
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEffective defect passivation with a designer ionic molecule for high-efficiency vapour-deposited inorganic phase-pure CsPbBr3 perovskite solar cells†
Ruxin
Guo‡
 ad,
Junmin
Xia‡
b,
Hao
Gu‡
b,
Xuke
Chu
a,
Yan
Zhao
a,
Xianghuan
Meng
a,
Zhiheng
Wu
ad,
Jiangning
Li
a,
Yanyan
Duan
a,
Zhenzhen
Li
a,
Zhaorui
Wen
b,
Shi
Chen
b,
Yongqing
Cai
b,
Chao
Liang
*c,
Yonglong
Shen
*ad,
Guichuan
Xing
*b,
Wei
Zhang
e and
Guosheng
Shao
*ad
ad,
Junmin
Xia‡
b,
Hao
Gu‡
b,
Xuke
Chu
a,
Yan
Zhao
a,
Xianghuan
Meng
a,
Zhiheng
Wu
ad,
Jiangning
Li
a,
Yanyan
Duan
a,
Zhenzhen
Li
a,
Zhaorui
Wen
b,
Shi
Chen
b,
Yongqing
Cai
b,
Chao
Liang
*c,
Yonglong
Shen
*ad,
Guichuan
Xing
*b,
Wei
Zhang
e and
Guosheng
Shao
*ad
aState Centre for International Cooperation on Designer Low-carbon & Environmental Materials (CDLCEM), School of Materials Science and Engineering, Zhengzhou University, Zhengzhou 450001, P. R. China. E-mail: shenyonglong@zzu.edu.cn; gsshao@zzu.edu.cn
bJoint Key Laboratory of the Ministry of Education, Institute of Applied Physics and Materials Engineering, University of Macau, Avenida da Universidade, Taipa, Macau 999078, P. R. China. E-mail: gcxing@um.edu.mo
cMOE Key Laboratory for Nonequilibrium Synthesis and Modulation of Condensed Matter, School of Physics, Xi'an Jiaotong University, Xi'an 710049, P. R. China. E-mail: chaoliang@um.edu.mo
dZhengzhou Materials Genome Institute (ZMGI), Building 2, Zhongyuanzhigu, Xingyang, Zhengzhou 450100, P. R. China
eAdvanced Technology Institute (ATI), University of Surrey, Guildford, Surrey, GU27XH, UK
First published on 9th December 2022
Abstract
While caesium lead bromide (CsPbBr3) is promising for highly stable perovskite solar cells (PSCs), the usual solution-based methods require tedious multistep spin coating processes, which imposes a practical barrier against scaling up to large areas for industrial exploitation. Although sequential vapour deposition (SVD) can meet commercial requirements, these films are limited by high trap density and impure phases, resulting in poor performance of PSCs. Here, we obtained low-trap density and effectively phase-pure CsPbBr3 films (grain size > 3 μm, trap density < 4 × 1015 cm−3) by systematic defect and phase management. With the identification of a molecular ionic liquid from theoretical simulation, we find that such a designer molecule can form multiple bonding interactions with the perovskite phase. This results in significantly enhanced crystallization of the CsPbBr3 phase, and more importantly, effective passivation of well recognized Cs- and Br-vacancy defects. CsPbBr3 PSCs with simplified architecture using carbon as electrodes without hole transport layer (HTL) achieved highest power conversion efficiency (PCE) of up to 11.21% for small area devices (0.04 cm2) and 9.18% for large area devices (1 cm2). The unencapsulated devices exhibited excellent long-term stability, maintaining over 91% of the initial PCE after 100 days in ambient air at a humidity of ∼55%. This work also provides a valuable approach to process phase-pure, low-defect, and large-area inorganic CsPbBr3 perovskite films for efficient and stable optoelectronic devices.
Introduction
The power conversion efficiency (PCE) of organic–inorganic hybrid perovskite solar cells (PSCs) has rapidly increased from 3.8% in 2009 to the latest certified 25.7%, which is comparable to that of silicon-based solar cells.1,2 However, the organic components in the perovskite layer, hygroscopic additives on the hole transport layer (HTL), and the use of precious metal electrodes have resulted in poor humidity and thermal instabilities as well as high cost of the organic–inorganic hybrid PSCs, which has severely hindered their application in the photovoltaic industry.3 Carbon-based HTL-free PSCs (C-PSCs) with an all-inorganic caesium lead bromide (CsPbBr3) as the light absorber is currently considered as a most promising approach to overcome the above-mentioned limitations, owing to the substitution of unstable organic cations with inorganic caesium ones and the replacement of organic HTLs as well as precious metal electrodes with low-cost carbon.4In 2016, a CsPbBr3 C-PSC with a PCE of 6.7% was first reported by Liang et al., which has attracted widespread attention because of its excellent environmental stability.5 For example, Duan et al. introduced Ln3+ ions into CsPbBr3 to increase the grain size, thereby prolonging the carrier lifetime and increasing the PCEs of photovoltaic devices to 10.14%.6 Subsequently, the PCEs of the state-of-the-art CsPbBr3 C-PSCs further increased to 11.08% with a high open-circuit voltage (Voc) of 1.702 V by incorporating a 2D Cl-terminated Ti3C2 (Ti3C2Clx) MXene into the bulk and surface of CsPbBr3 films.7 However, because of the low solubility of caesium bromide (CsBr), the CsPbBr3 film can only be prepared by a tedious multistep spin coating for the solution process, which is not conducive to industrial application in the future.7,8 Thus, vapour deposition has been utilized to achieve large-area perovskite films, which has been demonstrated to be the most prospective method for commercial optoelectronic thin-film devices.8–10 Unfortunately, CsPbBr3 films prepared by vapour deposition usually involve impurity phases of Cs4PbBr6 and CsPb2Br5, which not only introduces defects but also accelerates the decomposition of the films, thereby reducing the performance of PSCs.8,11 Although substantial efforts have been made in development of processing technology and interface engineering, the fabrication of low-defect and phase-pure CsPbBr3 films remains a bottleneck, hindering further development of the CsPbBr3 PSCs.
As demonstrated in our recent work, strong ionic coordination between ionic liquids (ILs) and the perovskite framework can induce the formation of phase-pure hybrid perovskite films.12 In addition, the incorporation of ILs into perovskite films significantly improves the performance and long-term stability of the devices, owing to defect passivation and suppression of compositional segregation.13–17 Herein, we have identified a novel 4-(dimethylamino)-1-(2,2,2-trifluoroacetyl)pyridin-1-ium 2,2,2-trifluoroacetate (DTPT) IL to achieve phase-pure CsPbBr3 films. X-ray diffraction (XRD), high-resolution transmission electron microscopy (HRTEM), and spectroscopic characterizations were used to evaluate the results. In addition, both simulations and experiments indicated that amino, acetyl, and acetate functional groups in DTPT comprehensively passivated the defects in the perovskite crystals, Cs (VCs) and Br (VBr) vacancies, effectively suppressing non-radiative recombination. As a result, the PSCs based on the device structure of FTO/TiO2/DTPT/CsPbBr3/DTPT/Carbon achieved a PCE of 11.21% for active area of 0.04 cm2, which is the highest efficiency reported for CsPbBr3-based PSCs. More importantly, we achieved a PCE of 9.18% for the device with an active area of 1 cm2, representing the most efficient large-area CsPbBr3-based PSCs. Furthermore, the optimized device retained ∼91% of its initial PCE in ambient air at a relative humidity (RH) of ∼55%, owing to the high-quality CsPbBr3 films and hydrophobic trifluoro (CF3–) groups in DTPT.
Results and discussion
The design of ILs play a key role in the defects and phase management of perovskite films. As shown in Fig. 1a, DTPT was designed as an ingenious interface layer owing to the comprehensive functional groups, including pyridine, amino, acetyl, acetate, and CF3–. In addition, a typical carbon-based HTL-free structure with FTO/TiO2/DTPT/CsPbBr3/DTPT/Carbon was employed in this study. It is well known that the electron transporting layer (ETL) is crucial for perovskite films and devices due to its surface morphology and roughness.18–20 Therefore, we first investigated the influence of DTPT modification on the TiO2 ETL, as shown in ESI, Fig. S1 and S2.† When the DTPT molecule was assembled onto the surface of a TiO2 film, root-mean-squared (RMS) roughness decreased from 36.5 to 27.8 nm. Additionally, owing to the existence of the hydrophobic CF3– functional group, the contact angle was enhanced on the TiO2 surface after DTPT modification (ESI, Fig. S3†), which could have decreased the hetero-nucleation of perovskite, thereby increasing the grain size of the CsPbBr3 phase in the film (ESI, Fig. S4†).21 Notably, the number of XRD diffraction peaks of Cs4PbBr6 in the CsPbBr3 film were markedly reduced from four down to one albeit without enhancement of the intensity of the only remnant peak of (321) plane (ESI, Fig. S5†), indicating that the phase purity of the CsPbBr3 film with DTPT on TiO2 substrate was effectively improved owing to suppressed nucleation of the impurity phases. The photovoltaic performance of PSCs based on TiO2 and TiO2/DTPT is shown in ESI, Fig. S6 and S7.† The current density–voltage (J–V) curves of PSCs based on TiO2 and TiO2/DTPT are shown in ESI, Fig. S6a,† and the corresponding photovoltaic performance data are summarized in ESI, Table S1.† Compared to the untreated, the device based on TiO2/DTPT has increased short-circuit current density (Jsc) from 8.22 mA cm−2 to 8.43 mA cm−2, Voc from 1.521 V to 1.535 V, FF from 78.66% to 79.87%, and PCE from 9.83% to 10.34%. The significant enhancement of Jsc and decrease of the hysteresis (ESI, Fig. S7†) may be attributed to the smooth surface of TiO2/DTPT, which is conducive to CsPbBr3 large grain growth and charge transport collection efficiency.21 As shown in ESI, Fig. S8,† the CsPbBr3 layer under the treated film has a high light absorption, which ensures more sunlight to be utilized by the CsPbBr3 layer for achieving high PCE.6 In addition, to demonstrate the reproducibility of PSCs based on TiO2 and TiO2/DTPT, the PCE distribution histogram (40 individual devices) is shown in ESI, Fig. S6b.† The result demonstrates that the efficiency of PSCs prepared based on TiO2/DTPT shows a narrower distribution, and the high-quality uniform growth of perovskite films may be the reason for the good reproducibility of the devices.8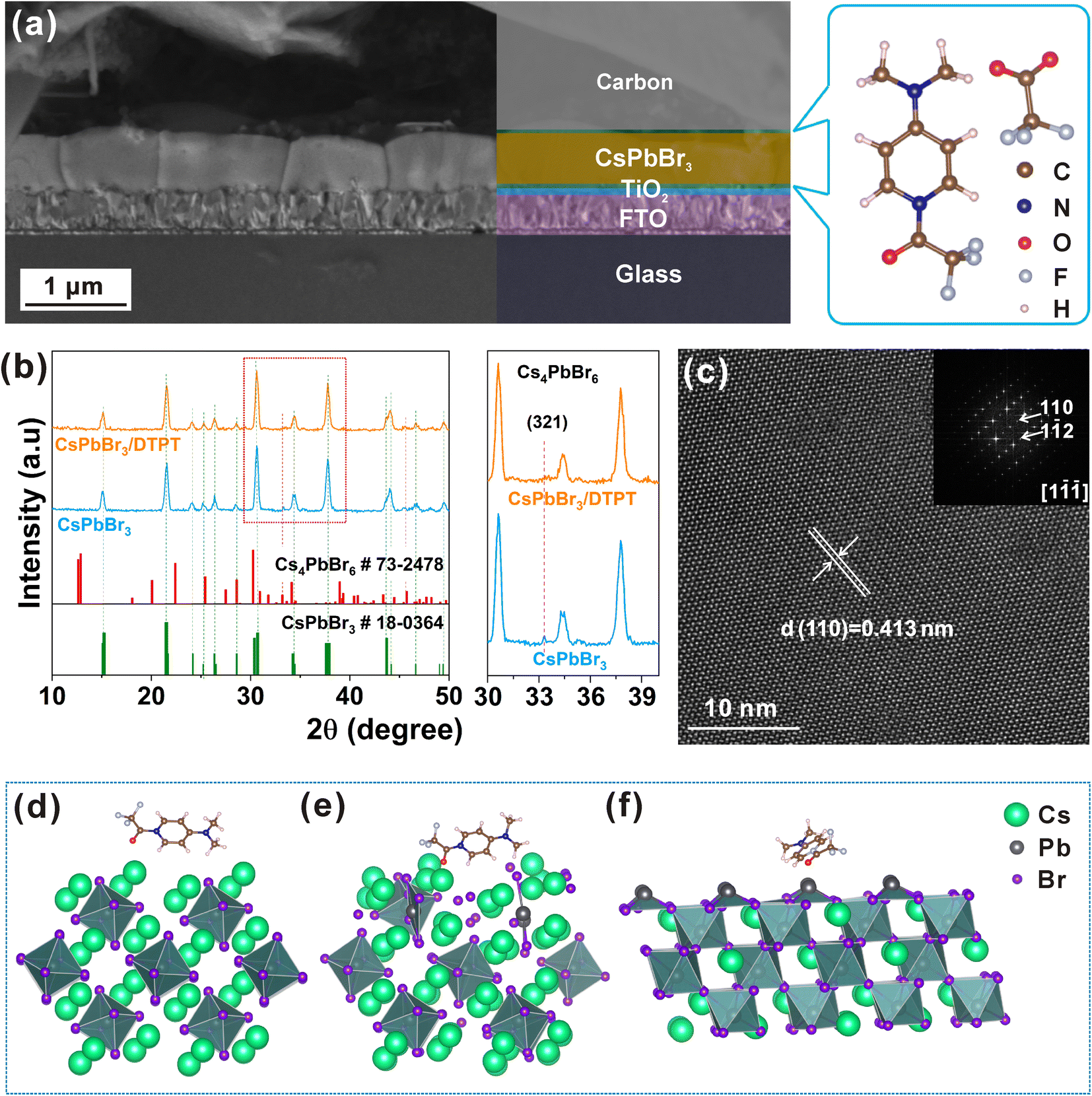 | ||
| Fig. 1 (a) Cross-sectional SEM image of CsPbBr3 PSC and chemical structure of the ionic liquid DTPT. (b) XRD patterns and Cs4PbBr6 for (321) planes of the CsPbBr3 and CsPbBr3/DTPT films. (c) HRTEM and FFT of the CsPbBr3/DTPT film. The structures of the Cs4PbBr6 with DTPT on the surface (d) before optimization and (e) after optimization. (f) The (110) phase structure of CsPbBr3 with DTPT on the surface. | ||
DTPT was then applied to treat the above CsPbBr3 film, (TiO2/DTFT/)CsPbBr3/DTFT. The XRD patterns of CsPbBr3 and CsPbBr3/DTPT are shown in Fig. 1b. All of the peaks at 15.2°, 21.5°, 26.4°, 30.7°, and 37.8° correspond to the (100), (110), (111), (200), and (−121) crystal planes of the phase of CsPbBr3 (PDF#18-0364), respectively. However, the diffraction peak for the (321) plane of the Cs4PbBr6 phase (PDF#73-2478) was not evident.
HRTEM and selected area electron diffraction (SAED) were used to investigate the effect of DTPT on the perovskite structure, as shown in Fig. 1c and ESI, Fig. S9–S11.† The lattice spacing of 0.413 nm in Fig. 1c corresponds to the (110) plane in the CsPbBr3 phase in the crystal direction of [1−1−1], which is consistent with the diffraction peak in the XRD pattern. The enhanced peak intensity for the (110) plane in the XRD pattern indicates the preferred orientation along its normal vector. As shown in ESI, Fig. S9,† the SAED pattern of the CsPbBr3/DTPT films is typically single crystalline, indicating no sub-grain boundaries within each fairly large crystal in the over micrometre grains. The SAED pattern (selected from the region shown in ESI, Fig. S9a†) of the CsPbBr3/DTPT films was used to demonstrate that the DTPT-modified CsPbBr3 film has a single orientation over a large range. In contrast, the Cs4PbBr6 phase was observed in the CsPbBr3 film without DTPT treatment (ESI, Fig. S10†). The lattice spacing of 0.269 nm corresponds to the (321) plane of the Cs4PbBr6 phase, which is consistent with the XRD results. Meanwhile, a small amount of PbBr2 phase exists in the CsPbBr3 film without DTPT treatment (ESI, Fig. S11†). Overall, the TEM results agree well with the XRD outcome, in that the impurity phases were only present in the sample without the top-surface DTPT treatment, but treatment with DTPT of the perovskite film helped eliminate PbBr2/Cs4PbBr6 impurities. Such DTPT effect is similar to previous finding that the presence of pyridine lowers the activation energy for the transformation of PbBr2/Cs4PbBr6 into the CsPbBr3 phase.22 The promoted phase purity is essential to benefit the performance and stability of perovskite PV cells.23
To further elaborate the possibility of the phase transition process from Cs4PbBr6 to CsPbBr3 by DTPT, we conducted density functional theory (DFT) simulation. The Cs4PbBr6 phase was found to be relatively stable when it was not modified by DTPT. The modifying molecule (Fig. 1d) will interact with the surface of the Cs4PbBr6 phase, resulting in local structural damage to the Cs4PbBr6. As shown in Fig. 1e, as the octahedron PbBr64− structure on the surface of the Cs4PbBr6 phase was undermined, it tends to restructure with neighbouring species to form the CsPbBr3 phase. Such a catalytic effect is to help facilitate reaction with nearby PbBr2, while the lower-nanoscale structures of impurity phases provide further leverage to enable such reaction at a moderate temperature during the after-treatment heating at 373 K.
In addition, we confirmed the preferred crystal orientation with modified molecules in the CsPbBr3 phase via simulation. In the presence of DTPT, it was found that the (110) plane had a lower formation energy than the (001) plane, −0.504 eV and −0.067 eV, respectively, as shown in Fig. 1f and ESI, Fig. S12.† This indicates that the DTPT molecule can promote the formation of the (110) plane as a higher orientation of CsPbBr3 than the (001) plane, which is consistent with the XRD and HRTEM results. From the enlarged XRD spectra of CsPbBr3 films deposited on TiO2 and TiO2/DTPT films in ESI, Fig. S13a,† it can be seen that the diffraction peak of the (110) plane of the CsPbBr3 film is significantly enhanced after DTPT modification of the TiO2 surface. However, as shown in ESI, Fig. S13b,† the diffraction peak of (110) plane before and after the DTPT modification of CsPbBr3 surface is not obvious (ESI, Fig. S13c†), since the prior DTPT treatment of the TiO2 substrate already delivered such an orientational benefit (Fig. S13a and c†), with the crystallization of Cs4PbBr6 phase largely prevented.
To investigate the surface chemical states of DTPT on the CsPbBr3 film, we conducted X-ray photoelectron spectroscopy (XPS). The high-resolution XPS spectra of Cs 3d, Pb 4f, Br 3d, and F 1s are presented in Fig. 2a–d, respectively. In pristine CsPbBr3 films, the binding energies of 724.34 and 738.27 eV correspond to the Cs 3d5/2 and Cs 3d3/2 peaks, respectively. After DTPT modification, the two typical peaks shifted to 724.58 and 738.51 eV, respectively, which may be attributed to the strong coupling effect between the Cs+ vacancies and amino functional group in DTPT.24 In addition, the 0.31 and 0.28 eV upshift of Pb 4f and Br 3d peaks, respectively, may be associated with the strengthened interaction between the O atom in the acetyl/acetate functional group and Pb species (or Br− vacancies). As expected, we also detected the F 1s peak at 688.28 eV in the DTPT-modified sample, which is consistent with the time-of-flight secondary-ion mass spectrometry (ToF-SIMS) results (ESI, Fig. S14†).16
To further elaborate the DTPT interactions with defects, we conducted DFT modelling on the most common ionic defects of Cs vacancies (VCs) and Br vacancies (VBr) in CsPbBr3.25 The ESI, Fig. S15† depicts the structures of CsPbBr3 with various vacancies and DTPT. Generally, the density of states (DOS) provides an intuitive description of the passivation effect. As shown in Fig. 2e, VCs caused the Fermi level to insert into the top of the valence band (VB) and bring an obvious trap state, which suppressed the separation of charge carriers and formed recombination centers.26 When the DTPT was anchored on the VCs site, we found that the Fermi level had a distinct blue shift and returned to the bandgap. Furthermore, the trap state at the VB was remarkably reduced, which was also evidenced by the electron localization function (ELF) results. As shown in ESI, Fig. S16,† the profile of ELF around VCs is similar to that of the perfect CsPbBr3 structure obtained after introducing DTPT, which indicated that the grafted amino functional group could imitate the role of Cs atoms to mitigate the delocalization of the wave function around Br ions caused by VCs.27 To further explore the mechanism, we calculated the differential charge density (DCD) of the system.16 Fundamentally, the passivation molecule had an apparent interaction with the peripheral atoms around the VCS, implying that DTPT can effectively modulate the defective structure.
 | ||
Fig. 2 High-resolution XPS analysis of (a) Cs 3d, (b) Pb 4f, (c) Br 3d, and (d) F 1s in CsPbBr3 and CsPbBr3/DTPT films. (e) Electronic partial density of state (PDOS) curves for Br of the CsPbBr3 with VCs passivated by DTPT. (f) Density of state (DOS) of CsPbBr3 with VBr passivated by the carboxylic acid group in CF3COO− and the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O in DTPT. The Fermi level (represented by a dashed line) has been set to zero. Differential charge density when DTPT fills into the vacancies of CsPbBr3. (g) VCs passivated by the DTPT; VBr passivated by (h) the CO in DTPT and (i) the carboxylic acid group in CF3COO−. The blue (yellow) isosurfaces represent the dissipation (accumulation) of electron density. O in DTPT. The Fermi level (represented by a dashed line) has been set to zero. Differential charge density when DTPT fills into the vacancies of CsPbBr3. (g) VCs passivated by the DTPT; VBr passivated by (h) the CO in DTPT and (i) the carboxylic acid group in CF3COO−. The blue (yellow) isosurfaces represent the dissipation (accumulation) of electron density. | ||
For VBr, as both acetyl/acetate and CF3– functional groups may coordinate with the exposed Pb atoms, we investigated the adsorption configurations of the molecules with different sites.28 First, we calculated the adsorption energies of the different systems to predict the appropriate passivation methods using the following eqn (1):
| Ead = Emolecule/CsPbBr3 − Emolecule − ECsPbBr3 | (1) |
O in DTPT (−2.03 eV) has a lower formation energy when passivating Br defects than CF3– group (−1.65 eV) in DTPT, so CO has a stronger interaction with surface with Br vacancy. Overall, DTPT has a comprehensive passivation effect on the CsPbBr3 films based on amino, acetyl, and acetate functional groups, which modulate the defective structures.31
To explore the photophysical mechanisms involved in the enhancement of DTPT modified devices, we used temperature-dependent photoluminescence (PL), time-resolved PL (TRPL), and femtosecond transient absorption (fs-TA) measurements. First, the temperature-dependent PL was conducted from 40 to 300 K for the CsPbBr3 and CsPbBr3/DTPT films, as shown in Fig. 3a and b, which indicated that the fluorescence intensity decreased with increasing temperature due to temperature-activated exciton dissociation.32,33 Notably, there was a significant red shift in the PL peak position after DTPT modification, which is related to the reduced quantum confinement in the phase-pure CsPbBr3 films.34 In addition, the PL spectrum of the CsPbBr3 film modified by DTPT indicated a narrower full width at half maximum (FWHM), further indicating that the DTPT modification can effectively reduce shallow- or deep-level defects and eliminate the Cs4PbBr6 phase.35 The exciton binding energy (Eb) is an important parameter for photovoltaic materials, which represents the energy required for the separation of bound excitons into free carriers. As shown in Fig. 3c, Eb can be obtained by fitting the relationship between the integrated PL intensity and temperature using the following Arrhenius equation:
 | (2) |
 | ||
| Fig. 3 Temperature-dependent PL spectra of (a) CsPbBr3 and (b) CsPbBr3/DTPT films. (c) Dependence of integrated PL intensity and reciprocal temperature (T−1). (d) The PL intensity versus photo-injected carrier density. (e) Time-resolved photoluminescence (TRPL) decay curves of CsPbBr3 and CsPbBr3/DTPT films. (f) Pseudo-colour femtosecond transient absorption (fs-TA) spectrum plots of CsPbBr3/DTPT film upon a pulsed fs-laser excitation at 400 nm. | ||
We conducted light-intensity-dependent J–V measurements and a series of electrochemical tests to profoundly investigate the charge recombination mechanism of the device. The relationship curves of logarithms of Jsc and intensity are plotted in Fig. 4a. According to the power law equation: Jsc ∝ Iα,4,17 the factor α related to bimolecular recombination can be calculated, and the value of α is 0.976 for the treated device and 0.959 for the control device, indicating that DTPT treatment can make the carriers transfer from the perovskite layer to the carbon faster and reduce the recombination of the charge at the CsPbBr3/carbon interface.6,23 Moreover, the factor n related to monomolecular recombination can be obtained by equation: Voc = nkT![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ln(I)/q + constant, where k, T and q represent Boltzmann constant, absolute temperature and elementary charge, respectively.11,17 As shown in Fig. 4b, the n value of the modified device is reduced from 1.67 to 1.38, demonstrating that the DTPT modification of the CsPbBr3 film significantly suppressed the trap assisted recombination process.7,41 To further evaluate the trap state density (ntrap) of the CsPbBr3 films, we performed space charge limited current (SCLC) method under the dark condition. The ntrap can be calculated with the trap-filled limit voltage (VTFL) by the equation: ntrap = 2εε0VTFL/qL2, where L is the thickness of the perovskite films, ε is the relative dielectric constant of CsPbBr3, ε0 is the vacuum permittivity, and VTFL is the kink point in the dark I–V curve.7,44,45 As shown in Fig. 4c, the VTFL is reduced from 1.12 V to 0.67 V after DTPT treatment, we then obtain the ntrap of CsPbBr3 and CsPbBr3/DTPT films as 1.54 × 1016 cm−3 and 9.24 × 1015 cm−3, respectively, which is consistent with the results of the previous theoretical model (Fig. 3d). It is proved once again that the treatment of DTPT can effectively passivate the traps in the CsPbBr3 layer, thereby improving the device performance through improved Jsc and FF.41 Besides, the dark J–V curves of devices based on CsPbBr3 and CsPbBr3/DTPT films indicate that the device based on CsPbBr3/DTPT film exhibited a smaller leakage current density under reverse bias in Fig. 4d, which indicates that the DTPT treatment can effectively reduce interface defects and suppress leakage current.6 To investigate the interfacial charge transfer characteristics of the devices, we performed electrochemical impedance spectroscopy (EIS) measurement at an applied bias of Voc under dark conditions with a frequency range of 0.1 Hz to 100 kHz and an amplitude of 5 mV and fitted with the equivalent circuit shown in the inset in Fig. 4e. The low-frequency arc in the Nyquist plots represents the recombination resistance (Rrec) at the interface, and the device after DTPT modification has a larger arc, which means that proper concentration of DTPT to treat the CsPbBr3 surface can effectively inhibit the recombination of interface charges, thereby contributing to enhance Jsc and FF of the device.4,41 Mott–Schottky analysis is often used as a measure to calculate the built-in potential (Vbi) of the device, and the equation is C−2 = 2(Vbi − V)/ε0εqA2NA, where C, ε0, ε, q, A and NA are the capacitance, vacuum dielectric constant, relative dielectric constant of CsPbBr3, elementary charge, active area of device and carrier concentration, static permittivity, dielectric constant.7 Therefore, we can obtain Vbi from the intercept of the linear region and the x-axis of the reciprocal of the square root of the capacitance and the voltage (C−2–V) curve. As shown in Fig. 4f, DTPT modification increased the Vbi from 1.21 to 1.35 V, which is consistent with the increased Voc of the devices. A higher Vbi means that the driving force for charge separation is stronger, and the charge accumulation at the perovskite/carbon interface is smaller, resulting in weaker hysteresis behaviour and improved performance. Therefore, modifying the surface of the CsPbBr3 film by DTPT can significantly reduce the energy level mismatch between CsPbBr3 and carbon, thereby increasing the Voc of the PCEs.4
ln(I)/q + constant, where k, T and q represent Boltzmann constant, absolute temperature and elementary charge, respectively.11,17 As shown in Fig. 4b, the n value of the modified device is reduced from 1.67 to 1.38, demonstrating that the DTPT modification of the CsPbBr3 film significantly suppressed the trap assisted recombination process.7,41 To further evaluate the trap state density (ntrap) of the CsPbBr3 films, we performed space charge limited current (SCLC) method under the dark condition. The ntrap can be calculated with the trap-filled limit voltage (VTFL) by the equation: ntrap = 2εε0VTFL/qL2, where L is the thickness of the perovskite films, ε is the relative dielectric constant of CsPbBr3, ε0 is the vacuum permittivity, and VTFL is the kink point in the dark I–V curve.7,44,45 As shown in Fig. 4c, the VTFL is reduced from 1.12 V to 0.67 V after DTPT treatment, we then obtain the ntrap of CsPbBr3 and CsPbBr3/DTPT films as 1.54 × 1016 cm−3 and 9.24 × 1015 cm−3, respectively, which is consistent with the results of the previous theoretical model (Fig. 3d). It is proved once again that the treatment of DTPT can effectively passivate the traps in the CsPbBr3 layer, thereby improving the device performance through improved Jsc and FF.41 Besides, the dark J–V curves of devices based on CsPbBr3 and CsPbBr3/DTPT films indicate that the device based on CsPbBr3/DTPT film exhibited a smaller leakage current density under reverse bias in Fig. 4d, which indicates that the DTPT treatment can effectively reduce interface defects and suppress leakage current.6 To investigate the interfacial charge transfer characteristics of the devices, we performed electrochemical impedance spectroscopy (EIS) measurement at an applied bias of Voc under dark conditions with a frequency range of 0.1 Hz to 100 kHz and an amplitude of 5 mV and fitted with the equivalent circuit shown in the inset in Fig. 4e. The low-frequency arc in the Nyquist plots represents the recombination resistance (Rrec) at the interface, and the device after DTPT modification has a larger arc, which means that proper concentration of DTPT to treat the CsPbBr3 surface can effectively inhibit the recombination of interface charges, thereby contributing to enhance Jsc and FF of the device.4,41 Mott–Schottky analysis is often used as a measure to calculate the built-in potential (Vbi) of the device, and the equation is C−2 = 2(Vbi − V)/ε0εqA2NA, where C, ε0, ε, q, A and NA are the capacitance, vacuum dielectric constant, relative dielectric constant of CsPbBr3, elementary charge, active area of device and carrier concentration, static permittivity, dielectric constant.7 Therefore, we can obtain Vbi from the intercept of the linear region and the x-axis of the reciprocal of the square root of the capacitance and the voltage (C−2–V) curve. As shown in Fig. 4f, DTPT modification increased the Vbi from 1.21 to 1.35 V, which is consistent with the increased Voc of the devices. A higher Vbi means that the driving force for charge separation is stronger, and the charge accumulation at the perovskite/carbon interface is smaller, resulting in weaker hysteresis behaviour and improved performance. Therefore, modifying the surface of the CsPbBr3 film by DTPT can significantly reduce the energy level mismatch between CsPbBr3 and carbon, thereby increasing the Voc of the PCEs.4
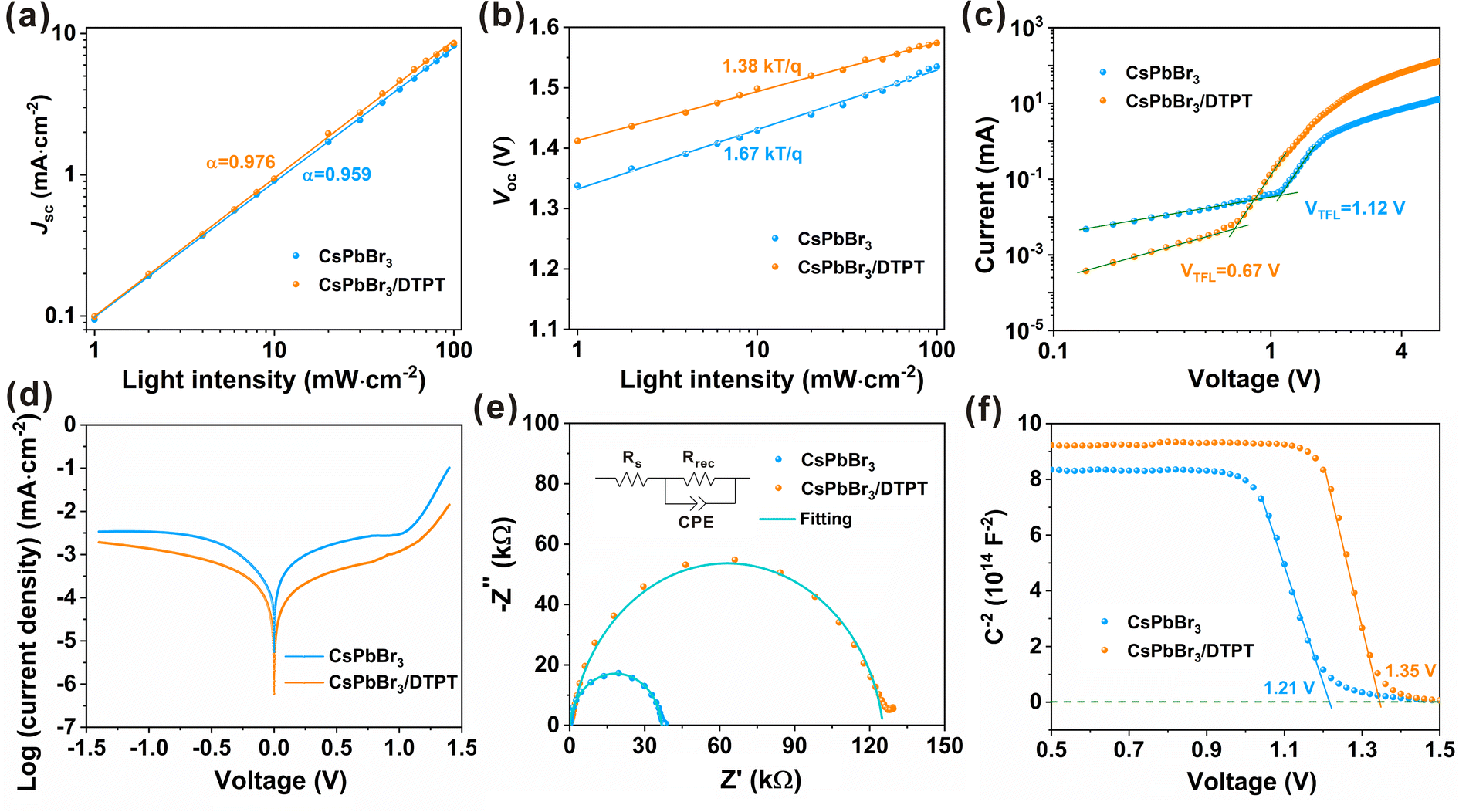 | ||
| Fig. 4 (a) Jsc and (b) Voc dependence on the light intensity. (c) Dark I–V curves with structure of FTO/TiO2/DTPT/CsPbBr3/PCBM/carbon or FTO/TiO2/DTPT/CsPbBr3/DTPT/PCBM/carbon. (d) Dark J–V curves, (e) Nyquist plots and the fitting curves, (f) Mott–Schottky (M–S) plots of the devices with and without DTPT. | ||
To investigate the effect of modifications in DTPT on device performance, we prepared 40 devices for each DTPT concentrations (0–6 mg mL−1), and the results are shown in ESI, Fig. S21.† Optimization revealed that the devices had the highest average PCE (11.14%) and lowest standard deviation (0.061) at 3 mg mL−1 of DTPT. We compared the device performance with that of the control and the optimal concentration of DTPT, and the corresponding J–V curve and specific photovoltaic parameters are shown in Fig. 5a and ESI, Table S2.† After modification, the PCE of the devices was significantly enhanced from 10.34% to 11.21% (Jsc = 8.52 mA cm−2, Voc = 1.574 V, and fill factor (FF) = 83.67%), which is the best performance reported for all CsPbBr3-based PSCs (ESI, Table S3†).7,8,11 Notably, the Voc of the devices increased from 1.535 to 1.574 V, possibly due to the reduction of the energy level offset and suppression of non-radiative recombination.45,46 To prove this hypothesis, ultraviolet photoemission spectroscopy (UPS) measurements (ESI, Fig. S22†) were used to investigate the influence of DTPT modification on the energy level alignment (ESI, Fig. S23†) of the CsPbBr3 films. The cut-off energy (Ecut-off) and valence band maximum (VBM) edges of control and modified CsPbBr3 films are shown in ESI, Fig. S22a and b,† where the VBM of CsPbBr3 and CsPbBr3/DTPT are −5.68 and −5.56 eV, respectively. This change in VBM is mainly attributed to a change in the work function, as the change in VBM with reference to the instrumental Fermi level was an order smaller (1.06 vs. 1.03 eV, approximately half the band gap value below the instrumental Fermi reference).23,47 Therefore, the energy offset between the VBM of CsPbBr3 and Fermi level (−5.0 eV) of the carbon electrode decreased from 0.68 to 0.56 eV after the DTPT modification, which corresponded to the improvement in Voc of the devices. The ESI, Fig. S24† displays the J–V curves of PSCs based on CsPbBr3 and CsPbBr3/DTPT under different scanning directions. The hysteresis index (H-index) can be obtained according to the formula: H-index = (PCEReverse − PCEForward)/PCEReverse, and the H-index of the device with DTPT modification (0.097) was significantly lower than that of the control device (0.157). We believe that the high FF and low hysteresis are attributed to the low-defect perovskite films and excellent interface of the CsPbBr3/carbon.45,46
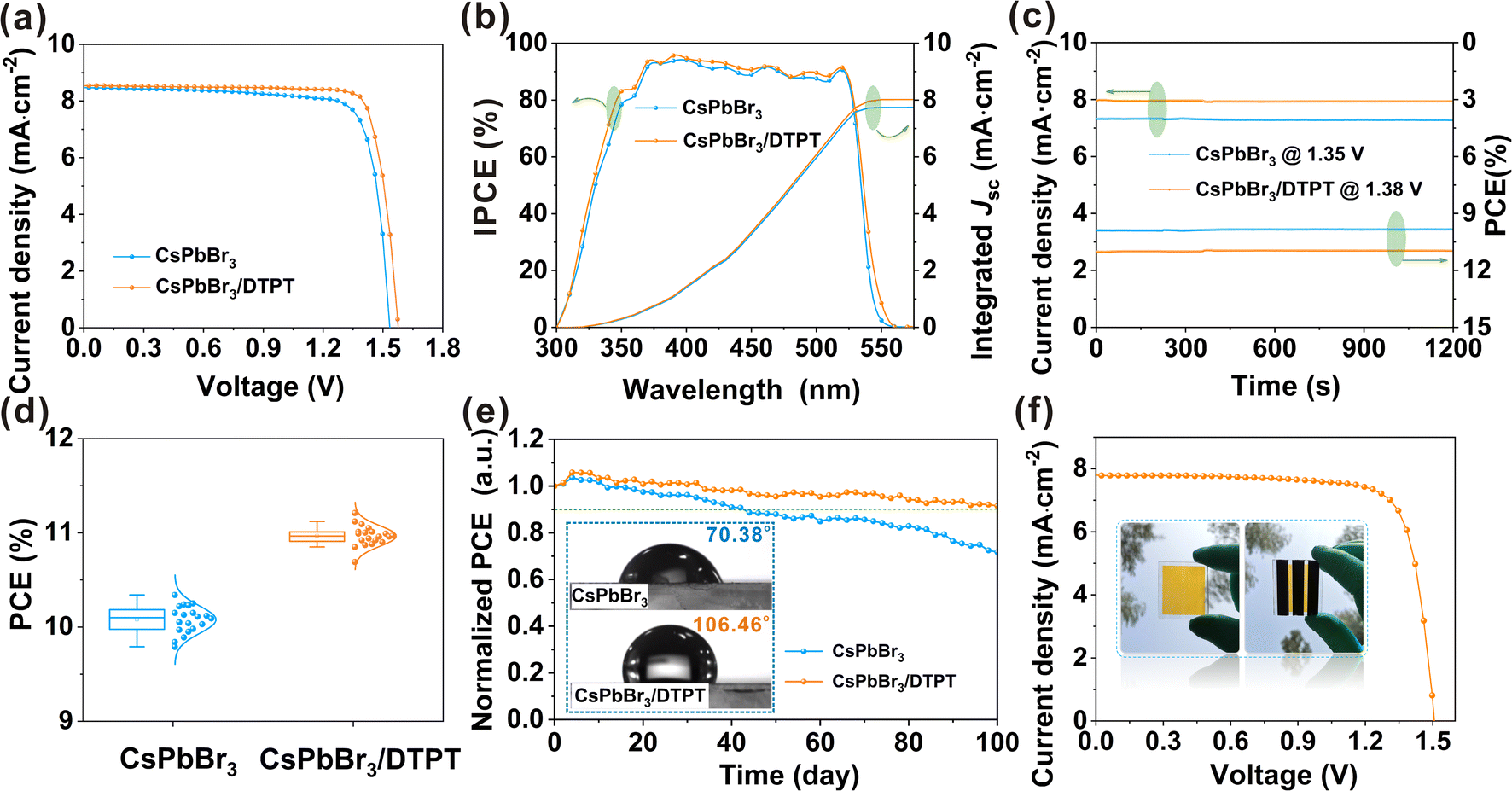 | ||
| Fig. 5 (a) J–V curves, (b) IPCE spectrum and corresponding integrated current, (c) stabilized power output, (d) PCEs of statistical and (e) long-term stability (∼25 °C and ∼55% RH) of the devices with and without DTPT; the water contact angle (illustration of (e)) of CsPbBr3 and CsPbBr3/DTPT films, (f) J–V curve of the large area device based on CsPbBr3/DTPT film. | ||
In addition, the incident photon-to-current efficiency (IPCE) spectrum (Fig. 5b) of the device based on CsPbBr3/DTPT was enhanced over the entire absorption region of 300–550 nm, and the light response edge of the device was slightly red-shifted as compared to that of the control, which agrees with the absorption spectra (ESI, Fig. S25†). As previously reported, the band gap of Cs4PbBr6 was ∼3.8 eV, which is wider than that of CsPbBr3 (2.3 eV). Thus, the red shift of the photo-response edge occurred due to the presence of DTPT.48 The integrated current densities obtained using these curves were 7.75 and 8.02 mA·cm−2 for the devices based on the control and CsPbBr3/DTPT films, respectively, which are consistent with the Jsc values obtained from the J–V curves. The steady-state photocurrent and PCE measured at the maximum power point (MPP) are shown in Fig. 5c. The device modified with DTPT obtained a stable PCE of 11.02%, which is higher than the value of the PCE based on original CsPbBr3 (9.91%), once again verifying the stability and reliability of the modified PSC. Fig. 5d and ESI, Fig. S26† depict the statistical analysis of PCEs, Voc, Jsc, and FF for the devices with and without DTPT. The DTPT-modified devices exhibited high reproducibility, which is conducive to large-scale production. In addition, the performance enhancement of modified PSCs is mainly due to the enhancement of Voc and FF, which can be attributed to the optimized energy level alignment, and the reduction of defect-related charge recombination.4
The long-term operational stability, a key parameter for PSCs, was also evaluated. Fig. 5e shows the PCE trend of the device placed in an ambient atmosphere of ∼25 °C and ∼55% RH. After 100 days, the DTPT-modified device exhibited excellent stability and maintained ∼91% of its initial performance, while the control device indicated a ∼30% reduction in efficiency. The high stability can be attributed to the phase-pure and low-defect CsPbBr3 films.49–51 In addition, owing to the introduction of the hydrophobic CF3– functional group in the DTPT molecule, the contact angle increased from 70.38° to 106.46°, which also played a significant role in enhancing the stability of the PSCs (Fig. 5e). Finally, to verify that this strategy can be applied to large-area devices, a device with an active area of 1 cm2 was prepared. Encouragingly, a PCE of 9.18% was achieved with a Voc of 1.509 V, Jsc of 7.81 mA·cm−2, and FF of 77.85% (Fig. 5f), which is also the highest efficiency reported thus far for large-area CsPbBr3-based PSCs (ESI, Table S4†).23,52
Experimental
Materials
PbBr2 and CsBr were purchased from the Xi'an Polymer Light Technology Corp. All solutions, carbon paste and 4-(dimethylamino)-1-(2,2,2-trifluoroacetyl)pyridin-1-ium 2,2,2-trifluoroacetate (DTPT) were purchased from Aladdin, the Shanghai MaterWin New Materials Co., Ltd, and TCI respectively.Device fabrication
Before preparing solar cells, the FTO glasses were cleaned by acetone, ethanol and deionized water for 15 min each and then treated in UV-ozone for 30 min. The treated FTO substrates were soaked in a 200 mM TiCl4 aqueous solution at 70 °C for 1 hour, in which the dilution of the TiCl4 solution (stored in the freezer) needs to be carried out at 0 °C. After washing with deionized water and ethanol, the substrates were annealed on a heating stage at 100 °C for 1 hour in ambient air.40 The CsPbBr3 film was fabricated by vapour deposition method, and the film thickness was controlled by a quartz crystal microbalance. Firstly, 100 nm PbBr2 film was deposited on the FTO/TiO2 substrate, and then removed from the vacuum chamber for 100 °C annealing for 60 min. Then 200 nm CsBr film were deposited on PbBr2 film, and removed from the vacuum chamber for 300 °C annealing for 60 min. Finally, 150 nm PbBr2 film were deposited on CsBr film, and then the PbBr2/CsBr/PbBr2 film was annealed at a heating table at 300 °C for 60 min. Both PbBr2 and CsBr films were deposited under a pressure of ∼8.0 × 10−4 Pa with a speed of 0.2 Å s−1 and all annealing processes were carried out in ambient air. The DTPT was dissolved in isopropyl alcohol with different concentrations and spin-coated on the surface of TiO2 and CsPbBr3, and then heating at 100 °C in ambient air for 10 min to remove residual solvent. At last, the conductive carbon paste was blade-coated on the CsPbBr3 film and heated at 100 °C for 10 min.Characterization
The scanning electron microscopy (SEM) images are taken by a field emission SEM (Sigma-500, Zeiss). X-ray diffraction (XRD) was carried out using the Cu Kα radiation (Empyrean, Netherlands). Transmission electron microscopy (TEM) was carried out using a FEI Talos F200X microscope with a field-emission source operating at 200 kV. The current density–voltage (J–V) curves of the devices were performed by using a Keithley 2400 system source meter under AM 1.5G simulated solar illumination. The incident photon-to-electron conversion efficiency (IPCE) was measured using Newport-74125 system (Newport Instrument). X-ray photoelectron spectroscopy (XPS) with Al Kα X-rays (1486.6 eV) and ultraviolet photoelectron spectroscopy (UPS) were performed using AXIS Supra (Kratos, England) under a vacuum of 1 × 10−9 Torr. The XPS spectra was calibrated using the C 1s binding energy of adventitious carbon (AdC) and the sample-work-function (ϕSA) method by Greczynski and Hultman.53 The UPS was carried out using the He I (hν = 21.22 eV), was calibrated with a standard Au film (ϕ = 5.3 eV) before starting work function measurements.54 UV-vis-NIR fluorescence spectrophotometer absorption spectra were performed by a Shimadzu UV 3600 spectrophotometer. Steady-state photoluminescence (PL) were tested by self-built fluorescence spectrometer, and the excitation wavelength was 400 nm. TRPL was obtained by an Optronis Optoscope streak camera by using 400 nm pulse laser. The broadband femtosecond TA spectra of the CsPbBr3 films were taken using the Ultrafast System HELIOS TA spectrometer. The 400 nm laser pulses were generated by passing the strong 800 nm femtosecond laser beam through a beta barium borate crystal. All electrochemical tests are performed by the CHI760E electrochemical station of Shanghai Chenhua.Computational details
DFT calculations are performed using the Vienna Ab Initio Simulation Packages (VASP) with the generalized gradient approximation (GGA) proposed by Perdew–Burke–Ernzerhof (PBE).55 The structure relaxations are carried out with a 450 eV plane-wave cutoff. The Brillouin zone integration was sampled following a gamma-centered Monkhorst–Pack scheme, using 1 × 1 × 1 k-point grids for the ionic optimization and 3 × 3 × 3 k-point grids for the density of state, respectively. The self-consistent total-energy difference and the convergence criterion for forces on atoms are set to 10−4 eV and 0.01 eV Å−1, respectively. In all of the calculations, the positions of the two atomic layers at the bottom of the slab were kept fixed at the relaxed bulk positions, while the first two layers of the top face, which was the surface under investigation, were fully relaxed. We used a vacuum over 10 Å to adequately separate images along the surface normal direction.Statistical analysis
Origin software (2018, OriginLab) was used for statistical analysis. Descriptive statistics was performed to evaluate efficiency of different devices, such as the efficiency statistics of devices with and without modification on TiO2 layer and the efficiency statistics of devices after different concentrations of ILs modified CsPbBr3 films. The Shapiro–Wilk test was used to test normality of the distribution (p > 0.05) by using 40 individual devices of each type.Conclusions
In this work, we have identified DTPT as dual-functional IL for both effective passivation of surficial/interfacial defects of the inorganic CsPbBr3 perovskite phase, and effective suppression of the nucleation of impurity phases. Such synergistic effects enable large-area vapour deposition of low-defect and phase pure CsPbBr3 films, and thus effectively mitigate the infamous high-defect problem of vapour deposited perovskite films.The highly improved material quality owing to DTPT treatment is fundamental to achieving the highest efficiencies even without the use of expensive organic hole-transfer layer and precious metal electrode. Over 14% improvement of the PCE (9.83% to the highest 11.21%) was achieved on usual laboratory cells (0.04 cm2), and more encouragingly, an average efficiency of 9.18% was also realized for large-area (1 cm2) PSC cells. In addition, the DTPT treatment also helps improve the environmental stability of devices because of its remarkable hydrophobicity, so that exposed devices could maintain 91% of its initial efficiency after being placed in air with a relative humidity about 55% for 100 days.
The current effort towards a much-simplified photovoltaic architecture and successful vapour deposition of low-defect inorganic perovskite films provide a significant step forward for low-cost production of inorganic perovskite solar cells with an established manufacturing facility.
Author contributions
G. X. and G. S. provided facilities, funding and supervised the overall research work. R. G. and C. L. designed and conducted the experiments, analysis and characterization. J. X. provided support in simulation and discussion. C. L. and H. G. provided optical characterization and analysis. Y. Z. participated in the preparation of CsPbBr3 films and devices. X. M. and Z. W. provided SEM images. J. L. and Y. D. participated in the debugging and maintenance of the instrument. C. L. and Y. S. provided with mechanism analysis and discussion. All authors discussed the results and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The work is supported by the National Natural Science Foundation of China (no. 51602290), The Key R&D and Promotion Project of Henan Province (212102210600), Science and Technology Development Fund, Macao SAR (file no. FDCT-0044/2020/A1, FDCT-014/2017/AMJ), UM's Research Fund (file no. MYRG2018-00148-IAPME, MYRG2020-00151-IAPME), Natural Science Foundation of Guangdong Province, China (2019A1515012186), Guangdong-Hong Kong-Macao Joint Laboratory of Optoelectronic and Magnetic Functional Materials (2019B121205002), and Shenzhen-Hong Kong-Macao Science and Technology Innovation Project (Category C) (SGDX2020110309360100).Notes and references
- A. Kojima, K. Teshima, Y. Shirai and T. Miyasaka, J. Am. Chem. Soc., 2009, 131, 6050–6051 CrossRef CAS PubMed.
- National Renewable Energy Laboratory (NREL), 2021, https://www.nrel.gov/pv/cell-efficiency.html.
- M. Shahiduzzaman, E. Y. Muslih, A. K. M. Hasan, L. Wang, S. Fukaya, M. Nakano, M. Karakawa, K. Takahashi, M. Akhtaruzzaman, J.-M. Nunzi and T. Taima, Chem. Eng. J., 2021, 411, 128461 CrossRef CAS.
- G. Zhang, P. Xie, Z. Huang, Z. Yang, Z. Pan, Y. Fang, H. Rao and X. Zhong, Adv. Funct. Mater., 2021, 31, 2011187 CrossRef CAS.
- J. Liang, C. Wang, Y. Wang, Z. Xu, Z. Lu, Y. Ma, H. Zhu, Y. Hu, C. Xiao, X. Yi, G. Zhu, H. Lv, L. Ma, T. Chen, Z. Tie, Z. Jin and J. Liu, J. Am. Chem. Soc., 2016, 138, 15829–15832 CrossRef CAS PubMed.
- J. Duan, Y. Zhao, X. Yang, Y. Wang, B. He and Q. Tang, Adv. Energy Mater., 2018, 8, 1802346 CrossRef.
- Q. Zhou, J. Duan, J. Du, Q. Guo, Q. Zhang, X. Yang, Y. Duan and Q. Tang, Adv. Sci., 2021, 8, 2101418 CrossRef CAS PubMed.
- G. Tong, T. Chen, H. Li, L. Qiu, Z. Liu, Y. Dang, W. Song, L. K. Ono, Y. Jiang and Y. Qi, Nano Energy, 2019, 65, 104015 CrossRef CAS.
- M. Liu, M. B. Johnston and H. J. Snaith, Nature, 2013, 501, 395–398 CrossRef CAS PubMed.
- G. Tong, H. Li, G. Li, T. Zhang, C. Li, L. Yu, J. Xu, Y. Jiang, Y. Shi and K. Chen, Nano Energy, 2018, 48, 536–542 CrossRef CAS.
- Y. Zhao, J. Duan, Y. Wang, X. Yang and Q. Tang, Nano Energy, 2020, 67, 104286 CrossRef CAS.
- C. Liang, H. Gu, Y. Xia, Z. Wang, X. Liu, J. Xia, S. Zuo, Y. Hu, X. Gao, W. Hui, L. Chao, T. Niu, M. Fang, H. Lu, H. Dong, H. Yu, S. Chen, X. Ran, L. Song, B. Li, J. Zhang, Y. Peng, G. Shao, J. Wang, Y. Chen, G. Xing and W. Huang, Nat. Energy, 2020, 6, 38–45 CrossRef.
- T. Niu, L. Chao, W. Gao, C. Ran, L. Song, Y. Chen, L. Fu and W. Huang, ACS Energy Lett., 2021, 6, 1453–1479 CrossRef CAS.
- S. Bai, P. Da, C. Li, Z. Wang, Z. Yuan, F. Fu, M. Kawecki, X. Liu, N. Sakai, J. T. Wang, S. Huettner, S. Buecheler, M. Fahlman, F. Gao and H. J. Snaith, Nature, 2019, 571, 245–250 CrossRef CAS PubMed.
- Y.-H. Lin, N. Sakai, P. Da, J. Wu, H. C. Sansom, A. J. Ramadan, S. Mahesh, J. Liu, R. D. J. Oliver, J. Lim, L. Aspitarte, K. Sharma, P. K. Madhu, A. B. Morales-Vilches, P. K. Nayak, S. Bai, F. Gao, C. R. M. Grovenor, M. B. Johnston, J. G. Labram, J. R. Durrant, J. M. Ball, B. Wenger, B. Stannowski and H. J. Snaith, Science, 2020, 369, 96–102 CrossRef CAS PubMed.
- J. Xia, C. Liang, S. Mei, H. Gu, B. He, Z. Zhang, T. Liu, K. Wang, S. Wang, S. Chen, Y. Cai and G. Xing, J. Mater. Chem. A, 2021, 9, 2919–2927 RSC.
- A. Wang, X. Deng, J. Wang, S. Wang, X. Niu, F. Hao and L. Ding, Nano Energy, 2021, 81, 105631 CrossRef CAS.
- C. Altinkaya, E. Aydin, E. Ugur, F. H. Isikgor, A. S. Subbiah, M. D. Bastiani, J. Liu, A. Babayigit, T. G. Allen, F. Laquai, A. Yildiz and S. D. Wolf, Adv. Mater., 2021, 33, 2005504 CrossRef CAS PubMed.
- Q. Jiang, L. Zhang, H. Wang, X. Yang, J. Meng, H. Liu, Z. Yin, J. Wu, X. Zhang and J. You, Nat. Energy, 2016, 2, 16177 CrossRef.
- W. Ke, G. Fang, Q. Liu, L. Xiong, P. Qin, H. Tao, J. Wang, H. Lei, Bo. Li, J. Wan, G. Yang and Y. Yan, J. Am. Chem. Soc., 2015, 137, 6730–6733 CrossRef CAS PubMed.
- Y. Zhang, X. Liu, P. Li, Y. Duan, X. Hu, F. Li and Y. Song, Nano Energy, 2019, 56, 733–740 CrossRef CAS.
- K. C. Tang, P. You and F. Yan, Sol. RRL, 2018, 2, 1800075 CrossRef.
- Y. Zhao, Q. Deng, R. Guo, Z. Wu, Y. Li, Y. Duan, Y. Shen, W. Zhang and G. Shao, ACS Appl. Mater. Interfaces, 2020, 12, 54904–54915 CrossRef CAS PubMed.
- X. Wang, Y. Wang, Y. Chen, X. Liu and Y. Zhao, Adv. Mater., 2021, 33, 2103688 CrossRef CAS PubMed.
- D. Yang, X. Li, W. Zhou, S. Zhang, C. Meng, Y. Wu, Y. Wang and H. Zeng, Adv. Mater., 2019, 31, 1900767 CrossRef PubMed.
- X. Li, Y. Wu, S. Zhang, B. Cai, Y. Gu, J. Song and H. Zeng, Adv. Funct. Mater., 2016, 26, 2435–2445 CrossRef CAS.
- Y.-H. Kim, J. S. Kim and T.-W. Lee, Adv. Mater., 2019, 31, 1804595 CrossRef CAS PubMed.
- W. Xu, Q. Hu, S. Bai, C. Bao, Y. Miao, Z. Yuan, T. Borzda, A. J. Barker, E. Tyukalova, Z. Hu, M. Kawecki, H. Wang, Z. Yan, X. Liu, X. Shi, K. Uvdal, M. Fahlman, W. Zhang, M. Duchamp, J.-M. Liu, A. Petrozza, J. Wang, L.-M. Liu, W. Huang and F. Gao, Nat. Photonics, 2019, 13, 418–424 CrossRef CAS.
- S. Yun, X. Zhou, J. Even and A. Hagfeldt, Angew. Chem., Int. Ed., 2017, 56, 15806–15817 CrossRef CAS PubMed.
- H. Huang, M. I. Bodnarchuk, S. V. Kershaw, M. V. Kovalenko and A. L. Rogach, ACS Energy Lett., 2017, 2, 2071–2083 CrossRef CAS PubMed.
- X. Zheng, Y. Hou, C. Bao, J. Yin, F. Yuan, Z. Huang, K. Song, J. Liu, J. Troughton, N. Gasparini, C. Zhou, Y. Lin, D.-J. Xue, B. Chen, A. K. Johnston, N. Wei, M. N. Hedhili, M. Wei, A. Y. Alsalloum, P. Maity, B. Turedi, C. Yang, D. Baran, T. D. Anthopoulos, Y. Han, Z.-H. Lu, O. F. Mohammed, F. Gao, E. H. Sargent and O. M. Bakr, Nat. Energy, 2020, 5, 131–140 CrossRef CAS.
- S. Tombe, G. Adam, H. Heilbrunner, D. H. Apaydin, C. Ulbricht, N. S. Sariciftci, C. J. Arendse, E. Iwuoha and M. C. Scharber, J. Mater. Chem. C, 2017, 5, 1714–1723 RSC.
- S. Sun, T. Salim, N. Mathews, M. Duchamp, C. Boothroyd, G. Xing, T. C. Sum and Y. M. Lam, Energy Environ. Sci., 2014, 7, 399–407 RSC.
- G. Mannino, I. Deretzis, E. Smecca, A. La Magna, A. Alberti, D. Ceratti and D. Cahen, J. Phys. Chem. Lett., 2020, 11, 2490–2496 CrossRef CAS PubMed.
- Y. Wei, Y. Zhao, C. Liu, Z. Wang, F. Jiang, Y. Liu, Q. Zhao, D. Yu and M. Hong, Adv. Funct. Mater., 2021, 31, 2106386 CrossRef CAS.
- D. Shi, V. Adinolfi, R. Comin, M. Yuan, E. Alarousu, A. Buin, Y. Chen, S. Hoogland, A. Rothenberger and K. Katsiev, Science, 2015, 347, 519–522 CrossRef CAS PubMed.
- J. Guo, Z. Shi, J. Xia, K. Wang, Q. Wei, C. Liang, D. Zhao, Z. Zhang, S. Chen, T. Liu, S. Mei, W. Hui, G. Hong, Y. Chen and G. Xing, Small, 2021, 2100560 CrossRef CAS PubMed.
- M. De Bastiani, I. Dursun, Y. Zhang, B. A. Alshankiti, X.-H. Miao, J. Yin, E. Yengel, E. Alarousu, B. Turedi, J. M. Almutlaq, M. I. Saidaminov, S. Mitra, I. Gereige, A. AlSaggaf, Y. Zhu, Y. Han, I. S. Roqan, J.-L. Bredas, O. F. Mohammed and O. M. Bakr, Chem. Mater., 2017, 29, 7108–7113 CrossRef CAS.
- G. Xing, N. Mathews, S. S. Lim, N. Yantara, X. Liu, D. Sabba, M. Gratzel, S. Mhaisalkar and T. C. Sum, Nat. Mater., 2014, 13, 476–480 CrossRef CAS PubMed.
- C. Liang, D. Zhao, P. Li, B. Wu, H. Gu, J. Zhang, T. W. Goh, S. Chen, Y. Chen, Z. Sha, G. Shao, T. C. Sum and G. Xing, Nano Energy, 2019, 59, 721–729 CrossRef CAS.
- S. Wang, Z. Li, Y. Zhang, X. Liu, J. Han, X. Li, Z. Liu, S. Frank Liu and W. C. H. Choy, Adv. Funct. Mater., 2019, 29, 1900417 CrossRef.
- C. Liang, K. M. M. Salim, P. Li, Z. Wang, T. M. Koh, H. Gu, B. Wu, J. Xia, Z. Zhang, K. Wang, T. Liu, Q. Wei, S. Wang, Y. Tang, G. Shao, Y. Song, N. Mathews and G. Xing, J. Mater. Chem. A, 2020, 8, 5874–5881 RSC.
- N. Yarita, H. Tahara, T. Ihara, T. Kawawaki, R. Sato, M. Saruyama, T. Teranishi and Y. Kanemitsu, J. Phys. Chem. Lett., 2017, 8, 1413–1418 CrossRef CAS PubMed.
- Q. Zhou, J. Duan, X. Yang, Y. Duan and Q. Tang, Angew. Chem., Int. Ed., 2020, 59, 21997–22001 CrossRef CAS PubMed.
- X. Zhu, M. Du, J. Feng, H. Wang, Z. Xu, L. Wang, S. Zuo, C. Wang, Z. Wang, C. Zhang, X. Ren, S. Priya, D. Yang and S. F. Liu, Angew. Chem., Int. Ed., 2021, 60, 4238–4244 CrossRef CAS PubMed.
- W. Zhang, X. Liu, B. He, Z. Gong, J. Zhu, Y. Ding, H. Chen and Q. Tang, ACS Appl. Mater. Interfaces, 2020, 12, 4540–4548 CrossRef CAS PubMed.
- G. Shao, Energy Environ. Mater., 2021, 4, 273–276 CrossRef CAS.
- G. Tong, T. Chen, H. Li, W. Song, Y. Chang, J. Liu, L. Yu, J. Xu, Y. Qi and Y. Jiang, Sol. RRL, 2019, 3, 1900030 CrossRef.
- N. Ahn, K. Kwak, M. S. Jang, H. Yoon, B. Y. Lee, J. K. Lee, P. V. Pikhitsa, J. Byun and M. Choi, Nat. Commun., 2016, 7, 13422 CrossRef CAS PubMed.
- Q. Wang, B. Chen, Y. Liu, Y. Deng, Y. Bai, Q. Dong and J. Huang, Energy Environ. Sci., 2017, 10, 516–522 RSC.
- X. Deng, L. Xie, S. Wang, C. Li, A. Wang, Y. Yuan, Z. Cao, T. Li, L. Ding and F. Hao, Chem. Eng. J., 2020, 398, 125594 CrossRef CAS.
- Y. Li, J. Duan, H. Yuan, Y. Zhao, B. He and Q. Tang, Sol. RRL, 2018, 2, 1800164 CrossRef.
- G. Greczynski and L. Hultman, Angew. Chem., Int. Ed., 2020, 132, 5034–5038 CrossRef.
- G. Greczynski and L. Hultman, Appl. Surf. Sci., 2018, 451, 99–103 CrossRef CAS.
- H. J. Monkhorst and J. D. Pack, Phys. Rev. B: Solid State, 1976, 13, 5188–5192 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ta06092b |
| ‡ The authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2023 |