
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCo-Al-CO3 layered double hydroxide: an efficient and regenerable catalyst for glycolysis of polyethylene terephthalate†
Deepthi
Thomas
 ab,
Rakesh
Ranjan
a and
Benny Kattikanal
George
*c
ab,
Rakesh
Ranjan
a and
Benny Kattikanal
George
*c
aAnalytical and Spectroscopy Division, Vikram Sarabhai Space Centre, Indian Space Research Organization, Thiruvananthapuram 695022, Kerala, India
bDepartment of Applied Chemistry, Cochin University of Science and Technology, Cochin 682022, India
cAnalytical, Spectroscopy and Ceramics Group, Vikram Sarabhai Space Centre, Indian Space Research Organization, Thiruvananthapuram 695022, India. E-mail: bkgeorge63@gmail.com
First published on 16th October 2023
Abstract
Polyethylene terephthalate (PET) is one among the common polymers we use in our day-to-day lives. Despite its wide range of applications, recycling of PET waste is a serious concern due to its non-biodegradability. This paper deliberates the use of Co-Al-CO3 layered double hydroxide (LDH) as a catalyst for the glycolysis of PET and the mechanistic aspects of catalysis. Co-Al-CO3 LDH showed superior properties compared to similar LDH materials. 100% PET conversion and 96% yield for bis hydroxy ethylene terephthalate (BHET) were achieved within a reaction time of 2 hours, at a reaction temperature of 180 °C and with a catalyst concentration of 1%. Glycolysis conditions such as reaction temperature, time, and ethylene glycol (EG)/PET ratio were optimized. The effect of M+2/M+3 on BHET yield was studied, and an increase in BHET yield was observed up to a ratio of 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. The replacement of Co2+, either completely or partially with another M2+, resulted in a significant decrease in BHET yield. The catalysis mechanism of Co-Al-CO3 LDH was explained by correlating the decarbonation temperature of the carbonate anion with catalytic performance. Magnetically separable CoAl31@Fe3O4 was prepared with a BHET yield of 99%. Regeneration was demonstrated up to 4 cycles and a BHET yield of 86% was achieved in the fourth cycle.
:1. The replacement of Co2+, either completely or partially with another M2+, resulted in a significant decrease in BHET yield. The catalysis mechanism of Co-Al-CO3 LDH was explained by correlating the decarbonation temperature of the carbonate anion with catalytic performance. Magnetically separable CoAl31@Fe3O4 was prepared with a BHET yield of 99%. Regeneration was demonstrated up to 4 cycles and a BHET yield of 86% was achieved in the fourth cycle.
Sustainability spotlightRecycling resources are essential for a carbon-neutral society. Especially non-biodegradable polymers such as Polyethylene terephthalate (PET) pose serious environmental concerns of plastic waste accumulation. Though glycolysis is a commercially viable route for PET recycling, the reaction is very sluggish in the absence of a catalyst. This work reports a reusable layered double hydroxide catalyst ‘Co-Al-LDH’ with a high bishydroxy ethylene terephthalate (BHET) yield of 96%. With the use of our new catalyst, we envisage glycolysis as a closed-loop reaction where along with the feedstock recycling of PET, the catalyst and reactant (ethylene glycol) can be regenerated and reused to realize the “Responsible Consumption and Production” of the Sustainable Development Goals (SDGs). |
1. Introduction
Polyethylene terephthalate contributes about 8% of the world's total plastic production and is the fourth most produced polymer after polyethylene, polypropylene, and polyvinyl chloride.1 Advantages like non-toxicity, transparency, and durability made it widely useful for producing mineral water bottles, soft drink bottles, packaging, fabrics, and films. As the major uses of PET are for disposable items, its recycling should be addressed seriously to reduce solid waste production. The non-biodegradable nature of PET aggravates the environmental pollution issues and currently, PET contributes about 12% by volume of the world's total solid waste production.2Mainly two approaches are used for recycling PET, mechanical recycling and chemical or feedstock recycling. Chemical recycling, where the polymer is chemically depolymerized into commercially valuable monomer/oligomeric molecules, is considered the most sustainable way of recycling.3–5 Glycolysis is an established method for the chemical recycling of PET, where the PET molecule is depolymerized using ethylene glycol to its monomer bis hydroxy ethylene terephthalate. BHET can be further utilized for PET production or other materials like polyurethanes and acrylate coatings.3
PET glycolysis is a slow process in the absence of a catalyst and often results in partially glycolyzed products instead of BHET. Catalysts reported for PET glycolysis can be categorized into metal derivatives,6–16 ionic liquid (IL) based,17–19 deep eutectic solvent (DES)20–24 and organic catalysts.25–30 Metal-based catalysts include metal salts,31,32 metal oxides,8,33–36 metal–organic frameworks (MOFs),37 metal nanoparticles,6,14–16,38 metal oxide doped graphene11,39, CNTs40 and layered double hydroxides (LDHs).41 Ionic liquids have emerged as green catalysts, but they suffer from low BHET yield and difficulty in catalyst regeneration.17,42,43 Nanocatalysts have received recent attention due to their intrinsic properties, promoting the catalysis process. Sodium and zinc titanate nanotubes,44,45 ultra-small cobalt nanoparticles,9 γ-Fe2O3/N-doped graphene,46 boron nitride nanosheets (h-BNNS) decorated with Fe3O4 nanoparticles,7 MnO2/graphene oxide nanosheets,11 magnetic Mg-Al-O@Fe3O4 microparticles47 and Fe3O4 nanodispersions15 are some of the nanocatalysts reported for PET glycolysis. 100% BHET yield is reported for (h-BNNS) decorated with Fe3O4 nanoparticles at 200 °C in an autoclave when reacted for 5 h.7 100% BHET gain was also achieved by γ-Fe2O3/N-doped graphene at 190 °C and 1.1 bar pressure.46 Magnetic Fe3O4 nanoparticles prepared by co-precipitation14 are the latest additions to this group and the authors have reported a BHET yield of 93% at 195 °C for 2 h. High BHET yields are reported for most nanocatalysts, but high reaction temperature and pressure, and complex synthesis methods are drawbacks for most of them.
Cobalt-containing catalysts such as ultra-small cobalt nanoparticles,9 cobalt oxide from spent lithium-ion batteries,48 magnetic nanoparticles Co Fe2O4,49 and cobalt-based ionic liquids on graphene support50 have been reported as promising catalysts for PET glycolysis with up to 95% BHET yield.
Layered double hydroxides (LDHs) are layered materials with M2+ and M3+ metal hydroxides in the main layers and interlayer spaces containing anionic species.51 LDH materials are promising entrants in the field of catalysis due to their simple synthesis methods, easily tunable properties, surface hydroxyl groups, presence of basic and Lewis acid sites, variability of intercalating anions, and biocompatibility.52–54 The major catalytic applications of LDH materials are as the precursor for mixed metal oxide catalysts. LDH materials are catalysts for aldol and Knoevenagel condensations, Michael reactions, and trans-esterification reactions.55,56 Recently LDH and exfoliated LDH layers gained a lot of scientific attention as photocatalysts for aerobic degradation of pollutants,57,58 water splitting,57,59,60 and CO2 photo-reduction.61 Though hydrotalcite-derived mixed metal oxides are explored as catalysts for PET glycolysis,34,47,62 only limited literature is available on the direct use of LDHs as a catalyst for glycolysis. Chen et al.34 studied the catalytic properties of hydrotalcite with different Mg/Al ratios and Mg–Al mixed oxides derived from them for PET glycolysis. A BHET yield of 66.4% was obtained for Mg-Al-CO3 with an Mg/Al ratio of 3. Eshaq et al.41 presented (Mg–Zn)–Al layered double hydroxide as a regenerable catalyst for PET glycolysis. They could achieve a BHET yield of 75% with 100% PET conversion and the reuse of the catalyst without appreciable loss in efficiency was also reported.
To the best of our knowledge, no reports are available on the utilization of Co-Al-LDH as a transesterification catalyst for PET. This work presents a systematic study on the catalytic activity of Co-Al-LDH for PET glycolysis. Different LDH materials were synthesized by the co-precipitation method and the synthesized materials were characterized and tested for PET glycolysis. Co-Al-LDH which gave the maximum BHET yield was selected for further studies. Glycolysis conditions were optimized and the BHET obtained was characterized by different analytical techniques. The effects of the M2+/M3+ ratio and replacement of Co2+ with other ions on the BHET yield were also explored.
2. Experimental
2.1 Materials
Co (NO3)2·6H2O (Alfa Aesar, 98%), Al(NO3)3·9H2O (Alfa Aesar, 98%), Mg(NO3)2·6H2O, Ni(NO3)2·6H2O (Aldrich, 98%), sodium hydroxide pellets (Alfa Aesar, 98%), Na2CO3 (Alfa Aesar, 99%) and ethylene glycol (Alfa Aesar, 99%) were used as such. BHET (Sigma Aldrich, >98%) was used after recrystallization. PET cut pieces (dimensions – 1–2 mm) were obtained from waste mineral water bottles. HPLC grade methanol (99.9%, Sigma Aldrich) was used for HPLC analysis of the glycolyzed products.2.2 Synthesis of LDH materials
2.3 PET glycolysis31,41
1.0 g of PET pieces obtained from mineral water bottles, 0.01 g (1%) LDH, and 10 ml ethylene glycol were refluxed in a 100 ml two-necked round bottom flask at 180° for 2 hours with a stirring rate of 600 rpm. After 2 hours the temperature was decreased to 100 °C and the unreacted PET that remained in the solution was separated. Then, hot water was added to the system and filtered to separate the catalyst. The filtrate was kept at 4 °C for 12 hours to precipitate BHET. Precipitated BHET was washed with water and dried at 80 °C for 12 Hrs. The dried BHET was characterized by FTIR, FTNMR, DSC, and HPLC techniques.The BHET yield was calculated using HPLC analysis. The products obtained after the glycolysis reaction were filtered to remove the catalyst and insoluble oligomers if any, and the filtrate was made up to 100 ml using HPLC grade methanol. This solution was diluted to a suitable concentration using methanol and injected into the HPLC. The concentration of BHET in the sample was estimated using the calibration graph generated using standard BHET. The BHET yield was calculated as follows:
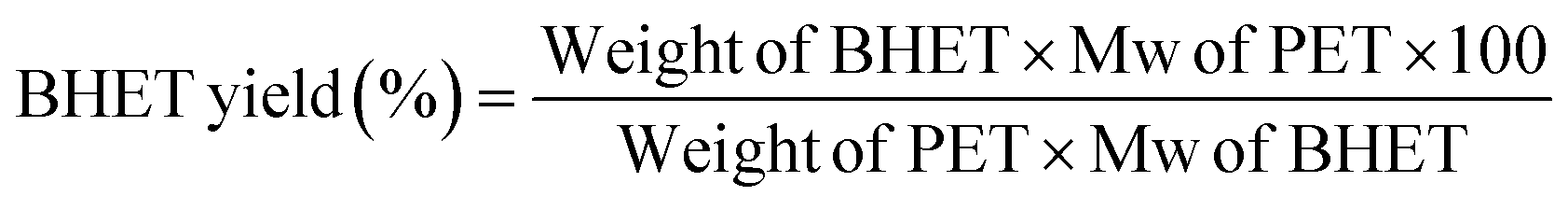
The PET conversion was calculated using the following equation.
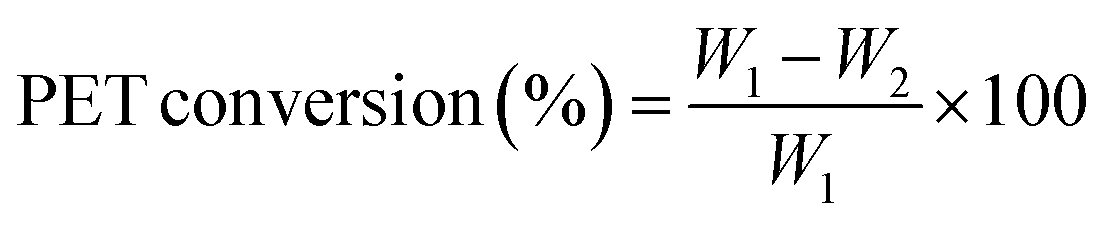
2.4 Characterization
FTIR spectra were recorded using a Nicolet iS50 FTIR spectrometer. The IR spectra of LDH materials and BHET were recorded in the region of 400–4000 cm−1 by pelletizing with KBr. The IR spectrum of PET was taken using an attenuated total reflectance (ATR) accessory. All spectra were recorded with a resolution of 4 cm−1 and 36 scans were accumulated. Scanning electron microscopy (SEM) imaging analysis was carried out using a Carl Zeiss Gemini 500 field emission scanning electron microscope (FESEM) with Bruker detectors. Before analysis samples were given a conductive coating of Au/Pd (80:20). The melting point was found using a TA instruments 2920 DSC. The sample was placed on an aluminum pan and then heated from 25 to 250 °C at a rate of 5 °C min−1 under a N2 flow of 10 ml min−1.
1H and 13C nuclear magnetic resonance (NMR) spectra and 1H NMR spectra were recorded using a Bruker Avance 400 MHz NMR spectrometer. CDCl3 was used as the solvent. X-ray diffraction (XRD) measurements were carried out using a Bruker D8-Discover X-ray diffractometer operating with a Cu anode (40 KV and 40 mA). A PerkinElmer OPTIMA 4300V was used for elemental analysis by inductively coupled plasma atomic emission spectrometry (ICP-AES). High-pressure liquid chromatography (HPLC) analyses were carried out with a PerkinElmer LC 300 UHPLC using methanol as the eluent at a flow rate of 0.3 ml min−1 and a C8-column.
3. Results and discussion
3.1 Characterization of the catalyst
Co-Al-CO3 LDH with a Co2+/Al3+ ratio of 3:1 was synthesized by the co-precipitation method and the material is denoted as Co-Al-31 for further reference. Co-Al-31 was characterized by FTIR spectroscopy, XRD, SEM, and ICP-AES. The XRD pattern shows Bragg reflections of basal planes (003) and (006) as shown in Fig. 1. The d003 spacing of 7.7 Å is characteristic of an LDH with carbonate as the interlayer anion. The unit cell parameters c and a, calculated from d003 and d010 values, are 22.98 Å and 3.08 Å, respectively. These values are typical of a hydrotalcite-type crystal structure. The crystallite size calculated using Scherrer's equation is 10 nm. The FTIR spectrum (Fig. S1†) has peaks at 752, 592, and 547 cm−1, which are attributed to the M–OH stretching modes of Co-OH, Co-(OH)-Al, and Al-OH, respectively. Peaks due to the asymmetric stretching of the carbonate interlayer anion are seen in the region 1354–1360 cm−1. A broad peak at 3400 cm−1 is assigned to the –OH of the LDH layers and a shoulder around 3000 cm−1 is attributed to the –OH stretching of water molecules hydrogen bonded to the intercalated carbonate anion. The surface area of Co-Al-31 evaluated using the BET method is 50.6 m2 g−1. The surface morphology of Co-Al-31 (Fig. 2) studied using SEM revealed that the material contains nanoplatelets of size <50 nm.
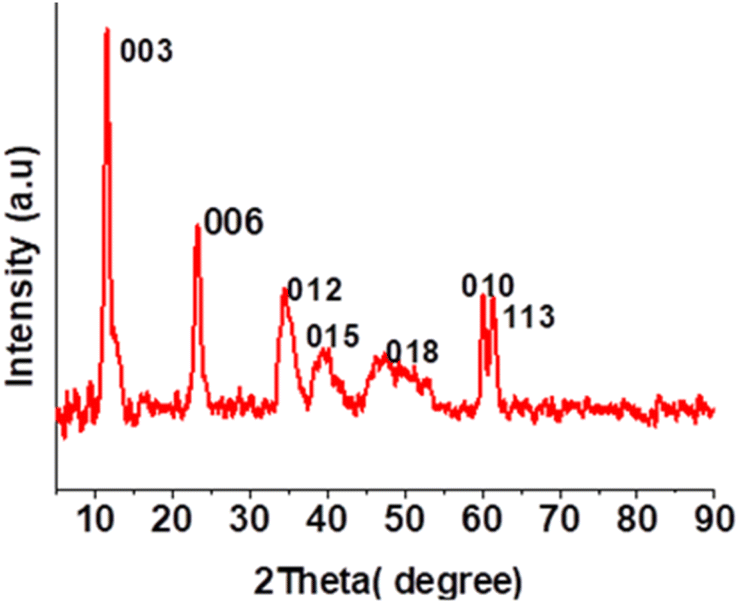 | ||
| Fig. 1 XRD of Co-Al-31. | ||
 | ||
| Fig. 2 FESEM image of Co-Al-31. | ||
3.2 Catalytic activity of Co-Al-31 in PET glycolysis
PET glycolysis was performed at 180 °C for 2 hours, with 1% catalyst and an EG/PET ratio of 10, and the degradation products were analyzed. BHET crystallized out from glycolysate was characterized by FTIR and NMR spectroscopy (Fig. S2–S4†). The signals at 61.3, 67.0, 166.0, 133.9, and 129.7 ppm in 13C NMR are characteristic of the BHET monomer. The absence of the signal at 63.0 ppm corresponding to the BHET dimer also confirmed the purity of the material. The overlaid FTIR spectra (Fig. S2†) of the PET sample used for glycolysis and the separated glycolysis product confirm that PET is converted to BHET. The purity of the separated BHET was verified by differential scanning calorimetry analysis. A sharp endotherm with peak onset at 109 °C is due to the melting of BHET (Fig. 3). HPLC analysis (Fig. 4) of the glycolysate gave a peak at a retention time of 3.5 minutes due to BHET and no dimer peaks are present in the chromatogram.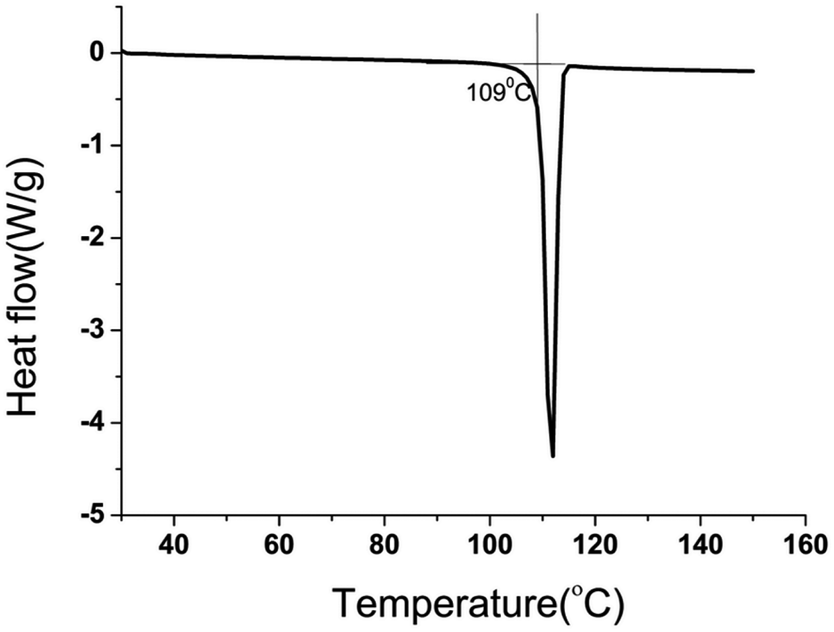 | ||
| Fig. 3 DSC of BHET from glycolysate. | ||
 | ||
| Fig. 4 HPLC of glycolysate and BHET standard. | ||
100% PET conversion and 96% BHET yield were obtained for an EG/PET ratio of 10 and a catalyst concentration of 1% when heated at 180 °C for 2 h. To the best of our knowledge, the BHET yield obtained for CoAl-31 is the highest reported among the hydrotalcite-type materials.
Eshaq et al. have reported a 75% yield for (Mg–Zn)–Al LDH41 and Chen et al. have achieved a 66% yield for Mg–Al hydrotalcites.62
3.3 Optimization of reaction parameters
The effects of different reaction parameters on BHET yield such as EG/PET ratio, temperature, and reaction time were studied. The EG/PET ratio was optimized by varying the ratio from 5 to 20, keeping the reaction temperature at 180 °C, reaction time at 2 hours, and catalyst concentration at 1%. The BHET yield increased with the increase in the EG/PET ratio and reached a maximum at a ratio of 10 (Fig. 5a). An increase in the ratio above 10 decreased the BHET yield. The PET conversion rate and BHET yield increased with an increase in reaction temperature (Fig. 5b). Increase in the BHET yield was observed up to a temperature of 180 °C and a further increase in temperature could cause only a slight increase in the BHET yield. The reaction time is another factor affecting the BHET yield and the BHET yield increased with the reaction time and reached a maximum value at 2 hours (Fig. 5C). Further increase in the reaction time led to a decrease in the BHET yield. This could be due to the re-polymerization from BHET.64 It is noteworthy that around 80% yield was obtained within a short time of one hour. | ||
| Fig. 5 Effect of the (a) EG/PET ratio, (b) reaction time, and (c) reaction temperature on the BHET yield. | ||
3.4 Effect of the M2+/M3+ ratio
Co-Al-LDH materials with –CO32− as the interlayer anion were prepared in different M2+/M3+ ratios (Fig. S5 and Table S1†) and their catalytic activities for the PET glycolysis reaction were studied. These studies revealed an increase in the BHET yield with an increase in the M2+/M3+ ratio (Fig. 6a). An increase in the M2+/M3+ ratio from 1 to 3 resulted in a rise in the BHET yield from 76% to 96%. Further increase in the M2+/M3+ ratio does not cause any considerable change in the BHET yield. | ||
| Fig. 6 Effect of the (a) M2+/M3+ ratio and (b) M2+ ion on the BHET yield. | ||
3.5 Effect of the M2+cation
Cobalt-based catalysts such as cobalt based nanoparticles,9,49,50 CoCl2·6H2O,65 Co3O4,48 mechanochemically synthesized CoFe2O4 (ref. 66) and other mixed metal oxides48,67 were reported for PET glycolysis. A comparison of the BHET yields of cobalt-based catalysts reported in the literature is given in Table S2.† The BHET yield of Co-Al-31 is in par or even better than those of recently reported catalysts (Table S2†). Comparatively low optimum reaction temperature and simple and energy efficient synthesis are additional advantages for Co-Al-31. The dependence of catalytic activity on the nature of the M2+cation was studied by changing M2+ cations while maintaining an M2+/M3+ ratio of 3. Mg-Al-LDH, Co-Mg-Al LDH, Ni-Al-LDH, Zn-Al-LDH and Cu-Al-LDH were prepared under similar experimental conditions used for Co-Al-LDH and characterized using XRD and FTIR (S6). Fig. 6b shows the BHET yields and PET conversion achieved. It is evident from the results that cobalt based LDH has a clear advantage over the rest of the catalysts. The activity is reduced to 88% when Co2+ ions are partially replaced by Mg2+ ions. Mg-Al-LDH also showed considerable activity as reported by Chen et al.34 Good catalytic efficiencies are reported in the literature for many Zn based catalysts36,38,67 and Zn-Al-LDH showed a good BHET yield of 88.6% as expected. Drastic decrease in the PET conversion and BHET yield was observed when Ni-Al-LDH and Cu-Al-LDH were employed as catalysts.3.6 Correlation between the thermal decomposition pattern and catalytic mechanism
The reported mechanism of LDH-catalyzed PET glycolysis involves the synergistic catalysis of Lewis acid and Brønsted basic sites.35 Metal ions act as Lewis acid sites and –OH and –CO32− will act as Brønsted basic sites. According to Liu et al.,68 basic sites in LDHs are the interlayer anions and hydroxyl groups in the layer. For LDH materials with the same interlayer anion, a large difference in catalytic activity was observed upon a change in M2+.In this scenario, an attempt was made to correlate BHET yield with the bonding strength of the interlayer carbonate. The thermal decomposition path of LDH carbonates involves mainly two mechanisms: loss of water by condensation of –OH (dehydroxylation) and loss of carbon dioxide by the decomposition of interlayer carbonates (decarbonation). The decarbonation temperature of the LDH carbonates can be correlated to the bonding strength of the interlayer carbonate.69 Carbonates strongly bonded to the cation layer will decarbonate at a higher temperature compared to weakly bonded carbonates. The decarbonation temperatures of the prepared LDH materials were obtained from TG-MS analysis (Fig. 7 and Table 1).
 | ||
| Fig. 7 CO2 evolution profile of LDHs. | ||
| Catalyst | Peak decarbonation temperature (°C) | |
|---|---|---|
| 1 | Co-Al-LDH | 294 |
| 2 | Co-Mg-Al-LDH | 342 |
| 3 | Ni-Al-LDH | 348 |
| 4 | Zn-Al-LDH | 337 |
| 5 | Mg-Al-LDH | 372 |
| 6 | Cu-Al-LDH | 595 |
The decarbonation temperatures of the prepared LDH materials are in the order Cu2+ > Mg2+ > Ni2+ > Zn2+ > Co2+ which matches with the trend in catalytic efficiency. Thus, a correlation can be established between the decarbonation temperature and the catalytic activity of LDH in glycolysis. Cobalt-based LDH materials having a lower decarbonation temperature showed maximum activity. When Co2+ is partially replaced by Mg2+ ions the peak due to decarbonation splits into two with peaks at 272 °C and 342 °C and the BHET yield decreased to 88%.
Mg-Al-LDH has shown a decarbonation temperature of 372 °C and a BHET yield of 80%. Zn-Al-LDH showed a decarbonation temperature of 337 °C and it also contains a minor amount of strongly bonded carbonates, which decarbonate around 692 °C. This observation justifies the low BHET yield (88.6%) of Zn-Al-LDH compared to Co-Al-LDH. Cu-Al-LDH has strongly bonded carbonates and showed the lowest BHET yield. Ni-Al-LDH is the only mismatch to this trend, where the BHET yield is very low considering its decarbonation temperature of 348 °C. Valente et al.69 also had a similar observation while he was correlating decarbonation temperature with the partial charge on oxygen. SEM analysis of the Ni-Al-LDHs (Fig. S7†) showed aggregation of particles compared to Co-Al-LDH. Even though surface areas are comparable (approx. 50 m2 g−1) for both materials, access to the interlayer carbonate may be denied by agglomeration. The combined effect of particle aggregation and strong bonding of interlayer carbonates might have led to a reduction in the activity.
Though there are many reports70,71 on the increase in catalytic activity with the M2+/M3+ ratio of hydrotalcite-derived mixed metal oxides, similar studies are not available for LDH materials. As per previous measurements by Yun72 and Di Cosimo,73 an increase of the c parameter with increasing Mg/Al molar ratio was observed, which can be correlated to the decreased attractive forces between the brucite-like layers and the interlayer. A decrease in binding energy with an increase in the our study also showed a similar trend for Co-Al-LDH. The M2+/M3+ ratio is also proved by modeling studies.52 As decarbonation temperature is related to the binding strength of the interlayer anion, TG-MS studies are conducted for LDH with varying M2+/M3+ ratios from 1 to 4. Fig. 8 shows the decarbonation profile of LDH samples with a change in the Co2+/Al3+ ratio. TG-MS studies revealed that as the Co2+/Al3+ ratio increases the decarbonation peak shifts towards lower temperature, which in turn increased the catalytic activity. It is interesting to note that, the trend obtained for decarbonation temperature is exactly inverse of that observed for the c-parameter (Fig. 9). This result again confirms the correlation of decarbonation temperature with the bonding strength of interlayer anions and cations in the layer.
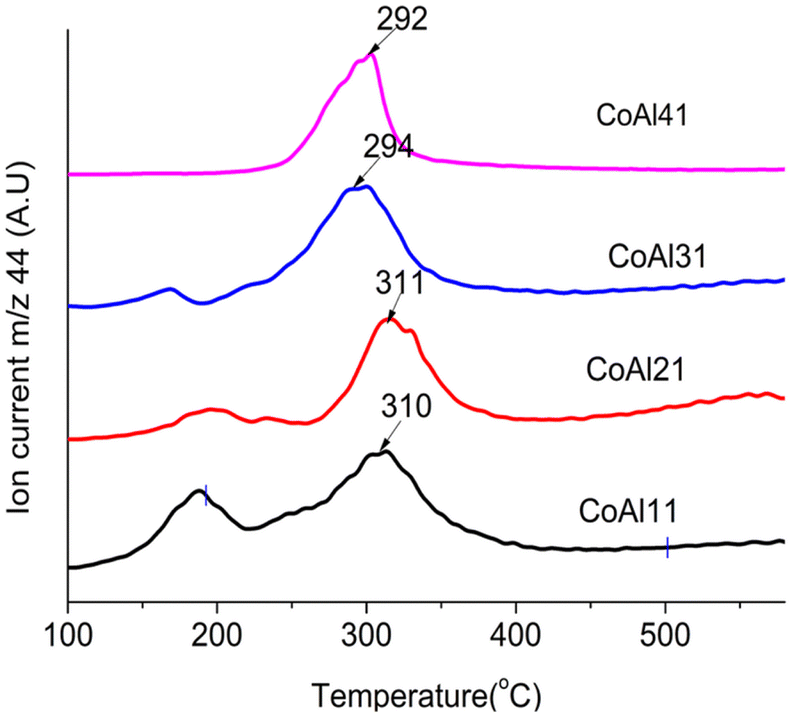 | ||
| Fig. 8 CO2 evolution profile of CoAlCO3-LDH of varying Co2+/Al3+ ratio. | ||
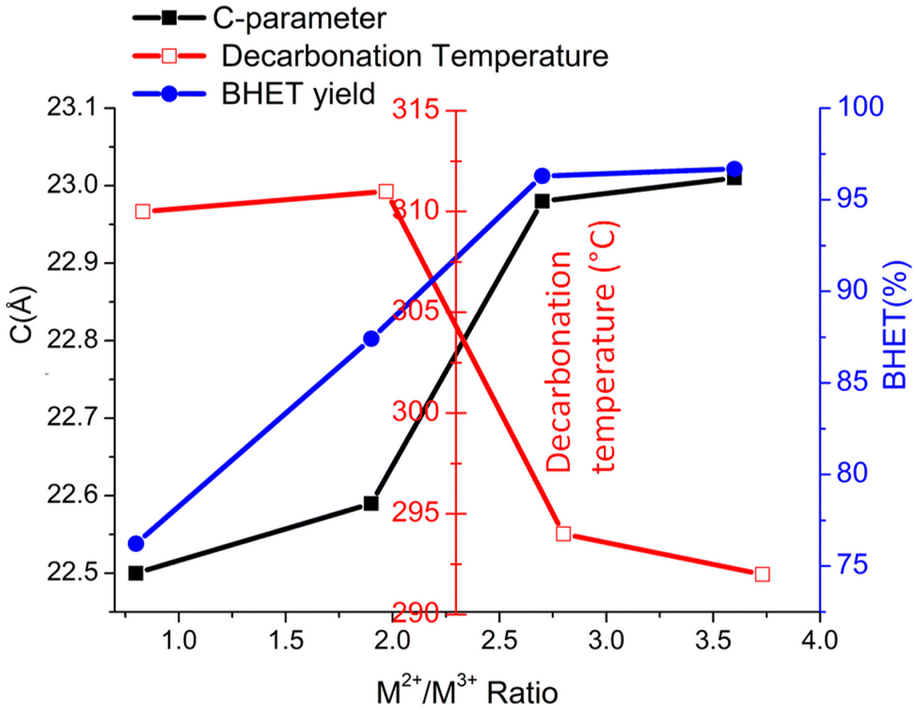 | ||
| Fig. 9 Variation in the decarbonation temperature and c-parameter of CoAlCO3-LDH of varying Co2+/Al3+ anion ratio. | ||
Glycolysis starts with the attack of deprotonated glycol on the electron-deficient carbonyl carbon of PET. Thus, a good catalyst should make the reaction site electron deficient by withdrawing electrons from the carbonyl bond and facilitating the deprotonation of EG. For LDH-CO3, the deprotonation of EG will occur by the abstraction of protons either by the interlayer carbonate or by layer hydroxyl. At the same time the interaction of metal cations in the LDH layers will make the carbonyl carbon electron deficient. If interlayer carbonates are strongly bonded to layer cations, the cation's interaction with the carbonyl group of PET will be weak. Adding to this, strongly bonded interlayer anions will be weak deprotonating agents for EG. Thus, an LDH with weakly bonded interlayer anions will be more efficient compared to a strongly bonded one. As discussed earlier, decarbonation temperature can be correlated with the bonding strength of interlayer anions, so LDH-CO3 with a low decarbonation temperature will be a better catalyst for PET glycolysis. Ethylene glycol can enter the interlayer spaces of LDH, and this enables effective interaction with the interlayer carbonate. A schematic of the mechanism envisaged for catalytic glycolysis is shown in Scheme 1.
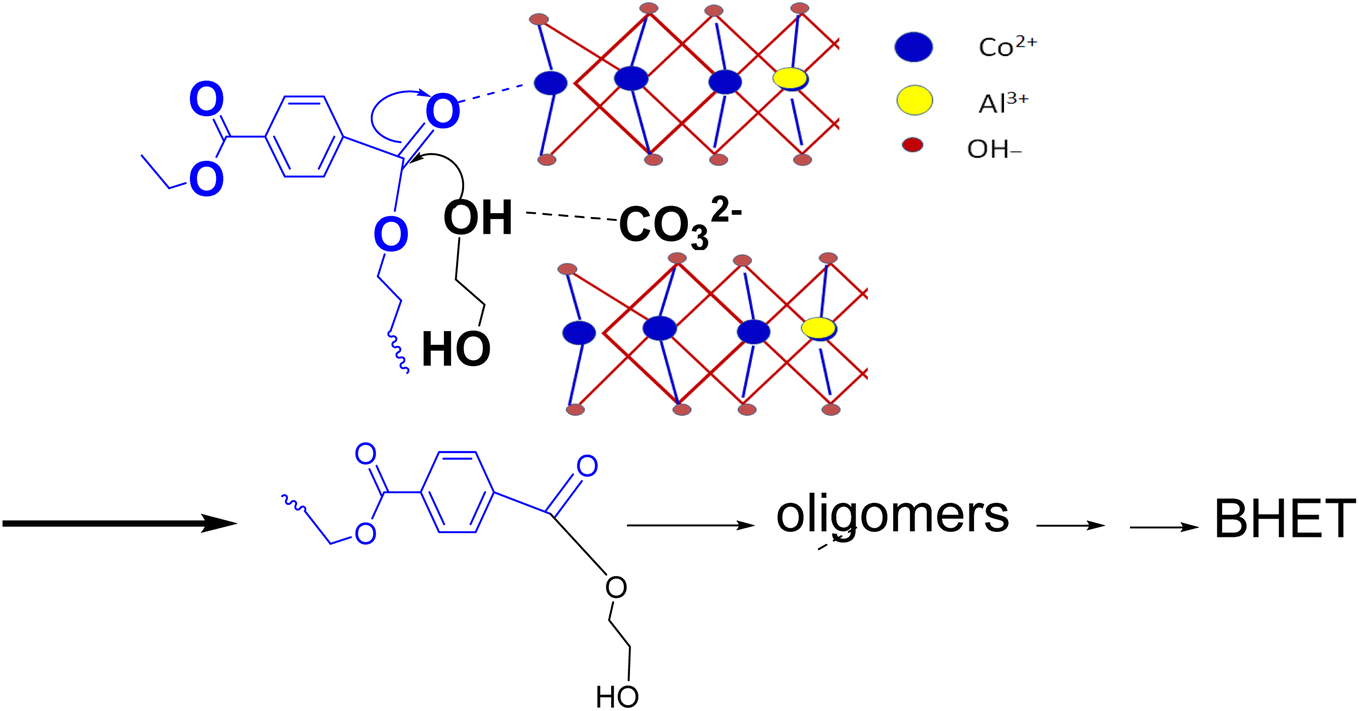 | ||
| Scheme 1 Schematic of the reaction mechanism. | ||
3.7 Regeneration of the catalyst
Co-Al-CO3 catalysts are regenerable and can be reused by filtration. Due to the fine nature of the catalyst direct separation by filtration creates practical difficulties. To overcome this, magnetically separable LDH catalysts were prepared by coprecipitation of cobalt and aluminum nitrates in a suspension of Fe3O4 (ref. 63) keeping a Co2+/Al3+ ratio of 3:1. The catalyst prepared by this method is named Co-Al31@Fe3O4.
Magnetically recoverable catalysts such as paramagnetic ionic liquid-coated SiO2@Fe3O4 nanoparticles,18 boron nitride nanosheets (h-BNNS) decorated with Fe3O4 nanoparticles,7 superparamagnetic γ-Fe2O3 nanoparticles, Fe3O4 nanosuspensions,15 Fe3O4 nanoparticles14 and Mg-Al-O@Fe3O4(ref. 47) are reported for PET glycolysis. In addition to easy separation, good catalytic efficiency was also obtained for most of them.
A comparison of the reported magnetically separable catalysts with Co-Al31@Fe3O4 is given in Table S3.†
Co-Al31@Fe3O4 was characterized by FTIR (Fig. 10A), XRD (Fig. 10B), SEM (Fig. 10C), and ICP analyses (Table S4†). Co-Al31@Fe3O4 has 18% iron as per ICP-AES analysis and the FTIR spectrum shows characteristic peaks of carbonate (1362 cm−1), M–OH (566 and 750 cm−1), and –OH (3500 cm−1). The XRD pattern of Co-Al31@Fe3O4 has characteristic planes of LDH and Fe3O4, but peaks are broadened due to the nanoparticle size of the LDH and ferric oxide.
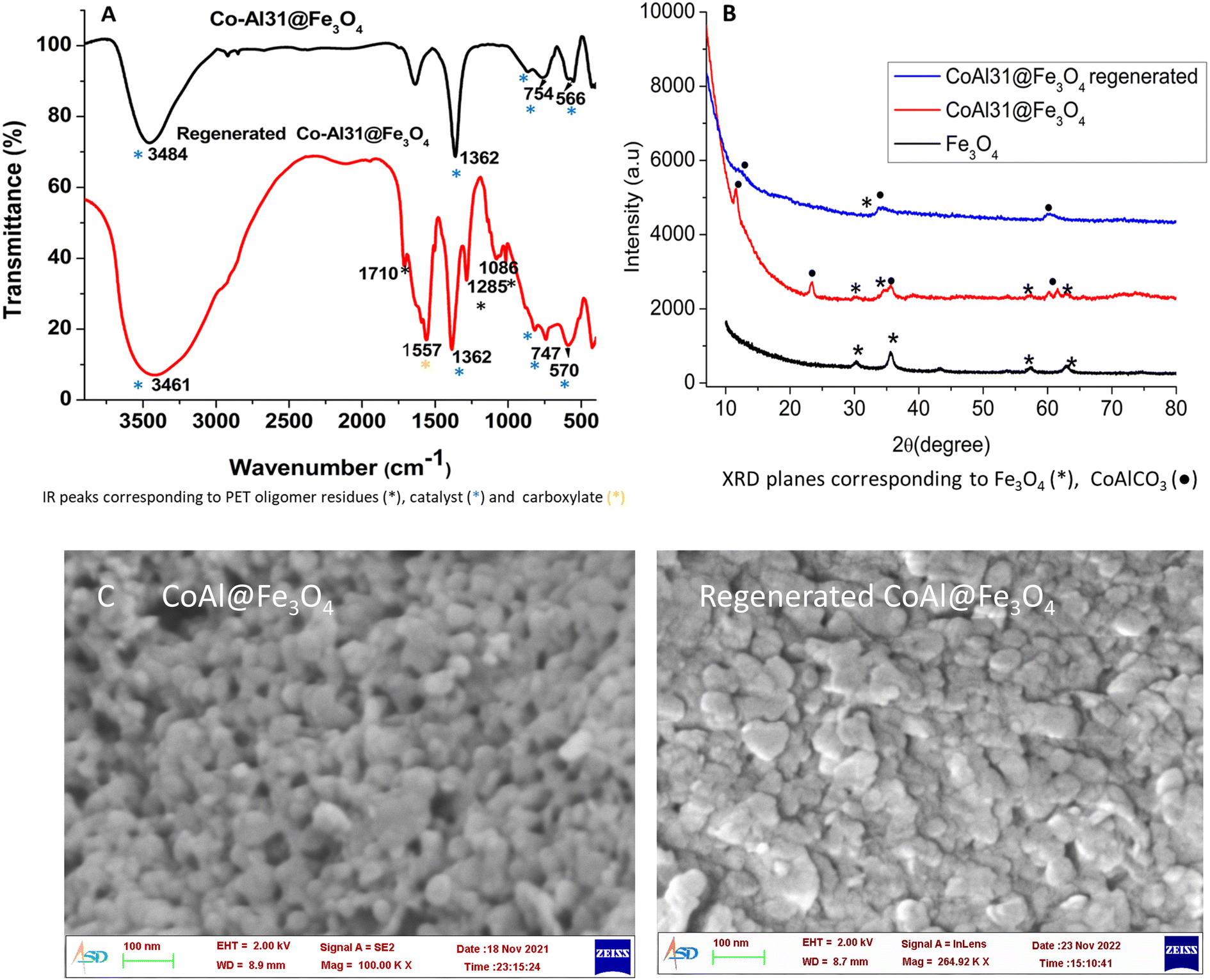 | ||
| Fig. 10 (A) FTIR spectra, (B) XRD, and (C) FESEM images of fresh and regenerated Co-Al31@Fe3O4. | ||
A BHET yield of 99% was obtained with Co-Al31@Fe3O4, which is marginally higher than that obtained for Co-Al-31. As Fe(III) based catalysts are reported to have good catalytic activity, the nano-Fe3O4 used for making Co-Al31@Fe3O4 was checked for its activity. Fe3O4 showed very low PET conversion (2%) and a BHET yield of only 1.6%. The increase in the BHET yield could be attributed to the increase in the surface area when LDH is dispersed on a support.
Four cycles of glycolysis reactions were carried out with Co-Al31@Fe3O4. BHET yields and PET conversion achieved in each cycle are depicted in Fig. 11. A good BHET yield of 99% was obtained for the first cycle and 86% for the 4th cycle. Though complete PET conversion was observed up to the 4th cycle, a steady decrease in the BHET yield was observed.
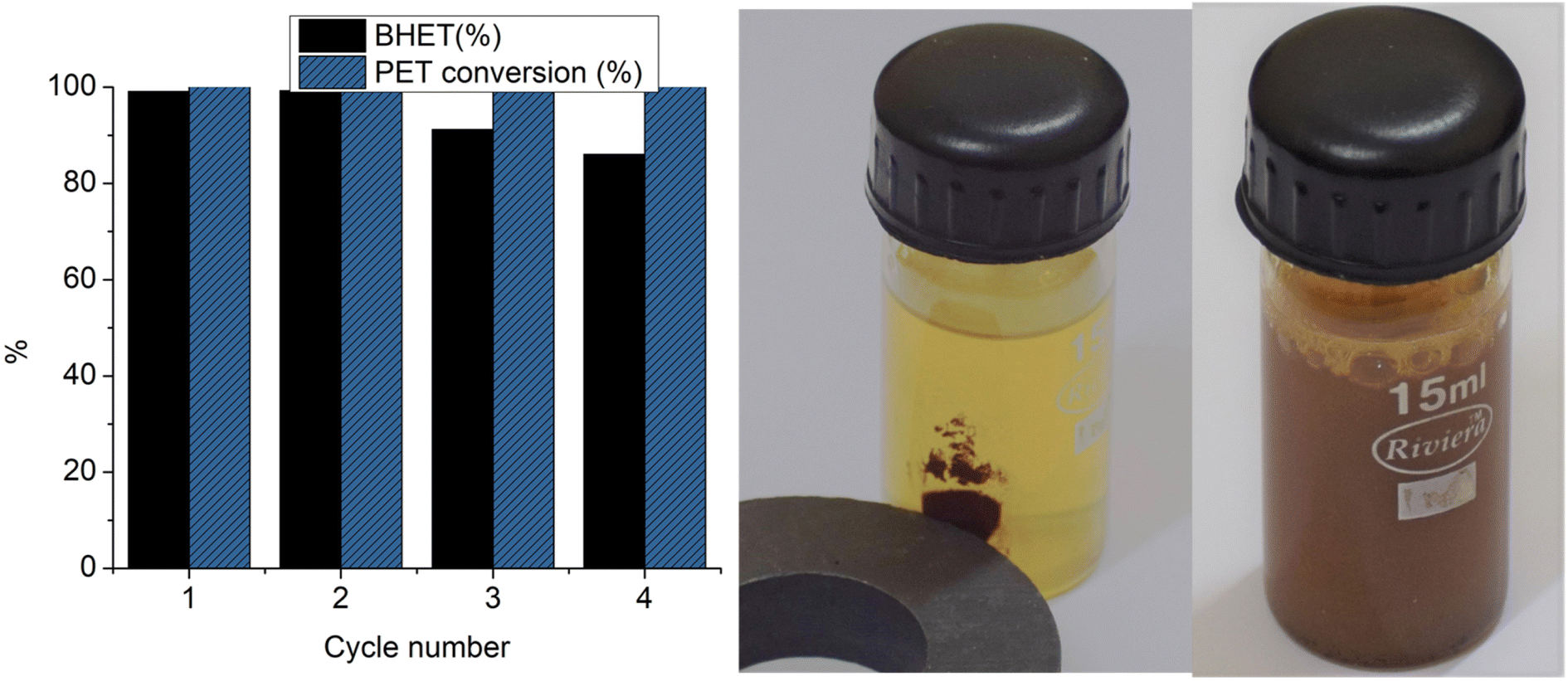 | ||
| Fig. 11 Regeneration cycles of Co-Al31@Fe3O4 and magnetic separation of the catalyst. | ||
The regenerated catalyst was characterized by FTIR, XRD, and SEM analyses (Fig. 10A). The XRD peaks of the regenerated catalyst are further broadened due to the presence of residual PET oligomers in the catalyst (Fig. 10B). The SEM image of the regenerated catalyst has the same morphology as that of the fresh one (Fig. 10C). The FTIR spectrum shows peaks characteristic of LDH at 1362 cm−1 due to interlayer carbonate, 566 and 750 cm−1 due to M–OH, and 3500 cm−1 due to –OH. A peak at 1712 cm−1 is due to residual PET oligomers present in the filtered catalyst. The peak at 1557 cm−1 is characteristic of the carboxylate anion. The presence of a carboxylate anion peak in the FTIR spectrum points towards the possible side reaction of ester hydrolysis. The hydrolysis product, terephthalic acid, can bond with the cations to form acid salts. This could be attributed to the reduction in the activity of recycled catalysts after a few cycles.
4. Conclusions
Co-Al-CO3 layered double hydroxide was reported as a promising catalyst for the recycling of PET by glycolysis. Glycolysis conditions were optimized and a BHET yield of 96% was obtained for CoAl-31 at a comparatively low temperature of 180 °C and reaction time of 2 h. BHET obtained from glycolysis was characterized using various analytical techniques. The catalytic activity decreases when Co2+ ions are completely or partially replaced by other M2+. The BHET yield can be correlated to the bonding strength of the interlayer carbonate, which is in turn related to the decarbonation temperature of LDH. Magnetic regeneration of the material was achieved by in situ formation of LDH in a suspension of nano-Fe3O4. 99% BHET yield and 100% PET conversion were achieved with the catalyst. Four cycles of glycolysis were demonstrated without a decline in PET conversion. However, the BHET yield has gradually decreased to 86% after the fourth cycle. Simple synthesis methods and magnetic regeneration make the catalyst a potential candidate for industrial use in the future.Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the Director, VSSC, for permission to publish this work. Motivation and guidance by Dr R Rajeev and analytical support given by colleagues in the Analytical and Spectroscopy Division, VSSC, are gratefully acknowledged.References
- I. A. Ignatyev, W. Thielemans and B. Vander Beke, ChemSusChem, 2014, 7, 1579–1593 CrossRef CAS PubMed.
- A. Rahimi and J. M. García, Nat. Rev. Chem, 2017, 1, 1–11 CrossRef.
- R. Francis, Recycling of Polymers: Methods, Characterization and Applications, John Wiley & Sons, 2016 Search PubMed.
- J. Aguado and D. P. Serrano, Feedstock Recycling of Plastic Wastes, Royal society of chemistry, 2007 Search PubMed.
- B. Geyer, G. Lorenz and A. Kandelbauer, eXPRESS Polym. Lett., 2016, 10, 559–586 CrossRef CAS.
- L. C. S. H. Rezende, J. H. de Oliveira, V. P. M. Zart, M. P. Moisés, G. P. Otto and S. L. Fávaro, Acta Sci., Technol., 2019, 41, e37303 CrossRef.
- M. R. Nabid, Y. Bide and M. Jafari, Polym. Degrad. Stab., 2019, 169, 108962 CrossRef CAS.
- L. Bartolome, M. Imran, K. G. Lee, A. Sangalang and J. K. Ahn, Green Chem., 2014, 16, 279–286 RSC.
- F. R. Veregue, C. T. Pereira da Silva, M. P. Moisés, J. G. Meneguin, M. R. r. Guilherme, P. A. Arroyo, S. L. Favaro, E. Radovanovic, E. M. Girotto and A. W. Rinaldi, ACS Sustain. Chem. Eng., 2018, 6, 12017–12024 CrossRef CAS.
- Z. Guo, M. Eriksson, H. de la Motte and E. Adolfsson, J. Cleaner Prod., 2021, 283, 124579 CrossRef CAS.
- S. B. Jin, J.-M. Jeong, S. G. Son, S. H. Park, K. G. Lee and B. G. Choi, Mater. Today Commun., 2021, 26, 101857 CrossRef CAS.
- J. Cao, Y. Lin, W. Jiang, W. Wang, X. Li, T. Zhou, P. Sun, B. Pan, A. Li and Q. Zhang, ACS Sustain. Chem. Eng., 2022, 10, 5476–5488 CrossRef CAS.
- S. Shirazimoghaddam, I. Amin, J. A. Faria Albanese and N. R. Shiju, ACS Eng. Au, 2023, 3, 37–44 CrossRef CAS PubMed.
- Y. Jo, E. J. Kim, J. Kim and K. An, Green Chem., 2023, 25, 8160–8171 RSC.
- Q. Sun, Y.-Y. Zheng, L.-X. Yun, H. Wu, R.-K. Liu, J.-T. Du, Y.-H. Gu, Z.-G. Shen and J.-X. Wang, ACS Sustain. Chem. Eng., 2023, 11, 7586–7595 CrossRef CAS.
- L.-X. Yun, Y. Wei, Q. Sun, Y.-T. Li, B. Zhang, H.-T. Zhang, Z.-G. Shen and J.-X. Wang, Green Chem., 2023, 25, 6901–6913 RSC.
- A. Al-Sabagh, F. Yehia, G. Eshaq and A. ElMetwally, Ind. Eng. Chem. Res., 2015, 54, 12474–12481 CrossRef CAS.
- I. Cano, C. Martin, J. A. Fernandes, R. W. Lodge, J. Dupont, F. A. Casado-Carmona, R. Lucena, S. Cardenas, V. Sans and I. de Pedro, Appl. Catal., B, 2020, 260, 118110 CrossRef CAS.
- M. Mohan, J. D. Keasling, B. A. Simmons and S. Singh, Green Chem., 2022, 24, 4140–4152 RSC.
- S. Choi and H.-M. Choi, Fibers Polym., 2019, 20, 752–759 CrossRef CAS.
- B. Liu, W. Fu, X. Lu, Q. Zhou and S. Zhang, ACS Sustain. Chem. Eng., 2018, 7, 3292–3300 CrossRef.
- E. Sert, E. Yılmaz and F. S. Atalay, J. Polym. Environ., 2019, 27, 2956–2962 CrossRef CAS.
- Q. Wang, X. Yao, Y. Geng, Q. Zhou, X. Lu and S. Zhang, Green Chem., 2015, 17, 2473–2479 RSC.
- R. Wang, T. Wang, G. Yu and X. Chen, Polym. Degrad. Stab., 2021, 183, 109463 CrossRef CAS.
- B. Liu, X. Lu, Z. Ju, P. Sun, J. Xin, X. Yao, Q. Zhou and S. Zhang, Ind. Eng. Chem. Res., 2018, 57, 16239–16245 CrossRef CAS.
- K. R. Delle Chiaie, F. R. McMahon, E. J. Williams, M. J. Price and A. P. Dove, Polym. Chem., 2020, 11, 1450–1453 RSC.
- Q. Wang, X. Yao, S. Tang, X. Lu, X. Zhang and S. Zhang, Green Chem., 2012, 14, 2559–2566 RSC.
- L. Wang, G. A. Nelson, J. Toland and J. D. Holbrey, ACS Sustain. Chem. Eng., 2020, 8, 13362–13368 CrossRef CAS.
- K. Fukushima, O. Coulembier, J. M. Lecuyer, H. A. Almegren, A. M. Alabdulrahman, F. D. Alsewailem, M. A. Mcneil, P. Dubois, R. M. Waymouth and H. W. Horn, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 1273–1281 CrossRef CAS.
- Z. Wang, Y. Jin, Y. Wang, Z. Tang, S. Wang, G. Xiao and H. Su, ACS Sustain. Chem. Eng., 2022, 10, 7965–7973 CrossRef CAS.
- R. López-Fonseca, I. Duque-Ingunza, B. de Rivas, L. Flores-Giraldo and J. I. Gutiérrez-Ortiz, Chem. Eng. J., 2011, 168, 312–320 CrossRef.
- S. Javed, D. Ropel and D. Vogt, Green Chem., 2023, 25, 1442–1452 RSC.
- Y. Zhao, M. Liu, R. Zhao, F. Liu, X. Ge and S. Yu, Res. Chem. Intermed., 2018, 44, 7711–7729 CrossRef CAS.
- F. Chen, G. Wang, W. Li and F. Yang, Ind. Eng. Chem. Res., 2013, 52, 565–571 CrossRef CAS.
- Y. Lin, D. Yang, C. Meng, C. Si, Q. Zhang, G. Zeng and W. Jiang, ChemSusChem, 2023, 16, e202300154 CrossRef CAS PubMed.
- Y.-W. Chiao, W. Liao, P. A. Krisbiantoro, B.-Y. Yu and K. C.-W. Wu, Appl. Catal., B, 2023, 325, 122302 CrossRef CAS.
- Q. Suo, J. Zi, Z. Bai and S. Qi, Catal. Lett., 2017, 147, 240–252 CrossRef CAS.
- Y. Li, J. Shen, Q. Liu, Y. Zhu, Z. Pang and M. Ge, J. Environ. Chem. Eng., 2023, 11, 109816 CrossRef CAS.
- G. Park, L. Bartolome, K. G. Lee, S. J. Lee and T. J. Park, Nanoscale, 2012, 4, 3879–3885 RSC.
- A. Al-Sabagh, F. Yehia, D. R. Harding, G. Eshaq and A. ElMetwally, Green Chem., 2016, 18, 3997–4003 RSC.
- G. Eshaq and A. ElMetwally, J. Mol. Liq., 2016, 214, 1–6 CrossRef CAS.
- A. Al-Sabagh, F. Yehia, A. Eissa, M. Moustafa, G. Eshaq, A. Rabie and A. ElMetwally, Polym. Degrad. Stab., 2014, 110, 364–377 CrossRef CAS.
- Q. Wang, Y. Geng, X. Lu and S. Zhang, ACS Sustain. Chem. Eng., 2015, 3, 340–348 CrossRef CAS.
- G. R. Lima, W. F. Monteiro, C. M. Scheid, R. A. Ligabue and R. M. C. Santana, Catal. Lett., 2019, 149, 1415–1426 CrossRef CAS.
- G. R. Lima, W. F. Monteiro, R. Ligabue and R. M. C. Santana, Mater. Res., 2017, 20, 588–595 CrossRef.
- M. R. Nabid, Y. Bide, N. Fereidouni and B. Etemadi, Polym. Degrad. Stab., 2017, 144, 434–441 CrossRef CAS.
- Z. Guo, E. Adolfsson and P. L. Tam, Waste Manage., 2021, 126, 559–566 CrossRef CAS PubMed.
- C. A. Fuentes, M. V. Gallegos, J. R. García, J. Sambeth and M. A. Peluso, Waste Biomass Valorization, 2019, 1–11 Search PubMed.
- T. Wang, Y. Zheng, G. Yu and X. Chen, Eur. Polym. J., 2021, 155, 110590 CrossRef CAS.
- S. Najafi-Shoa, M. Barikani, M. Ehsani and M. Ghaffari, Polym. Degrad. Stab., 2021, 192, 109691 CrossRef CAS.
- V. Rives, Layered Double Hydroxides: Present and Future, Nova Publishers, 2001 Search PubMed.
- H. Yan, X.-J. Zhao, Y.-Q. Zhu, M. Wei, D. G. Evans and X. Duan, The Periodic Table II, 2019, pp. 89–120 Search PubMed.
- F. Zhang, X. Xiang, F. Li and X. Duan, Catal. Surv. Asia, 2008, 12, 253–265 CrossRef CAS.
- D. P. Debecker, E. M. Gaigneaux and G. Busca, Chem. – Eur. J., 2009, 15, 3920–3935 CrossRef CAS PubMed.
- H.-y. Zeng, Z. Feng, X. Deng and Y.-q. Li, Fuel, 2008, 87, 3071–3076 CrossRef CAS.
- H. Y. Zeng, X. Deng, Y. J. Wang and K. B. Liao, AIChE J., 2009, 55, 1229–1235 CrossRef CAS.
- L. Mohapatra and K. Parida, J. Mater. Chem. A, 2016, 4, 10744–10766 RSC.
- L. Wang, Z. Zhu, F. Wang, Y. Qi, W. Zhang and C. Wang, Chemosphere, 2021, 130367 CrossRef CAS PubMed.
- M. Shao, M. Wei, D. G. Evans and X. Duan, Photofunctional Layered Materials, 2015, pp. 105–136 Search PubMed.
- H. Boumeriame, E. S. Da Silva, A. S. Cherevan, T. Chafik, J. L. Faria and D. Eder, J. Energy Chem., 2021, 64, 406–431 CrossRef.
- S. Ali, M. Asif, A. Razzaq and S.-I. In, Catalysts, 2020, 10, 1185–1195 CrossRef.
- F. Chen, F. Yang, G. Wang and W. Li, J. Appl. Polym. Sci., 2014, 131 Search PubMed.
- P. Koilraj and K. Sasaki, J. Environ. Chem. Eng., 2016, 4, 984–991 CrossRef CAS.
- H. Wang, R. Yan, Z. Li, X. Zhang and S. Zhang, Catal. Commun., 2010, 11, 763–767 CrossRef CAS.
- R. Esquer and J. J. García, J. Organomet. Chem., 2019, 902, 120972 CrossRef CAS.
- P. A. Krisbiantoro, Y.-W. Chiao, W. Liao, J.-P. Sun, D. Tsutsumi, H. Yamamoto, Y. Kamiya and K. C.-W. Wu, Chem. Eng. J., 2022, 450, 137926 CrossRef.
- M. Imran, W. A. Al-Masry, A. Mahmood, A. Hassan, S. Haider and S. M. Ramay, Polym. Degrad. Stab., 2013, 98, 904–915 CrossRef CAS.
- H.-M. Liu, X.-J. Zhao, Y.-Q. Zhu and H. Yan, Phys. Chem. Chem. Phys., 2020, 22, 2521–2529 RSC.
- J. S. Valente, F. Figueras, M. Gravelle, P. Kumbhar, J. Lopez and J.-P. Besse, J. Catal., 2000, 189, 370–381 CrossRef CAS.
- S. Arhzaf, M. N. Bennani, S. Abouarnadasse, H. Ziyat and O. Qabaqous, Mediterr. J. Chem., 2020, 10, 625–633 CrossRef CAS.
- A. Corma, V. Fornes, R. Martin-Aranda and F. Rey, J. Catal., 1992, 134, 58–65 CrossRef.
- S. K. Yun and T. J. Pinnavaia, Chem. Mater., 1995, 7, 348–354 CrossRef CAS.
- J. Di Cosimo, V. Dıez, M. Xu, E. Iglesia and C. Apesteguıa, J. Catal., 1998, 178, 499–510 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3su00304c |
| This journal is © The Royal Society of Chemistry 2023 |