
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThermo-switchable hydrophobic deep eutectic solvent for CuAAC†‡
Florence Charnay
Pouget
,
Jean-Michel
Andanson
* and
Arnaud
Gautier
 *
*
Université Clermont Auvergne, CNRS, ICCF, F-63000 Clermont–Ferrand, France. E-mail: J-Michel.Andanson@uca.fr; Arnaud.gautier@uca.fr
First published on 11th September 2023
Abstract
The use of a deep eutectic solvent with thermo-switchable hydrophobicity properties (lidocaine: decanoic acid) is presented. Upon a gentle thermal trigger, the solvent separated into aqueous and organic layers. This system is well adapted for the CuAAC reaction using an N-heterocyclic carbene copper catalyst. An alkyne–azide cycloaddition of hydrophilic compounds, such as carbohydrates, could be conducted with good yields in the aqueous layer after the thermal separation. The method presented herein causes low contamination of the products by metals and the solvent can be reused at least five times.
Sustainability spotlightA decrease in the impact of emissions from organic solvents during chemical synthesis is important. Therefore, the substitution of a traditional organic solvent by a renewable one offers a simple solution to improve sustainability. In this context, non-volatile solvents are of great importance, especially if smart properties are needed. We envisioned developing a protocol to improve the efficiency of the extraction phases in which a thermomorphic multiphase system could separate the product from the reaction mixture, including the metals of the catalyst. The unique properties of oleic or decanoic acid/lidocaine mixture toward water rendered them good candidates for such a separation. Remarkably, we achieved a sequence based on reaction, extraction, and separation during a one-pot process for the copper(I)-catalyzed alkyne–azide cycloaddition upon a small thermal trigger. Our work aligns with the following UN sustainable development goals: industry, innovation, and infrastructure (SDG 9) and sustainable consumption and production patterns (SDG 12). |
Introduction
Classical organic processes often involve the use of large solvents quantities (reaction, extraction, purification) for fine chemical and pharmaceutical sector applications.1 The UN has presented a sustainability plan entitled “Transforming Our World: The 2030 Agenda for Sustainable Development” which targets the need for greener and more sustainable chemistry.2 Environmental directives and legislation of the European Union focus on reducing solvent emissions and regulating the usage of potentially harmful or environmentally damaging chemical substances.3 When possible, the substitution of a traditional harmful organic solvent by a renewable and benign one offers a simple solution to improve the sustainability of the overall process. Alternative chemical procedures have appeared to decrease the E-factor. Among these procedures, mechanochemistry is perfect because it eliminates the usage of solvents, but it does not solve the issue of the purification step.4 One issue in using volatile solvents is the eventuality of releasing some into the atmosphere and in the environment. To solve this issue, it is possible to use ionic liquids that are practically non-volatile.5 More recently, deep eutectic solvents (DES) that are formed from mixtures of Brønsted or Lewis acids and bases have emerged as new solvents.6 The melting points of DES are lower than their individual components, and they are generally simply prepared by mixing two chemicals to form a liquid mixture. Different types of DES have been described, and most of them contain hydrogen-bond acceptors (HBAs) and hydrogen-bond donors (HBDs), such as choline chloride/urea. Like ionic liquids, they present very high enthalpies of vaporisation, and therefore, could be considered as non-volatile for most applications.6b Indeed, some of the ionic liquids have found applications in catalysis and extraction due to the thermomorphic multiphase systems (TMS) nature of their mixtures with organic solvents.7 By changing the temperature, the system can switch from one to two phases opening the possibility to perform a chemical reaction followed by a fast separation of the reacting materials (and the catalysts) upon a thermal trigger. However, most of these systems still contain volatile organic solvents. Recently, we reported the first example of a DES with switchable smart properties when using temperature as a trigger: a mixture of oleic acid/lidocaine (1/1 molar ratio) and water (50 wt%), which is homogeneous at room temperature, spontaneously separates into two phases (water and DES) at temperature higher than its lower critical solution temperature (LCST, 25 °C in this case).8 Although DES are mainly used for extraction, they have found applications in organic synthesis.9 Meyer et al. took advantage of this smart property to conduct biocatalysis in a homogeneous aqueous DES phase.8f After the reaction, the temperature increases, and the enzyme stays in the aqueous phase while the product is found in the DES. This separation of the two phases triggered by temperature perfectly fits to avoid the extraction step during the organic synthesis of aqueous soluble compounds.Therefore, we hypothesised that:
1- The separation of the two phases triggered by temperature perfectly replaces the extraction step during the organic synthesis of aqueous soluble compounds.
2- As DES are recognised to efficiently extract transition metals,10 we expect that the metal from a catalyst could easily separate from the reaction product as well.
We suspect that linking two partially hydrophilic compounds would produce an easier water-extractable compound, whereas the rest of the starting material would predominantly stay in the DES (Fig. 1). This approach could be of great importance in the synthesis of radiopharmaceuticals bearing metals or pharmaceutical compounds that are obtained through metal catalysis. In this context, the copper alkyne–azide cycloaddition (CuAAC) was chosen as a case study.11 This reaction, catalysed by Cu1+, furnishes exclusively 1,5 disubstituted 1,2,3-triazoles from the [3 + 2] cycloaddition of organic azides with terminal alkynes, while maintaining a perfect atom economy. Indeed, CuAAC is compatible with a broad range of solvents (from organic to aqueous) and accommodates many functional groups. In fact, a few publications reporting the application of CuAAC on different DES can already be found in the literature.12 Among these, the choline chloride/glycerol (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2) system is well-adapted for hydrophobic compounds that precipitate after the addition of a large volume of water into the reaction vessel. We have already reported the properties of lidocaine/oleic acid or decanoic acid (1/1 mol ratio, DES I and DES II, respectively) mixtures that separate in two aqueous and organic phases just above room temperature.8d This feature is due to the decrease in ion concentrations with an increase in the temperature ref. 8d. For the CuAAC reaction, a large set of ligands that strongly accelerate the process are available, ranging from highly hydrophilic (i.e., THPTA, bathophenanthroline disulfonic acid) to hydrophobic (i.e., TBTA, 1,10-phenanthroline, N-heterocyclic carbenes (NHCs)). Among these, the systems based on NHC ligands are particularly interesting because they do not require the addition of a reducing reagent (e.g., ascorbic acid) to keep copper at its +1 oxidation level. Thus, we selected the well-explored reagents, SIMesCuCl (A) and [SIMes(Cl2-Phen)CuCl] (B, generated in situ by the addition of 4,7-dichloro-1,10-phenanthroline to A) as catalysts.13
:2) system is well-adapted for hydrophobic compounds that precipitate after the addition of a large volume of water into the reaction vessel. We have already reported the properties of lidocaine/oleic acid or decanoic acid (1/1 mol ratio, DES I and DES II, respectively) mixtures that separate in two aqueous and organic phases just above room temperature.8d This feature is due to the decrease in ion concentrations with an increase in the temperature ref. 8d. For the CuAAC reaction, a large set of ligands that strongly accelerate the process are available, ranging from highly hydrophilic (i.e., THPTA, bathophenanthroline disulfonic acid) to hydrophobic (i.e., TBTA, 1,10-phenanthroline, N-heterocyclic carbenes (NHCs)). Among these, the systems based on NHC ligands are particularly interesting because they do not require the addition of a reducing reagent (e.g., ascorbic acid) to keep copper at its +1 oxidation level. Thus, we selected the well-explored reagents, SIMesCuCl (A) and [SIMes(Cl2-Phen)CuCl] (B, generated in situ by the addition of 4,7-dichloro-1,10-phenanthroline to A) as catalysts.13
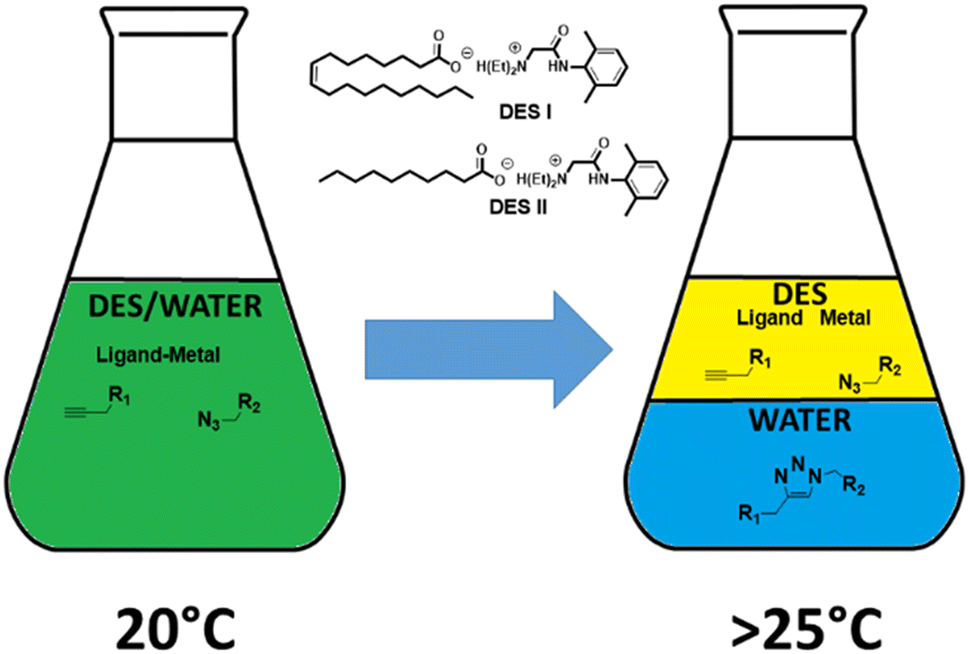 | ||
| Fig. 1 Scheme of the reaction and separation steps using thermal switchable aqueous DES. | ||
Results and discussion
We first examined the reaction of propargyl alcohol (1) with 1-azido propan-3-ol (2) furnishing triazole 3a (Table 1). Reactions were conducted at room temperature (typically 20 °C) for 24 h with 0.5 M solutions of 1 and 2. Conversions were measured by 1H NMR (10–15 μL of the DES/water mixture dissolved in dmso_d6) by comparing the triazole signal (8 ppm) and the propyne proton (2.4 ppm).|
|
|||
|---|---|---|---|
| Entry | Solventa | Cat. (mol%) | Conversionb |
| a DES/water (v/v). b Taken by 1H NMR. c Isolated yield. | |||
| 1 | DES I (1/0) | A (2.5) | 0% |
| 2 | DES I (1/0) | B (2.5) | 100% |
| 3 | DES I (1/1) | A (2.5) | 0% |
| 4 | DES I (1/1) | B (2.5) | 0% |
| 5 | DES I (2/1) | A (2.5) | 0% |
| 6 | DES I (2/1) | B (2.5) | 100% |
| 7 | DES II (1/0) | A (2.5) | 0% |
| 8 | DES II (1/0) | B (2.5) | 100% (93%)c |
| 9 | DES II (1/1) | A (2.5) | 0% |
| 10 | DES II (1/1) | B (2.5) | 0% |
| 11 | DES II (2/1) | A (2.5) | 0% |
| 12 | DES II (2/1) | B (2.5) | 100% |
| 13 | DES II (2/1) | B (1.0) | 100% |
| 14 | DES II (2/1) | B (0.5) | 100% |
| 15 | DES II (2/1) | B (0.25) | 24% |
| 16 | Water | B (2.5) | 0% |
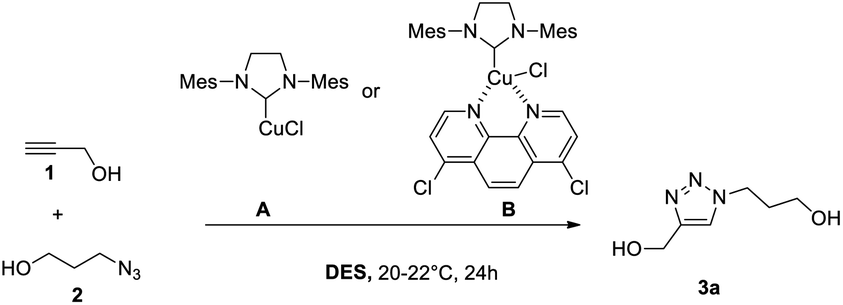
No reaction occurs between DES I (Table 1, entry 1) and catalyst A, but a complete conversion takes place after the addition of the N-donor (entry 2, catalyst B). The addition of an equal volume of water has a deleterious effect on both catalysts (entries 3–4), probably due to the precipitation of the catalyst. However, decreasing the ratio to 2/1 (v/v) allows the reaction to proceed with catalyst B (entry 6), whereas the catalyst A stays inactive. The same tendency was observed with DES II: A is inactive (entries 7, 9, and 11), which definitively rules out the possibility of using this catalyst, whereas B performs well in the pure solvent or in the 2/1 (v/v) mixture (entries 8, 12 versus entry 10). The triazole 3a was extracted well (see general procedures), furnishing the product with 93% yield (entry 8). With the 2/1 volumetric ratio, the loading of B could be decreased to 0.5 mol% (entries 13, 14 versus 15) without altering the conversion.
The blank experiment (entry 16) shows that no reaction takes place in pure water. Although it has been reported that Cu–NHCs can catalyse ‘on-water’ cycloadditions even at ppm-scale loadings,14 our result can be explained by the total solubility of the starting material in water, which helps avoid the formation of an organic layer on the water in which the catalyst dissolves. Our results outline the necessary presence of the N-donor heterocycle (catalyst B) for the reaction to proceed. This is explained by the breaking of the copper-chloride bond to free the active catalyst. Therefore, pure DES (I and II) do not seem to have strong hydrolytic properties. It is important to mention that the presence of a double bond in DES I does not interact with the catalyst. However, it could potentially create troubles with other transition metal catalysis (Pt2+, Pd2+), therefore, DES II was preferred for the rest of this study. To assess that the reaction is catalysed and does not occur spontaneously in the DES, especially during the extraction process during which the temperature increases, the azide and alkyne were mixed at 45 °C for 24 h in different DES II/water mixtures (Table 2). Entries 1–3 indicate that uncatalyzed cycloaddition occurs to a lesser extent at 45 °C in DES II, in water or in the 2/1 mixture (two phases in this case as the reaction temperature is above 25 °C) after 24 h. As expected, a substantial proportion of 1,5 regioisomer 3b is observed in each reaction. This is in sharp contrast with the high yield and the total selectivity for the 1,4-regioisomer 3a observed in the presence of catalyst B (entry 4).
|
|
||||
|---|---|---|---|---|
| Entry | Ratio (DES II/water) | Catalyst Ba | Yield | 3a/3b |
| a Mol%. b Reaction conducted in a 2-phase system because reaction temperature is superior to the LCST (25 °C). c Determined by 1H qNMR in the crude mixture. d Isolated yield. | ||||
| 1 | 1/0 | 0 | <10%c | 1/1 |
| 2 | 0/1 | 0 | <10%c | 2/1 |
| 3 | 2/1b | 0 | <10%c | 1/1 |
| 4 | 2/1b | 0.7 | 93%d | 1/0 |
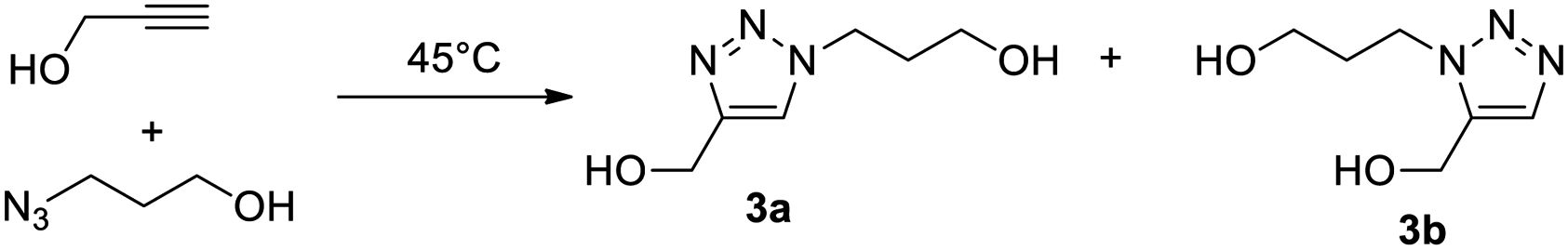
Inspection of the advancement shows that the reaction progresses slowly in pure DES II (24 h), whereas the presence of water strongly increases the reaction rate (1 h 30 min to 2 h – it should be noted that the addition of water to the DES II induces a decrease in the viscosity that could be involved in the improvement of the kinetics of the reaction). Interestingly, the strongly exothermic reaction causes an increase in the temperature (up to 40 °C with 10 mol% of B) in DES II that causes phase separation (temperature superior to the LCST, 25 °C). This interesting result indicates that the solvent itself is responsive to triggers from the reaction and paves the way to smart solvents that would change their properties in case of run-away reactions (therefore, in the current study, the vials were placed at 20 °C with a water bath). Another concern was the fate of the catalyst B after the reaction. Due to the sensitivity of Cu+1 toward oxygen, the reaction usually ends with unliganded Cu2+ and the protonated ligand (1,3-dimesitylimidazolinium). ICP-OES indicates that the mass contamination of the product by copper represents 0.05 wt% (copper vs. product). 1H quantitative NMR was used to determine the partition of the imidazolium and 4,7-dichloro-1,10-phenanthroline. The compounds stay essentially in the DES II layer (>99%). In all cases, after copper catalysis of the test reaction, we observed a small amount of lidocaine in the aqueous phase after extraction. This is consistent with the results reported by Kroon et al.10 which studied the extraction of transition metals (including copper) from water with the hydrophobic mixture of decanoic acid and lidocaine (2/1 molar ratio) forming a biphasic liquid system with water, unlike our (1/1) mixture that separated upon heating. The transition metal stays bound to the organic acid in the DES phase, thereby forming Cu(Deca)2 and releasing two protonated lidocaine ions in the aqueous layer. We therefore determined the amount of lidocaine in the water (using 1H q-NMR) and found that it represents twice the amount of catalyst added which agrees well with Kroon's observations.§ Before exemplifying our system, we examined the extractability of the compounds in the model reaction (the triazole was synthesised separately, purified, and re-engaged in an extraction protocol). The extraction consists of the dissolution of the compound at 0.5 M in a 2/1 (v/v) DES II/water mixture at room temperature (<25 °C), followed by an increase in temperature to 40–45 °C for 20 minutes to separate the two phases. The number of compounds was determined by quantitative 1H NMR (3-(trimethylsilyl)propane-1-sulfonic acid sodium salt for the aqueous phase and 1,4-dimethyl benzene for the organic layer as internal standards). We found that propargyl alcohol has a strong propensity to stay in the DES II layer with a partition ratio of 9/1. Azido propanol splits quasi-equally between the two phases (partition of 6/4). However, the extraction of triazole 3a occurs more efficiently albeit with the introduction of a hydrophobic heterocycle (partition 8/2). A second extraction (same volume ratio, 1/2 DES/water) affords a total transfer in the aqueous layer. Fig. 2 shows a typical example of the process. As can be seen, the reaction starts with a reddish colour, which is typical of the catalyst B (copper-to-ligand charge transfer). At the end of the reaction, oxidation by molecular oxygen furnishes a greenish colour from a copper2+ carboxylate. Upon a thermal trigger, a clean-phase separation occurs leaving the copper in the DES II layer (top phase).
 | ||
| Fig. 2 Reaction process. Left: reaction at t = 0, T = 20 °C. Middle: end of the reaction, T = 20 °C. Right: phase separation immediately after a thermal trigger, T = 45°. | ||
Therefore, we suppose that the “diol effect” could be applied to other compounds, such as carbohydrates. This protocol was then applied to exemplify the system; Table 3 summarises the isolated yields and conversions obtained for 11 polyols.
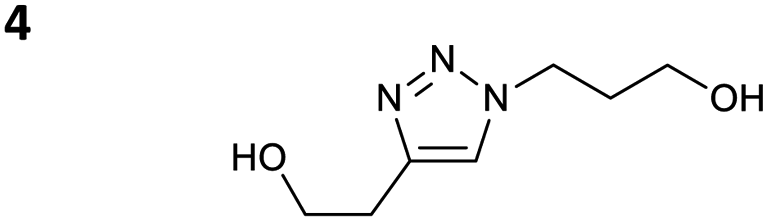
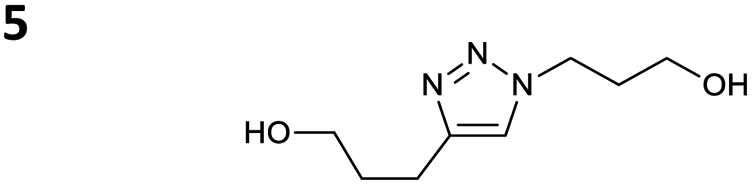
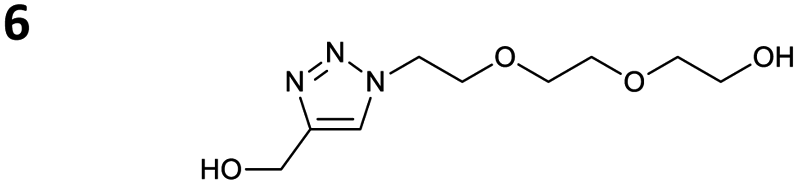
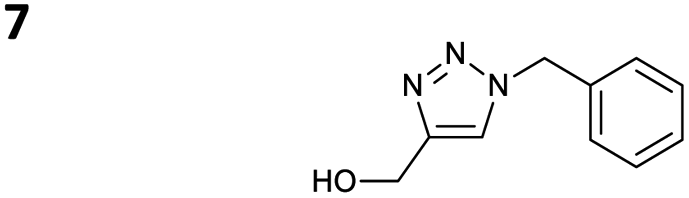
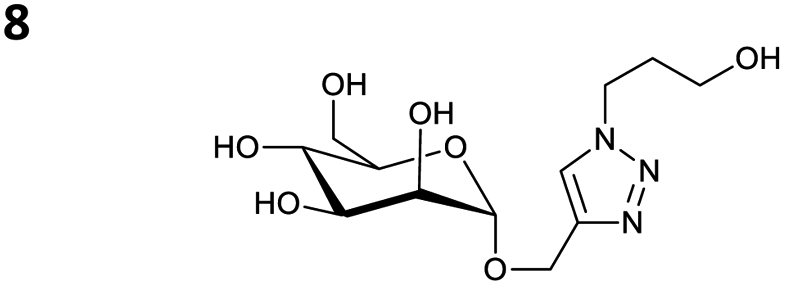
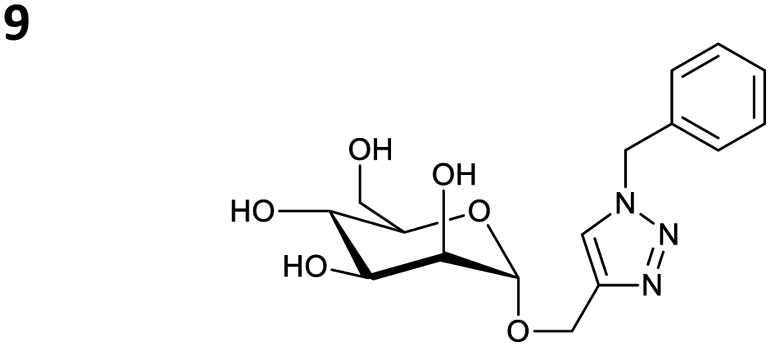
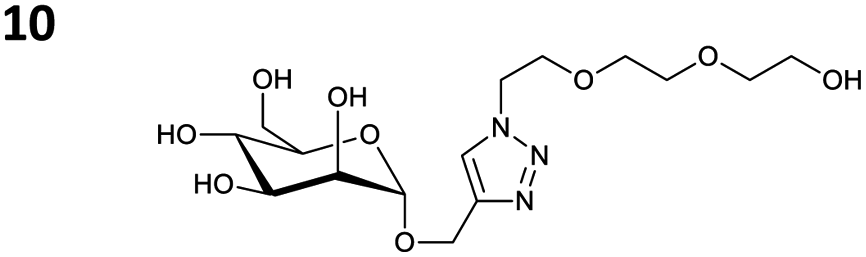
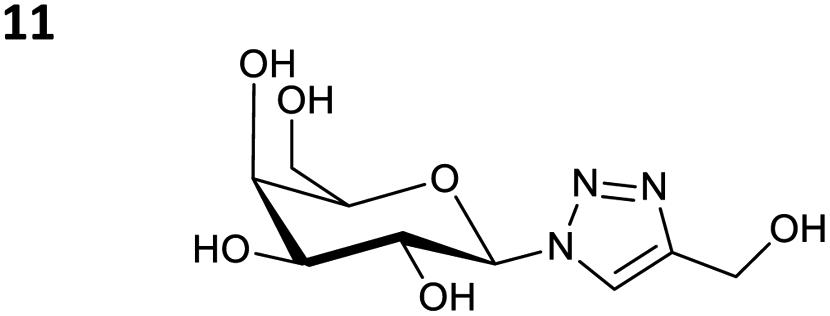

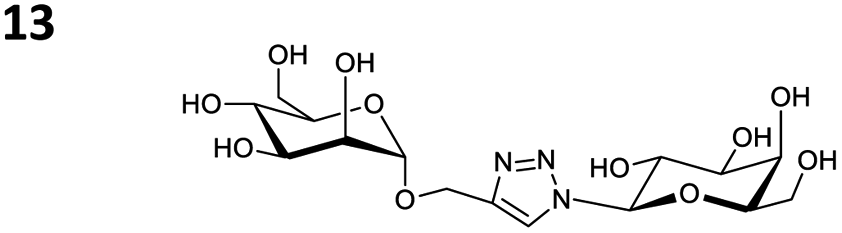
The reactions of propargyl and homopropargyl alcohols with azido-3-propanol (compounds 3a and 4 entries 1–2) occur with a total conversion; after two aqueous extractions, isolated yields are excellent. Here again, contamination by copper from the aqueous extract was determined by ICP-OES and represented 0.05% of the mass product. A lower yield is observed when switching to pentyne-1-ol (5, entry 3), albeit a total conversion is observed. 1H NMR inspection of the DES layer reveals that a large amount of the product stays inside, highlighting the poor efficiency of the extraction in this case. The reaction of the PEG azidoethoxy-ethoxy-ethanol (6, entry 4) allows an averaged isolated yield. 1H NMR of the crude aqueous extract shows that it is contaminated by 10–20% of the DES II mixture, probably because of the amphiphilic properties of the product. Stirring the aqueous extract with tert-butyl methyl or di-isopropyl ether allows the removal of the DES (the very pale green colour of the ether layer indicates the presence of a trace amount of copper). As an alternative, a simple purification through a C18 column affords the pure compound. The reaction of benzyl azide (7, entry 5) affords a low isolated yield although the reaction reaches completion, indicating that several hydrophilic functions are needed to conduct the extraction step efficiently. Therefore, the reactions of highly hydrophilic carbohydrates were explored. The reaction of α-D-galactopyranosyl bearing a propargyl function at the anomeric centre with azido propanol occurs efficiently (8, entry 6). Similarly, the reaction with benzyl azide presented total completion (9, entry 7, 85% isolated yield), although it was still contaminated by 10–20% of DES. This successful extraction proves that the lipophilic moiety is well counterbalanced by the carbohydrate. Using the same strategy, the reaction of the azido-PEG affords an isolated yield of 84% after further purification (10, entry 8, the crude extract is still contaminated by the DES). We also installed the azide function on the carbohydrate (β-D-galactopyranosyl), which was reacted with propargyl alcohol in 86% isolated yield (11, entry 9). Notably, the reaction is totally inhibited for phenylacetylene (entry 10). Finally, the two carbohydrates were efficiently linked affording 85% of isolated yield (13, entry 11). Finally, we examined the reusability of the solvent after extraction.¶Table 4 indicates the yield of triazole 3a and copper contamination after each reaction cycle (DES II was not purified between the two experiments). We also examined the effects of generating the catalyst by adding A and 4,7-dichloro-1,10-phenanthroline (Table 4, upper results); as the phenanthroline additive is left untouched after the reaction, the addition of only A takes place only after the first run (results at the bottom).
| Experiment | 1 | 2 | 3 | 4 | 5 |
|---|---|---|---|---|---|
| a Measured by 1H NMR with 3-trimethylsilyl-1-propanesulfonic acid sodium salt as internal standard. b Contamination: mass Cu/total mass. | |||||
| Yield (catalyst)a | 95% (B) | 99% (B) | 95% (B) | 95% (B) | 95% (B) |
| Copperb | 0.03% | 0.04% | 0.02% | 0.05% | 0.02% |
| Yield (catalyst)a | 97% (B) | 95% (A) | 97% (A) | 80% (A) | 59% (A) |
| Copperb | 0.03% | 0.2% | 0.07% | 0.18% | 0.37% |
The iterative experiments show that the solvent could be used at least 5 times with catalyst B without a decrease in the yield of the reaction or copper contamination of the product. The second method shows that the addition of 4,7-dichloro-1,10-phenanthroline is necessary as the yield decreases and the metal contamination increases after the third cycle.
The process presented here allows a reaction/extraction sequence in a ‘one pot’ process with good separation of the metal and the catalyst's ligand; the main contamination occurred due to the presence of lidocaine salt in small amounts (2%). Regarding the E-factor, the overall process comprises the DES that could be used 5 times and a small amount of di-isopropyl ether. With this smart solvent that switches from hydrophilic to hydrophobic, it is possible to obtain an E-factor below 5.
Conclusion
In conclusion, we have reported that the azide–alkyne cycloaddition catalyzed by NHC–CuI takes place effectively in the aqueous deep eutectic solvents made of lidocaine and fatty acid. Using the thermal trigger, hydrophilic compounds are extracted efficiently in water, permitting an efficient separation of the product. However, for compounds possessing amphiphilic properties, a substantial amount of DES is produced within the aqueous layer, and an additional extraction step with ether solvent is necessary because of the relatively high concentrations of reactants used. Notably, the DES acts as a metal trap providing products with low copper contamination. The solvent can be reused at least 5 times, still providing a good yield and low metal content. Investigations of the properties of other DES for catalysis are under evaluation in our laboratory.Experimental section
General information
Commercial grade reagents and solvents (tert-butyl and di-isopropyl ether, 4,7-dichloro,1,10-phenanthroline) were used as received from Sigma-Aldrich. Lidocaine, oleic, and decanoic acids were purchased from TCI. Mono distilled water was used. Purifications for analytical purposes were performed on a spot II Flash chromatography ARMEN instrument with a C18 Büchi column (40 μm irregular) using H2O/MeOH/TFA (v/v = 10/0 to 6/4, 0.1% TFA). The nuclear magnetic resonance (NMR) spectra were recorded with a 400 MHz spectrometer (Bruker Advance III). 1H NMR and 13C{1H} NMR chemical shifts (δ, relative to tetramethylsilane for 1H and relative to the solvent for 13C{1H}) are reported in ppm (part per million). Coupling constants (J) are reported in hertz (Hz). Infrared spectra were measured using a PerkinElmer (spectrum two, ATR mode) and are reported in cm−1. High-resolution mass spectra (HRMS) were obtained using time-of-flight (TOF) and orbitrap MS. Copper contamination was determined using an ICP-OES Agilent 5800 equipment. 1-Azido-propane-3-ol,15 2,3,4,6-tetra-O-acetyl-β-D-galactopyranosyl azide,16 1-O-propargyl-D-mannose,17 and SIMesCuCl18 were synthesised from procedures reported in literature.DES preparation
The two components of the DES (1 to 1 molar ratio) were added together and then heated at 80 °C in a closed vial; this temperature was above the melting temperatures of both HBD and HBA. DES mixtures became perfectly transparent liquid after a few minutes. DES was then left overnight to cool down at room temperature before further use.General procedure
In a conical reaction vial (surrounded by a water batch at room temperature, typically 20–22 °C) that contained 1.3 mL of DES, 6.5 mg of SIMesCuCl (0.016 mmol, 1.6 mol%) and 4.7 mg of 4,7-dichloro-1,10-phenanthroline (0.016 mmol, 1.6 mol%) was introduced. The mixture was heated at 45 °C, while stirring until all the solids were dissolved and a red colour was developed. After cooling to room temperature, a mixture of 101 mg of 1-azido propanol (1 mmol, 1.0 equivalent) and 56 mg of propargyl alcohol (1 mmol, 1.0 equivalent) in 0.7 mL of water was added. After 24 h, the solution was heated to 45 °C for 20 minutes to produce a phase separation (in case of a poor separation, 1 mL of water was added). The lower aqueous layer was separated using a syringe. Two millilitres of water were added and the reaction mixture was cooled to room temperature under stirring until it turned limpid. The liquid–liquid separation was repeated at 45 °C for 20 minutes, and the two aqueous phases were mixed. The rest of DES II (in compounds 3a, 4, 5, and especially 6) was removed from the aqueous layers by extraction with 0.5–0.7 mL of di-isopropyl ether. For analytical purposes, the products containing sugars were purified on C18 using 0 to 20% methanol gradient (0.1% TFA).Quantitative NMR
Trimethylsilyl propane-1-sulfonic acid sodium (1/9 of the expected number of mol) was added to the aqueous extracts and 20% (v/v) of D2O. An aliquot of the aqueous layer was analysed by quantitative 1H NMR.19 Dosage was performed by comparing signals at −0.2 ppm (TMS) and 8.0 ppm (triazole).059, 1003, 963, 908, 916.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research project was supported by the Université Clermont Auvergne (CATHY project) and CNRS.Notes and references
- D. J. C. Constable, C. Jimenez-Gonzalez and R. K. Henderson, Perspective on Solvent Use in the Pharmaceutical Industry, Org. Process Res. Dev., 2007, 11(1), 133–137, DOI:10.1021/op060170h.
- United Nation Home Page, ‘United Nations General Assembly. Transforming Our World: The 2030 Agenda for Sustainable Development; 2015’, https://sdgs.un.org/2030agenda, accessed 2023–2024 Search PubMed.
- (a) EU Home Page, ‘On the Limitation of Emissions of Volatile Organic Compounds Due to the Use of Organic Solvents in Certain Activities and Installations’, https://eur-lex.europa.eu/legal-content/EN/TXT/?uri=celex%3A31999L0013, accessed 2023–2024 Search PubMed; (b) F. Kerton and R. Marriott, Chapter 2. Green Solvents – Legislation and Certification, RSC Green Chemistry, The Royal Society of Chemistry, UK, 2013; pp. 31–50 Search PubMed.
- J. L. Do and T. Friščić, ACS Cent. Sci., 2017, 3(1), 13–19 CrossRef CAS PubMed.
- M. J. Earle, J. M. S. S. Esperança, M. A. Gilea, J. N. Canongia Lopes, L. P. N. Rebelo, J. W. Magee, K. R. Seddon and J. A. Widegre, Nature, 2006, 439, 831–834 CrossRef CAS PubMed.
- (a) B. B. Hansen, S. Spittle, B. Chen, D. Poe, Y. Zhang, J. M. Klein, A. Horton, L. Adhikari, T. Zelovich, B. W. Doherty, B. Gurkan, E. J. Maginn, A. Ragauskas, M. Dadmun, T. A. Zawodzinski, G. A. Baker, M. E. Tuckerman, R. F. Savinell and J. R. Sangoro, Chem. Rev., 2021, 121, 1232–1285 CrossRef CAS PubMed; (b) K. Xin, I. Roghair, F. Gallucci and M. van Sint Annaland, J. Mol. Liq., 2021, 325, 115227–115235 CrossRef CAS.
- Y. Qiao, W. Ma, N. Theyssen, C. Chen and Z. Hou, Chem. Rev., 2017, 117, 6881–6928 CrossRef CAS PubMed; Y. Kohno and H. Ohno, Chem. Commun., 2012, 48, 7119–7130 RSC; J. Luo, T. Xin and Y. Wang, New J. Chem., 2013, 37, 269–273 RSC.
- (a) A smart solvent can have his properties changed reversibly depending of the application of a trigger, P. J. Jessop, D. J. Heldebrant, X. Li, C. A. Eckert and C. L. Liotta, Nature, 2005, 436, 1102 CrossRef CAS PubMed; (b) As example, the addition/removal of CO2 into some liquid mixtures can switch from hydrophobic to hydrophilic, J. J. Jessop, L. Phan, A. Carrier, S. Robinson, C. J. Dürra and J. R. A Harjania, Green Chem., 2010, 12, 809–814 RSC; (c) Temperature can also be use as trigger, few aqueous amine mixtures have a Lower Critical Solution Temperature (LCST), M. Goral, D. G. Shaw, A. Maczynski, B. Wiśniewska-Gocłowska and P. Oracz, J. Phys. Chem. Ref. Data, 2012, 41, 043107 CrossRef; (d) O. Longeras, A. Gautier, K. Ballerat-Busserolles and J.-M. Andanson, ACS Sustainable Chem. Eng., 2020, 8(33), 12516–12520 CrossRef CAS; (e) J. Castaneda Corzo, K. Ballerat-Busserolles, J. Y. Coxam, A. Gautier and J. M. Andanson, J. Mol. Liq., 2023, 377, 121468 CrossRef CAS; (f) L.-E. Meyer, M. B. Andersen and S. Kara, Angew. Chem., Int. Ed., 2022, 61, e2022038 Search PubMed.
- Selected examples: (a) P. H. Tran, H. T. Nguyen, P. E. Hansen and T. E. Le, RSC Adv., 2016, 6, 37031–37038 RSC; (b) R. Martínez, L. Berbegal, G. Guillena and D. J. Ramón, Green Chem., 2016, 18, 1724–1730 RSC; (c) S. A. Padvi and S. P. Dalal, Synth. Commun., 2017, 47, 779–787 CrossRef CAS; (d) X. Marset, J. M. Péreza and D. J. Ramón, Green Chem., 2016, 18, 826–833 RSC; (e) C. Vidal, J. García-Álvarez, A. Hernán-Gómez, A. R. Kennedy and E. Hevia, Angew. Chem., Int. Ed., 2016, 55, 16145–16148 CrossRef CAS PubMed.
- D. J. G. P. van Osch, D. Parmentier, C. H. J. T Dietz, A. van den Bruinhorst, R. Tuinier and M. C. Kroon, Chem. Commun., 2016, 52, 11987–11990 RSC.
- (a) C. W. Tornøe, C. Christensen and M. Meldal, J. Org. Chem., 2002, 67, 3057–3064 CrossRef PubMed; (b) H. C. Kolb, M. G. Finn and K. B Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 CrossRef CAS.
- (a) S. Sethi, N. C. Jana, S. Behera, R. R. Behera and B. Bag, ACS Omega, 2023, 8, 868–878 CrossRef CAS PubMed; (b) S. V. Giofrè, M. Tiecco, A. Ferlazzo, R. Romeo, G. Ciancaleoni, R. Germani and D. Iannazzo, Eur. J. Org Chem., 2021, 34, 4777–4789 CrossRef; (c) A. Kafle and S. T. Handy, Tetrahedron, 2017, 73, 7024–7029 CrossRef CAS.
- (a) S. Díez-González, A. Correa, L. Cavallo and S. P. Nolan, Chem.–Eur. J., 2006, 12, 7558–7564 CrossRef PubMed; (b) M.-L. Teyssot, A. Chevry, M. Traïkia, M. El-Ghozzi, D. Avignant and A. Gautier, Chem.–Eur. J., 2009, 15, 6322–6326 CrossRef CAS PubMed.
- S. Díez-González and S. P. Nolan, Angew. Chem., Int. Ed., 2008, 47, 8881–8884 CrossRef PubMed.
- S. Kalhor-Monfared, C. Beauvineau, D. Scherman and C. Gurard, Eur. J. Med. Chem., 2016, 122, 436–441 CrossRef CAS PubMed.
- S. Wang, J. A. Cuesta-Seijo, D. Lafont, M. M. Palcic and S. Vidal, Chem.–Eur. J., 2013, 19, 15346–15357 CrossRef CAS PubMed.
- I. Krabicová, B. Dolenský and M. Rezanka, Molecule, 2022, 27, 1483–1493 CrossRef PubMed.
- C. Gibard, H. Ibrahim, A. Gautier and F. Cisnetti, Organometallics, 2013, 32(15), 4279–4283 CrossRef CAS.
- (a) G. F. Pauli, S.-N. Chen, C. Simmler, D. C. Lankin, T. Gödecke, B. U. Jaki, J. B. Friesen, J. B. McAlpine and J. G. Napolitano, J. Med. Chem., 2014, 57, 9220–9231 CrossRef CAS PubMed; (b) Y. Nishizaki, D. C. Lankin, S.-N. Chen and G. F. Pauli, Anal. Chem., 2021, 93, 2733–2741 CrossRef CAS PubMed; (c) A. Akhdar, J. M. Andanson, S. Faure, M. Traïkia and A. Gautier, J. Organomet. Chem., 2021, 950, 121991–212995 CrossRef CAS.
- A. Palmioli, A. Aliprandi, D. Septiadi, M. Mauro, A. Bernardi, L. De Colab and M. Panigati, Org. Biomol. Chem., 2017, 15, 1686–1699 RSC.
- C. Girard, E. Önen, M. Aufort, S. Beauvière, E. Samson and J. Herscovici, Org. Lett., 2006, 8(8), 1689–1692 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3su00249g |
| ‡ Indeed, the pKa of lidocaine is ∼8 at room temperature, but it decreases rapidly with temperature. As the ionic form of lidocaine is much more soluble than the molecular one, the increase in temperature reduces the solubility of organics in the aqueous phase, until separation of the two liquid phases occurs. |
| § See ESI for lidocaine determination. |
| ¶ Alternatively, it has been reported that sodium oxalate could be used to extract the metal from similar DES, see ref. 9. |
| This journal is © The Royal Society of Chemistry 2023 |