Al Kα source. The peaks fitting was done with Casa XPS software (version 2.3.18) on a Shirley background using the Gaussian–Lorentzian function.
Results and discussion
Pyrolysis of cobalt hexacyanocobaltate
The primary goal was to test the hypothesis that pyrolysis of the cobalt analogue of PB will yield a material with catalytic properties emulate those of well characterized materials formed by electrochemical conversion of the PB.46 Initially, the thermal conversion of CoHCNCo@rGO was monitored by TGA, and the characteristics of the resulting material were elucidated by spectroscopy. Fig. S2† illustrates the mass vs. temperature profile of the starting material, CoHCNCo@rGO. An initial mass loss occurred below 180 °C. This loss, ∼11%, was attributed to desorption of water. Increasing the temperature to 280 °C resulted in an additional mass decrease of 1 wt%, which was ascribed to the elimination of coordinated water. It is noteworthy that the CoHCNCo core was thermal stabile up to 280 °C. At 300 °C, pyrolysis of the CoHCNCo occurred. The product, which is labelled NCoC@rGO, was thermally stable up to 800 °C. Its mass was 45% of that of the starting material. Unless otherwise stated, an intermediate temperature of 500 °C was used to pyrolyze the CoHCNCo@rGO in subsequent experiments.The general characterization of the structure of the pyrolyzed material was by XRD. The crystallinity of the starting material, CoHCNCo, was compared to that of the NCoC@rGO formed at 500 °C (Fig. 1). The XRD pattern of CoHCNCo (Fig. 1a) gave the following diffraction peaks: 17.37, 24.68, 35.21, 39.54, 43.55, 50.54, 53.94, and 56.95° 2θ. They were attributed to (200), (202), (400), (402), (422), (404), (600), and (602) reflections, respectively. These results validated the presence of CoHCNCo nanostructures (JCPDS no. 77-1161).
 | ||
| Fig. 1 X-ray diffraction patterns for (a) CoHCNCo and (b) NCoC@rGO. | ||
The diffractograms for the pyrolysis product NCoC@rGO (Fig. 1b), were different from those of the precursor. Its XRD pattern has a strong, broad peak at 25.0° 2θ that is from the (111) reflection plane of graphene sheets. In addition, the spectrum had peaks at 37.15, 40.95, and 45.34, and 56.74 2θ; they correspond to the (100), (002), (101), and (102) lattice planes of the Co2N phase (COD 96-152-8415). The small peaks at 36.92, 40.56, 42.68, 44.53, and 58.80° 2θ are related to (101), (020), (111), (210), and (121) planes of the Co2C lattice (JCPDS 65-1457).63 The XRD pattern of NCoC@rGO also gave peaks at 29.85, 30.91 and 43.54 2θ, which correspond to the reported values of K2SO4 (COD 96-210-1320), a salt present in the solution in which CoHCNCo was prepared. They also are observed with non-pyrolyzed samples. Overall, the XRD spectrum of NCoC@rGO confirmed the formation of Co2C and Co2N bonds during pyrolysis. The XRD spectra are consistent with a face-centered cubic structure of CoHCNCo. When the cobalt species were in the form used in the electrocatalysis experiments, NCoC, the peaks were broader than those of CoHCNCo, which suggests a decrease in particle size upon heat treatment. In summary, pyrolysis at 500 °C directly affected the structure and crystallite size of the chemically prepared CoHCNCo.
Given their importance to the active area of the electrode surface in the electrochemical application, the specific surface area and the pore size distribution of NCoC@rGO and of its separate components were investigated using N2 adsorption/desorption isotherms (Fig. S3†). All of the plots had a hysteresis loop, which is consistent with Brauner's type II isotherms. According to the IUPAC classification, such shapes further indicate type-H3, slit-shaped pore geometries.64 Perhaps more important, the BET data show that pyrolyzing CoHCNCo@rGO at 500 °C increases the specific surface area by more than five-fold relative to the starting material. In this regard, the CoHCNCo (dried at temperatures below 50 °C) has a specific surface area of 24.31 ± 0.04 m2 g−1; for rGO alone, it was 34.16 ± 0.1 m2 g−1; and for NCoC@rGO, it was 141.77 ± 0.4 m2 g−1. Pyrolyzing the CoHCNCo and rGO separately resulted in specific active surface area of 112.43 ± 0.06 m2 g−1 and 54.13 ± 0.07 m2 g−1, respectively. Hence, the increased specific surface area of NCoC@rGO relative to the starting materials is predominantly due to the thermal effect on the CoHCNCo component rather than on the rGO. The surface area of CoHCNCo increases more upon pyrolysis when it is on rGO than when it is a single component.
From N2 adsorption/desorption experiments, pore size and pore volumes distributions varied over a wide range for the sample types in Fig. S4.† The pore size ranges for NCoC@rGO were 0.8–0.9 nm and 2–4 nm. For CoHCNCo there were three ranges, namely 0.6–0.9 nm, 2–3 nm, and 20–40 nm. Pore volumes ranges for NCoC@rGO and CoHCNCo were determined likewise. They were 0.295 and 0.097 cm3 g−1 for NCoC@rGO whereas the values were 0.0456 and 0.0073 cm3 g−1, respectively, for CoHCNCo. The pore diameters were 1.7–300 nm for CoHCNCo and <1.7 nm for NCoC@rGO. Because the signal-to-noise ratio decreases as the pore diameter increase, the upper level of 300 nm is an estimate with the instrumentation used. Based on these BET data, the average grain size for NCoC@rGO was 21 nm and that for CoHCNCo 120 nm. Overall, the BET results demonstrated that the pore characteristics of NCoC@rGO relative to the non-pyrolyzed material are more favorable to electrochemical cell performance because the structure provides more pathways for mass transport within the layer on the electrode and greater populations of catalytic centers for a given geometric area of the electrode.
The morphology of NCoC@rGO also was examined also using HR-TEM and STEM (Fig. 2). These images indicated that NCoC@rGO contained sheets of rGO co-existing with NCoC nanostructures (dark spots). Fig. 2a also shows that the NCoC nanostructures are spherical and highly dispersed. The average size of NCoC particles estimated from HR-TEM images was 17–28 nm, which agrees with the BET results.
 | ||
| Fig. 2 Typical HR-TEM images of NCoC@rGO. (a) High magnification, (b) low magnification, (c) HAADF pattern and (d–f) corresponding elemental mappings. | ||
Spectroscopic characterization of the pyrolysis product
The physicochemistry of NCoC and rGO was further studied by Raman spectroscopy (Fig. 3). The spectra of bare rGO (Fig. 3a) depicted two well-defined peaks at 1338 cm−1 and 1574 cm−1, which correspond to its D and G bands.65 They relate to vibrations at the edges of graphene sheets and vibrations inside a graphene layer, respectively. That is, the D band is associated with defects in the graphitic structure, whereas the G band corresponds to orderly arranged graphite skeletons in rGO. The ratio of the D to G band intensities, ID/IG, reflects the degree of surface defects in graphene-based materials. The ID/IG ratios for rGO and NCoC@rGO were 1.32 and 1.19, respectively. The similar of these ratios indicates that the pyrolysis process does not significantly affect the basic structure of rGO, but the pyrolytic formation of NCoC@rGO does cause some interaction between the rGO and the cobalt species. In this regard, the D and G band positions show a red shift in the peak positions for rGO as a result of the pyrolysis. The Raman spectra of NCoC@rGO (Fig. 3c) do not display strong vibrational bands in the range 2050 cm−1 to 2200 cm−1; however, bands in this region are clearly visible in the spectra of CoHCNCo (Fig. 3b). This set of bands is due to CN stretching.66,67 Their presence supports the model of a mixed oxidation state of cobalt in CoHCNCo.68,69 In addition, bands at 456 cm−1 and 489 cm−1 in the spectrum of NCoC@rGO are observed, they probably originate from the CoC (A1g and Eg) and Co–CN (F2g) vibration bands.70 Finally, in the lower energy region, a band is present at 227 cm−1. This signal is likely related to the F2g symmetry mode and C–Co–C deformation. These assignments agree with the XRD analysis. Overall, the Raman data support the hypothesis that pyrolysis of this Prussian blue analogue yields a material structurally related to that prepared by the electrochemical procedure.47,71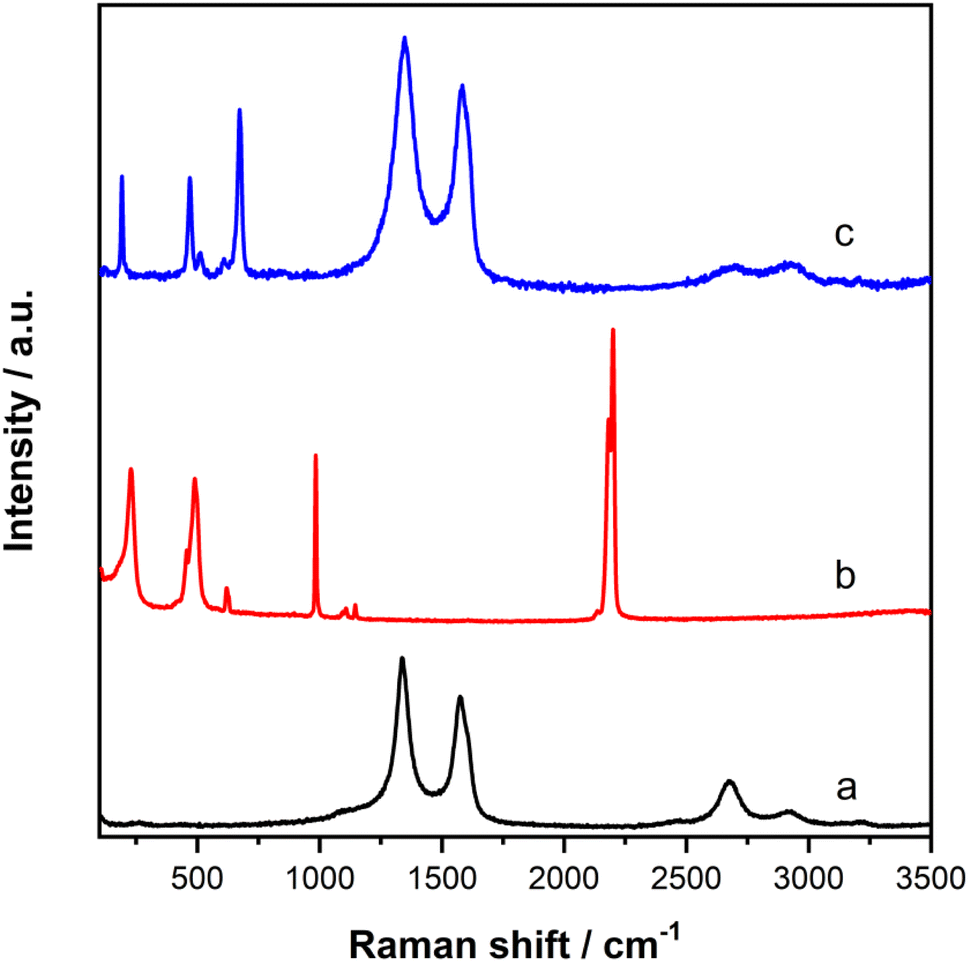 | ||
| Fig. 3 The Raman spectra of (a) rGO, (b) CoHCNCo and (c) NCoC@rGO. | ||
Further investigation of the influence of pyrolysis on CoHCNCo was by ex situ XPS surface analysis. First, spectra of NCoC@rGO and of CoHCNCo in the absence of rGO were obtained (Fig. S5A†). The initial step in the interpretation was to correct for shifts in the position of the binding energy due to sample charging; the shifts were quantified using the 2s potassium signal (NIST X-ray photoelectron spectroscopy database) as the standard. The survey spectrum (Fig. S5A†) of NCoC@rGO and CoHCNCo powders indicated the presence of cobalt (793 eV), nitrogen (N 395 eV), carbon (C 281 eV) and oxygen (O 528 eV). Second, the effect of pyrolysis at 500 °C, was determined. The data were consistence with the evidence of pyrolytic changes of the chemical structure and related bonding of cobalt discussed above. The XPS peaks related to Co 2p are shifted to a lower binding energy by the thermal treatment. Comparing the Co 2p region (Fig. 4a and b) for NCoC@rGO and CoHCNCo shows a distinctive spin–orbital splitting of Co 2p2/3 and Co 2p1/2 bands with the centers at 779 eV and 794 eV, respectively. The XPS spectrum of NCoC@rGO has shake-up satellites peaks at the high binding energy side. These peaks are consistent with the above hypothesis of mixed oxidation states of cobalt that resulted from Raman spectroscopy (Fig. 3c). From Fig. 4, XPS results are consistent with the presence of Co(II), but there also is evidence of Co(III). In this regard, the suggested formation of Co(III) during pyrolysis is supported by the low intensity of satellites peaks for Co 2p3/2 when the final product was the interrogated sample. The mixed oxidation states of Co after the pyrolysis step are analogous to the those of material formed by electrochemical processing of the ruthenium analogue of PB.47 It should be noted that when the materials is used in electrochemical experiments, the oxidation states of cobalt sites are dependent on the applied potentials.
 | ||
| Fig. 4 XPS analysis of the Co 2p region of (a) NCoC@rGO and (b) CoHCNCo. Also shown are (c) N 1s and (d) C 1s spectra for NCoC@rGO catalyst sample. | ||
Whereas the finding of mixed oxidation states of cobalt in NCoC@rGO is the XPS result of most importance for hypothesizing its electrocatalytic properties, two other structural features were identified. The peak at 779.5 eV is related to Co–C bonding and is indicative of formation of Co2C,9,61,72 and the 778.4 eV peak is consistent with the presence of a cobalt nitride component.73–77 The latter, mainly Co–N bonding, has an almost identical atomic structure to metallic cobalt; the difference is a slightly greater distance between the cobalt atoms due to the presence of nitrogen in the center of the unit cell.78–80 The N 1s band (Fig. 4c) was to attributed to the following species: pyridinic N (399.1 eV), Co–N (399.9 eV), graphitic-N (401.4 eV), and oxidized graphitic-N (404.1 eV). The 399.1, 399.9, and 401.4 eV peaks confirm the linking of cobalt with nitrogen, which is important in the ORR process.81 The presence of pyridine-related nitrogen is important because it can interact with Co(II) and Co(III).82,83 The C 1s spectrum of NCoC@rGO (Fig. 4d) is characterized by peaks that are centered at 284.7 (C–C), 286.2 (C–O), 288.8 (O–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), and 283.7 (Co–C) eV.84 Confirmation of the presence of carbide and nitride forms is supported by the results in Fig. 4a and c, namely CoCx at 283.6 eV (ref. 85) CoNy at 399.9 eV.74,75,86,87 These data agree with the XRD results. It is noteworthy that the existence of MNy (where M is Co or Fe) previously has been postulated for the various N-coordinated systems after pyrolysis.88–93
O), and 283.7 (Co–C) eV.84 Confirmation of the presence of carbide and nitride forms is supported by the results in Fig. 4a and c, namely CoCx at 283.6 eV (ref. 85) CoNy at 399.9 eV.74,75,86,87 These data agree with the XRD results. It is noteworthy that the existence of MNy (where M is Co or Fe) previously has been postulated for the various N-coordinated systems after pyrolysis.88–93
Electrocatalysis of the oxygen reduction reaction
Electrochemical studies confirm that pyrolysis is a practical method for preparation of a material, NCoC@rGO, for electrocatalysis of the ORR. Fig. 5 illustrates a comparison of ORR currents obtained at a rotating glassy carbon (GC) disk electrode modified in separate experiments with rGO, CoHCNCo, and NCoC@rGO. The modifications were performed by evaporation of suspensions of these materials in Nafion-containing liquids on the GC surface. The voltammetry was performed in O2-saturated 0.1 mol per dm KOH. The plateau in the current–voltage curve for the ORR at the NCoC@rGO-modified electrode was higher by a factor 10 at 0.4 V relative to that with CoHCNCo@rGO as the electrode modifier. At 0.4 V, modification of the glassy carbon with the suspension of rGO alone did not yield a surface at which oxygen reduction occurred. These data support the proposition that mediation of the ORR by the Co(II)/Co(III) couple of NCoC@rGO occurs.94 | ||
| Fig. 5 The RDE voltammograms of rGO, CoHCNCo, and NCoC@rGO recorded in O2-saturated 0.1 mol per dm KOH solution. Scan rate: 10 mV s−1. Rotation rate: 1600 rpm. | ||
The onset potential, Eonset, is a measure of electrocatalytic efficiency for application to fuel cells and batteries. The Eonset-values of the ORR was at an overpotential lower by about 400 mV with NCoC@rGO than with CoHCNCo@rGO as the modifier (Fig. 5). These values were determined by the method proposed by Di Noto,95 namely the potential where the current density is 5% of that of the limiting current. The capacitance currents are high in both cases because the effective surface areas of porous films on electrodes are much greater than their geometric areas. This limitation of potential-scanning methodology is obviated when potentiostatic methodology is employed because the capacitive current is proportional to scan rate.
A comparison was made of the Eonset with the NCoC@rGO modifier to that when commercially available film of Pt nanoparticle supported on Vulcan, Pt/C, served as the modifier (Fig. 6). At potentials more negative than 0.5 V, the underpotential formation of adsorbed hydrogen species on Pt/C is observed, which is typical for the electrochemistry of Pt in alkaline media.96,97 The similarity of the onset potential with P/C, which can serve as a benchmark, and with NCoC@rGO as electrocatalysts of the ORR is indicative of the utility of the latter. A comparison to literature results shown Table S1† further exemplifies the utility of NCoC@rGO as a catalyst for the electrochemical reduction of oxygen.
 | ||
| Fig. 6 Cyclic voltammetric characterization of Pt/C nanoparticles recorded in O2-saturated (dashed line) and deaerated (solid line) 0.1 mol per dm KOH. Scan rate: 50 mV s−1. | ||
Voltammetry at a RRDE was used to further investigate the efficacy of NCoC@rGO as an electrochemical catalysts for the ORR. In this case, the objective was to determine how closely the number of electrons transferred was to the ideal 4-electron reduction to water. Here, two methods were used. The relative currents at the disk electrodes was compared to that when Pt/C was the catalyst was one method. The second was monitoring the product of the ORR at the disk electrode by oxidation at the ring electrode. Here, the relevant fraction of the solution reaching the ring of the RRDE originates at the disk. These currents were obtained by scanning the potential of the disk electrode while the ring electrode that was held at a static potential.57Fig. 7a exhibits background-corrected voltammograms at the disk electrode; it was rotated at 1600 rpm and scanned at 10 mV s−1. The electrolyte was O2-saturated 0.1 mol per dm KOH. The currents at Pt/C and NCoC@rGO-modified disk electrodes yielded similar values. However, comparing these cathodic currents is not definitive because the active areas of the NCoC@rGO- and the Pt/C-modified disks can differ.
 | ||
| Fig. 7 (a) Cathodic current–potential RRDE curves at the disk (background subtracted) for oxygen reduction at NCoC@rGO (solid line), and Pt/C nanoparticles (dashed line). Solution, O2-saturated 0.1 mol per dm KOH; scan rate 10 mV s−1; rotation rate, 1600 rpm. (b) Anodic currents at the ring are measured at 1.2 V for NCoC@rGO (solid line), and Pt/C nanoparticles (dashed line). | ||
The second of the above methods is based on comparison of the ring and disk currents using eqn (1). During the voltammetry at the disk electrodes (Fig. 7a), the side product of the ORR, namely HO2−, was oxidized at the ring electrode, which was held at 1.2 V (Fig. 7b). Ideally, for the four-electron reduction of O2 to water, this background-corrected current is zero. The result in Fig. 7 show that with the NCoC@rGO-modified disk at 0.6 V, the ring current is 25 μA. The corresponding ring current with the Pt/C catalyst is 5.1 μA. A qualitative comparison of these values shows that whereas the Pt/C electrode more closely approaches the ideal result for the ORR, the NCoC@rGO electrocatalyst for the ORR in the alkaline medium has merit.
A more quantitative comparison of these catalysts was made from the RRDE data shown in Fig. 8. The conditions were those in Fig. 7. The relative concentration of peroxide, X (expressed as % HO2−), formed during the ORR at the NCoC@rGO-modified disk was determined by eqn (1).
 | (1) |
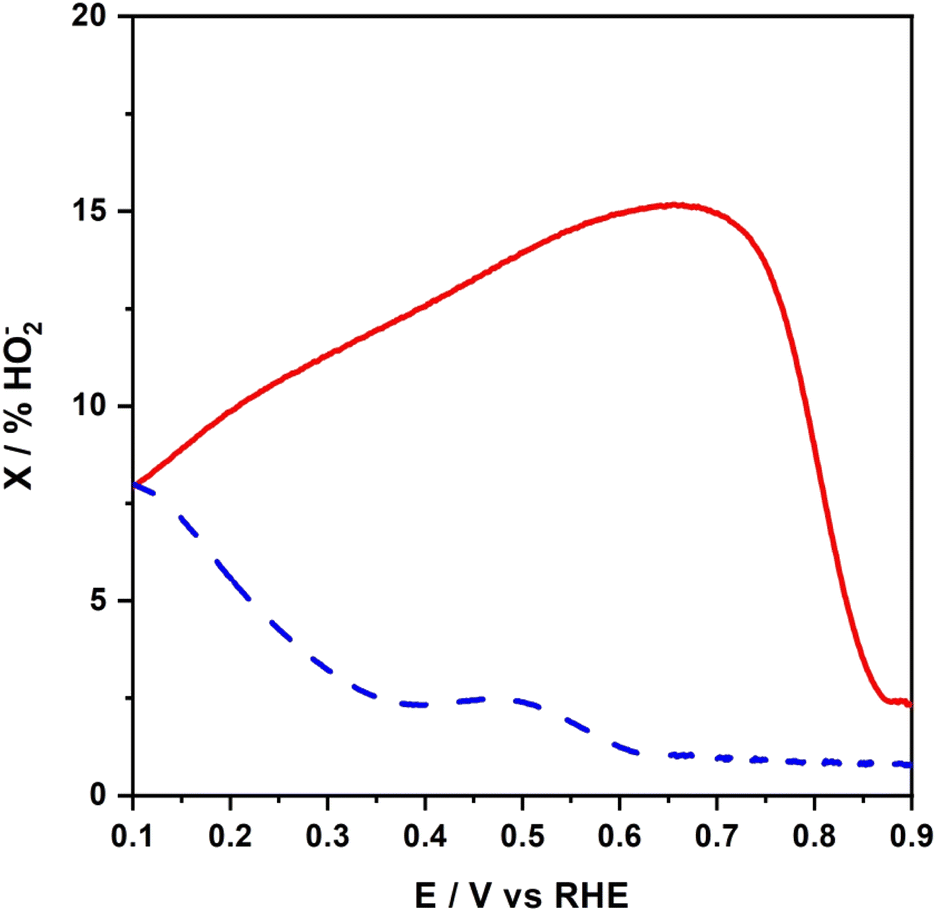 | ||
| Fig. 8 Fraction of peroxide anion (intermediate species) expressed as percent of HO2− formed during the ORR at NCoC@rGO (solid curve), and Pt/C nanoparticles (dashed line). | ||
Here, Ir and Id are the ring and disk currents, respectively, and N is the collection efficiency.98 The current values used in eqn (1) were with the disk electrode at either 0.80 V or 0.65 V and the ring electrode at 1.2 V. When the disk electrode was modified with NCoC@rGO, the residual amount of HO2− represented by the ring electrode current were the following: with the NCoC@rGO-modified disk electrode held at 0.8 V to 0.65 V, the respective X% values were 9% and 15%. With the Pt/C-modified disk, the analogous values were 1% and 1.3%.
The number of electrons transferred, n, were calculated with eqn (2).99
 | (2) |
Over the disk electrode potential range, 0.8 V to 0.1 V (Fig. S6†), the value of n over with the NCoC@rGO catalyst, varied from 3.7 to 3.8, which is about 10% lower than the analogous results with the Pt/C surface, 3.8–3.9. Whereas these data are evidence of a somewhat greater side reaction at NCoC@rGO than at Pt/C, they do not preclude utility of NCoC@rGO as a catalyst of the ORR; however, further study is needed.
The identification of the electrocatalytically active sites produced by the pyrolysis of cobalt hexacyanocobaltate was explored by voltammetry at a rotating disk electrode. The test ample was 2 mmol per dm HO2− in argon-saturated 0.1 mol per dm KOH, and the electrode was a GC disk that was modified with either rGO, CoHCNCo@rGO, or the test catalyst, NCoC@rGO (Fig. S7†). The results were consistent with the working hypothesis that the current yielded at the ring electrode in the above experiments with a RRDE was from the oxidation of HO2− that was produced by incomplete reduction of O2 (i.e., n < 4) at the disk electrode. The NCoC@rGO-modified electrode exhibited electrocatalytic activity toward HO2− oxidation at 0.8 V. This catalytic activity was much higher than that of the CoHCNCo film in terms of onset potential and anodic current. Such behavior may be related to the formation of CoN sites during pyrolysis, which have been shown to electrochemically catalyze H2O2 oxidation.100–102 In this regard, the formation of CoN during pyrolysis was demonstrated by spectroscopic data (Fig. 4).
Voltammetry of an oxygen saturated solution with RDE was performed to elucidate the dynamics of the ORR when the modifier was NCoC@rGO. The polarization curves as a function of rotation rate, ω, from 400–2500 rpm are shown in Fig. S8A.† The onset potential, 0.92 V, of the ORR was independent of ω over the range investigated. The plateau current, j, increased with ω; therefore, mass transport at least in part determines its value. To test whether diffusion is the current-limiting factor, a plot of j vs. ω1/2 was made with the disk electrode at 0.55 V (Fig. S8B†). At 0.55, linear responses with the same slopes were observed with both Pt/C and NCoC@rGO modifiers. With the latter modifier, linear least squares analysis of the plot gave a slope of 0.46 ± 0.02 at an applied potential of 0.55 V, which compares favorably with the theoretical value 0.5 for a diffusion-limited process.
When 0.8 V was applied potential, a departure from linearity was observed at the high end of the rotation rate. That is, at this potential, the current was limited by a mixed mass transport and charge-transfer kinetic process, thereby permitting charge-transfer kinetic information to be elucidated using the Koutecky–Levich eqn (3). The behavior in this mixed diffusion-kinetic region as a function of ω when fitted to eqn (3) is shown in Fig. 9. Here, jlim, is the experimental current density, jL and jkin are the respective diffusion and kinetic current densities; khet, is the heterogeneous rate constant for the catalytic reaction; and CO2 is the bulk reactant concentration (for oxygen in the employed electrolyte, 1.2 × 10−3 mol dm−3).103 The other symbols have their usual meaning.
 | (3) |
 | ||
| Fig. 9 Koutecky–Levich reciprocal plots for the electroreduction of oxygen at NCoC@rGO, and at Pt/C nanoparticles with applied potentials, 0.8 V and 0.55. | ||
The conditions for use of this approach were met because charge propagation within the layer of NCoC@rGO is fast, and the oxygen reactant has facile transport to the active sites, as demonstrated by the development of diffusion-limited currents at less positive potentials. Moreover, we utilized minimal amounts of Nafion for binding the catalytic material to the GC surface to avoid its hindrance of mass transfer.104,105 With these conditions, we assumed that only mass transport of O2 and the kinetics of the electrochemical reaction serve as components of the current-limiting steps. Utilizing reciprocal coordinates (Fig. 9) yielded non-zero intercepts, which allows the determination of the kinetic current density values with eqn (3).
The plots of eqn (3) (Fig. 9) approach linearity. At 0.55 V, the results with NCoC@rGO and Pt/C as modifiers nearly superimpose. These data are consistent with a process that approaches the four-electron transfer mechanism of the ideal ORR. In contrast, at 0.8 V the kinetics of the electrode reaction has a marked influence on the current density; here, the kinetic parameter, khet, for the heterogeneous charge transfer can be determined. The khet value of the NCoC@rGO and the Pt/C modified GC electrodes are both in the range, 1–2·× 10−1 cm s−1. These results are further evidence of that these modifiers are comparable in their promotion of the ORR.
To further study the ORR mechanisms at NCoC@rGO and at Pt/C, the data set used in Fig. 7 was analyzed by Tafel plot (Fig. 10). At potentials where the current densities are low, the Tafel slopes with the NCoC@rGO- and Pt/C-modified films on the working electrodes were −63 mV dec−1 and −59 mV dec−1, respectively. The similarity of these values is consistent with the conclusion that the rate-determining step with these systems are the same. Moreover, the small Tafel slope means that the kinetics of the electrochemical reduction of O2 at the NCoC@rGO film are sufficiently facile to support its practical use as an alternative to Pt/C; in fact, the energy barrier at NCoC@rGO is slightly lower. At high current densities, the Tafel slopes for NCoC@rGO- and Pt/C-modified GC are −130 mV dec−1 and −120 mV dec−1, respectively. These slopes agree closely with the theoretical Tafel value, 120 mV dec−1, which suggests that transfer of the first electron is the rate-determining step of the ORR within the potential range where kinetics primarily limits the current.
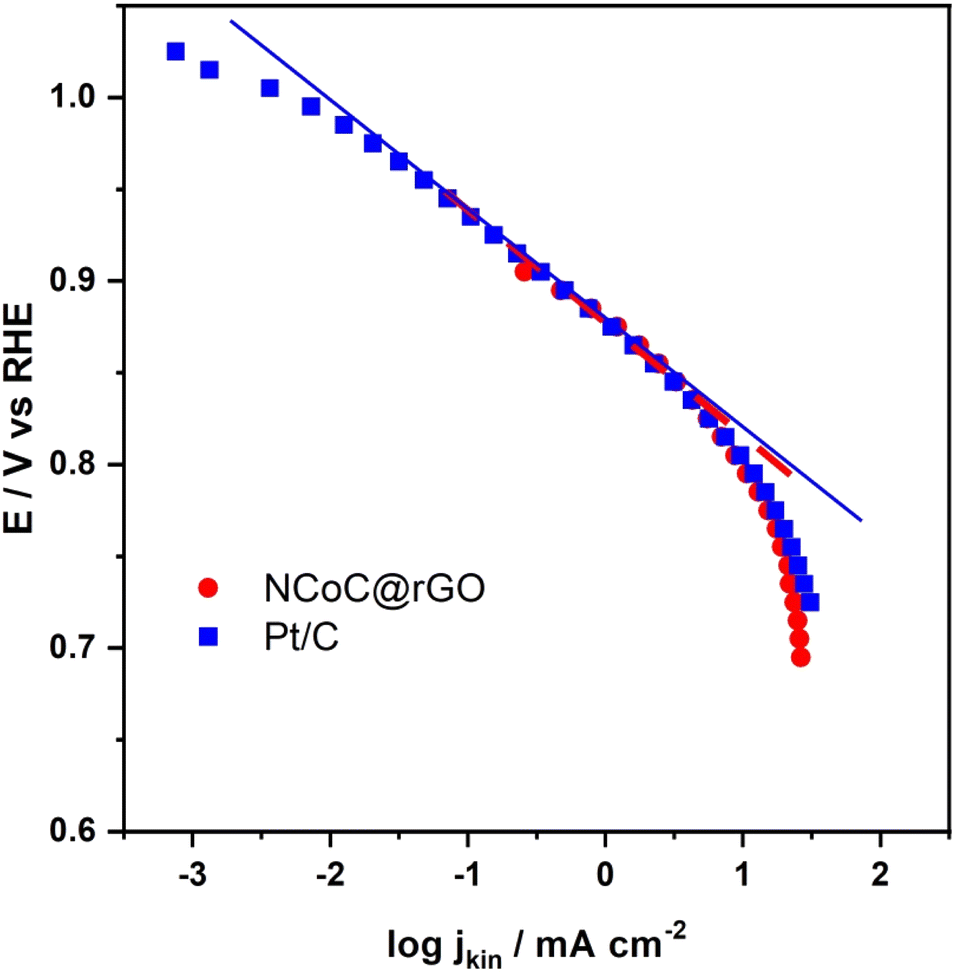 | ||
| Fig. 10 Tafel plots of NCoC@rGO and Pt/C catalysts in O2-saturated, 0.1 mol per dm KOH. | ||
Stability of the cobalt catalyst
The stability of NCoC@rGO is a key issue in its practical applications. It was tested preliminarily by cyclic voltammetry in oxygen-saturated 0.1 mol per dm KOH in the following manner: (1) a voltammetric scan at a RDE at a NCoC@rGO modified surface was recorded, (2) at this electrode in static solution, 3000 CV scans were applied in the potential range, 0.6–1.0 V, at 0.1 V s−1, and (3) the initial RDE measurement was repeated. This sequence was repeated with a Pt/C-modified electrode. The results are shown in Fig. 11A. With the NCoC@rGO modifier, the current at 0.6 V decreased by 3.5%, and Eonset had a shift of −15 mV over this series of cycles. The corresponding Eonset shift was −30 mV at Pt/C. These results further support the case that NCoC@rGO is a promising alternative to commercial Pt/C for applications involving the ORR in alkaline medium. | ||
| Fig. 11 (A) Test of the stability of the NCoC@rGO-modified electrode by linear scan voltammetry (3000 potential cycles at 50 mV s−1) at a RDE in O2-saturated 0.1 mol per dm KOH. (a) Initial scan and (b) final scan. (B) Test of the effect of methanol. Dashed line, 0.1 mol per dm KOH and 0.5 mol per dm methanol; solid line, absence of methanol. Rotation rate, 1600 rpm; scan rate, 10 mV s−1. | ||
An additional stability test was to determine the effect of methanol on the ORR at the NCoC@rGO-modified disk. In Fig. 11B, it is shown that the current densities and potentials for the electrochemical reduction of oxygen are practically unchanged when 0.5 mol per dm methanol is included in the O2-saturated alkaline electrolyte. In contrast, poisoning of Pt/C by methanol is well known.106 This tolerance is a potentially important attribute of the NCoC@rGO catalyst.
Conclusion
A non-precious metal material that was prepared by pyrolyzing cobalt hexacyanocobaltate at 500 °C and immobilizing the product on reduced graphene oxide was demonstrated to electrochemically catalyze the reduction of oxygen in alkaline media. The presence of cobalt in mixed oxidation states as well as C- or N-bridging of the pyrolysis product, NCoC@rGO, are factors that serve to increase electrocatalytic activity of the mediated reduction of O2 by Co(II) over that of the non-pyrolyzed material. In this regard, spectroscopic analysis of the surface state of the NCoC@rGO exhibits the presence of CoCx and CoNy. Nanoparticulate platinum supported on carbon (commercially available as Pt/Vulcan), which is known as a highly efficient electrocatalyst of the ORR, was use as a reference with which to evaluate NCoC@rGO. In general, formation of HO2− during the ORR causes departure from the ideal four-electron process. The formation of HO2− in 0.1 mol per dm KOH at a NCo@rGO-modified electrode was comparable to that at a Pt/C electrode, as determined by voltammetry at a RRDE. Specifically, this side reaction only reduced the n-value from the ideal, a four-electron reduction of O2 to H2O by about 10%. Moreover, the NCoC@rGO had comparable stability to Pt/C in a test involving 3000 potential cycles between 0.6 V and 1.0 V, a range that covers mixed kinetic-diffusion to diffusion-limited control of the ORR. These properties support our current strategy of producing enhanced electrocatalysts for oxygen reduction based on pyrolysis of metal hexacyanometallates composited with rGO. On mechanistic grounds, it is reasonable to expect that, as reported earlier for various pyrolyzed N-coordinated cobalt or iron systems,89,93,107,108 the adsorption of O2 on Co(II) precedes fast transfer of the first electron. Further research to confirm the potential utility of these materials as catalysts in fuel cells will be expanded, especially factors involving long term stability. Applications involving other nonprecious metals centers and direct comparisons to analogous materials formed electrochemically will be explored. Additional studies aimed at testing additives (co-catalysts) capable of further diminishing formation of the hydrogen peroxide intermediates would be helpful as well.Author contributions
B. Z.: investigation, methodology, data curation, visualization; A. J.: investigation, data curation; L. A.: investigation, methodology, formal analysis; B. D.: formal analysis, methodology; A. K.: formal analysis, methodology; M. S.: investigation, formal analysis; I. A. R.: formal analysis, methodology; P. J. K. formal analysis, methodology, writing – review & editing; M. M. formal analysis, methodology writing – review & editing; J. A. C. formal analysis, methodology, writing – review & editing; K. M. conceptualization, methodology, formal analysis, investigation, writing – original draft, supervision, funding acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work has been supported by the National Science Center (Poland) under Opus Project No. 2015/19/B/ST4/03758. Moreover, this work was implemented as a part of Operational Project Knowledge Educational Development 2014-2020 co-financed by European Social Fund, Project No POWR.03.02.00-00-I007/16-00 (POWR 2014-2020). This work was also supported in part by the National Science Center (Poland) under Opus Lap Project 2020/39/I/ST5/03385.References
- R. Bashyam and P. Zelenay, Nature, 2006, 443, 63–66 CrossRef CAS PubMed.
- G. Wu, K. L. More, C. M. Johnston and P. Zelenay, Science, 2011, 332, 443 CrossRef CAS PubMed.
- A. Morozan, S. Campidelli, A. Filoramo, B. Jousselme and S. Palacin, Carbon, 2011, 49, 4839–4847 CrossRef CAS.
- N. Mihara, Y. Yamada, H. Takaya, Y. Kitagawa, S. Aoyama, K. Igawa, K. Tomooka and K. Tanaka, Chem.–Eur. J., 2017, 23, 7508–7514 CrossRef CAS PubMed.
- S. Zoladek, I. A. Rutkowska, M. Blicharska, K. Miecznikowski, W. Ozimek, J. Orlowska, E. Negro, V. D. Noto and P. J. Kulesza, Electrochim. Acta, 2017, 233, 113–122 CrossRef CAS.
- E. Luo, Y. Chu, J. Liu, Z. Shi, S. Zhu, L. Gong, J. Ge, C. H. Choi, C. Liu and W. Xing, Energy Environ. Sci., 2021, 14, 2158–2185 RSC.
- S. Zhao, L. Zhang, B. Johannessen, M. Saunders, C. Liu, S.-Z. Yang and S. P. Jiang, Adv. Mater. Interfaces, 2021, 8, 2001788 CrossRef CAS.
- J. Liu, H.-Y. Yu, T.-H. Zhang, W.-T. Wang, X.-F. Han, Y.-X. Yuan, J.-L. Yao, R. Yang and J.-H. Tian, ACS Appl. Energy Mater., 2021, 4, 2522–2530 CrossRef CAS.
- Y. Li, J. Gao, F. Zhang, Q. Qian, Y. Liu and G. Zhang, J. Mater. Chem. A, 2018, 6, 15523–15529 RSC.
- A. Friedman, M. Mizrahi, N. Levy, N. Zion, M. Zachman and L. Elbaz, ACS Appl. Mater. Interfaces, 2021, 13, 58532–58538 CrossRef CAS PubMed.
- X. Wu, C. Tang, Y. Cheng, X. Min, S. P. Jiang and S. Wang, Chem.–Eur. J., 2020, 26, 3906–3929 CrossRef CAS PubMed.
- A. Sokka, M. Mooste, M. Marandi, M. Käärik, J. Kozlova, A. Kikas, V. Kisand, A. Treshchalov, A. Tamm, J. Leis, S. Holdcroft and K. Tammeveski, ChemElectroChem, 2022, 9, e202200161 CrossRef CAS.
- Y. Kumar, E. Kibena-Põldsepp, J. Kozlova, M. Rähn, A. Treshchalov, A. Kikas, V. Kisand, J. Aruväli, A. Tamm, J. C. Douglin, S. J. Folkman, I. Gelmetti, F. A. Garcés-Pineda, J. R. Galán-Mascarós, D. R. Dekel and K. Tammeveski, ACS Appl. Mater. Interfaces, 2021, 13, 41507–41516 CrossRef CAS PubMed.
- X. Ge, A. Sumboja, D. Wuu, T. An, B. Li, F. W. T. Goh, T. S. A. Hor, Y. Zong and Z. Liu, ACS Catal., 2015, 5, 4643–4667 CrossRef CAS.
- B. Zakrzewska, B. Dembinska, S. Zoladek, I. A. Rutkowska, J. Żak, L. Stobinski, A. Małolepszy, E. Negro, V. Di Noto, P. J. Kulesza and K. Miecznikowski, J. Electroanal. Chem., 2020, 875, 114347 CrossRef CAS.
- J. H. Zagal and M. T. M. Koper, Angew. Chem., Int. Ed., 2016, 55, 14510–14521 CrossRef CAS PubMed.
- A. A. Gewirth, J. A. Varnell and A. M. DiAscro, Chem. Rev., 2018, 118, 2313–2339 CrossRef CAS PubMed.
- G. Li, W. Deng, L. He, J. Wu, J. Liu, T. Wu, Y. Wang and X. Wang, ACS Appl. Mater. Interfaces, 2021, 13, 28324–28333 CrossRef CAS PubMed.
- W. Kiciński, J. P. Sęk, E. Matysiak-Brynda, K. Miecznikowski, M. Donten, B. Budner and A. M. Nowicka, Appl. Catal., B, 2019, 258, 117955 CrossRef.
- M. Wang, W. Yang, X. Li, Y. Xu, L. Zheng, C. Su and B. Liu, ACS Energy Lett., 2021, 6, 379–386 CrossRef CAS.
- H. Zhong, L. A. Estudillo-Wong, Y. Gao, Y. Feng and N. Alonso-Vante, ACS Appl. Mater. Interfaces, 2020, 12, 21605–21615 CrossRef CAS PubMed.
- N. Zion, J. C. Douglin, D. A. Cullen, P. Zelenay, D. R. Dekel and L. Elbaz, Adv. Funct. Mater., 2021, 31, 2100963 CrossRef CAS.
- N. Zion, D. A. Cullen, P. Zelenay and L. Elbaz, Angew. Chem., Int. Ed., 2020, 59, 2483–2489 CrossRef CAS PubMed.
- X. Xie, C. He, B. Li, Y. He, D. A. Cullen, E. C. Wegener, A. J. Kropf, U. Martinez, Y. Cheng, M. H. Engelhard, M. E. Bowden, M. Song, T. Lemmon, X. S. Li, Z. Nie, J. Liu, D. J. Myers, P. Zelenay, G. Wang, G. Wu, V. Ramani and Y. Shao, Nat. Catal., 2020, 3, 1044–1054 CrossRef CAS.
- L. Yang, S. Jiang, Y. Zhao, L. Zhu, S. Chen, X. Wang, Q. Wu, J. Ma, Y. Ma and Z. Hu, Angew. Chem., Int. Ed., 2011, 50, 7132–7135 CrossRef CAS PubMed.
- C. Zhang, L. Hou, C. Cheng, Z. Zhuang, F. Zheng and W. Chen, ChemElectroChem, 2018, 5, 1891–1898 CrossRef CAS.
- Y. Lv, L. Yang and D. Cao, ChemElectroChem, 2019, 6, 741–747 CrossRef CAS.
- B. Dembinska, K. Brzozowska, A. Szwed, K. Miecznikowski, E. Negro, V. Di Noto and P. J. Kulesza, Electrocatalysis, 2019, 10, 112–124 CrossRef CAS.
- X. Zhang, X. Wen, C. Pan, X. Xiang, C. Hao, Q. Meng, Z. Q. Tian, P. K. Shen and S. P. Jiang, Chem. Eng. J., 2022, 431, 133216 CrossRef CAS.
- K. Veske, A. Sarapuu, M. Käärik, A. Kikas, V. Kisand, H.-M. Piirsoo, A. Treshchalov, J. Leis, A. Tamm and K. Tammeveski, Catalysts, 2022, 12, 568 CrossRef CAS.
- J. Lilloja, E. Kibena-Põldsepp, A. Sarapuu, M. Käärik, J. Kozlova, P. Paiste, A. Kikas, A. Treshchalov, J. Leis, A. Tamm, V. Kisand, S. Holdcroft and K. Tammeveski, Appl. Catal., B, 2022, 306, 121113 CrossRef CAS.
- A. Sokka, M. Mooste, M. Käärik, V. Gudkova, J. Kozlova, A. Kikas, V. Kisand, A. Treshchalov, A. Tamm, P. Paiste, J. Aruväli, J. Leis, A. Krumme, S. Holdcroft, S. Cavaliere, F. Jaouen and K. Tammeveski, Int. J. Hydrogen Energy, 2021, 46, 31275–31287 CrossRef CAS.
- R. Z. Snitkoff-Sol, A. Friedman, H. C. Honig, Y. Yurko, A. Kozhushner, M. J. Zachman, P. Zelenay, A. M. Bond and L. Elbaz, Nat. Catal., 2022, 5, 163–170 CrossRef CAS.
- N. Zion, L. Peles-Strahl, A. Friedman, D. A. Cullen and L. Elbaz, ACS Appl. Energy Mater., 2022, 5, 7997–8003 CrossRef CAS.
- S. Zoladek, M. Blicharska-Sobolewska, A. A. Krata, I. A. Rutkowska, A. Wadas, K. Miecznikowski, E. Negro, K. Vezzù, V. Di Noto and P. J. Kulesza, J. Electroanal. Chem., 2020, 875, 114694 CrossRef CAS.
- D. Wang, X. Pan, P. Yang, R. Li, H. Xu, Y. Li, F. Meng, J. Zhang and M. An, ChemSusChem, 2021, 14, 33–55 CrossRef CAS PubMed.
- X. Song, S. Song, D. Wang and H. Zhang, Small Methods, 2021, 5, 2001000 CrossRef CAS PubMed.
- R. Zhou and S. Z. Qiao, Chem. Commun., 2015, 51, 7516–7519 RSC.
- C. Deng, K.-H. Wu, J. Scott, S. Zhu, X. Zheng, R. Amal and D.-W. Wang, ACS Appl. Mater. Interfaces, 2019, 11, 9925–9933 CrossRef CAS PubMed.
- Y.-N. Hou, Z. Zhao, H. Zhang, C. Zhao, X. Liu, Y. Tang, Z. Gao, X. Wang and J. Qiu, Carbon, 2019, 144, 492–499 CrossRef CAS.
- K. Hurlbutt, F. Giustino, M. Pasta and G. Volonakis, Chem. Mater., 2021, 33, 7067–7074 CrossRef CAS.
- Z. Guo, D. K. Panda, K. Maity, D. Lindsey, T. G. Parker, T. E. Albrecht-Schmitt, J. L. Barreda-Esparza, P. Xiong, W. Zhou and S. Saha, J. Mater. Chem. C, 2016, 4, 894–899 RSC.
- H. Yi, R. Qin, S. Ding, Y. Wang, S. Li, Q. Zhao and F. Pan, Adv. Funct. Mater., 2021, 31, 2006970 CrossRef CAS.
- B. Singh and A. Indra, Mater. Today Energy, 2020, 16, 100404 CrossRef.
- D. Zhao, J. Dai, N. Zhou, N. Wang, X. Peng, Y. Qu and L. Li, Carbon, 2019, 142, 196–205 CrossRef CAS.
- J. A. Cox and P. J. Kulesza, Anal. Chem., 1984, 56, 1021–1025 CrossRef CAS.
- P. J. Kulesza, J. Electroanal. Chem. Interfacial Electrochem., 1987, 220, 295–309 CrossRef CAS.
- J. A. Cox and T. J. Gray, Anal. Chem., 1989, 61, 2462–2464 CrossRef CAS PubMed.
- J. A. Cox, R. Jaworski and P. J. Kulesza, Electroanalysis, 1991, 3, 869–877 CrossRef CAS.
- X. Song, S. Song, D. Wang and H. Zhang, Small Methods, 2021, 5, 2001000 CrossRef CAS PubMed.
- J.-P. Xuan, N.-B. Huang, J.-J. Zhang, W.-J. Dong, L. Yang and B. Wang, J. Solid State Chem., 2021, 294, 121788 CrossRef CAS.
- K. Sawai and N. Suzuki, Chem. Lett., 2004, 33, 1540–1541 CrossRef CAS.
- K. Sawai and N. Suzuki, J. Electrochem. Soc., 2004, 151, A2132 CrossRef CAS.
- S. Choi, C. Kim, J. M. Suh and H. W. Jang, Carbon Energy, 2019, 1, 85–108 CrossRef CAS.
- P. J. Kulesza, J. K. Zak, I. A. Rutkowska, B. Dembinska, S. Zoladek, K. Miecznikowski, E. Negro, V. D. Noto and P. Zelenay, Curr. Opin. Electrochem., 2018, 9, 257–264 CrossRef CAS.
- S. Hussain, N. Kongi, A. Treshchalov, T. Kahro, M. Rähn, M. Merisalu, A. Tamm, V. Sammelselg and K. Tammeveski, Nanoscale Adv., 2021, 3, 2261–2268 RSC.
- W. John. A. Hitchman and L. Michael, Ring-disc Electrodes, Clarendon Press, London, 1971 Search PubMed.
- A. Ratuszna and G. Małecki, Mater. Sci. Forum, 2000, 321–324, 947–953 CAS.
- J. Clarke and K. H. Jack, Chem. Ind., 1951, 46, 1004–1005 Search PubMed.
- F. Song, W. Li, J. Yang, G. Han, T. Yan, X. Liu, Y. Rao, P. Liao, Z. Cao and Y. Sun, ACS Energy Lett., 2019, 4, 1594–1601 CrossRef CAS.
- H. Qiao, J. Yu, J. Lu, H. Bai, H. Liu, J. Hu, H. Huang and B. Wen, ACS Sustainable Chem. Eng., 2021, 9, 1373–1382 CrossRef CAS.
- P. Zhou, F. Xue and S. C. F. Au-Yeung, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1998, 54, IUC9800062 CrossRef.
- Q. Lin, B. Liu, F. Jiang, X. Fang, Y. Xu and X. Liu, Catal. Sci. Technol., 2019, 9, 3238–3258 RSC.
- K. S. W. Sing, Pure Appl. Chem., 1982, 54, 2201–2218 CrossRef.
- L. Stobinski, B. Lesiak, A. Malolepszy, M. Mazurkiewicz, B. Mierzwa, J. Zemek, P. Jiricek and I. Bieloshapka, J. Electron Spectrosc. Relat. Phenom., 2014, 195, 145–154 CrossRef CAS.
- L. Xia and R. L. McCreery, J. Electrochem. Soc., 1999, 146, 3696–3701 CrossRef CAS.
- L. Xu, G. Zhang, J. Chen, Y. Zhou, G. Yuan and F. Yang, J. Power Sources, 2013, 240, 101–108 CrossRef CAS.
- S. F. A. Kettle, E. Diana, E. M. C. Marchese, E. Boccaleri and P. L. Stanghellini, J. Raman Spectrosc., 2011, 42, 2006–2014 CrossRef CAS.
- M. L. Ríos, J. Rodríguez-Hernández, L. F. Del Castillo and J. Balmaseda, Crystals, 2017, 7, 16 CrossRef.
- J. Roque, E. Reguera, J. Balmaseda, J. Rodríguez-Hernández, L. Reguera and L. F. del Castillo, Microporous Mesoporous Mater., 2007, 103, 57–71 CrossRef CAS.
- M. Ciabocco, M. Berrettoni, S. Zamponi and J. A. Cox, J. Solid State Electrochem., 2016, 20, 1323–1329 CrossRef CAS.
- Q. Xu, H. Jiang, Y. Li, D. Liang, Y. Hu and C. Li, Appl. Catal., B, 2019, 256, 117893 CrossRef CAS.
- M. C. Biesinger, B. P. Payne, A. P. Grosvenor, L. W. M. Lau, A. R. Gerson and R. St. C. Smart, Appl. Surf. Sci., 2011, 257, 2717–2730 CrossRef CAS.
- X. X. Wang, D. A. Cullen, Y.-T. Pan, S. Hwang, M. Wang, Z. Feng, J. Wang, M. H. Engelhard, H. Zhang, Y. He, Y. Shao, D. Su, K. L. More, J. S. Spendelow and G. Wu, Adv. Mater., 2018, 30, 1706758 CrossRef PubMed.
- S. Chao, Z. Bai, Q. Cui, H. Yan, K. Wang and L. Yang, Carbon, 2015, 82, 77–86 CrossRef CAS.
- X. Li, Q. Jiang, S. Dou, L. Deng, J. Huo and S. Wang, J. Mater. Chem. A, 2016, 4, 15836–15840 RSC.
- S. Xie, S. Huang, W. Wei, X. Yang, Y. Liu, X. Lu and Y. Tong, ChemElectroChem, 2015, 2, 1806–1812 CrossRef CAS.
- X. Zhao, L. Ke, C.-Z. Wang and K.-M. Ho, Phys. Chem. Chem. Phys., 2016, 18, 31680–31690 RSC.
- R. Gupta, N. Pandey, A. Tayal and M. Gupta, AIP Adv., 2015, 5, 097131 CrossRef.
- P. Chen, K. Xu, Y. Tong, X. Li, S. Tao, Z. Fang, W. Chu, X. Wu and C. Wu, Inorg. Chem. Front., 2016, 3, 236–242 RSC.
- Y. Hou, T. Huang, Z. Wen, S. Mao, S. Cui and J. Chen, Adv. Energy Mater., 2014, 4, 1400337 CrossRef.
- X. Wang, J. Zhou, H. Fu, W. Li, X. Fan, G. Xin, J. Zheng and X. Li, J. Mater. Chem. A, 2014, 2, 14064–14070 RSC.
- S. You, X. Gong, W. Wang, D. Qi, X. Wang, X. Chen and N. Ren, Adv. Energy Mater., 2016, 6, 1501497 CrossRef.
- M. D. Meganathan, S. Mao, T. Huang and G. Sun, J. Mater. Chem. A, 2017, 5, 2972–2980 RSC.
- T. Zhang, J. Wu, Y. Xu, X. Wang, J. Ni, Y. Li and J. W. Niemantsverdriet, Catal. Sci. Technol., 2017, 7, 5893–5899 RSC.
- C. Wu, D. Liu, H. Li and J. Li, Small, 2018, 14, 1704227 CrossRef PubMed.
- X. Zhang, Z. Yang, Z. Lu and W. Wang, Carbon, 2018, 130, 112–119 CrossRef CAS.
- J. H. Zagal, S. Griveau, J. F. Silva, T. Nyokong and F. Bedioui, Coord. Chem. Rev., 2010, 254, 2755–2791 CrossRef CAS.
- C. Zúñiga, C. Candia-Onfray, R. Venegas, K. Muñoz, J. Urra, M. Sánchez-Arenillas, J. F. Marco, J. H. Zagal and F. J. Recio, Electrochem. Commun., 2019, 102, 78–82 CrossRef.
- R. Venegas, K. Muñoz-Becerra, C. Candia-Onfray, J. F. Marco, J. H. Zagal and F. J. Recio, Electrochim. Acta, 2020, 332, 135340 CrossRef CAS.
- F. Jaouen, M. Lefèvre, J.-P. Dodelet and M. Cai, J. Phys. Chem. B, 2006, 110, 5553–5558 CrossRef CAS PubMed.
- U. I. Kramm, I. Herrmann-Geppert, J. Behrends, K. Lips, S. Fiechter and P. Bogdanoff, J. Am. Chem. Soc., 2016, 138, 635–640 CrossRef CAS PubMed.
- W. Orellana, C. Z. Loyola, J. F. Marco and F. Tasca, Sci. Rep., 2022, 12, 8072 CrossRef CAS PubMed.
- D. J. Wasylenko, C. Ganesamoorthy, J. Borau-Garcia and C. P. Berlinguette, Chem. Commun., 2011, 47, 4249–4251 RSC.
- V. Di Noto, G. Pagot, E. Negro, K. Vezzù, P. J. Kulesza, I. A. Rutkowska and G. Pace, Curr. Opin. Electrochem., 2022, 31, 100839 CrossRef CAS.
- N. M. Marković, H. A. Gasteiger and P. N. Ross, J. Phys. Chem., 1996, 100, 6715–6721 CrossRef.
- S. Gottesfeld, Fuel Cell Catalysis: A Surface Science Approach, John Wiley & Sons, INC., 2009 Search PubMed.
- W. J. Albery, Ring-disc Electrodes [by] W. J. Albery and M. L. Hitchman, Clarendon Press, Oxford, 1971 Search PubMed.
- R. Zhou, Y. Zheng, M. Jaroniec and S.-Z. Qiao, ACS Catal., 2016, 6, 4720–4728 CrossRef CAS.
- B. Yan, S. Gu and Y. Shen, Analyst, 2021, 146, 2313–2320 RSC.
- Y. Hu, C. Bai, M. Li, M. Hojamberdiev, D. Geng and X. Li, J. Mater. Chem. A, 2022, 10, 3190–3200 RSC.
- Z. Li, R. Liu, C. Tang, Z. Wang, X. Chen, Y. Jiang, C. Wang, Y. Yuan, W. Wang, D. Wang, S. Chen, X. Zhang, Q. Zhang and J. Jiang, Small, 2020, 16, 1902860 CrossRef CAS PubMed.
- B. B. Blizanac, P. N. Ross and N. M. Marković, J. Phys. Chem. B, 2006, 110, 4735–4741 CrossRef CAS PubMed.
- M. Watanabe, H. Igarashi and K. Yosioka, Electrochim. Acta, 1995, 40, 329–334 CrossRef CAS.
- M. Chojak, A. Kolary-Zurowska, R. Wlodarczyk, K. Miecznikowski, K. Karnicka, B. Palys, R. Marassi and P. J. Kulesza, Electrochim. Acta, 2007, 52, 5574–5581 CrossRef CAS.
- C. Domínguez, K. M. Metz, Md. K. Hoque, M. P. Browne, L. Esteban-Tejeda, C. K. Livingston, S. Lian, T. S. Perova and P. E. Colavita, ChemElectroChem, 2018, 5, 62–70 CrossRef.
- N. Ramaswamy, U. Tylus, Q. Jia and S. Mukerjee, J. Am. Chem. Soc., 2013, 135, 15443–15449 CrossRef CAS PubMed.
- S. Fletcher, J. Solid State Electrochem., 2009, 13, 537–549 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ta08682d |
| This journal is © The Royal Society of Chemistry 2023 |