
In situ surface-trap passivation of CuBi2O4 photocathodes for unbiased solar water splitting†
Yingfei
Hu
 ab,
Jun
Wang
a,
Huiting
Huang
ac,
Jianyong
Feng
*ac,
Wangxi
Liu
a,
Hangmin
Guan
b,
Lingyun
Hao
b,
Zhaosheng
Li
*ac and
Zhigang
Zou
a
ab,
Jun
Wang
a,
Huiting
Huang
ac,
Jianyong
Feng
*ac,
Wangxi
Liu
a,
Hangmin
Guan
b,
Lingyun
Hao
b,
Zhaosheng
Li
*ac and
Zhigang
Zou
a
aCollaborative Innovation Center of Advanced Microstructures, National Laboratory of Solid State Microstructures, School of Physics, Nanjing University, 22 Hankou Road, Nanjing 210093, China. E-mail: fengjianyong@nju.edu.cn; zsli@nju.edu.cn
bSchool of Materials Engineering, Jinling Institute of Technology, 99 Hongjing Avenue, Nanjing 211169, China
cCollege of Engineering and Applied Sciences, Nanjing University, Nanjing 210093, China
First published on 30th November 2022
Abstract
Passivating surface traps plays a crucial role in mitigating the efficiency loss of solar water-splitting electrodes. However, the associated surface-trap passivation approaches require the introduction of an overlayer, complicating the fabrication process and increasing the capital cost of photoelectrodes. Herein, using CuBi2O4 as a prototype, an in situ surface-trap passivation strategy is developed, which yields a beneficial 90 mV anodic shift in hydrogen-evolution onset. Detailed mechanism investigations prove that the intentionally added Mg2+ ions in the precursor gradually segregate as MgO and enrich at the grain boundaries/surface of the CuBi2O4 multicrystalline, porous film during annealing, via which surface traps stemming from dangling bonds are spontaneously passivated; measurements of photovoltage generation characteristics and carrier lifetime validate the favorable roles of the MgO passivator in CuBi2O4 photocathodes. A bias-free water-splitting device is assembled using MgO-passivated CuBi2O4 and Mo-doped BiVO4 as the photocathode and photoanode respectively in a tandem configuration, delivering a solar-to-hydrogen conversion efficiency of approximately 0.41%.
Introduction
Photoelectrochemical (PEC) water splitting, which directly captures and converts renewable solar energy to green hydrogen, is a promising approach to mitigate the increasingly urgent energy crisis and severe environmental pollution stemmed from excessive consumption of fossil fuels.1–7 To enable large-scale application of PEC water splitting, numerous efforts have been devoted to seeking and developing high-efficiency, earth abundant and stable semiconductor electrodes in the past few decades.8–13 Metal oxides have attracted much attention due to their high crustal abundance and stability in ambient environments.14–18 However, most of the metal oxides, such as TiO2 (∼3.0 eV),19,20 WO3 (∼2.6 eV),21,22 BiVO4 (∼2.4 eV),23–28 and Fe2O3 (∼2.1 eV),29–34 show n-type semiconducting characteristics and thereby could serve as water-oxidation photoanodes. Comparatively, metal oxides with p-type conductivity and suitable for construction of water-reduction photocathodes are relatively limited, prohibiting the development of high-performance all oxide-based bias-free solar water splitting systems/devices.Recently, a p-type metal oxide of CuBi2O4 was identified as a promising photocathode candidate, due to its favorable bandgap of about 1.8 eV, sufficiently positive flat band potential at about 1.2 V vs. the reversible hydrogen electrode (RHE) and air processibility.35–37 In principle, CuBi2O4 could produce a water-reduction photocurrent of 19 mA cm−2 under AM 1.5 G 1-sun conditions, yet the reported performances of CuBi2O4 photocathodes are far from the theoretical value.38,39 This phenomenon can be rationalized by the highly mismatched light penetration depths (about 280 nm for the 550 nm photon) and charge carrier diffusion lengths (10–50 nm) of CuBi2O4.40–43 By shortening the migration lengths of minority carriers and increasing the volume ratio of the space-charge region to the electrode bulk, nanostructuring could be an available strategy to address the above issue for CuBi2O4 photocathodes.44 However, unfavorable surface traps/states with energy levels in the band gap may develop for CuBi2O4 photocathodes upon nanostructuring, due to the increased densities of dangling bonds and defects, and contribute to Fermi level pinning thereby enhancing charge carrier recombination. Therefore, the beneficial effects of nanostructuring are generally cancelled out by the detrimental influences of surface traps/states, making nanostructured CuBi2O4 photocathodes less efficient for water reduction.
The above challenge faced by nanostructured CuBi2O4 photocathodes could be circumvented by depositing a thin passivation overlayer, as has been extensively investigated for α-Fe2O3 photoanodes.45,46 Although this surface-trap passivation scheme is effective, it usually involves the use of complicated post-treatment processes such as atomic layer deposition, chemical bath deposition and physical vapor deposition. If the surface traps/states on nanostructured CuBi2O4 photocathodes could be in situ passivated/ameliorated during the electrode fabrication/formation procedure, it would be more intriguing and cost-effective. Herein, by introducing appropriate amounts of Mg2+ ions into the precursor solution, in situ developed MgO segregations during the annealing step locate at the surface/grain boundaries of CuBi2O4 and serve as effective surface-trap passivators. The resulting MgO-passivated CuBi2O4 photocathode shows improved H2 evolution activity at more anodic potentials when compared to the pristine one, and meanwhile allows combination with the Mo-doped BiVO4 photoanode to drive bias-free overall water splitting under light illumination.
Results and discussion
All CuBi2O4 photocathodes were synthesized via a metal–organic decomposition method; varied amounts of Mg(NO3)2·6H2O were added into the precursor solution to form Mg-modified CuBi2O4 photocathodes. The fabrication process of Mg-modified CuBi2O4 photocathodes is schematically illustrated in Fig. 1a. Fig. 1b shows the PEC water reduction performances of CuBi2O4 and Mg-modified CuBi2O4 (with 1%, 3%, 5%, 7%, 10% of Mg) photocathodes in a KBi electrolyte (pH 9.2) under AM 1.5 G simulated sunlight irradiation. As depicted by the J–V characteristics, when the introduced amounts of Mg are less than 3%, the water reduction activities of CuBi2O4 and Mg-modified CuBi2O4 photocathodes are essentially identical. Adding more Mg (5%, 7% and 10%) offers CuBi2O4 photocathodes with largely improved performances; among them, 5% and 7% of Mg-modified CuBi2O4 photocathodes deliver the highest onset potentials at about 1.15 V vs. RHE, and the latter electrode also shows the highest saturation photocurrent density of –0.2 mA cm−2 at 0.7 V vs. RHE. The photocurrent densities (at 0.7 and 1 V vs. RHE) and photocurrent onsets of CuBi2O4 and Mg-modified CuBi2O4 photocathodes are summarized in Fig. 1c and d. As 7% of introduced Mg affords the CuBi2O4 photocathode the highest photocurrent density along with a 90 mV positive shift in onset potential, it is denoted as Mg-CuBi2O4 hereafter and subjected to further characterization studies.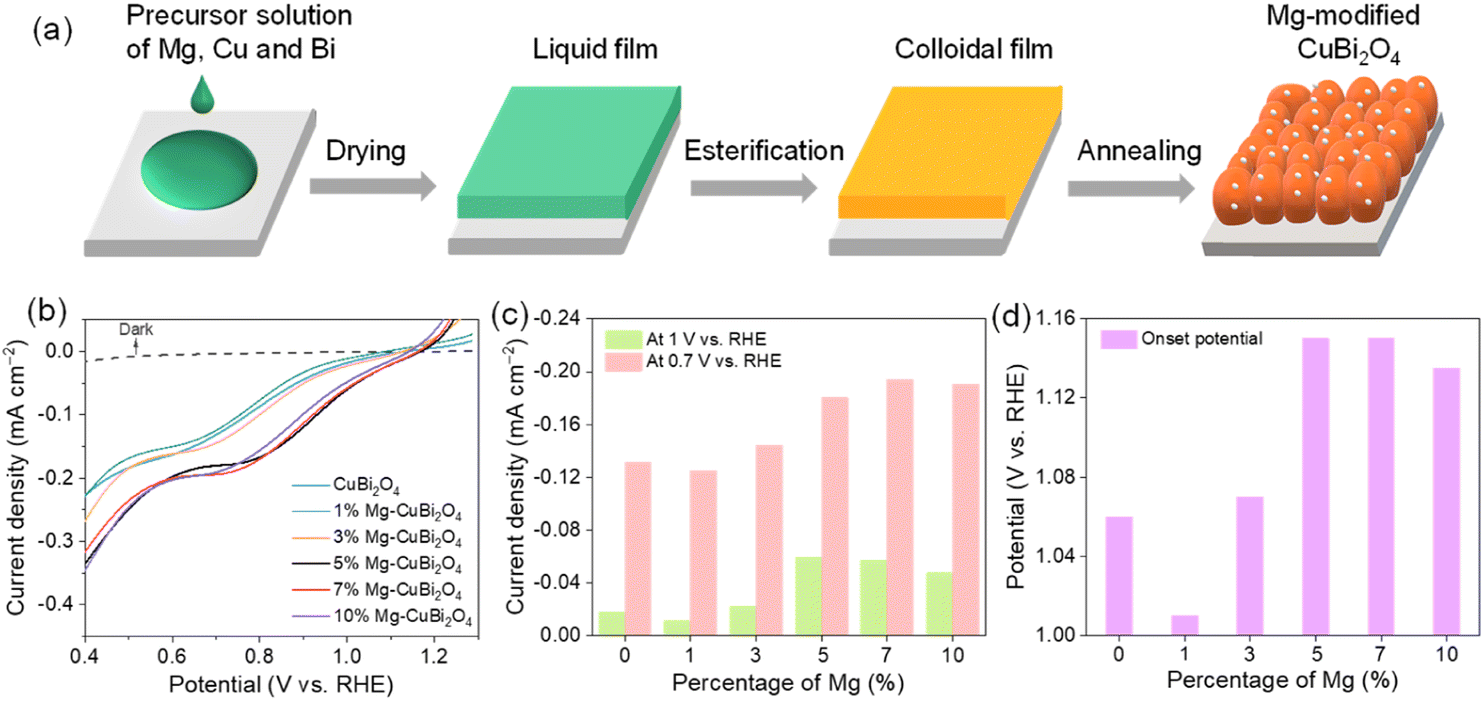 | ||
| Fig. 1 (a) Schematic illustration of the synthetic process of Mg-modified CuBi2O4 photocathodes. (b) J–V curves of CuBi2O4 and Mg-modified CuBi2O4 photocathodes in KBi (pH = 9.2); the corresponding (c) photocurrent densities at 0.7 and 1 V vs. RHE and (d) onset potentials. | ||
XRD patterns collected on CuBi2O4 and Mg-CuBi2O4 electrodes are shown in Fig. 2a and S1.† Besides signals from FTO substrates, all other diffraction peaks are readily assigned to tetragonal CuBi2O4 (JCPDS 42-0334). Interestingly, no observable shift of XRD peaks can be detected in Mg-CuBi2O4 electrodes compared to the pristine CuBi2O4 film, indicating that introduced Mg does not occupy Cu or Bi sites in the lattice of CuBi2O4. On the other hand, no MgO or its related phase/component can be resolved by XRD, possibly due to its low content in the Mg-CuBi2O4 composite electrode (Fig. S2†). Raman spectra of both CuBi2O4 and Mg-CuBi2O4 electrodes show sharp characteristic peaks of CuBi2O4 at 134, 261, 400, and 577 cm−1 (Fig. 2b), confirming the formation of well crystalline CuBi2O4 films; again, no Raman peaks belonging to MgO can be probed in the Mg-CuBi2O4 electrode. Therefore, it is hypothesized that the introduced low-content Mg in the Mg-CuBi2O4 electrode may exist in the form of MgO nanoparticles and distribute uniformly in the matrix of the CuBi2O4 film. Light harvesting efficiency measurements on CuBi2O4 and Mg-CuBi2O4 electrodes reveal their almost identical photon capturing abilities in the tested wavelength range, suggesting a minor contribution of MgO to the light absorption process of CuBi2O4 (Fig. 2c). In addition, both CuBi2O4 and Mg-CuBi2O4 electrodes display an absorption edge of ca. 682 nm, corresponding well to the bandgap of tetragonal CuBi2O4 (1.85 eV, Tauc plot in inset of Fig. 2c).
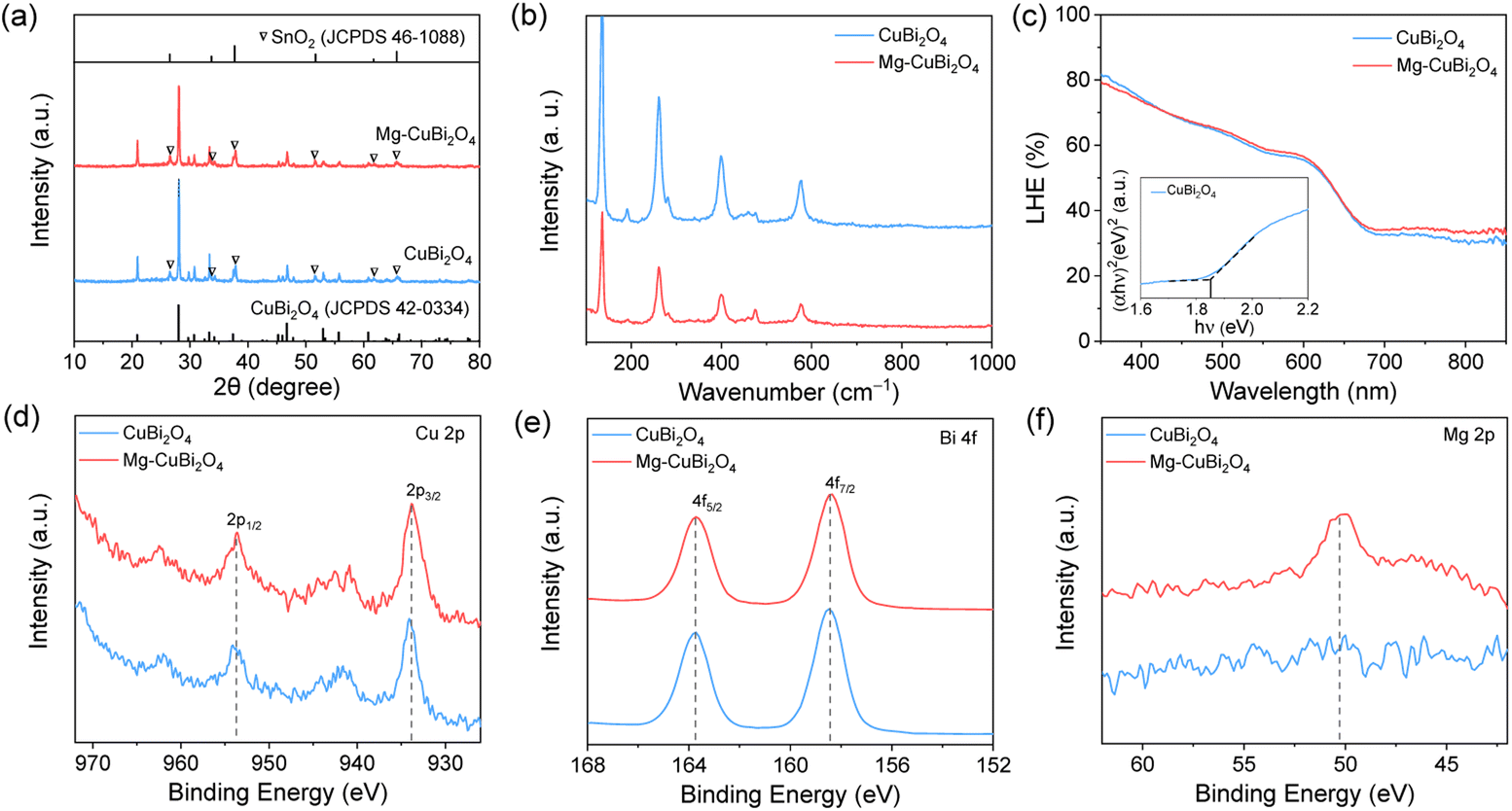 | ||
| Fig. 2 (a) XRD patterns (triangles: peaks of FTO), (b) Raman spectra and (c) UV-vis absorption spectra (inset: Tauc plots, (αhν)2 as a function of hν) of CuBi2O4 and Mg-CuBi2O4 photocathodes. XPS spectra of (d) Cu 2p, (e) Bi 4f and (f) Mg 2p orbits in CuBi2O4 and Mg-CuBi2O4 samples. | ||
XPS characterization studies were carried out to investigate the compositions and chemical bond information of CuBi2O4 and Mg-CuBi2O4 electrodes. As shown in Fig. 2d, both electrodes display spin–orbit doublets at 953.7 and 933.8 eV along with two shake-up satellite peaks in the Cu 2p spectra, suggesting an almost identical environment and oxidation state (+2) of Cu in them. In the Bi 4f region (Fig. 2e), both electrodes exhibit binding energies of 163.7 and 158.4 eV, which can be assigned to 4f5/2 and 4f7/2 of the Bi element with a valence state of +3. The O 1s spectra of the two electrodes can be deconvoluted into two peaks at 531.3 and 529.3 eV (Fig. S3†), which correspond to adsorbed hydroxyl species and lattice oxygen, respectively. As expected, a Mg signal is only detected on the Mg-CuBi2O4 electrode (Fig. 2f), the position of which (at ∼50.6 eV) points to the presence of MgO. The above XPS analyses demonstrate that the chemical states of Cu, Bi and O in CuBi2O4 are not altered upon the introduction of Mg, and these Mg species form MgO instead of dopants in the CuBi2O4 film matrix.
As revealed by SEM observations, both CuBi2O4 and Mg-CuBi2O4 electrodes have porous structures, with particle sizes in the range of 100–300 nm (Fig. 3a and b and S4†). Such a film morphology is favorable for the collection of minority charge carriers. The firmly interconnected film particles along with high crystallinity, on the other hand, facilitate efficient transport of majority carriers in CuBi2O4 and Mg-CuBi2O4 electrodes.47–49 In sharp contrast to the pristine CuBi2O4 electrode which exhibits a relatively smooth particle surface, a large quantity of white spots appear on the Mg-CuBi2O4 electrode. These nanoparticles could be in situ generated MgO segregations, the formation and distribution of which are confirmed by the energy dispersive X-ray spectroscopy (EDS) elemental mapping characterization studies.
 | ||
| Fig. 3 SEM images of (a) CuBi2O4 and (b) Mg-CuBi2O4 films (several MgO particles are marked by red circles). (c) TEM (inset: SAED) and (d) HR-TEM images of Mg-CuBi2O4. (e) EDS elemental mappings of the Mg-CuBi2O4 sample (scale bar: 200 nm). | ||
To further investigate the characteristics of CuBi2O4 and Mg-CuBi2O4 electrodes, and probe the nature and distribution of the segregated nanoparticles in Mg-CuBi2O4, HR-TEM and EDS analyses were performed on CuBi2O4 and Mg-CuBi2O4 particles peeled from the corresponding electrodes. As depicted by Fig. S5†, sphere-like CuBi2O4 particles with a smooth surface are observed, and the measured lattice spacing of 0.190 nm agrees well with the (4 2 0) plane of tetragonal CuBi2O4. Bright diffraction spots are obtained in the selected area electron diffraction pattern, demonstrating the high crystallinity of CuBi2O4 particles, which is also consistent with XRD and Raman characterization studies. In terms of Mg-CuBi2O4, particles with sizes of several to several tens of nm are observed to distribute randomly on the surface of Mg-CuBi2O4 particles (Fig. 3c), and the calculated interplanar spacing of 0.243 nm accords well with the (1 1 1) plane of MgO (Fig. 3d). Therefore, the introduced Mg in the Mg-CuBi2O4 electrode exists in the form of MgO nanoparticles, which distribute uniformly on the Mg-CuBi2O4 particle surface as demonstrated by EDS elemental mapping analyses (Fig. 3e and S6†).
To understand how the presence of segregated MgO nanoparticles affects the water reduction performance of the CuBi2O4 photocathode, charge separation efficiencies (ηsep) of CuBi2O4 and Mg-CuBi2O4 photocathodes were measured. As shown in Fig. 4a, the ηsep values of the Mg-CuBi2O4 photocathode are higher than those of the pristine CuBi2O4 photocathode at all applied potentials. Specially, ηsep of the Mg-CuBi2O4 photocathode increases from 0.5% at 1.1 V vs. RHE to 5.4% at 0.6 V vs. RHE, while those for the CuBi2O4 photocathode are 0.1% at 1.1 V vs. RHE to 3.2% at 0.6 V vs. RHE, respectively. The larger ΔOCP (difference of open-circuit potential in the dark and under light) of the Mg-CuBi2O4 photocathode (0.213 V) than that of the pristine CuBi2O4 photocathode (0.086 V) further confirms the enhanced charge separation and band bending of Mg-CuBi2O4 compared to CuBi2O4 (Fig. 4b). Furthermore, the measurement of ΔOCP also provides a means to extract charge carrier recombination lifetimes and thereby understand the quality of the electrode/electrolyte junction. Under open-circuit conditions, upon switching from the quasi-equilibrium of illumination to the dark equilibrium, the charge carrier recombination is largely dominated by the built-in electric field (band bending) of the space charge layer. The larger the band bending is, the faster the charge carrier recombination occurs at the transient of light-off. The charge carrier recombination lifetime is quantified using the following equation:
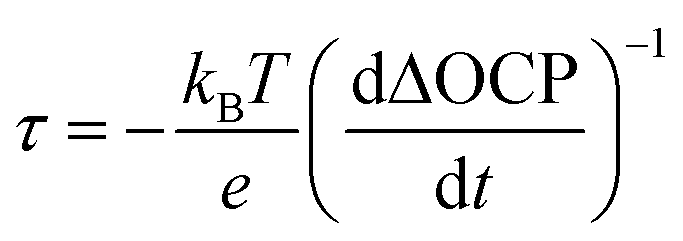
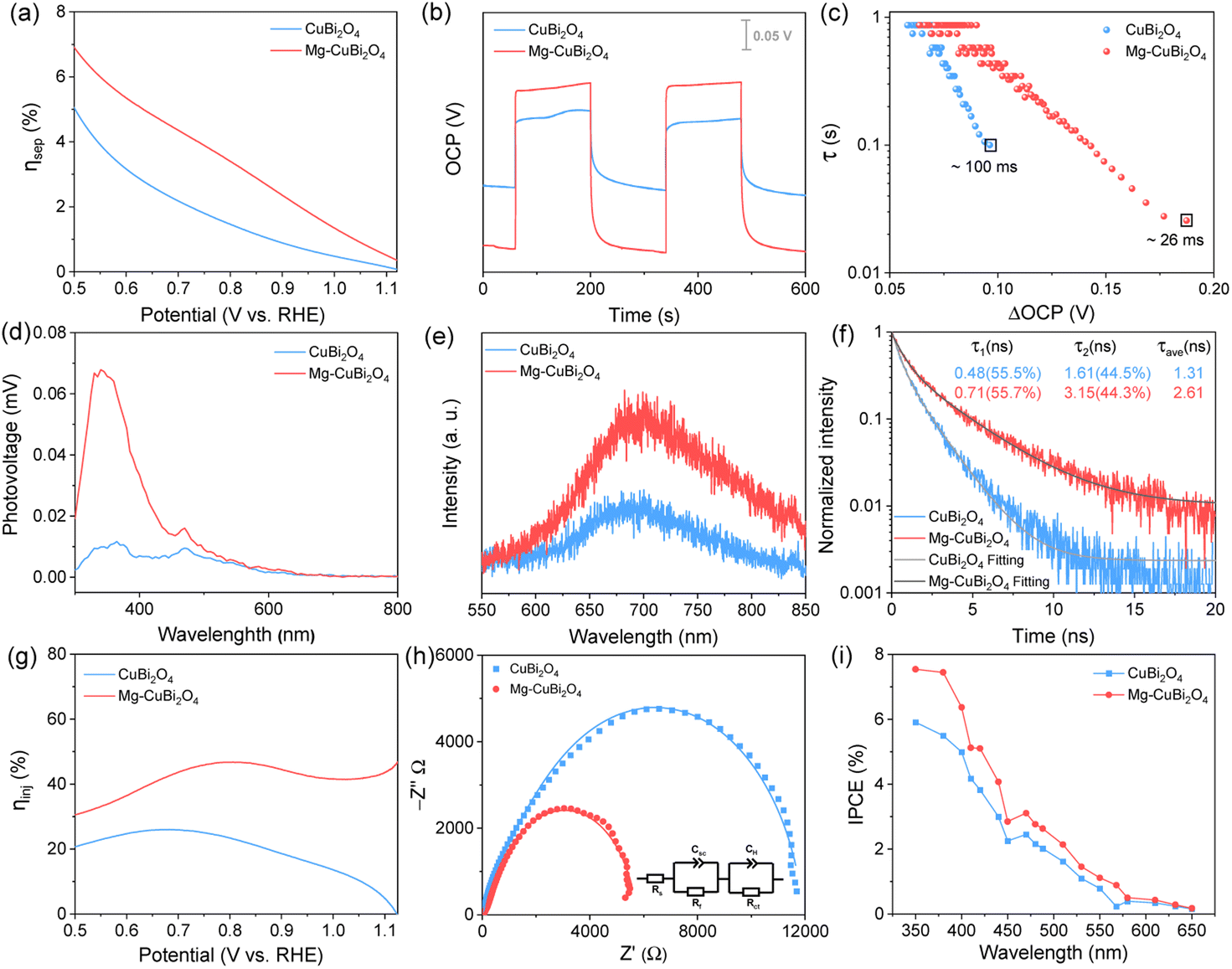 | ||
| Fig. 4 (a) Charge separation efficiency (ηsep), (b) open-circuit potential (OCP) values, (c) carrier lifetime as a function of ΔOCP originated from OCP-decay curves when turning off light, (d) surface photovoltage curves, (e) steady-state PL spectra, (f) TRPL spectra, (g) surface charge injection efficiency (ηinj), (h) Nyquist plots (inset: the equivalent circuit for the samples), and (i) IPCE spectra of the CuBi2O4 and Mg-CuBi2O4 photocathodes. | ||
Using Mott–Schottky analysis, the possible effects of Mg as an acceptor-type dopant have been excluded (Fig. S7†). It is therefore reasonable to propose that these segregated MgO nanoparticles may play a role in surface-trap passivation, via which Fermi level pinning is lessened and band bending (built-in electric field) in the Mg-CuBi2O4 photocathode is enhanced. Photoluminescence (PL) and time-resolved PL (TRPL) measurements were conducted to investigate the defects and trap states in CuBi2O4 and Mg-CuBi2O4 electrodes. Both CuBi2O4 and Mg-CuBi2O4 samples exhibit a PL emission peak at 692 nm, which agrees well with the bandgap of tetragonal CuBi2O4. Compared to pristine CuBi2O4, enhanced PL intensity is obtained on the Mg-CuBi2O4 sample (Fig. 4e). Meanwhile, the carrier lifetime of Mg-CuBi2O4 (2.61 ns) extracted from its exponentially decayed TRPL signal is about twice that of pristine CuBi2O4 (1.31 ns), as shown in Fig. 4f. The greatly enhanced PL intensity and prolonged PL lifetime of Mg-CuBi2O4 over CuBi2O4 suggest that the presence of segregated MgO nanoparticles indeed leads to a reduced density of trap states and a suppressed carrier recombination rate in the Mg-CuBi2O4 photocathode.
The surface water reduction behaviors of CuBi2O4 and Mg-CuBi2O4 photocathodes were evaluated by probing their charge injection efficiency (ηinj) curves (Fig. 4g). Substantially increased ηinj values are observed on the Mg-CuBi2O4 photocathode compared with the pristine CuBi2O4 photocathode, especially at more positive potentials. Specifically, at an applied potential of 0.6 V vs. RHE, the ηinj values for CuBi2O4 and Mg-CuBi2O4 photocathodes are 24.8% and 36.5%, respectively; at 0.8 V vs. RHE, the ηinj value for the Mg-CuBi2O4 photocathode is almost double that of the pristine CuBi2O4 photocathode (46.7% vs. 23.4%). Photo-assisted electrochemical impedance spectroscopy (PEIS) measurements were also performed to analyze the interfacial charge transfer behaviors of CuBi2O4 and Mg-CuBi2O4 photocathodes. The collected Nyquist curves shown in Fig. 4h reveal that the charge transfer resistance (Rct) of the pristine CuBi2O4 photocathode is ca. 2 times as large as that of the Mg-CuBi2O4 photocathode. The above ηsep, ΔOCP, ηinj and PEIS results together suggest that surface trap states stemming from surface dangling bonds of CuBi2O4 not only cause Fermi level pinning thereby suppressing charge carrier separation in the CuBi2O4 film bulk, but also retard water reduction kinetics at the CuBi2O4 surface, as schemed in Fig. S8a†. Using in situ developed MgO segregations, surface traps of the CuBi2O4 electrode are effectively passivated, giving rise to enhanced charge separation in the bulk and accelerated charge transfer kinetics at the surface of the CuBi2O4 electrode simultaneously, as illustrated in Fig. S8b.†
IPCE spectra of CuBi2O4 and Mg-CuBi2O4 electrodes were measured at 0.6 V vs. RHE in KBi solution. As shown in Fig. 4i, both IPCE curves of CuBi2O4 and Mg-CuBi2O4 electrodes exhibit a monotonic decrease trend from 350 to 650 nm, and the onset wavelength of ca. 650 nm matches closely with their light absorption curves. Compared to that on the pristine CuBi2O4 photocathode, the overall profile of the photocurrent action spectrum on the CuBi2O4 photocathode improves obviously, consistent with the J–V measurements in Fig. 1b. The i–t curves displayed in Fig. S9† reveal that the water reduction performance of the Mg-CuBi2O4 photocathode is always higher than that of pristine CuBi2O4 across the entire test, demonstrating the positive roles of segregated MgO nanoparticles in the Mg-CuBi2O4 photocathode; the rapid decrease of photocurrents at the initial stage may result from poor water reduction kinetics at the CuBi2O4 surface. In addition, the results of XRD and SEM demonstrated that the structure and morphology remain stable during the reaction (Fig. S10 and S11†).
Based on the above analyses, the roles played by spontaneously segregated MgO nanoparticles in the Mg-CuBi2O4 photocathode can be understood as follows. In an ideal case, the photogenerated electrons in the conduction band of p-type CuBi2O4 would migrate to the electrode surface to drive the water reduction reaction under the action of the built-in electric field; the photogenerated holes in the valence band would transport to the counter electrode to oxidize water. In practice, to address the issue of mismatched light penetration depths and charge carrier diffusion lengths, nanostructured CuBi2O4 photocathodes are usually employed and excess dangling bonds are then created on the CuBi2O4 photocathode surface and form trap states. Under this condition, the built-in electric field (band bending) is deteriorated and photogenerated electrons are easily captured by surface trap states, which lead to increased carrier recombination, delayed photocurrent onset and suppressed saturation photocurrents on nanostructured CuBi2O4 photocathodes (Fig. 5a). By introducing Mg2+ ions in the precursor solution, MgO nanoparticles segregate and enrich at the grain boundaries/surface of the CuBi2O4 photocathode during the single-pass fabrication procedure. These spontaneously segregated MgO nanoparticles would eliminate (part of) the dangling bonds thereby reducing the trap-state density on the CuBi2O4 photocathode surface. Under this condition, an enhanced built-in electric field (band bending) and accelerated charge transfer kinetics are achieved on the Mg-CuBi2O4 photocathode, leading to increased saturation photocurrents and a positively shifted onset potential (Fig. 5b). When used as the passivator, MgO has the following advantages. First, its large bandgap (about 7.8 eV) ensures that MgO will not interfere with the light absorption of CuBi2O4. Second, MgO exhibits good chemical and electrochemical stability, which would sustain its functions during long-term operation. Third, as an oxyphilic element, Mg would be strongly bonded to oxygen, thus efficiently reducing the density of surface dangling bonds on the CuBi2O4 photocathode.
 | ||
| Fig. 5 Schematic diagram of the role of MgO in the PEC water reduction on CuBi2O4 photocathodes: carrier transport and energy bands of (a) CuBi2O4 and (b) Mg-CuBi2O4 photocathodes. | ||
Due to its favorable hydrogen-evolution onset potential, the Mg-CuBi2O4 photocathode was series-connected with a classical Mo-doped BiVO4 photoanode to assemble a bias-free tandem water-splitting device (Fig. 6a). Overlap between the J–V characteristics of Mg-CuBi2O4 and Mo:BiVO4 electrodes yields a maximum reachable photocurrent of 0.33 mA cm−2 (Fig. 6b), which is increased by 37.5% compared to that of the CuBi2O4‖Mo:BiVO4 device (Fig. S12†). Under AM 1.5 G simulated sunlight and bias-free conditions, a photocurrent of ca. 0.28 mA cm−2 is generated on the Mg-CuBi2O4‖Mo:BiVO4 device, demonstrating its ability to drive unbiased overall water splitting (Fig. 6c).
 | ||
| Fig. 6 Performances of the p–n tandem cell: (a) schematic diagram of the Mg-CuBi2O4‖Mo:BiVO4 tandem cell in operation; (b) J–V curves of a single Mo:BiVO4 photoanode and a single Mg-CuBi2O4 photocathode; (c) i–t curve of the p–n tandem cell consisting of Mo:BiVO4 and Mg-CuBi2O4 under continuous illumination at zero bias voltage. | ||
Conclusions
In summary, an in situ surface-trap passivation approach for the CuBi2O4 photocathode is proposed. It is demonstrated that spontaneously segregated MgO nanoparticles on the CuBi2O4 photocathode surface partially eliminate surface-trap states, resulting in increased bulk charge separation and improved interfacial charge transfer characteristics. The as-derived MgO(7%)-passivated CuBi2O4 photocathode yields a photocurrent onset of 1.15 V vs. RHE, which is ca. 90 mV anodic shift of the pristine CuBi2O4 photocathode. When the MgO-passivated CuBi2O4 photocathode is combined with a Mo-doped BiVO4 photoanode to construct a bias-free tandem water-splitting device, a solar-to-hydrogen conversion efficiency of 0.41% is achieved, 37.5% greater than that assembled with a pristine CuBi2O4 photocathode. This study identifies MgO as a novel yet effective large-bandgap material for passivating surface traps thereby boosting the performances of water-splitting photoelectrodes. Moreover, the concept of in situ surface-trap passivation would flourish the passivating strategies for semiconductor-based devices and inspire cost-effective device manufacturing processes.Experimental
Materials
Bismuth(III) nitrate pentahydrate (Bi(NO3)3·5H2O, ≥99.99%, Aladdin Chemistry Co., Ltd); copper(II) nitrate trihydrate (Cu(NO3)2·3H2O, ≥99%, Sinopharm Chemical Reagent Co., Ltd); magnesium(II) nitrate hexahydrate (Mg(NO3)2·6H2O, ≥99%, Xilong Chemical Co., Ltd); concentrated nitric acid (HNO3, 65–68%, Sinopharm Chemical Reagent Co., Ltd); glacial acetic acid (CH3COOH, ≥99.5%, Sinopharm Chemical Reagent Co., Ltd); absolute ethanol (C2H6O, ≥99.7%, Sinopharm Chemical Reagent Co., Ltd); boric acid (H3BO3, ≥99.5%, Nanjing Chemical Reagent Co., Ltd); potassium hydroxide (KOH, ≥99.7%, Sinopharm Chemical Reagent Co., Ltd) and deionized water were used without further purification.Preparation of CuBi2O4 and Mg-modified CuBi2O4 photocathodes
CuBi2O4 and Mg-modified CuBi2O4 photocathodes were synthesized via the metal–organic decomposition method (Fig. 1a). Bi(NO3)3·5H2O in glacial acetic acid (0.1 mol L−1), Cu(NO3)2·3H2O in absolute ethanol (0.05 mol L−1) and Mg(NO3)2·6H2O in absolute ethanol (0.05 mol L−1) were used as precursor solutions. For the preparation of the CuBi2O4 film, a certain amount of Cu and Bi solutions were mixed according to a stoichiometric ratio of Cu![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Bi = 1:2. Then the mixed solution was dropped on FTO substrates (1 cm × 2 cm), dried at 45 °C and then calcined at 600 °C in a muffle furnace for 60 min to form CuBi2O4. Mg-modified CuBi2O4 electrodes were synthesized by adding 1%, 3%, 5%, 7% and 10% of magnesium (with respect to Bi) to the precursor solutions.
:Bi = 1:2. Then the mixed solution was dropped on FTO substrates (1 cm × 2 cm), dried at 45 °C and then calcined at 600 °C in a muffle furnace for 60 min to form CuBi2O4. Mg-modified CuBi2O4 electrodes were synthesized by adding 1%, 3%, 5%, 7% and 10% of magnesium (with respect to Bi) to the precursor solutions.
Preparation of Mo-doped BiVO4 photoanodes
Mo-doped BiVO4 photoanodes were prepared by a sol–gel method. 1.25 mmol Bi2O3 and 1.5 mL HNO3 were dissolved in 23.5 mL ethylene glycol to obtain 0.1 mol L−1 Bi. 2.5 mmol NH4VO3 and 1.5 mL HNO3 were dissolved in 23.5 mL ethylene glycol to form 0.1 mol L−1 V. 0.357 mmol (NH4)6Mo7O24·4H2O was added in 23.5 mL ethylene glycol to produce 0.1 M Mo. The precursor solution of Mo-doped BiVO4 was prepared by mixing 0.2 mmol anhydrous citric acid, 1 mL precursor solution of Bi, and 1 mL precursor solution of V. The mixed solution was dropped on FTO substrates (exposed 1 cm × 1 cm), dried at 90 °C for 20 min, then heated at 120 °C for 20 min, and then calcined at 520 °C for 1 h to synthesize Mo:BiVO4 photoanodes. The NiFeOx cocatalyst was loaded on Mo:BiVO4 photoanodes by a photo-assisted electrodeposition method. Electrolyte for deposition was potassium borate buffer solution (KOH: 0.2 M; H3BO3: 0.4 M) containing 1.5 mmol L−1 FeSO4·7H2O and 1.5 mmol L−1 Ni(CH3COO)2·4H2O. The applied potential was at −0.14 V vs. Ag/AgCl by the i–t testing method. The total time of deposition was 6 min, during which the light was turned off for 20 s after every 30 seconds of illumination.Characterization of photoelectrodes
The crystal structures of all the photoelectrodes were detected by X-ray diffraction (XRD, Rigaku Ultima III) with Cu Kα radiation (λ = 1.54056 Å). The light absorption spectra of films were recorded on a UV-visible (UV-vis) spectrophotometer (Shimadzu, UV-vis 2550; PerkinElmer, Lambda 950). The structure of the material and Raman vibrational spectra were measured using a LabRAM ARAMIS Raman spectrometer (HORIBA Scientific). The chemical states of elements were analyzed by X-ray photoelectron spectroscopy (XPS, Thermo ESCALAB 250). The morphologies and elemental distribution of photocathodes were surveyed using a scanning electron microscope (FEI Nova NanoSEM 230 and ZEISS ULTRA 55). High resolution transmission electron microscopy (HRTEM) and selected area electron diffraction (SAED) analysis were investigated using a transmission electron microscope (JEOL 2100F TEM and an FEI double-aberration corrected Titan G2 60-300S/TEM). Surface photovoltage was surveyed using a surface photovoltage spectrometer (CEL-SPS1000). Photoluminescence (PL) and time-resolved photoluminescence (TRPL) decay spectra were observed using a fluorescence spectrometer (Andor-SR-500i).Electrochemical characterization studies
PEC performances were measured in a three-electrode cell using an electrochemical workstation (CHI-760E, Shanghai Chenhua). The PEC cell consists of a working electrode (photocathode), a reference electrode (Ag/AgCl electrode) and a counter electrode. The electrolyte was a potassium borate buffer solution (KOH: 0.2 M; H3BO3: 0.4 M). The potential vs. Ag/AgCl was converted to that vs. the reversible hydrogen electrode (RHE). Photocurrents were tested under AM 1.5 G simulated sunlight (100 mW cm−2), from a simulator (Newport Sol3A Class AAA; CEL-AAAS50, Beijing Zhongjiao Jinyuan Technology Co., Ltd). The light intensity was adjusted using a standard silicon cell (Newport 91150).Author contributions
Yingfei Hu: investigation, methodology, validation, formal analysis, writing – original draft. Jun Wang: methodology, formal analysis, writing – review & editing. Huiting Huang: data curation, methodology, formal analysis, writing – review & editing. Jianyong Feng: formal analysis, supervision, project administration, writing – original draft. Wangxi Liu: data curation, methodology, formal analysis. Hangmin Guan: resources, formal analysis. Lingyun Hao: resources, formal analysis. Zhaosheng Li: supervision, project administration, funding acquisition, writing – review & editing. Zhigang Zou: supervision, project administration, funding acquisition.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 22109059, 51902153, and U1663228), National Key Research and Development Program of China (No. 2021YFA1502100 and 2018YFA0209303), Natural Science Foundation of the Jiangsu Higher Education Institutions of China (No. 20KJB430025), Natural Science Foundation of Anhui Provincial (No. 1708085MB42), and Foundations Support from the Jinling Institute of Technology (No. JIT-B-201833, JIT-FHXM-201920, and JIT-B-201903). We thank Yue Xu (Beijing Zhongjiao Jinyuan Technology Co., Ltd) for the surface photovoltage experiment.References
- Y. Fang, Y. Zheng, T. Fang, Y. Chen, Y. Zhu, Q. Liang, H. Sheng, Z. Li, C. Chen and X. Wang, Sci. China: Chem., 2020, 63, 149–181 CrossRef CAS.
- X. Li, Y. Chen, Y. Tao, L. Shen, Z. Xu, Z. Bian and H. Li, Chem Catal., 2022, 2, 1315–1345 CrossRef.
- X. Liu, Y. Zhao, X. Yang, Q. Liu, X. Yu, Y. Li, H. Tang and T. Zhang, Appl. Catal., B, 2020, 275, 119144 CrossRef CAS.
- J. Li, Z. Lou and B. Li, Chin. Chem. Lett., 2022, 33, 1154–1168 CrossRef CAS.
- G. Zhang, Z. Guan, J. Yang, Q. Li, Y. Zhou and Z. Zou, Sol. RRL, 2022, 6, 2200587 CrossRef CAS.
- L. Pei, H. Cai, H. Jin, T. Li, H. Zhu, Y. Yuan, J. Zhong, S. Yan and Z. Zou, ChemCatChem, 2021, 13, 180–184 CrossRef CAS.
- M. Wang, G. Zhang, Z. Guan, J. Yang and Q. Li, Small, 2021, 17, e2006952 CrossRef.
- J. Feng, H. Huang, S. Yan, W. Luo, T. Yu, Z. Li and Z. Zou, Nano Today, 2020, 30, 100830 CrossRef CAS.
- W. Zhang, Y. Tian, H. He, L. Xu, W. Li and D. Zhao, Natl. Sci. Rev., 2020, 7, 1702–1725 CrossRef CAS PubMed.
- H. Huang, J. Feng, Z. Li and Z. Zou, Sci. Bull., 2022, 67, 226–228 CrossRef CAS.
- X. Wu, C. Y. Toe, C. Su, Y. H. Ng, R. Amal and J. Scott, J. Mater. Chem. A, 2020, 8, 15302–15318 RSC.
- M. A. Lumley, A. Radmilovic, Y. J. Jang, A. E. Lindberg and K. S. Choi, J. Am. Chem. Soc., 2019, 141, 18358–18369 CrossRef CAS PubMed.
- J.-B. Pan, S. Shen, L. Chen, C.-T. Au and S.-F. Yin, Adv. Funct. Mater., 2021, 31, 2104269 CrossRef CAS.
- X. Li, M. Kan, T. Wang, Z. Qin, T. Zhang, X. Qian, Y. Kuwahara, K. Mori, H. Yamashita and Y. Zhao, Appl. Catal., B, 2021, 296, 120387 CrossRef CAS.
- R.-T. Gao, L. Wu, S. Liu, K. Hu, X. Liu, J. Zhang and L. Wang, J. Mater. Chem. A, 2021, 9, 6298–6305 RSC.
- W. Li, L. Du, Q. Liu, Y. Liu, D. Li and J. Li, Chem. Eng. J., 2020, 384, 123323 CrossRef CAS.
- J. Feng, H. Huang, W. Guo, X. Xu, Y. Yao, Z. Yu, Z. Li and Z. Zou, Chem. Eng. J., 2021, 417, 128095 CrossRef CAS.
- W. Zhou, J.-K. Guo, S. Shen, J. Pan, J. Tang, L. Chen, C.-T. Au and S.-F. Yin, Acta Phys.-Chim. Sin., 2020, 36, 1906048 Search PubMed.
- H. Zhu, Q. Yang, D. Liu, Y. Du, S. Yan, M. Gu and Z. Zou, J. Am. Chem. Soc., 2021, 143, 9236–9243 CrossRef CAS PubMed.
- H. Zhu, S. Xiao, W. Tu, S. Yan, T. He, X. Zhu, Y. Yao, Y. Zhou and Z. Zou, J. Phys. Chem. Lett., 2021, 12, 10815–10822 CrossRef CAS PubMed.
- H. Quan, Y. Gao and W. Wang, Inorg. Chem. Front., 2020, 7, 817–838 RSC.
- J. Feng, X. Zhao, B. Zhang, Z. Chen, Z. Li and Y. Huang, J. Energy Chem., 2022, 71, 20–28 CrossRef CAS.
- K. Zhang, Y. Lu, Q. Zou, J. Jin, Y. Cho, Y. Wang, Y. Zhang and J. H. Park, ACS Energy Lett., 2021, 6, 4071–4078 CrossRef CAS.
- H. Xu, W. Fan, Y. Zhao, B. Chen, Y. Gao, X. Chen, D. Xu and W. Shi, Chem. Eng. J., 2021, 411, 128480 CrossRef CAS.
- Y. Song, X. Zhang, Y. Zhang, P. Zhai, Z. Li, D. Jin, J. Cao, C. Wang, B. Zhang, J. Gao, L. Sun and J. Hou, Angew. Chem., Int. Ed., 2022, 61, e202200946 CAS.
- Q. Wang, L. Wu, Z. Zhang, J. Cheng, R. Chen, Y. Liu and J. Luo, ACS Appl. Mater. Interfaces, 2022, 14, 26642–26652 CrossRef CAS PubMed.
- X. Hu, Y. Li, X. Wei, L. Wang, H. She, J. Huang and Q. Wang, Adv. Powder Mater., 2022, 1, 100024 CrossRef.
- J.-B. Pan, B.-H. Wang, J.-B. Wang, H.-Z. Ding, W. Zhou, X. Liu, J.-R. Zhang, S. Shen, J.-K. Guo, L. Chen, C.-T. Au, L.-L. Jiang and S.-F. Yin, Angew. Chem., Int. Ed., 2021, 60, 1433–1440 CrossRef CAS PubMed.
- Y. Zhao, C. Deng, D. Tang, L. Ding, Y. Zhang, H. Sheng, H. Ji, W. Song, W. Ma, C. Chen and J. Zhao, Nat. Catal., 2021, 4, 684–691 CrossRef CAS.
- D. Cao, J. Zhang, A. Wang, X. Yu and B. Mi, J. Mater. Sci. Technol., 2020, 56, 189–195 CrossRef.
- P. Liu, C. Wang, L. Wang, X. Wu, L. Zheng and H. G. Yang, Research, 2020, 2020, 1–8 Search PubMed.
- N. Zhang, X. Wang, J. Feng, H. Huang, Y. Guo, Z. Li and Z. Zou, Natl. Sci. Rev., 2020, 7, 1059–1067 CrossRef PubMed.
- Y. Li, N. Zhang, C. Liu, Y. Zhang, X. Xu, W. Wang, J. Feng, Z. Li and Z. Zou, Chin. J. Catal., 2021, 42, 1992–1998 CrossRef CAS.
- J.-B. Pan, X. Liu, B.-H. Wang, Y.-A. Chen, H.-Y. Tan, J. Ouyang, W. Zhou, S. Shen, L. Chen, C.-T. Au and S.-F. Yin, Appl. Catal., B, 2022, 315, 121526 CrossRef CAS.
- C. Li, J. He, Y. Xiao, Y. Li and J.-J. Delaunay, Energy Environ. Sci., 2020, 13, 3269–3306 RSC.
- S. P. Berglund, F. F. Abdi, P. Bogdanoff, A. Chemseddine, D. Friedrich and R. van de Krol, Chem. Mater., 2016, 28, 4231–4242 CrossRef CAS.
- D. Kang, J. C. Hill, Y. Park and K.-S. Choi, Chem. Mater., 2016, 28, 4331–4340 CrossRef CAS.
- A. Song, P. Plate, A. Chemseddine, F. Wang, F. F. Abdi, M. Wollgarten, R. van de Krol and S. P. Berglund, J. Mater. Chem. A, 2019, 7, 9183–9194 RSC.
- D. Huang, K. Wang, L. Li, K. Feng, N. An, S. Ikeda, Y. Kuang, Y. Ng and F. Jiang, Energy Environ. Sci., 2021, 14, 1480–1489 RSC.
- C. Ma, D.-K. Ma, W. Yu, W. Chen and S. Huang, Appl. Surf. Sci., 2019, 481, 661–668 CrossRef CAS.
- N. Xu, F. Li, L. Gao, H. Hu, Y. Hu, X. Long, J. Ma and J. Jin, ACS Sustainable Chem. Eng., 2018, 6, 7257–7264 CrossRef CAS.
- A. K. Shah, T. K. Sahu, A. Banik, D. Gogoi, N. R. Peela and M. Qureshi, Sustainable Energy Fuels, 2019, 3, 1554–1561 RSC.
- S. Pulipaka, N. Boni, G. Ummethala and P. Meduri, J. Catal., 2020, 387, 17–27 CrossRef CAS.
- Y. Hu, H. Huang, J. Feng, W. Wang, H. Guan, Z. Li and Z. Zou, Sol. RRL, 2021, 5, 2100100 CrossRef CAS.
- F. Le Formal, N. Tetreault, M. Cornuz, T. Moehl, M. Gratzel and K. Sivula, Chem. Sci., 2011, 2, 737–743 RSC.
- F. Le Formal, K. Sivula and M. Grätzel, J. Phys. Chem. C, 2012, 116, 26707–26720 CrossRef CAS.
- J. Brillet, M. Grätzel and K. Sivula, Nano Lett., 2010, 10, 4155–4160 CrossRef CAS PubMed.
- K. Sivula, F. Le Formal and M. Gratzel, ChemSusChem, 2011, 4, 432–449 CrossRef CAS PubMed.
- Z. Li, J. Feng, S. Yan and Z. Zou, Nano Today, 2015, 10, 468–486 CrossRef CAS.
- M. Zhong, T. Hisatomi, Y. Kuang, J. Zhao, M. Liu, A. Iwase, Q. Jia, H. Nishiyama, T. Minegishi, M. Nakabayashi, N. Shibata, R. Niishiro, C. Katayama, H. Shibano, M. Katayama, A. Kudo, T. Yamada and K. Domen, J. Am. Chem. Soc., 2015, 137, 5053–5060 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ta07117g |
| This journal is © The Royal Society of Chemistry 2023 |