
A band-to-band transition visible-light-responsive anatase titania photocatalyst by N,F-codoping for water splitting and CO2 reduction†
Juhong Lian‡
a,
Kengo Shibata‡b,
Yejun Xiao‡c,
Shiwen Dua,
Toshiya Tanakab,
Yu Qia,
Osamu Ishitani b,
Kazuhiko Maeda*b,
Zhaochi Fenga and
Fuxiang Zhang*a
b,
Kazuhiko Maeda*b,
Zhaochi Fenga and
Fuxiang Zhang*a
aState Key Laboratory of Catalysis, iChEM, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian National Laboratory for Clean Energy, Dalian 116023, Liaoning, China. E-mail: fxzhang@dicp.ac.cn
bDepartment of Chemistry, School of Science, Tokyo Institute of Technology, 2-12-1-NE-2 Ookayama, Meguro-ku, Tokyo 152-8550, Japan
cState Key Laboratory of Molecular Reaction Dynamics and Dynamics Research Center for Energy and Environmental Materials, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian 116023, Liaoning, China
First published on 25th November 2022
Abstract
To efficiently utilize solar energy and catalyze the water splitting reaction under visible-light, a novel strategy of first nitridation and subsequent calcination post-treatment methods is developed to prepare N,F-codoped TiO2 (TNOF) photocatalysts in this work. The prepared sample exhibits obvious band-to-band absorption with an edge up to ca. 580 nm. After calcinating the nitrided sample at low temperature in air, the Ti3+ defects, which could trap photo-generated electrons during photocatalysis, would be significantly reduced. Under visible-light irradiation, the TNOF photocatalyst presents excellent water oxidation performance in AgNO3 aqueous solution or Fe3+ reversible electron acceptor. Based on the obtained TNOF sample, an efficient Z-scheme water overall splitting system is successfully constructed combining with a known Ru/SrTiO3:Rh H2-evolution photocatalyst. Moreover, the TNOF photocatalysts also show good visible-light-driven water reduction and CO2 reduction activities. This work presents a novel strategy to prepare nitrogen-incorporated oxide photocatalysts with low defect concentrations for photocatalytic applications.
Introduction
The utilization of solar energy to produce hydrogen (H2) directly from water splitting and to reduce carbon dioxide (CO2) into valuable chemicals can effectively solve the fossil energy and environmental issues.1,2 The key to photocatalysis is to develop efficient photocatalysts.3 Up to now, some progress has been made,4 among which titania (TiO2) has remained one of the top research hot spots since the first discovery of the Fujishima–Honda effect in 1972.5 Meanwhile, anatase titania has been commonly believed to have much better charge separation efficiency with respect to the rutile one.6 However, anatase TiO2 merely responds to the ultraviolet light that accounts for only 4% of the solar spectrum owing to the wide band gap (3.2 eV), severely limiting the utilization of solar energy. Therefore, narrowing the band gap to enhance visible-light absorption capacity is still highly desirable for TiO2-based photocatalysis.Doping of foreign elements, containing metal element doping (Fe, Cu, Rh, etc.),7–9 non-metallic element doping (such as N, F, C, S, I, B, etc.),10–15 metal and non-metallic element codoping (Fe + N, Ta + N, Nb + N, etc.),16–18 or non-metallic element doping (N + F, N + C, N + S, etc.),19–21 has been widely adopted to narrow the band gap of TiO2. For example, since Asahi and coworkers reported nitrogen (N) doped TiO2,10 extensive investigations have been made to broaden the light absorption edge of TiO2. However, most of them can only create weak shoulder absorption at 400–550 nm wavelength rather than strong band-to-band absorption.22,23 Recently, codoping of two different anions has been demonstrated to favor band-to-band visible-light absorption of TiO2. For instance, Domen et al. reported that N,F-codoped anatase TiO2 (TiNxOyFz) has an efficient band-to-band visible-light absorption and shows excellent water oxidation performance compared to single N-doped TiO2.24 Maeda and coworkers synthesized a N,F-codoped rutile TiO2 photocatalyst (R-TiO2:N,F), which is active for photocatalytic water oxidation in a reversible electron acceptor.25,26 Liu et al. prepared an unusual red TiO2 photocatalyst (using B and N as codopants), which can harvest the full visible light spectrum (the absorption edge up to 700 nm).27 Although the codoped TiO2 with band-to-band absorption properties could well drive the photocatalytic or photoelectrochemical water oxidation activities, their photocatalytic water reduction and CO2 reduction under visible-light are extremely difficult. Accordingly, it is still desirable to develop novel strategies to address their structure as well as functionalities.
In this work, a novel strategy containing first nitridation and subsequent calcination post-treatment is reported to fabricate N,F-codoped anatase TiO2 (TNOF) photocatalysts with obvious band-to-band visible-light absorption properties and low defect concentrations, based on which the functionalities of photocatalytic water oxidation, water reduction and CO2 reduction under visible-light illumination can be observed. Moreover, a visible-light-driven Z-scheme overall water splitting system could be successfully constructed using RuO2/TNOF as an O2-evolving photocatalyst, together with employing Ru/SrTiO3:Rh28 and Fe3+/Fe2+ as a H2-evolving photocatalyst and shuttle mediator, respectively.
Experimental section
Synthesis of TNOF photocatalysts
Visible-light-responsive photocatalyst N,F-codoped TiO2 (TNOF) was prepared by first nitridation and subsequent calcination post-treatment. The starting chemical, TiF3 (Aladdin, 98%), was dried at 80 °C overnight and kept in a desiccator prior to use. TiO2 (anatase, Beijing Yinuokai Technology Co., Ltd, 99.9%) and TiF3 were mixed in an agate mortar with a molar ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 using an appropriate amount of methanol. The mixture was then placed on a Ni plate to prevent contamination. Before starting the heat up, air was purged with ammonia gas for 30 min, and then the mixture was heated to 450 °C (ramp rate: 5 °C min−1) and kept at the target temperature for 15 h under an ammonia atmosphere of 150 mL min−1. To reduce the Ti3+ defects originating from the long-term nitridation, the obtained N,F-codoped TiO2 sample was post-treated by calcining it in an air atmosphere at 350, 450, and 500 °C, respectively. The obtained samples are correspondingly named TNOF-n (n represents the temperature of post-calcination). Additionally, the N-doped TiO2 (N–TiO2) and F-doped TiO2 (F–TiO2) samples were also prepared for discussion. For preparation of N–TiO2, some appropriate amount of TiO2 was nitrided at 500 °C (ramp rate: 5 °C min−1) for 15 h under an ammonia flow of 250 mL min−1. As for preparation of the F–TiO2 sample, the mixture of TiO2 and TiF3 was calcined at 450 °C (ramp rate: 5 °C min−1) for 15 h under an argon flow of 150 mL min−1.
:1 using an appropriate amount of methanol. The mixture was then placed on a Ni plate to prevent contamination. Before starting the heat up, air was purged with ammonia gas for 30 min, and then the mixture was heated to 450 °C (ramp rate: 5 °C min−1) and kept at the target temperature for 15 h under an ammonia atmosphere of 150 mL min−1. To reduce the Ti3+ defects originating from the long-term nitridation, the obtained N,F-codoped TiO2 sample was post-treated by calcining it in an air atmosphere at 350, 450, and 500 °C, respectively. The obtained samples are correspondingly named TNOF-n (n represents the temperature of post-calcination). Additionally, the N-doped TiO2 (N–TiO2) and F-doped TiO2 (F–TiO2) samples were also prepared for discussion. For preparation of N–TiO2, some appropriate amount of TiO2 was nitrided at 500 °C (ramp rate: 5 °C min−1) for 15 h under an ammonia flow of 250 mL min−1. As for preparation of the F–TiO2 sample, the mixture of TiO2 and TiF3 was calcined at 450 °C (ramp rate: 5 °C min−1) for 15 h under an argon flow of 150 mL min−1.
Synthesis of a SrTiO3:Rh photocatalyst
SrTiO3:Rh was obtained by the solid state reaction method according to the previous reports.29,30 In a typical synthesis, SrTiO3 (99.9%, Kojundo Chemical Laboratory Co., Ltd), TiO2 (99.9%, Kojundo Chemical Laboratory Co., Ltd) and Rh2O3 (99.9%, Alfa Aesar) were mixed with a small amount of methanol in a ratio of Sr:Ti:Rh = 1.03:1:0.01. After grinding in the agate mortar fully, the mixture was calcined in air at 1150 °C for 10 h.
Modification of cocatalysts on TNOF and SrTiO3:Rh photocatalysts
Loading of a reduction cocatalyst of Rh on TNOF samples was carried out by photodeposition. 0.15 g TNOF photocatalyst and a certain amount of Na3RhCl6 (Rh: 17.1%, Alfa Aesar) aqueous solution containing 0.3 wt% of Rh to TNOF were dispersed in triethanolamine aqueous solution (100 mL, 10 vol%), and then irradiated with a 300 W Xe lamp (full spectrum) for 5 h to obtain Rh-loaded TNOF.Water oxidation cocatalyst CoPi was loaded on the surface of the TNOF-350 sample by the photodeposition method. 0.2 g TNOF-350 powder and a certain amount of Co(NO3)2 (Co: 1 mg mL−1) were dispersed and sonicated for 5 min in phosphate buffer solution (100 mL, 0.1 M, PBS, pH = 6.8). A 300 W Xe lamp (I = 15 A, full spectrum) was employed to deposit cobalt phosphate (CoPi) for 3 h, and the resultant powder was centrifuged and washed with deionized water 3 times.
The RuO2-modified TNOF-350 sample was prepared using the impregnation method. TNOF-350 powder was dispersed with stirring in RuCl3 aqueous solution (Ru: 0.5 mg mL−1), followed by evaporation of the solvent at 80 °C and heating at 350 °C for 1 h in air.
The Ru cocatalyst (0.7 wt%) on the SrTiO3:Rh photocatalyst was deposited by the photodeposition method. SrTiO3:Rh was dispersed in methanol aqueous solution (100 mL, 10 vol%) containing an appropriate amount of RuCl3 and then irradiated with visible light (300 W Xe lamp, λ > 420 nm) for 3 h to obtain the Ru-loaded SrTiO3:Rh sample.
Modification of the TNOF-450 sample with Ag nanoparticles and a binuclear ruthenium(II) complex (the structure shown in Chart 1, ESI,† named RuRu′): According to previous reports, Ag nanoparticles that work as promoters of interfacial electron transfer from the semiconductor to RuRu′ during Z-scheme CO2 reduction were loaded onto the as-prepared TNOF-450 by an impregnation method.31–33 An appropriate amount of TNOF-450 powder was dispersed in AgNO3 (99.8%, Kanto Chemicals Co) aqueous solution containing 1.5 wt% Ag. After stirring for 2 h at room temperature, the solution was distilled under reduced pressure to remove H2O. Finally, the resulting sample was subjected to heating under an H2 stream (20 mL min−1) at 200 °C for 1 h.
RuRu′ (3.0 μmol g−1), synthesized according to the previous report,34 was adsorbed on the as-prepared Ag/TNOF-450 as described in the previous reports.31–33 The Ag/TNOF-450 sample was dispersed in an acetonitrile solution of RuRu′. The suspension was stirred at room temperature in the dark overnight to allow for adsorption/desorption equilibrium. The obtained powder RuRu′/Ag/TNOF-450 was collected by filtration, washed with acetonitrile and used as the photocatalyst for CO2 reduction.
Photocatalytic reaction
Photocatalytic water splitting reactions were carried out in a Pyrex top-irradiation type reaction vessel connected to a closed gas circulation system. Before irradiation, the system was evacuated to remove air completely, and then irradiated from the top side using a 300 W Xe lamp (λ > 420 nm). A flow of cooling water was used to maintain the reaction suspension at 15 °C. The evolved gases were analyzed by gas chromatography (Shimadzu, GC-2014, MS-5A column, TCD, Ar carrier). AgNO3 (10 mM) and triethanolamine aqueous solution (100 mL, 10 vol%) were used as the sacrificial reagents for O2 and H2 evolution reactions, respectively. The overall water splitting reaction was performed using a Z-scheme system, which was composed of a Ru/SrTiO3:Rh H2-evolution photocatalyst, a RuO2/TNOF-350 O2-evolution photocatalyst, and an Fe3+/Fe2+ electron mediator.The apparent quantum efficiency (AQE) for the O2 evolution reaction at 420 nm was calculated using the equation AQE = (AR/I) × 100%, where A represents a coefficient (4 for O2 evolution), R represents the evolution rate of O2 in the initial 1 h irradiation in a proton reduction reaction and I represents the absorption rate of incident photons. The total number of incident photons at a monochromatic wavelength of 420 nm was measured to be 1.50 × 1020 photons per h and the evolution rate of O2 under the 420 nm wavelength was tested to be 4.54 μmol h−1.
As for the photocatalytic CO2 reduction reaction, acetonitrile (MeCN, >99.5%) was dehydrated using a 3 Å molecular sieve (Kanto Chemicals Co.) and then distilled over CaH2 prior to use. Triethanolamine (TEOA, >98%) was distilled under reduced pressure (<1 torr) and then stored under N2 prior to use. The RuRu′/Ag/TNOF-450 photocatalyst powder (4 mg) was suspended in a MeCN/TEOA mixed solution (4:1 v/v, 4 mL) in a test tube (8 mL capacity). Prior to irradiation, the reaction suspension was purged with CO2 gas (>99.9995%, Taiyo Nippon Sanso Co.) for 20 min. A 400 W high pressure Hg lamp (SEN) with an aqueous NaNO2 solution filter was used as the light source for visible light irradiation. After 15 h of reaction, products in liquid and gas phases were quantified by capillary electrophoresis (Otsuka Electronics Co. Agilent 7100) and gas chromatography (GL science, GC323), respectively. The amounts of products were calculated by subtracting blank portions. The turnover number (TON) for HCOOH generation was calculated as:
| TON = [HCOOH]/[RuRu′] |
Isotopic tracer experiment
RuRu′/Ag/TNOF-450 (10 mg) was suspended in MeCN/TEOA mixed solution (4:1 v/v, 2 mL). The suspension was purged with unlabelled CO2 (Taiyo Nippon Sanso Co., >99.995%, saturated) or 13CO2 (Aldrich Co., >99%, 650 torr) and then irradiated at 370–500 nm (λmax = 410 and 460 nm) using a merry-go-round-type apparatus with an LED light (Iris-MG, CELL System Co.). After irradiation for 20 h, the suspension was filtered. The filtrate was added to MeCN-d3 (>99.9%, Kanto Chemical) and analyzed by 1H NMR (JEOL ECA400II).
Results and discussion
Characterization studies of photocatalysts
Fig. 1a shows X-ray diffraction (XRD) patterns of all the TNOF-n samples with post-calcination treatment, which exhibit a similar crystal structure to the precursor of anatase TiO2, matching well with its standard card (JCPDS No. 21-1272). The impurity phase of Ti3O5 (Ti3+ defects, JCPDS No. 09-0309) resulting from the time-consuming nitridation under the ammonia flow,35 was detected for TNOF-0 (without calcination post-treatment), which could be reduced once the TNOF-0 sample was calcined under air conditions, suggesting that calcination post-treatment can lower the Ti3+ defect concentration. It is worth noting that the main diffraction peak of TiO2 located at 25.4° is slightly shifted towards the lower angle side after the N,F-codoping (Fig. 1a), which can be reasonably ascribed to the incorporation of N atoms with a larger ionic radius (1.32 Å) into the crystal lattice of TiO2 to substitute the O atoms with a smaller ionic radius (1.24 Å).36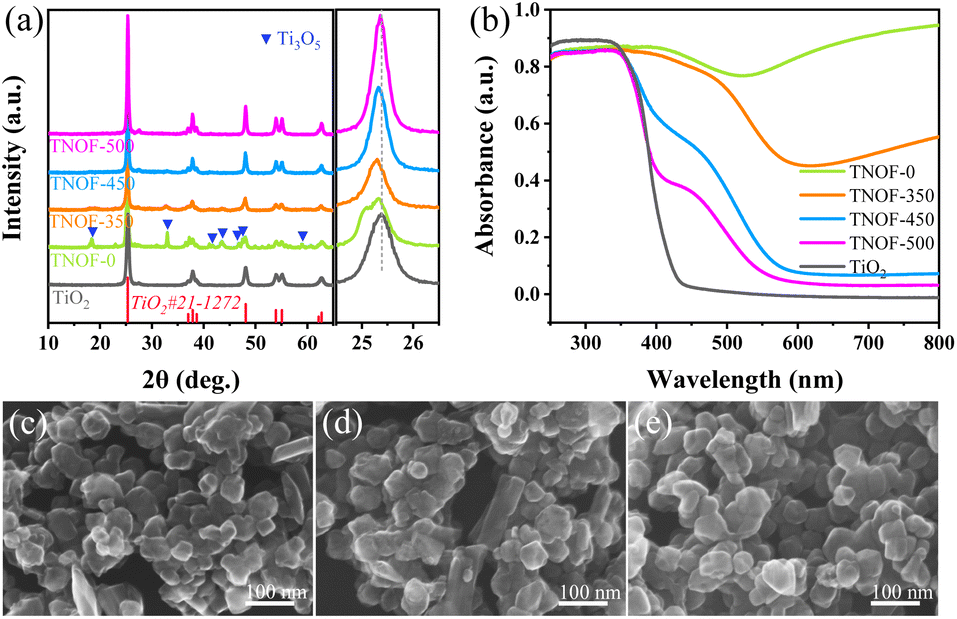 | ||
| Fig. 1 XRD patterns (a) and UV-vis absorption spectra (b) of the obtained TNOF-n samples; and their typical HRSEM images: (c) TNOF-0, (d) TNOF-350, and (e) TNOF-500. | ||
As seen in Fig. 1c–e, similarly aggregated morphology with particle sizes of 50–100 nm can be observed in the typical HRSEM images of TNOF-0, TNOF-350 and TNOF-500 samples, demonstrating that the post-treatment has a minor or no obvious effect on their morphology. Furthermore, elemental analysis of different selected regions in TNOF-0 was performed to validate the distribution form of doped N and F elements in the synthesized sample. As shown in Fig. S1,† all elements of Ti, N, O, and F in TNOF-0 can be detected, and the doping amounts of N and F elements in the selected regions are almost identical (Table S1†), revealing the similar distribution of N and F elements in the TNOF-0 sample.
The TNOF-n samples display remarkable band-to-band transition visible-light absorption (Fig. 1b). In contrast, single N-doping or F-doping does not change the intrinsic band-edge of TiO2, except that only shoulder absorption within the visible-light region can be observed (Fig. S2†), similar to previous reports.35,37 The red shift of the absorption edge should be primarily ascribed to the enhanced content of the N dopant due to the F codoping to compensate for the charge imbalance between N3− and O2− ions. The proposal can be verified by the N element analysis (Table 1), where the N,F-codoped TNOF-0 shows five times the N element amount with respect to the single N-doped TiO2 (N–TiO2). Moreover, the amount of the N dopant decreased by 12.7% for the TNOF-350 sample under air conditions, while a 94.9% amount loss was observed for N–TiO2 with the same post-treatment. This phenomenon demonstrates that the N, F co-doping not only increases the amount of the N dopant, but also stabilizes the doped N atoms, consistent with previous theoretical calculations.38 However, the N,F-codoped sample oppositely increases the mass amount of Ti3+ defects according to the increased absorption at 600–800 nm on the TNOF-0 sample. Noteworthily, the presence of these defects is normally detrimental to the photocatalytic activity. Fortunately, when the TNOF-0 sample is calcined in air, the characteristic absorption of Ti3+ will be continuously decreased by an increase in temperature, indicating the positive effects of the post-treatment. It's worth noting that the absorption edge of TiO2 would reappear when the calcination temperature was higher than 450 °C, which may be derived from the partial decomposition of the TNOF sample.
| Unexecuted | Executed | Δmol% | |
|---|---|---|---|
| TNOF (N% m m−1) | 5.98% | 5.22% | 12.7% |
| N–TiO2 (N% m m−1) | 1.17% | 0.06% | 94.9% |
The chemical forms of doped N and F elements in the TNOF-0, N–TiO2 and F–TiO2 samples were investigated by X-ray photoelectron spectroscopy (XPS). The binding energies of F 1s centered at 684.4 eV and 686.9 eV for the F–TiO2 sample (Fig. 2a) can be attributed to surface substitutional (or isolated surface terminal) Ti–F species and interstitial F insertion, respectively.37,39,40 In this work, only the former Ti–F species is present in the TNOF-0 sample. Fig. 2b presents two peaks located at 399.7 eV and 395.8 eV for N 1s in the N–TiO2 sample. The former can be assigned to surface-doped or interstitial N species,41 which contributes less to visible-light absorption. The latter is generally considered as the evidence for the presence of Ti–N bonds (bulk doping) formed when N atoms replace the O atoms in the TiO2 crystal lattice.42 The N 1s spectrum of TNOF-0 only exhibits one bulk-doped N peak, indicating that the F incorporation can assist N atoms in replacing the O atoms in the crystal lattice of TiO2 by compensating for the overall charge balance. The Ti3+ defects in TNOF-0 can be observed directly from the XPS analysis of Ti 2p3/2 as shown in Fig. S3a,† which can be deconvoluted into two peaks centered at around 458.7 eV (Ti4+ species) and 457.6 eV (Ti3+ species), respectively. When TNOF-0 is calcined in air, the peak ascribed to Ti3+ species will be gradually decreased, in accordance with the UV-vis absorption analysis mentioned above. However, the calcination post-treatment should be executed moderately, as it can also result in the loss of doped N and F elements and the reappearance of the lost lattice oxygen in TiO2 (Fig. S3b–d†). Additionally, compared with those of TNOF-0, the XPS peaks (e.g., N, O, etc.) of TNOF-n move to lower binding energies slightly, which may be originated from the decreased content of codoped F ions with the highest electronegativity. Shortly, the XPS results well reveal that the F cooping can facilitate the incorporation of N atoms into the lattice of TiO2, leading to improved visible-light absorption.
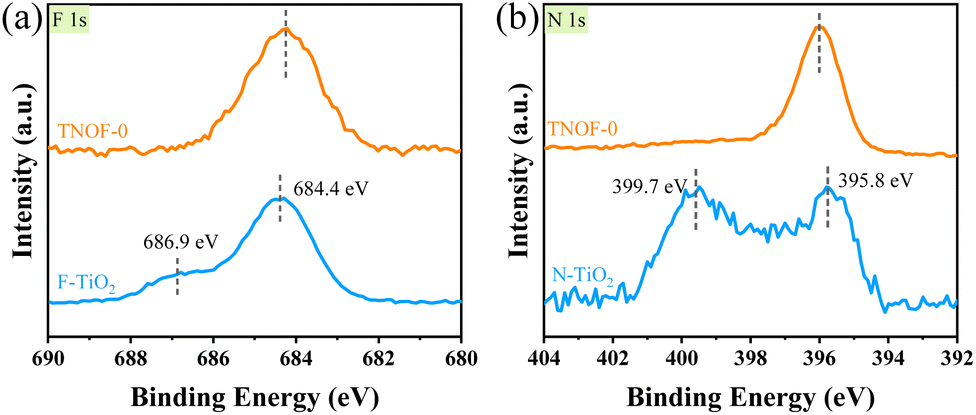 | ||
| Fig. 2 XPS spectra of (a) F 1s and (b) N 1s for TNOF-0, N–TiO2, and F–TiO2 samples. | ||
In order to estimate band potentials of the TNOF photocatalysts, Mott–Schottky plots of TNOF-350 were selectively measured in Na2SO4 aqueous solution. As shown in Fig. 3a, N,F-codoping does not change the intrinsic characterization of the n-type semiconductor of TiO2. The flat band potential of TNOF-350 is deduced to be ca. 0.07 V vs. RHE under the test conditions, based on which the conduction band minimum (CBM) of TNOF-350 is estimated to be −0.13 eV according to the fact that the bottom of the conduction band (CB) of an n-type semiconductor is more negative by ca. 0.2 eV than flat band potential.43 Combining with the band gap value of 2.16 eV in Fig. 3b, the valence band maximum (VBM) of TNOF-350 should be located at 2.03 eV. Therefore, the schematic diagram of the CB and valence band (VB) positions of TNOF can be described as shown in Fig. 3c, where TNOF-350 shows a remarkable upward shift in the VBM compared with pristine TiO2. To clarify the underlying reasons for the upward shift, partial density of states (DOS) calculations were implemented. As shown in Fig. 3d, the CBM of TNOF-350 is constituted by the Ti 3d orbital, while the top of the VB is sufficiently overlapped by N 2p and O 2p orbitals, which should be responsible for the shift of the VBM in TNOF-350 in consideration of the lower electronegativity of the N element with respect to the O element. It should be pointed out that both the CB and VB of TNOF-350 possess sufficient potentials for the reduction of water/carbon dioxide and oxidation, respectively.
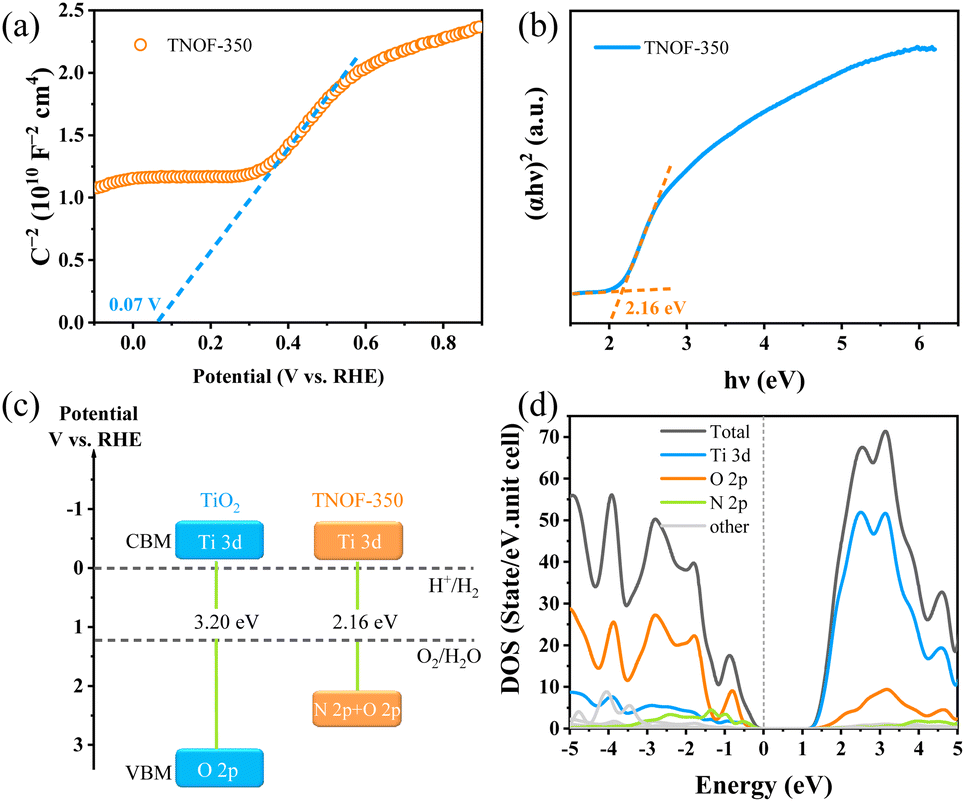 | ||
| Fig. 3 Band characterization studies of TNOF-350: (a) Mott–Schottky plots, electrolyte: 0.5 M Na2SO4 aqueous solution (pH = 8.5, adjusted by NaOH); frequency: 1 kHz; (b) Tauc plots, (c) the estimated band positions of the referenced TiO2 and TNOF-350 samples; (d) partial density of states (PDOS) for TNOF (the Fermi level is set to zero). | ||
Photocatalytic water splitting and CO2 reduction
Encouraged by the appropriate thermodynamic CB and VB positions of TNOF samples, the photocatalytic performances of TNOF for water reduction or oxidation reactions were first examined under visible-light irradiation in the presence of triethanolamine or AgNO3 sacrificial agent. Rh was loaded as the reduction cocatalyst using the photodeposition method and the amount of the Rh cocatalyst was optimized at 0.3 wt% (Fig. S4a†). No obvious H2 can be detected on the TNOF-0 sample without post-calcination, as this should be greatly affected by the existence of massive Ti3+ defects to trap the photo-generated electrons, resulting in charge recombination or few photogenerated electrons could transfer to the surface of TNOF-0 to take part in the water reduction reactions. This deduction can be confirmed by our experimental results which show that the inhibition of Ti3+ defects by post-calcination can obviously promote the H2 evolution rate. Similar phenomena are observed for the O2 evolution on the TNOF-n samples. However, it should be mentioned that the optimized sample is different for the H2 and O2 evolutions, primarily because of the different amounts of Ti3+ defects in different samples. It is worth noting that the O2 evolution rate of TNOF-350 is pretty good (69.9 μmol h−1), which can be promoted to 234.8 μmol h−1 after modifying the CoPi cocatalyst with an apparent quantum efficiency (AQE) of 7.2% at 420 ± 10 nm. Additionally, although many visible-light-responsive TiO2 have been reported for extended visible-light utilization,19,27,44 most of them, especially the N,X-codoped (X = other non-metallic elements) ones, cannot drive the water reduction to produce H2 under visible light irradiation, and their O2 evolution rates are not as good as that of TNOF here (Table S2†), either.To gain insight into the effect of defect levels in TNOF samples, transient absorption spectroscopy was conducted. As displayed in Fig. 4, the TNOF-350 sample shows a typical absorption band from 420 nm to 665 nm while the TNOF-500 sample does not. Generally, the transient absorption spectra in the wavelength region of 3000 nm to 10000 nm are assigned to free electrons or shallowly trapped electrons in the CB, while those in the region of 500–3000 nm are attributed to the deeply trapped electrons in the mid-gap.17,45 Therefore, the strong absorption within 420–665 nm for TNOF-350 can be assigned to the deeply trapped electrons, indicating that few photogenerated electrons could be transferred to the surface of the photocatalysts to take part in the reduction reaction. This can be proved by the moderate H2 evolution activity of the TNOF-350 sample. Meanwhile, TNOF-350 shows superior O2 evolution activity to other samples as shown in Table 2, which should result from the deeply trapped electrons prolonging the life of photo-generated holes.46 By further increasing the postcalcination temperature to 500 °C, although the deeply trapped defects can be eliminated (Fig. 4b), the N dopant will be lost voluminously and lead to the weaker visible-light absorption of TNOF samples, which is not conducive to the efficient utilization of solar energy.
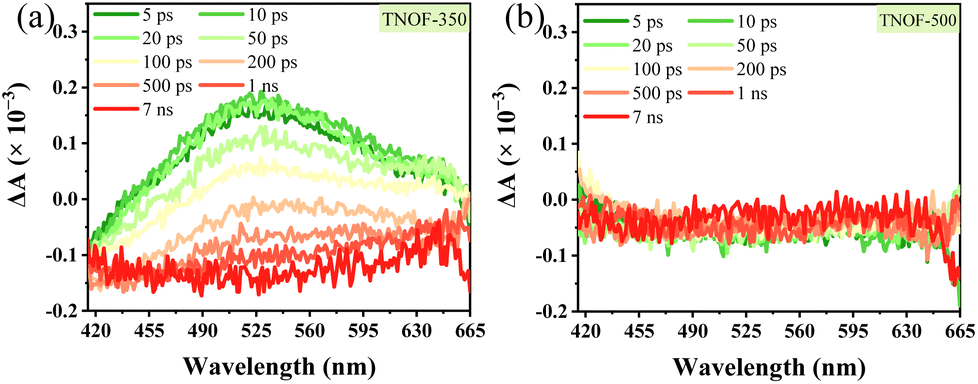 | ||
| Fig. 4 Transient absorption spectra of (a) TNOF-350 and (b) TNOF-500 with a 400 nm laser pulse. | ||
| Reaction | N–TiO2 | TNOF-0 | TNOF-350 | TNOF-450 | TNOF-500 | ||
|---|---|---|---|---|---|---|---|
| H2 evolution reaction conditions: 150 mg 0.3 wt%-Rh/TNOF photocatalysts, 100 mL 10 vol% triethanolamine aqueous solution. O2 evolution reaction conditions: 100 mg TNOF photocatalysts, 150 mg La2O3, 150 mL 10 mM AgNO3 aqueous solution. Pyrex top irradiation type, 300 W xenon lamp (λ > 420 nm).a Photocatalysts: 100 mg sample modified with 1 wt%-CoPi. | |||||||
| H2 evolution (μmol h−1) | 0.8 | 0.0 | 0.6 | 2.9 | 3.7 | ||
| O2 evolution (μmol h−1) | 2.9 | (9.4a) | 5.0 | 69.9 | (234.8a) | 10.0 | 2.3 |
Encouraged by its excellent water oxidation activity, the TNOF-350 sample was employed for assembly of Z-scheme overall water splitting (OWS) under visible-light irradiation without any sacrificial agent combined with Ru/SrTiO3:Rh as the H2-evolving photocatalyst,25,47 and Fe3+/Fe2+ as the redox electron mediator. Fig. S5† shows that the TNOF-350 sample modified with RuO2 can well drive the water oxidation by using Fe3+ as an electron acceptor under visible-light irradiation. Fig. 5 shows the time course of gas evolution from the Z-scheme OWS system under visible-light irradiation, in which the stoichiometric H2 and O2 gases can be detected without noticeable degradation even after 24 h of irradiation. It should be mentioned that no obvious H2 and O2 will be detected if the photocatalytic system is operated without RuO2/TNOF-350, Ru/SrTiO3:Rh, Fe(NO3)3 or light. These results indicate that the OWS reaction occurs via photocatalysis instead of a possible dark reaction.
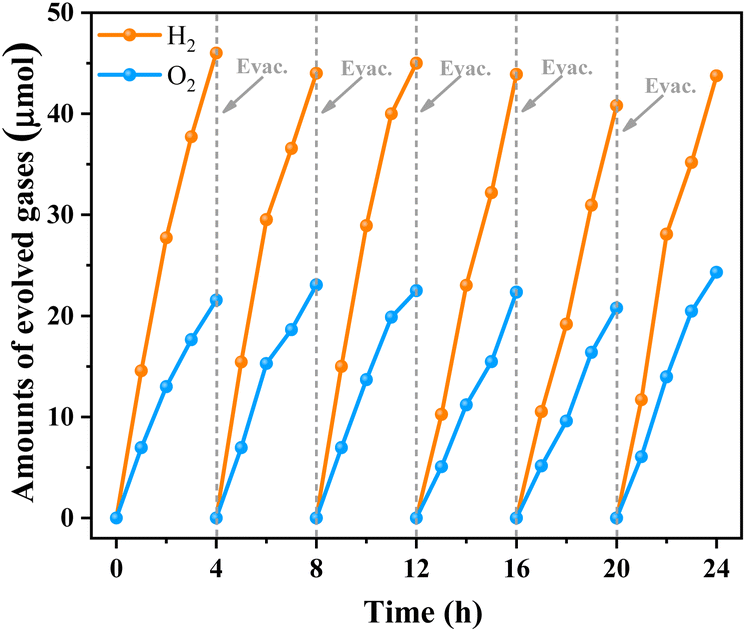 | ||
| Fig. 5 Time courses of gas evolution from the Z-scheme water splitting system using RuO2/TNOF-350 as the O2-evolving photocatalyst. Reaction conditions: 25 mg O2-evolution photocatalyst (RuO2/TNOF-350), 100 mg H2-evolution photocatalyst (Ru/SrTiO3:Rh), 100 mL 0.5 mM Fe(NO3)3 aqueous solution; light source: 300 W Xe lamp (λ > 420 nm). | ||
Apart from photocatalytic water splitting, photocatalytic CO2 reduction into valuable chemicals has also attracted much attention.48 Considering that the TNOF sample has excellent activity for photocatalytic water reduction, it is expected that it can photo-reduce CO2 to more valuable chemicals with high activity and high selectivity. Based on this speculation, a CO2 fixation system was constructed with TNOF-450 and a binuclear Ru(II) complex (named RuRu′, the details are presented in the ESI†), which works via a two-step photoexcitation mechanism49 (Fig. S6†). The reaction was conducted in a mixed acetonitrile (MeCN)/triethanolamine (TEOA) solution (4:1, v/v), in which TEOA worked as a sacrificial electron donor. Table 3 shows that the RuRu′/Ag/TNOF-450 catalyst has excellent CO2 reduction activity towards HCOOH with a turnover number (TONHCOOH) of 220 evaluated based on the amount of RuRu′. Moreover, TNOF showed a high HCOOH selectivity of up to 97.2% with negligible production of other products such as CO. These values for TONHCOOH and selectivityHCOOH are superior (or comparable) to those for most visible-light-responsive photocatalysts, such as foreign atom-doped TiO2 and g-C3N4, etc. (Table S3†). To investigate the origin of the HCOOH produced during the reaction, isotope-labelling experiments with 13CO2 were conducted subsequently. As shown in Fig. S7,† a singlet peak appears at 8.42 ppm in the 1H NMR spectrum in the case when unlabelled CO2 was used as a reactant, while a doublet peak (J13CH = 188 Hz) appears separately at 8.67 and 8.20 ppm in the case when 13CO2 was used. This signal is attributed to protons bound to 13C in formate.50,51 Considering the fact that RuRu′ cannot induce the reduction of CO2 without a photocatalyst,52,53 the above results indicate the visible-light-responsive TNOF-450 can drive the photoreduction of CO2 to produce HCOOH.
| Sample | HCOOH/μmol | CO/μmol | H2/μmol | TONHCOOH |
|---|---|---|---|---|
| a Reaction conditions: catalyst: 4 mg RuRu′ (3 μmol g−1)/Ag (1.5 wt%)/TNOF-450, solvent: MeCN/TEOA (4:1, 4 mL), lamp: 400 W high pressure Hg lamp with a NaNO2 solution filter (λ > 400 nm), 15 h. Ru = [Ru(dmb)2{bpy(CH2PO3H2)2}]2+, Ru′ = [Ru{bpy(CH2PO3H2)2}(CO)2Cl2]. |
||||
| TNOF-450 | 2.6 ± 0.2 | N.d. | 0.15 ± 0.01 | 2.2 × 102 |
Conclusions
In summary, a band-to-band visible-light-responsive N,F-codoped TiO2 (TNOF) photocatalyst was prepared by a novel nitridation combined with a calcination post-treatment method. The resultant TNOF photocatalyst shows an extended visible-light absorption edge of 580 nm, which is ascribed to the doped N content increase caused by F codoping to compensate for the charge imbalance between N3− and O2− anions. Meanwhile, the codoped F element can stabilize the doped N element to prevent its loss during calcination post-treatment in air. Moreover, the Ti3+ defects caused by doping could be decreased significantly after calcination post-treatment at low temperatures. Finally, the photocatalysts demonstrate excellent water splitting and CO2 reduction to produce HCOOH. Application of this novel strategy developed here would be expected to motivate the development of other visible-light-responsive (oxy)nitrides with low defect concentrations for photocatalytic applications.Author contributions
J. L., K. S. and Y. X. contributed equally to this work. J. L. performed the sample preparation and most characterization studies as well as the photocatalytic water splitting reaction with help from Y. Q. K. S. and T. T. performed the photocatalytic CO2 reduction reaction. K. M. supervised the reduction of CO2. Y. X. carried out the characterization of transient absorption spectra and analyzed the result. S. D. performed the partial density of states (DOS) calculations, analyzed the result and revised the manuscript. F. Z. conceived the idea, designed the project and the experiment, and was in charge of planning. The manuscript was drafted by J. L. and revised by F. Z. and K. M., and all co-authors contributed to the analysis and discussion.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The authors are thankful for the financial support from the National Natural Science Foundation of China (No. 21925206 and 22279138), the “Transformational Technologies for Clean Energy and Demonstration”, Strategic Priority Research Program of the Chinese Academy of Sciences, Grant No. XDA 21000000. K. M. acknowledges the support by the Grants-in-Aid for Scientific Research for Transformative Research Areas (A) “Supra-ceramics” (JP22H05148) and a Core-to-Core Program (JPJSCCA20200004) from JSPS. The authors thank Dr Yoshinobu Kamakura for assistance in the 13CO2 isotope experiment.Notes and references
- S. Chen, T. Takata and K. Domen, Nat. Rev. Mater., 2017, 2, 17050 CrossRef CAS.
- X. Jiao, K. Zheng, L. Liang, X. Li, Y. Sun and Y. Xie, Chem. Soc. Rev., 2020, 49, 6592–6604 RSC.
- B. Li, W. Wang, J. Zhao, Z. Wang, B. Su, Y. Hou, Z. Ding, W. Ong and S. Wang, J. Mater. Chem. A, 2021, 9, 10270–10276 RSC.
- Q. Wang and K. Domen, Chem. Rev., 2020, 120, 919–985 CrossRef CAS PubMed.
- A. Fujishima and K. Honda, Nature, 1972, 238, 37–38 CrossRef CAS PubMed.
- J. Schneider, M. Matsuoka, M. Takeuchi, J. Zhang, Y. Horiuchi, M. Anpo and D. W. Bahnemann, Chem. Rev., 2014, 114, 9919–9986 CrossRef CAS PubMed.
- G. Cheng, X. Liu, X. Song, X. Chen, W. Dai, R. Yuan and X. Fu, Appl. Catal., B, 2020, 277, 119196 CrossRef CAS.
- V. Kumaravel, S. Mathew, J. Bartlett and S. C. Pillai, Appl. Catal., B, 2019, 244, 1021–1064 CrossRef CAS.
- S. Ida, K. Sato, T. Nagata, H. Hagiwara, M. Watanabe, N. Kim, Y. Shiota, M. Koinuma, S. Takenaka, T. Sakai, E. Ertekin and T. Ishihara, Angew. Chem., Int. Ed., 2018, 57, 9073–9077 CrossRef CAS PubMed.
- R. Asahi, T. Morikawa, T. Ohwaki, K. Aoki and Y. Taga, Science, 2001, 293, 269 CrossRef CAS PubMed.
- Q. Han, C. Wu, H. Jiao, R. Xu, Y. Wang, J. Xie, Q. Guo and J. Tang, Adv. Mater., 2021, 33, 2008180 CrossRef CAS PubMed.
- T. Li, A. Abdelhaleem, W. Chu, S. Pu, F. Qi and J. Zou, Chem. Eng. J., 2021, 411, 128450 CrossRef CAS.
- J. Wang, D. N. Tafen, J. P. Lewis, Z. Hong, A. Manivannan, M. Zhi, M. Li and N. Wu, J. Am. Chem. Soc., 2009, 131, 12290–12297 CrossRef CAS PubMed.
- E. B. Simsek, Appl. Catal., B, 2017, 200, 309–322 CrossRef.
- W. Yuan, L. Cheng, Y. An, S. Lv, H. Wu, X. Fan, Y. Zhang, X. Guo and J. Tang, Adv. Sci., 2018, 5, 1700870 CrossRef PubMed.
- H. Jiang, J. Liu, M. Li, L. Tian, G. Ding, P. Chen and X. Luo, Chin. J. Catal., 2018, 39, 747–759 CrossRef CAS.
- A. Nakada, S. Nishioka, J. J. M. Vequizo, K. Muraoka, T. Kanazawa, A. Yamakata, S. Nozawa, H. Kumagai, S. Adachi, O. Ishitani and K. Maeda, J. Mater. Chem. A, 2017, 5, 11710–11719 RSC.
- H. Lin and C. Shih, J. Mol. Catal. A: Chem., 2016, 411, 128–137 CrossRef CAS.
- T. Wu, P. Niu, Y. Yang, L. C. Yin, J. Tan, H. Zhu, J. T. S. Irvine, L. Wang, G. Liu and H. M. Cheng, Adv. Funct. Mater., 2019, 29, 1901943 CrossRef.
- Q. Hu, Y. Liu, W. Li, Y. Wang, W. Liao, H. Zou, J. Li and X. Huang, Chem. Eng. J., 2023, 451, 138670 CrossRef CAS.
- Y. Yang, B. Zhu, L. Wang, B. Cheng, L. Zhang and J. Yu, Appl. Catal., B, 2022, 317, 121788 CrossRef CAS.
- G. Liu, X. Wang, L. Wang, Z. Chen, F. Li, G. Q. Lu and H. M. Cheng, J. Colloid Interface Sci., 2009, 334, 171–175 CrossRef CAS PubMed.
- T. Wang, X. Yan, S. Zhao, B. Lin, C. Xue, G. Yang, S. Ding, B. Yang, C. Ma, G. Yang and G. Yang, J. Mater. Chem. A, 2014, 2, 15611–15619 RSC.
- K. Nukumizu, J. Nunoshige, T. Takata, J. N. Kondo, M. Hara, H. Kobayashi and K. Domen, Chem. Lett., 2003, 32, 196–197 CrossRef CAS.
- A. Miyoshi, J. Jhon, M. Vequizo, S. Nishioka, Y. Kato, M. Yamamoto, S. Yamashita, T. Yokoi, A. Iwase, S. Nozawa, A. Yamakata, T. Yoshida, K. Kimoto, A. Kudo and K. Maeda, Sustainable Energy Fuels, 2018, 2, 2025–2035 RSC.
- A. Miyoshi, K. Kato, T. Yokoi, J. J. Wiesfeld, K. Nakajima, A. Yamakara and K. Maeda, J. Mater. Chem. A, 2020, 8, 11996–12002 RSC.
- G. Liu, L. C. Yin, J. Wang, P. Niu, C. Zhen, Y. Xie and H. M. Cheng, Energy Environ. Sci., 2012, 5, 9603–9610 RSC.
- Y. Sasaki, H. Kato and A. Kudo, J. Am. Chem. Soc., 2013, 135, 5441–5449 CrossRef CAS PubMed.
- R. Konta, T. Ishii, H. Kato and A. Kudo, J. Phys. Chem. B, 2004, 108, 8992–8995 CrossRef CAS.
- H. Kato, Y. Sasaki, N. Shirakura and A. Kudo, J. Mater. Chem. A, 2013, 1, 12327–12333 RSC.
- F. Yoshitomi, K. Sekizawa, K. Maeda and O. Ishitani, ACS Appl. Mater. Interfaces, 2015, 7, 13092–13097 CrossRef CAS PubMed.
- K. Muraoka, H. Kumagai, M. Eguchi, O. Ishitani and K. Maeda, Chem. Commun., 2016, 52, 7886–7889 RSC.
- R. Kuriki, H. Matsunaga, T. Nakashima, K. Wada, A. Yamakata, O. Ishitani and K. Maeda, J. Am. Chem. Soc., 2016, 138, 5159–5170 CrossRef CAS PubMed.
- K. Sekizawa, K. Maeda, K. Domen, K. Koike and O. Ishitani, J. Am. Chem. Soc., 2013, 135, 4596–4599 CrossRef CAS PubMed.
- C. Foo, Y. Li, K. Lebedev, T. Chen, S. Day, C. Tang and S. C. E. Tsang, Nat. Commun., 2021, 12, 661 CrossRef CAS PubMed.
- S. Du, H. Zou, Y. Bao, Y. Qi, X. Xin, S. Wang, Z. Feng and F. Zhang, Nano Res., 2022 DOI:10.1007/s12274-022-4529-6.
- Y. Wang, Y. Zhang, X. Zhu, Y. Liu and Z. Wu, Appl. Catal., B, 2022, 316, 121610 CrossRef CAS.
- A. Miyoshi, A. Kuwabara and K. Maeda, Inorg. Chem., 2021, 60, 2381–2389 CrossRef CAS PubMed.
- H. G. Yang, C. H. Sun, S. Z. Qiao, J. Zou, G. Liu, S. C. Smith, H. M. Cheng and G. Q. Lu, Nature, 2008, 453, 638–641 CrossRef CAS PubMed.
- H. Seo, L. R. Baker, A. Hervier, J. Kim, J. L. Whitten and G. A. Somorjai, Nano Lett., 2011, 11, 751–756 CrossRef CAS PubMed.
- N. Kumar, U. Maitra, V. I. Hegde, U. V. Waghmare, A. Sundaresan and C. N. R. Rao, Inorg. Chem., 2013, 52, 10512–10519 CrossRef CAS PubMed.
- X. Zong, Z. Xing, H. Yu, Z. Chen, F. Tang, J. Zou, G. Q. Lu and L. Wang, Chem. Commun., 2011, 47, 11742–11744 RSC.
- Y. Matsumoto, J. Solid State Chem., 1996, 126, 227–234 CrossRef CAS.
- X. Hong, J. Tan, H. Zhu, N. Feng, Y. Yang, J. T. S. Irvine, L. Wang, G. Liu and H. M. Cheng, Chem.–Eur. J., 2019, 25, 1787–1794 CrossRef CAS PubMed.
- A. Yamakata, H. Yeilin, M. Kawaguchi, T. Hisatomi, J. Kubota, Y. Sakata and K. Domen, J. Photochem. Photobiol., A, 2015, 313, 168–175 CrossRef CAS.
- J. Kegel, F. Laffir, I. M. Povey and M. E. Pemble, Phys. Chem. Chem. Phys., 2017, 19, 12255–12268 RSC.
- Q. Jia, A. Iwase and A. Kudo, Chem. Sci., 2014, 5, 1513 RSC.
- R. Wang, P. Yang, S. Wang and X. Wang, J. Catal., 2021, 402, 166–176 CrossRef CAS.
- K. Sekizawa, K. Maeda, K. Domen, K. Koike and O. Ishitani, J. Am. Chem. Soc., 2013, 135, 4596–4599 CrossRef CAS PubMed.
- R. Kuriki, H. Matsunaga, T. Nakashima, K. Wada, A. Yamakata, O. Ishitani and K. Maeda, J. Am. Chem. Soc., 2016, 138, 5159–5170 CrossRef CAS PubMed.
- R. Kuriki, T. Ichibha, K. Hongo, D. Lu, R. Maezono, H. Kageyama, O. Ishitani, K. Oka and K. Maeda, J. Am. Chem. Soc., 2018, 140, 6648–6655 CrossRef CAS PubMed.
- A. Nakada, H. Kumagai, M. Robert, O. Ishitani and K. Maeda, Acc. Mater. Res., 2021, 2, 458–470 CrossRef CAS.
- K. Muraoka, M. Eguchi, O. Ishitani, F. Cheviré and K. Maeda, J. Energy Chem., 2021, 55, 176–182 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ta08076a |
| ‡ The authors equally contributed to this work. |
| This journal is © The Royal Society of Chemistry 2023 |