
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHeterogeneous biocatalytic reduction of 5-(hydroxy)methyl furfural using two co-immobilised alcohol dehydrogenases†
Jakub F.
Kornecki
 abc,
André
Pick
*b,
Pablo
Dominguez de María
d and
Fernando
López-Gallego
*ae
abc,
André
Pick
*b,
Pablo
Dominguez de María
d and
Fernando
López-Gallego
*ae
aCenter for Cooperative Research in Biomaterials (CIC biomaGUNE), Basque Research and Technology Alliance (BRTA), Paseo de Miramon 194, 20014, Donostia-San Sebastián, Spain. E-mail: flopez@cicbiomagune.es
bCASCAT GmbH, Europaring 4, 94315, Straubing, Germany. E-mail: pick@cascat.de
cUniversidad del País Vasco – UPV/EHU, Barrio Sarriena s/n, Leioa 48940, Bizkaia, Spain
dSustainable Momentum SL, Av. Ansite 3, 4-5, 35011, Las Palmas de Gran Canaria, Spain
elKERBASQUE, Basque Foundation for Science, 48009 Bilbao, Spain
First published on 21st September 2023
Abstract
Biocatalyst heterogenisation may enable robust processes that can be applied in biorefineries to selectively valorise highly functionalised platform chemicals. In this work, we co-immobilise two dehydrogenases and successfully apply them in the selective reduction of 5-hydroxymethylfurfural (HMF) to 2,5-bis(hydroxymethyl) furan (BHMF) with efficient in situ cofactor regeneration. First, we select the best enzyme candidates (an alcohol dehydrogenase from Escherichia coli together with a thermostable glucose dehydrogenase from Bacillus subtilis) and then screen a variety of carriers and chemistries to find the optimal individual immobilisation protocols for each dehydrogenase. As a result, methacrylate carriers (Purolite™) functionalised with either aldehydes or with epoxy and cobalt-chelate groups co-immobilise both enzymes in high yields with a sufficient activity recovery (>20%). These optimal heterogeneous biocatalysts enable the quantitative bio-reduction of HMF to BHMF with >99% selectivity in only fifteen minutes, exhibiting an outstanding reusability of >15 batch cycles with a total volumetric productivity of ∼5 g L−1 h−1 of BHMF. Preliminary experiments on a semipreparative scale with HMF loadings of 40 mM also reach high product conversions (86%). Overall, the judicious selection of enzymes, carriers and reaction conditions enables the design of robust biocatalysts that may contribute to paving the way to the valorisation of highly functionalised chemicals in biorefineries.
Sustainability spotlightThe chemical industry heavily relies on fossil resources for materials and energy, but their depletion necessitates alternatives. 5-Hydroxymethylfurfural (HMF) has emerged as a problem-solving building block, obtained from biomass, particularly lignocellulosic biomass, through fructose dehydration. In our research, we have successfully valorised HMF into 2,5-bis(hydroxymethyl) furan (BHMF) by developing highly active and robust biocatalysts as an environmentally friendly alternative to processes that depend on noble metals. By doing so, our technology aligns with the UN Sustainable Development Goals, specifically goals 9, 12, and 13, which focus on industry, responsible consumption and production, and climate action, respectively. |
Introduction
Renewable biogenic resources represent a promising feedstock to produce chemicals. In the field of plastics and polymers, the need for alternative manufacturing processes and monomers that can be sustainably delivered is particularly encouraged. Starting from fructose, and ultimately from lignocellulosic hydrolysates, 5-hydroxymethyl-furfural (HMF) can be produced as a platform molecule to access a variety of chemicals.1–3 In particular, the reduction of HMF to 2,5-bis(hydroxymethyl)furan (BHMF) is gaining momentum as it may serve as a monomer for a new generation of bio-based furan-based plastics.4,5 Moreover, BHMF can also be blended in industrial lubricants, resins, and adhesives, or be used as drug intermediates.5–8 Given this potential, different chemical processes to afford BHMF have been disclosed using (heterogeneous) catalysts and nanocatalysts, using raw materials like fructose (from glucose isomerisation), or lignocellulose, and operating in cascade-like fashion.5–9 Typically, these synthetic alternatives present selectivity issues and often require noble metals that may suffer from shortages and hazardous operational conditions.6,10–13Due to these challenges, and also due to the inherent instability and cross-reactivity of furans under severe reaction conditions, biocatalysis has emerged in the last years as a promising alternative for furan valorisation.3,14 In fact, the use of mild processing conditions often applied in enzymatic reactions, together with the high chemo, regio, and enantioselectivity that biocatalysts exhibit, may represent a valuable alternative to efficiently valorise highly functionalised platform chemicals of biorefineries, while reducing by-product formation and maximizing the carbon upgrade (i.e. through cascade-like processing obtaining several products). Concerning the reduction of HMF to BHMF, few recent examples using whole-cells,15 and more rarely, plant tissues,16 have been reported. The use of whole-cell biocatalysts may have advantages for practical use (i.e. straightforward production, storage and disposal), but they often display low productivity due to the inhibitory effects of HMF on cell growth and stability.15 Although these hurdles have been recently ameliorated, further improvements are demanded.10,15,17 Conversely, the use of isolated enzymes may overcome the above-mentioned selectivity issues of whole-cells and tissue bio-transformations. However, isolated enzymes need a redox cofactor recycling system, and (semi-)pure enzyme extracts are often too labile and expensive for their implementation in bioprocesses under industrially relevant conditions (i.e. need for high substrate loadings, high process temperatures, use of organic solvents, etc.). In this context, enzyme immobilisation contributes to adapting enzymes for industrial constraints.18–20 First, an immobilised enzyme can be easily separated from the reaction media, simplifying the downstream processing. Second, immobilisation can enhance the enzyme stability and minimise its inhibition, but only if the immobilisation protocol has been carefully designed and characterised.21 Last but not least, enzyme immobilisation is a key enabling technology to perform biocatalysis in flow. The development of immobilisation protocols that synthesize more active and stable heterogeneous biocatalysts has opened new operational windows for enzymatic processes towards their industrial implementation.22,23
In the case of redox multi-enzyme systems where NAD cofactors must be shuttled from one enzyme (oxidoreductase) active site to the other, enzyme co-immobilisation on porous carriers enhances the cofactor recycling efficiency.24,25 As the oxidoreductases are confined inside a porous space, the effective local concentration of the two nicotinamide cofactor species (NAD+ and NADH) is higher, enhancing the activity of the co-immobilised dehydrogenases, and leading to a largely optimised performance.
This paper explores the potential of biocatalyst heterogenisation by co-immobilizing two enzymes for a redox process with a focus on biorefineries, where crude effluents and highly-functionalised and inherently reactive platform chemicals are used. Thus, a new NADH-dependent multi-enzyme system is assembled to perform the selective reduction of HMF to BHMF with the in situ recycling of the cofactor. A zinc-dependent alcohol dehydrogenase from Escherichia coli (EcADHZ3) engineered to accept NADH as a cofactor26,27 and a thermostable glucose dehydrogenase from Bacillus subtilis designed by Figueroa et al.28 (BsGDH-2M) as the main dehydrogenase and NADH recycling system are selected, respectively. For the immobilisation, a diversity of chemistries and materials as enzyme carriers is screened to find a consensus immobilisation protocol that results in efficient and robust heterogeneous biocatalysts for the selective reduction of HMF to BHMF. Finally, an HMF bioreduction process is implemented with optimal bi-functional heterogeneous biocatalysts, achieving excellent results in terms of titers and volumetric productivity even at high substrate loads.
Results and discussion
Identification and characterisation of NADH recycling enzymes
First of all, enzymes able to reduce HMF were searched for. In that respect, EcADHZ3 was selected due to its previously reported capability to reduce analogous furfural.26,29 Once we selected EcADHZ3 as the main dehydrogenase, we proceeded to select the NADH recycling enzyme. Several NAD+ dependent dehydrogenases were considered as recycling enzymes using different electron donors (Table S1†); thermostable glucose dehydrogenase from Bacillus subtilis developed by Figueroa et al. (BsGDH-2M),28 phosphite dehydrogenase from Pseudomonas stutzeri (PsPDH) and formate dehydrogenase from Candida boidinii (CbFDH). BsGDH-2M outperformed the rest of the candidates with a specific activity of 77.5 U mg−1 (Table S1†). Once screened for their activity, the kinetic characterisation of the three dehydrogenases from different microbial sources was conducted, using different substrates (Table S2†). The Michaelis–Menten kinetic parameters confirm that BsGDH-2M outperforms any of the dehydrogenases herein studied for cofactor recycling using glucose as an ancillary electron donor. Furthermore, the stability of the NADH recycling enzymes in media containing HMF was subsequently analysed to validate the biocatalyst's performance under real processing conditions (Fig. 1). As observed, PsPDH performance is severely inhibited by HMF, while CbFDH and BsGDH-2M presented a similar furan tolerance. At 50 mM HMF, these two enzymes maintain more than 60% of their activity in the absence of HMF. Although CbFDH shows a slightly higher tolerance to HMF than BsGDH-2M (22% loss of initial activity for CbFDH vs. 40% loss for BsADH), the latter is selected due to its >150-fold higher specific activity (60 U mg−1 for BsADH vs. 0.39 U mg−1 for CbFDH). Furthermore, BsGDH-2M also tolerates BHMF similarly to HMF, which provides further arguments for its selection as a catalyst (Fig. S1†).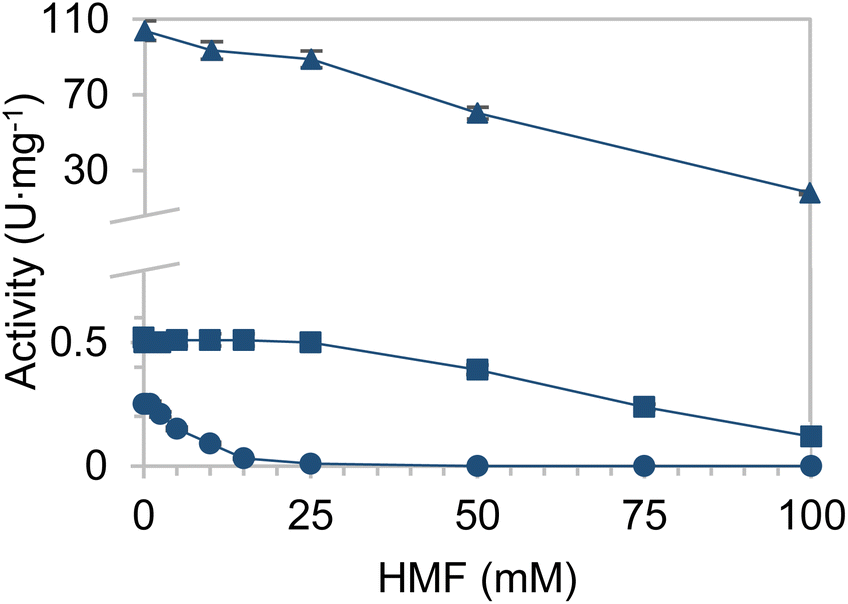 | ||
| Fig. 1 Recovered activity of BsGDH-2M (triangles), FDH (squares) and PDH (circles) in the presence of increasing concentrations of HMF. Tris 50 mM, pH 7.0, 0.5 mM NAD+, and 25 mM of the corresponding substrate at 25 °C. Glucose, sodium formate and sodium phosphite were used as substrates for BsGDH-2M (0.2 μg mL−1), CbFDH (0.01 mg mL−1) and PsPDH (3.8 mg mL−1), respectively. | ||
Besides, we determined the kinetic parameters of BsGDH-2M in the presence of both a substrate and product (Table 1). These values evidence that neither HMF nor BHMF influence the Km of BsGDH-2M; however, these reagents significantly decrease the Vmax of the enzyme, presumably due to a non-competitive inhibition with KI = 16 and 38 mM, respectively. However, we cannot discard the irreversible enzyme inactivation promoted by these two reagents as observed in the discontinuous use of this enzyme in consecutive batch cycles (vide infra).
| mM | HMF | BHMF | ||
|---|---|---|---|---|
| K m | V max | K m | V max | |
| 0 | 1.34 | 109.14 | 1.34 | 109.14 |
| 10 | 1.16 | 94.89 | 1.44 | 108.24 |
| 25 | 1.85 | 93.53 | 1.02 | 95.60 |
| 50 | 1.45 | 51.17 | 1.21 | 73.08 |
| 100 | 1.54 | 15.38 | 2.89 | 29.84 |
Immobilisation screening for BsGDH-2M and EcADHZ3
Upon enzyme selection, we performed a systematic assessment of different immobilisation procedures. A total of twenty-seven different procedures were tested to identify the consensus conditions to co-immobilise the two dehydrogenases with the ultimate goal of fabricating an efficient and robust bifunctional heterogeneous biocatalyst for the bio-reduction of HMF to BHMF. To that end, six different types of carriers functionalised with up to four different reactive groups and combinations thereof were screened. The materials were subjected to thirteen different immobilisation conditions to promote both reversible and irreversible attachment between enzymes and the carriers.As materials, biopolymers like agarose beads, organic polymer carriers like acrylic beads, as well as some inorganic particles like yttrium stabilised zirconia (YSZ), and porosity-controlled glass-based carriers like EziG™ were selected. All of them were functionalised with a plethora of reactive groups like cobalt chelates, epoxide, aldehydes and amines. Table 2 summarises the type of materials as well as the immobilisation chemistry and conditions herein utilised. EcADHZ3 and BsGDH-2M were individually immobilised following these different immobilisation protocols, resulting in a total of twenty-seven different immobilised biocatalysts for each dehydrogenase. Both enzymes were immobilised on most of the carriers with immobilisation yields higher than 50% (Table S3†). Regarding the recovered activity upon the immobilisation protocol, Table 2 shows that most of the immobilisation protocols led to higher recovered activity for immobilised BsGDH-2M than for immobilised EcADHZ3. The higher tolerance to immobilisation exhibited by the recycling enzyme may be due to its high thermostability. In general, thermostable enzymes (i.e. thermophilic ones) may better resist the conformational changes induced by enzyme attachments to solid carriers, leading to higher recovered activities upon the immobilisation processes.30,31 Among all carriers herein tested, acrylic ones, specifically Purolite™ (entries 22–27) presented the highest recovered activity for the two dehydrogenases using a wider range of immobilisation chemistries. For example, when both BsGDH-2M and EcADHZ3 were immobilised on heterofunctional carriers activated with epoxides and cobalt(II)-chelates, the recovered activity was one of the highest in the screening (Table 2, entries 26 and 27). This family of carriers enables enzyme immobilisation through a two-step mechanism where a first directing group (cobalt(II)-chelate) interacts rapidly and selectively with the enzyme (His-tag) through reversible bonds. Then, a second type of groups (epoxides) establishes a multivalent irreversible attachment with the nucleophilic amino acids (i.e. Lys, Tyr, and Cys) exposed on the enzyme surface. These carriers have been successfully exploited for site-directed immobilisation and rigidification of enzymes through oriented multivalent attachment.32,33 This immobilisation approach also allows the one-pot purification and immobilisation of His-tagged enzymes.34 On these heterofunctional carriers, once the enzyme is immobilised, an additional blocking step is necessary for the inertisation of the remaining epoxy groups on the support, neutralizing their reactivity upon the immobilisation protocol, and allowing the safe storage of the heterogeneous biocatalysts.
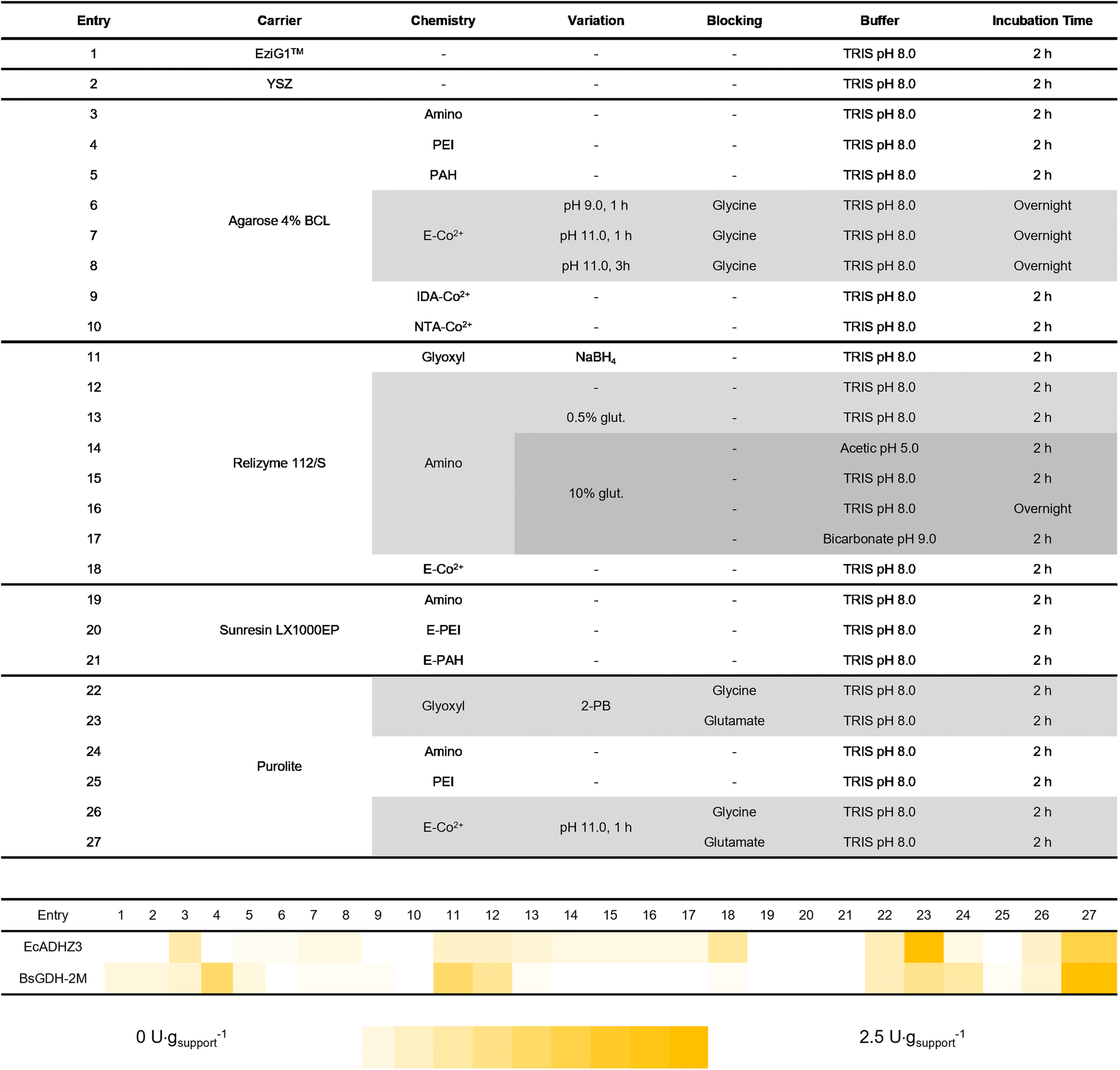
|
To this aim, we tested two different amino acids, glycine and glutamate, as blocking agents. The amine group of these amino acids efficiently reacts with the remaining epoxy groups.35 In our system case, glutamate was chosen as the blocking agent because the recovered activity was significantly higher (Table 2, entry 27) than when glycine is used (entry 26). Finally, from the immobilisation screening, we could also recover high activities upon the immobilisation of these two dehydrogenases on carriers functionalised with aliphatic aldehydes (glyoxyl groups). The oligomeric nature of these enzymes allowed their immobilisation through aldehyde chemistry at neutral pH as previously reported.36 In this carrier, the ε-NH2 of the exposed lysine residues of the enzyme acts as a nucleophile, attacking the aldehydes and forming imine bonds that must be further reduced to make the enzyme-carrier bonds irreversible and the carrier surface inert. As an alternative to the reduction step, which often deactivates the enzymes, amino acids in combination with 2-picoline borane are employed for the same purpose.37 Like in the carriers activated with epoxides (previous case), the amino acids here also act as blocking agents to neutralise the reactivity of the remaining aldehydes of the support that cannot be directly reduced by picoline borane. For glyoxyl immobilisation chemistry followed by the reduction step, we observed that reduction with 2-picoline borane/amino acids (entries 22 and 23) led to higher recovered activity of EcADHZ3 than that obtained with the reduction with NaBH4 as the reducing agent (entry 11). Out of the twenty-seven immobilisation protocols and according to the volumetric recovered activity (U g−1), Purolite™ ECR8204F was selected as the carrier, combined with either glyoxyl (entries 22 and 23), or the pair epoxide/cobalt(II)-chelates (entries 26 and 27) as reactive groups to functionalise the surface of this acrylic material. Upon immobilisation through these two types of chemistries, we selected glycine (entries 22 and 26) and glutamate (entries 23 and 27) as blocking agents.
Carrier screening for enzyme co-immobilisation
After the above-discussed exhaustive immobilisation screening, the selected carrier (Purolite™ ECR8204F) functionalised with either glyoxyl groups (Pu-G), or epoxy and cobalt(II)-chelates (Pu-E/Co2+) was used to co-immobilise the two dehydrogenases for the BHMF bio-redox cascade. We tested four different immobilisation protocols evaluating how the nature of the blocking agent affects the recovered activity of the co-immobilised enzymes. Table 3 shows the immobilisation parameters of the two enzymes co-immobilised on the same carriers using either glycine or glutamate in the blocking step. Importantly, the co-immobilisation of the two enzymes negligibly affected the trend of recovered activity observed for the different carriers when they were individually immobilised (previous section). The data reported in Table 3 further support that glutamate is a better blocking agent for both Pu-G and Pu-E/Co2+ carriers upon co-immobilisation, as the recovered activity of both dehydrogenases were from two to eight times higher depending on the enzyme and the immobilisation chemistry.| Mod | Block | EcADH | BsGDH-2M | |||
|---|---|---|---|---|---|---|
| Y | RA | Y | RA | |||
| Carrier | Glyoxyl | Gly | 96 | 0.3 | 88 | 0.52 |
| Glut | 96 | 1.4 | 88 | 0.8 | ||
| E-CO2+ | Gly | 99 | 0.3 | 98 | 0.24 | |
| Glut | 99 | 1.4 | 98 | 2.14 | ||
Hence, the optimal co-immobilisation protocol is co-immobilizing EcADHZ3 and BsGDH-2M on Pu-E/Co2+ at pH 8.0 and then blocking with 1 M glutamate at pH 9.0. This multifunctional heterogeneous biocatalyst exhibits a volumetric activity of 1.2 and 2.14 U g−1 for EcADHZ3 and BsGDH-2M, respectively. The layer of negative charges resulting from the blocking of the electrophilic groups (epoxides and aldehydes) may create a beneficial microenvironment for the immobilised enzymes, allowing their substrates to access their active sites more easily. Similar results have been previously reported for the immobilisation of individual hydrolases.38,39 Therefore, a negative environment seems to improve the properties of these dehydrogenases when they are immobilised on acrylic carriers. The higher polarity of glutamate compared to that of glycine might increase the hydrophilicity of the Purolite™ surface, creating a more enzyme-compatible environment.
Thermal stability and enzyme leakage assays
Next, the thermal stabilities of the dehydrogenases immobilised on Pu-G and Pu-E/Co2+ and blocked with glutamate were analyzed. Fig. 2 and S3† show the kinetic thermal deactivation of the two dehydrogenases individually immobilised and co-immobilised on the carriers described in Table 3 (Table 2, entries 23 and 26). As a reference, the soluble enzymes were also inactivated under the same conditions. For both dehydrogenases, most immobilisation protocols increased their thermal stability although they followed a different stability trend depending on the selected support. While EcADHZ3 was similarly stabilised on both carriers (Fig. 2A) (t1/2 > 120 min), BsGDH-2M presented a higher half-life time (t1/2) when immobilised on Pu-E/Co2+ (t1/2 = 23 min) than on Pu-G (t1/2 < 5 min) (Fig. 2B). Furthermore, we observed that the thermostability of EcADHZ3 and BsGDH-2M slightly increases when they are co-immobilised on Pu-E/Co2+ (Fig. S3 and Table S4†).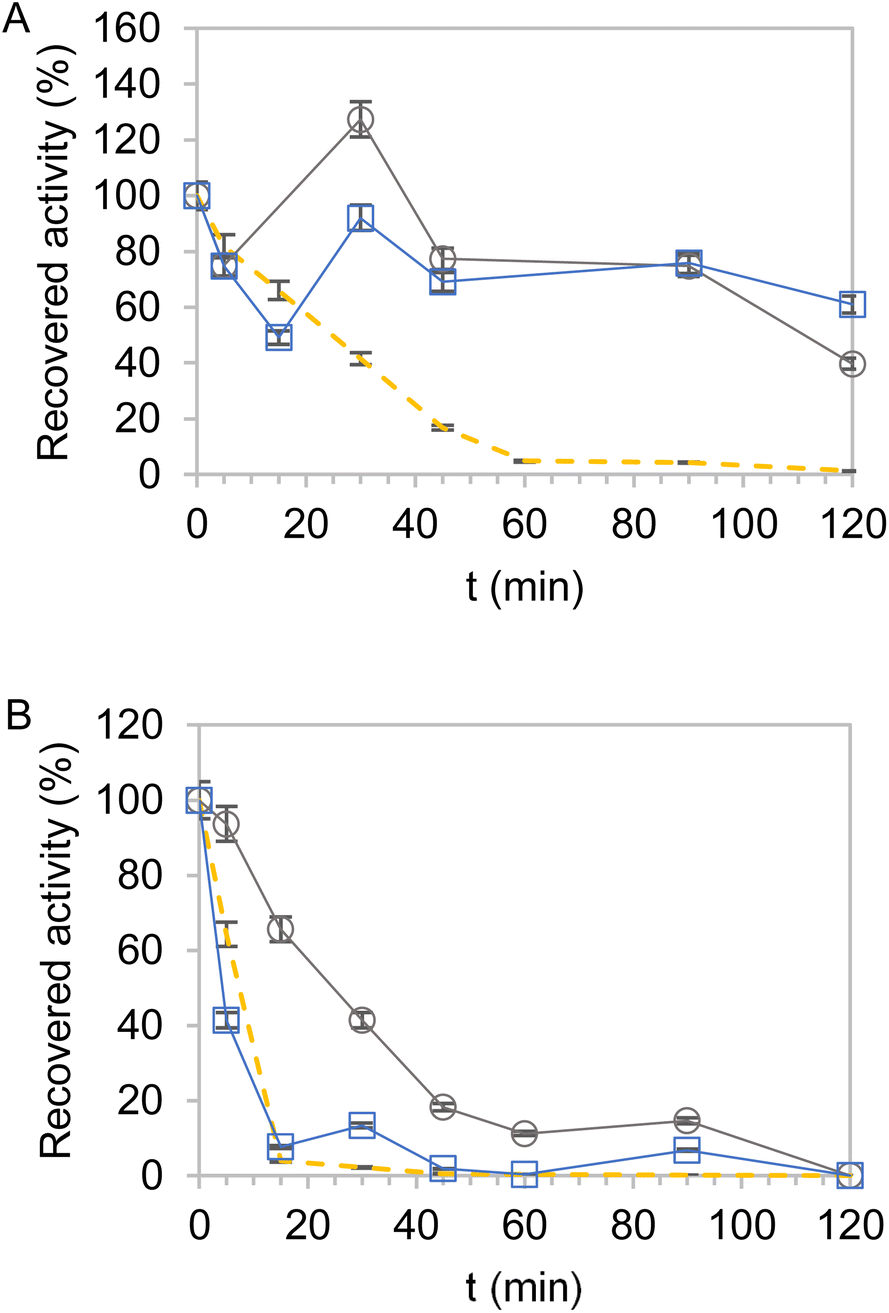 | ||
| Fig. 2 Recovered activity of EcADHZ3 at 70 °C (A) and BsGDH-2M at 80 °C (B) co-immobilised on Pu-E/Co2+ (grey) and Pu-G (blue) and compared against that of the free counterpart (orange dashed). All heterogeneous biocatalysts were blocked with glutamic acid and inactivated in 50 mM Tris–HCl a pH 7.0. 100% activity of EcADHZ3 corresponds to 1.3 (circles) and 1.8 (squares) U g−1 in panel (A). 100% activity of BsGDH-2M corresponds to 2.2 (circles) and 1 (squares) U g−1 in panel (B). 100% activity of free enzymes (dash lines) corresponds to 0.1 U mL−1 in both panels. | ||
Besides the thermal stability of the heterogeneous biocatalysts, we also studied their capacity to irreversibly attach the two multimeric dehydrogenases upon thermal shocks. First, we incubated the two dehydrogenases immobilised on Pu-G and Pu-E/Co2+ blocked with glutamate at 25 and 75 °C, and the supernatants were analysed by SDS-PAGE to assess the amount of each enzyme released after thermal incubation (Fig. S7†). The immobilisation on Pu-E/Co2+ prevented the dissociation of the enzyme subunits forming EcADHZ3 and BsGDH-2M upon incubation at 25 °C, demonstrating a high stability of the two multimeric dehydrogenases upon the immobilisation process. Even when the heterogeneous biocatalysts were incubated at a much higher temperature (75 °C), no leakage of any of the two enzymes was observed. In contrast, some subunits of BsGDH-2M were lixiviated from Pu-G at 25 °C, indicating a partial reversible immobilisation of this enzyme through the glyoxyl chemistry. When this heterogeneous biocatalyst was subsequently incubated at 75 °C, no further leakage of BsGDH-2M subunits was observed, demonstrating that the enzyme molecules remaining in the carrier were irreversibly attached to it. The leakage studies support a good geometric congruence of dimeric EcADHZ3 in the two carriers. In contrast, the geometric congruence of BsGDH-2M with the surface of Pu-G is suboptimal, and thus, its quaternary structure cannot be fully stabilised, explaining why this immobilisation chemistry stabilises this enzyme to a lower extent than E/Co2+ (Fig. 2B).
HMF conversion using co-immobilised solid systems EcADHZ3/BsGDH-2M
Subsequently, the most active and thermostable multi-functional heterogeneous biocatalysts were evaluated for their operational performance in the bio-reduction of HMF to BHMF in batch, using 10 mM HMF as the substrate load and 0.1 equivalents of NADH. For comparison, we also studied the effect of the two blocking strategies during co-immobilisation (glycine and glutamate) on the two Pu-G and Pu-E/Co2+ carriers on the operational performance of the biocatalysts. Furthermore, as a reference for a suboptimal heterogeneous biocatalyst, we utilised the two enzymes co-immobilised on Purolite™ functionalised with primary amine groups and further cross-linked with 0.5% glutaraldehyde, which significantly inactivated EcADHZ3 upon the immobilisation protocol (Table 2, entry 24). Fig. 3 demonstrates that the two co-immobilised systems on both Pu-G and Pu-E/Co2+ outperformed the co-immobilised system on aminated carriers crosslinked with glutaraldehyde as expected from the recovered activities reported in Table 3. Moreover, the heterogeneous biocatalysts blocked with glutamate produced BHMF 20% faster than the ones blocked with glycine, with this difference being more significant in the case of Pu-E/Co2+. Hence the maximum volumetric productivity of BHMF (5.2 g L−1 h−1) was achieved with the system co-immobilised on Pu-G and Pu-E/Co2+ blocked with glutamate (Table S5†). This value is higher than the volumetric productivity reported for most HMF-tolerant whole-cell biocatalysts reported until now, which perform at 0.5–2.2 g L−1 h−1.15,40 Remarkably, these two heterogeneous biocatalysts were able to quantitatively reduce 10 mM HMF in only fifteen minutes with a heterogeneous biocatalyst loading of 10% (w/v) and a EcADHZ3 total turnover number (TTN) of 1.1 × 103. Another remarkable result is the exquisite selectivity of co-immobilised systems that led to >99% chromatographic yield of BHMF, without by-product detection. Although our cell-free system still presents some HMF tolerance limits, during the reviewing process of this work, Wu et al. reported that EcADHZ3 overexpressed in E. coli and used as a permeabilised whole-cell biocatalyst tolerates concentrations as high as 1 M HMF and produces BMHF with a volumetric productivity of 15 g L−1 h−1 in a gram-scale synthesis.29 This work together with our reported results, support that EcADHZ3 is an excellent biocatalyst to perform selective HMF reduction in different reaction configurations.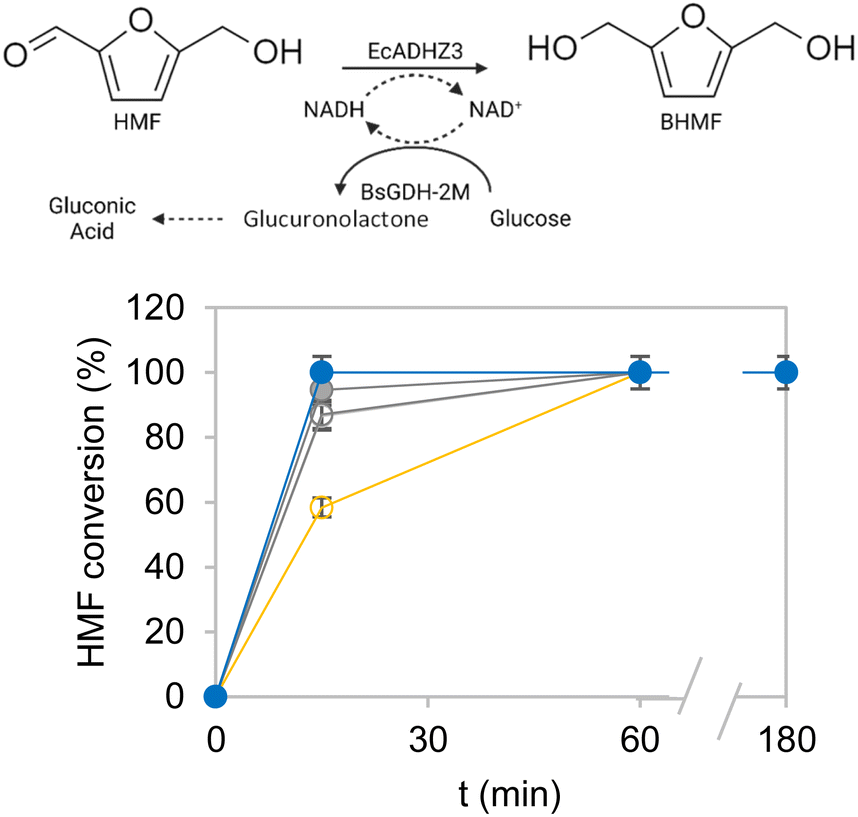 | ||
| Fig. 3 HMF conversion catalyzed by EcADHZ3 and BsGDH-2M co-immobilised on different carriers. Reaction scheme (top) and reaction time courses (bottom) catalyzed by the two enzymes co-immobilised on Purolite™ ECR8204F chemically activated with an amino group (orange empty), on Pu-E/Co2+ blocked with glycine (empty grey) and with glutamate (filled grey), and on Pu-G blocked with glycine (empty blue) and blocked with glutamate (filled blue). HMF 10 mM, NADH 1 mM, glucose 25 mM and Tris pH 7.0, 50 mM. 0.1 gcatalyst mL−1. | ||
Reusability of the optimal bifunctional heterogeneous biocatalysts
To further assess the robustness of the most productive heterogeneous biocatalysts, discontinuous operation in consecutive batch cycles was conducted, evaluating the product yield. After each cycle, the heterogeneous biocatalysts were separated by centrifugation, and fresh reaction media were added. Each reaction cycle was performed using 10 mM HMF, 25 mM glucose and 1 mM NADH and operated for 1 h at 25 °C and pH 7.0. As observed (Fig. 4), the two most active bifunctional heterogeneous catalysts immobilised on Pu-G and Pu-E/Co2+ (blocked with glutamate), could be re-used over fifteen cycles, maintaining their productivity and maximum product yield after each cycle. These two heterogeneous biocatalysts showed similar operational stabilities despite exhibiting different thermal stabilities as shown in Fig. 2. As a result, the accumulated TTN of immobilised EcADHZ3 was 1.7 × 104 after fifteen reaction cycles using 10 mM HMF as the substrate load.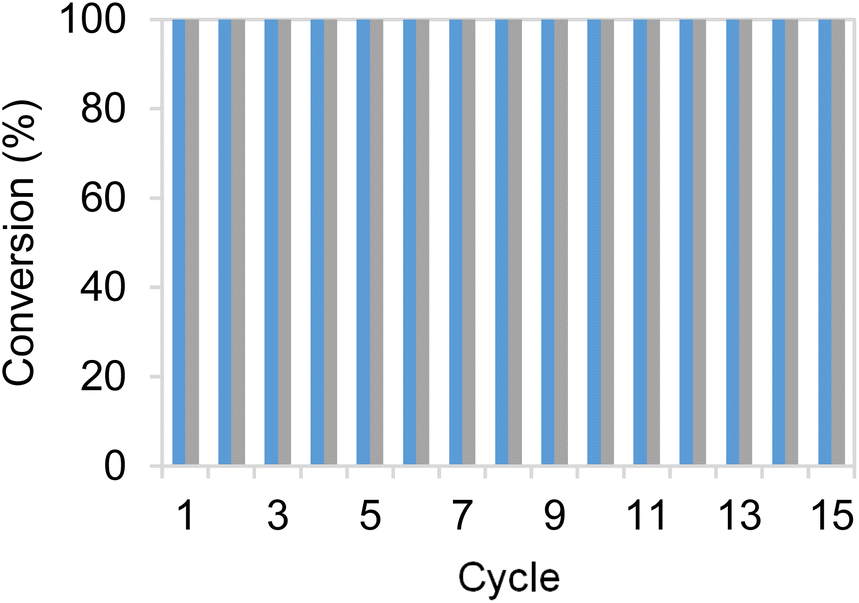 | ||
| Fig. 4 Cycles of HMF conversion by co-immobilised systems of EcADHZ3 and BsGDH-2M on Pu-E/Co2+ (grey) and Pu-G (blue). Reaction mixture contains HMF 10 mM, 25 mM glucose, and 1 mM NADH in 50 mM Tris–HCl buffer at pH 7.0, with a catalyst load of 0.1 gcatalyst mL−1. Each reaction cycle is performed at 25 °C for 15 minutes. | ||
Process intensification and sustainability assessment
Encouraged by the excellent results obtained with the bi-functional heterogeneous biocatalysts using both types of carriers Pu-G and Pu-E/Co2+, a pre-scale up was assessed by increasing the substrate loadings, decreasing the biocatalyst load, decreasing the excess of glucose as the ancillary substrate, and increasing the reaction volume. First, HMF concentration was increased from 10 to 50 mM keeping constant the excess of 2.5 equivalents of glucose in 1 mL of reaction (Fig. 5). Using both bifunctional heterogeneous biocatalysts, the optimal balance between substrate loadings and product yields was achieved at 40 mM HMF, reaching a 91% BHMF yield in 2 h. However, under these conditions the heterogeneous biocatalysts could only be reused in two consecutive cycles, being dramatically inactivated in the third one (Fig. S8†). A plausible explanation may be that the pH drop underlying the gluconic acid production occurred during the NADH recycling and triggered the inactivation of the immobilised enzyme. At the beginning of the reaction the pH was 7.0, registering a value as low as 4.5 after 2 hours of reaction. However, the HMF driven enzyme inactivation cannot be discarded as another plausible reason to explain the lower operational performance of the co-immobilised systems at high substrate concentrations.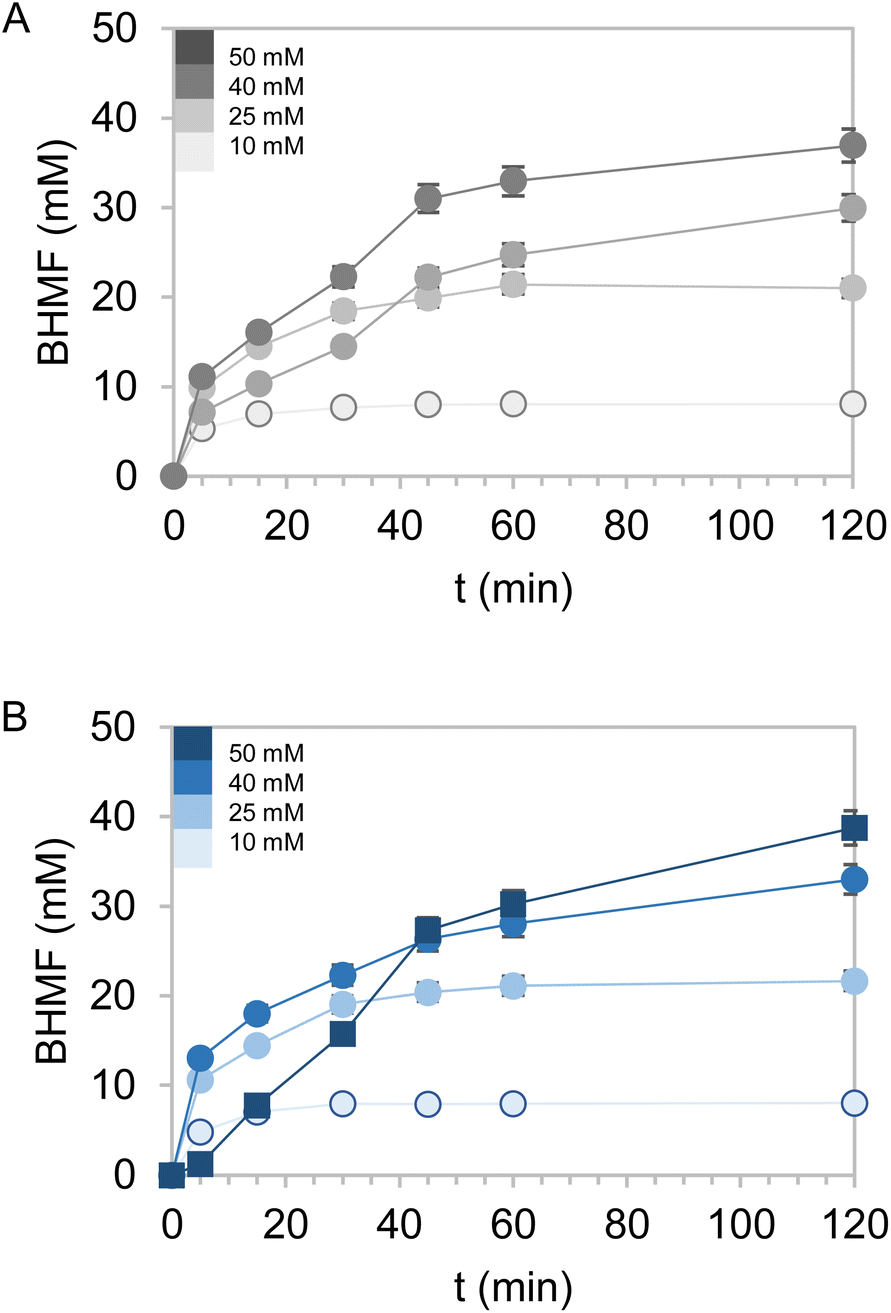 | ||
| Fig. 5 HMF conversion by co-immobilised systems of EcADHZ3 and BsGDH-2M over Pu-E/Co2+ (A) and Pu-G (B) at different substrate concentrations: 10 mM, 25 mM, 40 mM and 50 mM. Reaction mix contains equimolar amounts of glucose relative to HMF and 1 mM NADH in 100 mM Tris–HCl at pH 7.0 and 25 °C. Catalyst load 0.1 gcatalyst mL−1. In all cases, no by-products were detected, only the produced BHMF and the remaining HMF. | ||
To overcome this issue, the buffer concentration was increased up to 200 mM. Under these conditions (Fig. 6) both heterogeneous biocatalysts resulted more operationally stable, reaching their maximum performance for three consecutive cycles. After the third cycle, however, a steady inactivation of both heterogeneous biocatalysts was observed, regardless of the carrier used for their preparation. Nonetheless, the system co-immobilised on Pu-E/Co2+ tolerated its consecutive reuse slightly better than its counterpart immobilised on Pu-G. It must be noted that free enzymes failed to reduce HMF when 40 mM HMF was added, supporting the need for developing robust immobilisation systems. Subsequently, the reaction volume was increased to 125 mL using a biocatalyst load of 0.4% (w/v) with 10 mM HMF and 25 mM glucose, reaching an 86 and 69% HMF conversion in 24 h using the dehydrogenases co-immobilised on Pu-E/Co2+ and Pu-G, respectively, both blocked with glutamate (Fig. 7). In the second cycle of this scaled biotransformation, the substrate conversion decreases to 15%, indicating the inactivation of the two heterogeneous biocatalysts under the intensified conditions (10 mM HMF, 25 mM glucose, and 1 mM NADH with a biocatalyst load of 0.004 gcatalyst mL−1). Enzyme co-immobilisation on Pu-E/Co2+ promotes higher operational stability in the bi-enzymatic system than that on Pu-G.
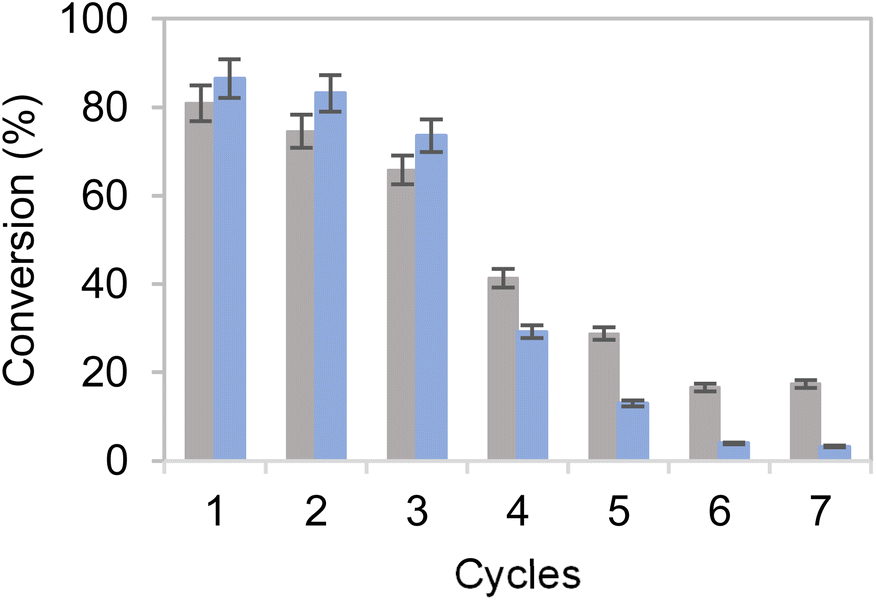 | ||
| Fig. 6 Cycles of HMF conversion by co-immobilised systems of EcADHZ3 and BsGDH-2M over Pu-E/Co2+ (grey) and Pu-G (blue). Reaction mixtures contain 40 mM HMF, 40 mM glucose, 1 mM NADH and 200 mM Tris–HCl buffer at pH 7.0, with a load of 0.1 gcatalyst mL−1. Each reaction cycle is performed at 25 °C for 24 h. | ||
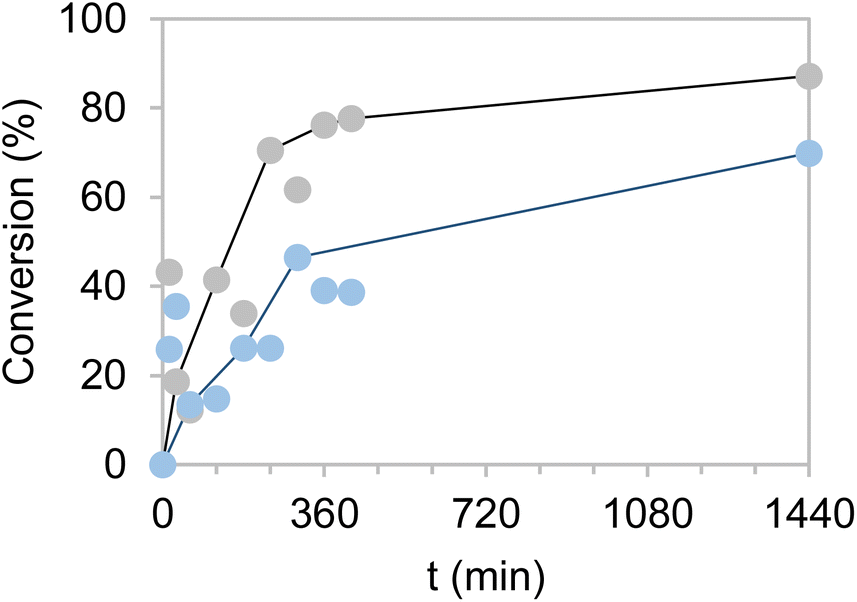 | ||
| Fig. 7 HMF conversion by co-immobilised systems of EcADHZ3 and BsGDH-2M over Pu-E/Co2+ (grey) and Pu-G (blue) with shifted w/v proportions (0.5 g in 125 mL). Reaction mixture contains 10 mM HMF, 25 mM glucose, NADH 1 mM, and 50 mM Tris–HCl buffer at pH 7.0, with a load of 0.004 gcatalyst mL−1. The reaction is performed at 25 °C for 24 h. | ||
The promising results observed during the pre-scale-up, both in terms of substrate loading and reaction volume, encouraged us to conduct a preliminary estimation of the E-factor for the upstream section (the biocatalytic reaction) (Fig. 8). As observed, the E-factor (kgwaste kgproduct−1 (BHMF)) diminished significantly when higher substrate loadings were set, consistent with previous literature.41,42 As expected, the major contribution to the E-factor was provided by water (as a reaction medium). The subsequent set-up of mild wastewater treatment devices may lead to significantly diminished environmental footprints (in terms of CO2 production), giving support for biocatalytic processes in aqueous media. Apart from the upstream unit, an extractive downstream would be possibly needed in this reaction, whereby the use of biogenic solvents (i.e. ethyl acetate) that could be recycled may lead to environmentally acceptable downstream units as well.42
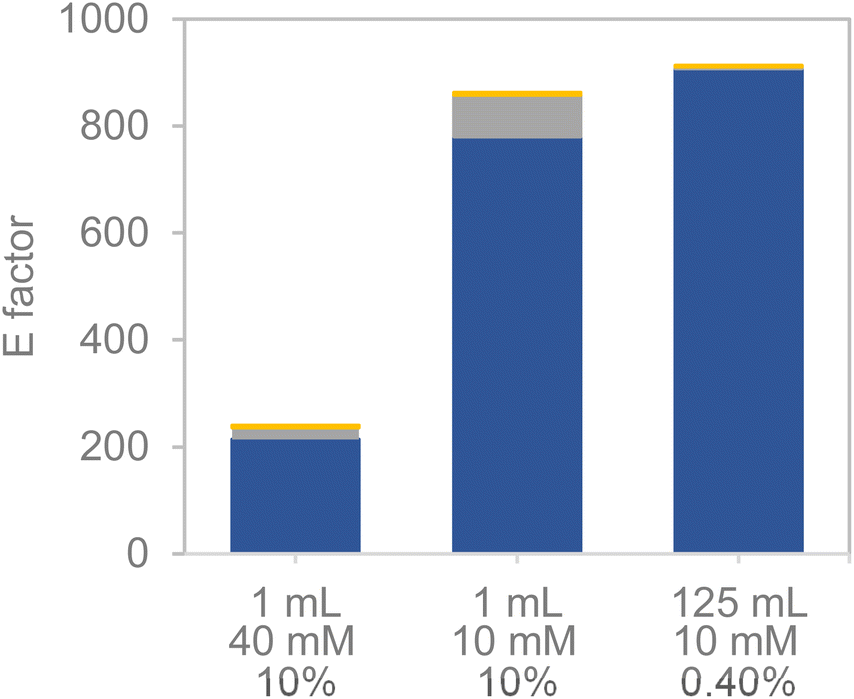 | ||
| Fig. 8 E-factor contribution of water (blue), catalyst (grey) and reagents (orange) towards the E-factor metrics under different reaction conditions. Below the bars, the reaction volume (mL) (top), substrate concentration (mM) (middle) and heterogeneous biocatalyst load (v/w) (bottom) are provided. | ||
Conclusions
Enzymes can find promising uses in future biorefineries when highly functionalised and inherently unstable and reactive platform chemicals such as furans are used. The mild reaction conditions and the exquisite selectivity of enzymes can enable these new valorisation routes. However, the use of free enzymes is often not sufficient to warrant a competitive biotransformation since inhibitions and deactivations may occur. Herein, we have designed a bifunctional heterogeneous biocatalyst with outstanding productivity and operational stability for the reduction of HMF into BHMF, integrating an in situ recycling system at the expense of glucose as an ancillary electron donor. This was possible through a sequential approach that first involved a database-driven enzyme screening to find the optimal dehydrogenase pair, followed by an intensive immobilisation screening that allowed us to find the best immobilisation protocol to efficiently co-immobilise the two-enzyme system. As a result, two highly robust and productive heterogeneous biocatalysts were developed, based on a commercially available carrier (Purolite™) functionalised by us with either glyoxyl or the pair of epoxy groups and metal chelates. Using these heterogeneous systems, high yields of BHMF starting with up to 40 mM HMF were afforded, overcoming the inhibition issues undergone by most of the bioprocesses reported nowadays for this biotransformation. Furthermore, we found an exquisite selectivity of the heterogeneous biocatalysts herein studied as >99% of HMF was reduced to BHMF without detecting other sub-products via HPLC. Finally, the recyclability of the heterogeneous biocatalysts was demonstrated, paving the way for their implementation in continuous processes in future program activities.Experimental
Materials
Epoxy methacrylate microbeads ECR8204F were kindly donated by Purolite™, YSZ was kindly donated by Dr. Jonas Guralakis (AENEAM, Zaragoza, Spain), 4% crosslinked agarose beads (AG) were purchased from Agarose Bead Technologies (Madrid, Spain), and TALON was bought at Takara Bio (Kusatsu, Japan), Sunresin LX1000EP was kindly donated by Sunresin (Xi'an, China), EziG1™ was kindly donated by EnginZyme (Solna, Sweden) and acrylic ECR8204F resin was kindly donated by Purolite™ (King of Prussia, USA). Relizyme 112/S was kindly donated by Resindion SRL (Binasco, Italy). A C18 Poroshell EC-C18 (4.6 × 100 mm × 2.7 μm) column was purchased fromAgilent (Santa Clara, USA).Enzyme expression and purification
The same protocol for protein expression was used for the two enzymes. Briefly, E. coli BL21(DE3) containing the plasmid encoding the enzyme of interest were grown in 50 mL autoinduction media for efficient protein expression.43 The preculture was incubated in 4 mL of LB medium with 100 μg mL−1 kanamycin at 37 °C overnight on a rotary shaker (180 rpm). The expression culture was inoculated with a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :100 dilution of the preculture. Incubation was performed at 37 °C for 24 h. Cells were harvested by centrifugation and resuspended in 50 mM sodium phosphate buffer (pH 8.0, 20 mM imidazole, 500 mM NaCl and 10% glycerol).
:100 dilution of the preculture. Incubation was performed at 37 °C for 24 h. Cells were harvested by centrifugation and resuspended in 50 mM sodium phosphate buffer (pH 8.0, 20 mM imidazole, 500 mM NaCl and 10% glycerol).
Crude extracts were prepared by the use of an ultrasonic liquid processer Sonicator XL2020 (Misonix Incorporated) and subsequent addition of MgCl2 to attain a final concentration of 2.5 mM in combination with DNaseI (10 μg mL−1), followed by incubation for 20 min at room temperature for successful DNA degradation. The insoluble fraction of lysate was removed by centrifugation at 16130g for 20 min at 4 °C. The supernatant was filtered through a 0.45 μm syringe filter and applied to an IMAC affinity resin column, 5 mL Protino Ni-NTA, equilibrated with the resuspension buffer using an ÄKTA purifier system. The enzyme was washed with 20 mL of resuspension buffer and eluted with 50 mM sodium phosphate buffer (pH 8.0, 500 mM imidazole, 500 mM NaCl and 10% glycerol). Aliquots of each eluted fraction were subjected to 12% SDS-PAGE gel electrophoresis. The fractions containing the eluted protein were pooled and the protein was desalted using a HiPrep™ 26/10 desalting column, which had been pre-equilibrated with 50 mM Tris–HCl pH 8.0. Protein concentrations were determined using a Bradford assay (Bradford Reagent, HiMedia).44 Specific activities of the obtained pure enzymes were 55 and 53 U mg−1 for EcADHZ3 and BsGDH-2M, respectively.
Chemical activation of solid carriers
| IDA, mM | pH | Incubation time (h) | Activation degree (μmol g−1) | |
|---|---|---|---|---|
| IDA | Epoxy | |||
| 500 | 9.0 | 1 | 5.2 | 33.5 |
| 500 | 11.0 | 1 | 20.2 | 18.5 |
| 500 | 11.0 | 3 | 26.0 | 12.7 |
After the incubation, the carrier was washed with abundant water. Then, 10 mL 30 mg mL−1 CoCl2 solution were added to 1 g of carrier and incubated at room temperature for 1 h under mild agitation. The IDA and epoxy densities were determined by titrating the remaining epoxy groups after each functionalisation step following a stepwise hydrolysis/oxidation route.50 After incubation the carriers were washed with abundant water and stored at 4 °C.
EcADHZ3 activity was followed via spectrophotometry, using the decrease of absorbance produced by the decrease of NADH54 in the reaction media at 340 nm. The reactions were performed in the presence of 0.5 mM NADH, 10 mM HMF and Tris buffer 50 mM, pH 7.0 at 25 °C.
GDH activity was followed via spectrophotometry, using the increase of absorbance produced by the increase of NADH in the reaction media at 340 nm. The reactions were performed in the presence of 0.5 mM NAD+, 25 mM glucose and 50 mM Tris buffer at pH 7.0 and 25 °C. To determine the Michaelis–Menten parameters of BsGDH-2M, the same reaction mixture was used but by varying the glucose concentration (1–100 mM) and fixing the NAD+ concentration (0.5 mM) at different concentrations of HMF (0–100 mM) and BHMF (0–100 mM). The initial rate of each measurement was plotted against the glucose concentration and the apparent kinetic parameters were calculated with a simplified Michaelis–Menten equation assuming that NAD+ saturates the enzyme under any condition as follows.
| V = Vmax[NAD+]/(KM + [NAD+]) |
The BsGDH-2M inhibition constants towards HMF and BHMF were calculated as follows:
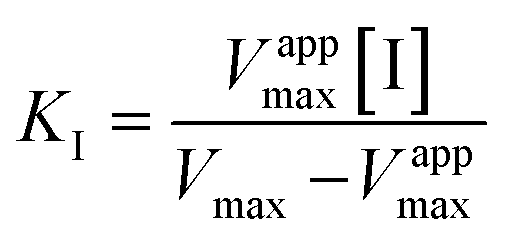
FDH activity was followed via spectrophotometry, using the increase in absorbance produced by the increase in NADH in the reaction media at 340 nm. The reactions were performed in the presence of 0.5 mM NAD+, 25 mM sodium formate and Tris buffer 50 mM, pH 7.0 at 25 °C.
PDH activity was followed via spectrophotometry, using the increase in absorbance produced by the increase in NADH in the reaction media at 340 nm. The reactions were performed in the presence of 0.5 mM NAD+, 25 mM sodium phosphite and Tris buffer 50 mM, pH 7.0 at 25 °C.
All activity measurements are performed in duplicate for the free enzymes and quadruplicate for the immobilised enzymes. Each measurement contained between 5 and 20 μL of sample with an additional 200 μL of reaction mixture described for each enzyme.
For inactivation experiments of GDH, FDH and PDH, these activity assays were performed under the same conditions but adding 0–100 mM HMF and BHMF to the reaction mixture.
:10 (w/v) suspension of the immobilised enzymes on Pu-G was incubated with either 1 mg mL−1 NaBH4 at pH 10.0, or 30 mM 2-methylpyridine borane, 20% DMSO and 1 M glycine or glutamic acid at pH 9.0.55,56 We offered 50 U g−1 (0.7 mgenzyme g−1) of each enzyme, incubating 1 mL of 5 U mL−1 enzyme solution with 0.1 g of each corresponding carrier under the specific immobilisation conditions. The resulting suspensions were gently stirred during the immobilisation process. To follow the process, samples of the suspension and supernatant were withdrawn, and enzyme activity was analyzed as described before. For blocking the suspension of the immobilised enzymes on Pu-E/Co2+ see above. When immobilisation was finished, all supernatants were removed and the immobilisates were stored at 4 °C for further assays.
For each immobilisation experiment, immobilisation yield (Y), recovered activity (RA) and effectiveness (see in Table S3†) are defined and calculated as follows:
Immobilisation yield (Y) is the percentage of the offered enzyme that is immobilised on the support. The free enzymes were incubated under the immobilisation conditions without the carriers as blank samples. In these blanks, no activity loss was observed during the immobilisation time. Ψ is defined by using the following formula
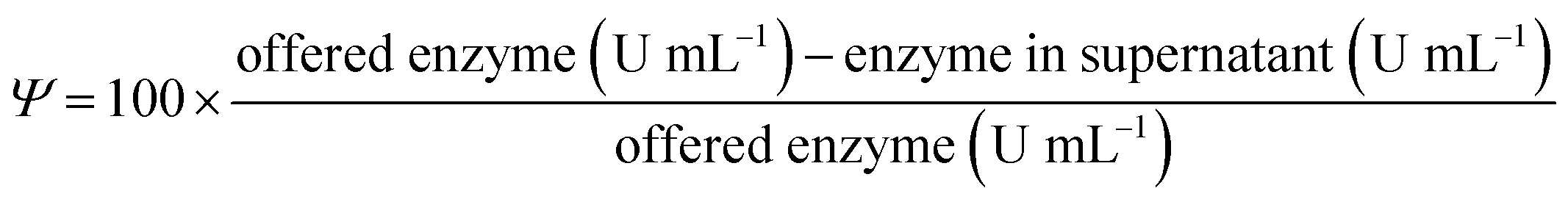
Recovered activity (RA) is defined as the measured enzyme activity per gram of carrier and is expressed in U g−1. 10 μL of a 1:10 (w/v) suspension of the heterogeneous biocatalysts were placed in a microtiter plate with the reaction mixture as described in the enzyme activity assays.
Immobilisation effectiveness or efficiency is defined as the fraction of offered activity per mass of carrier recovered upon immobilisation. This parameter is defined by using the following formula:
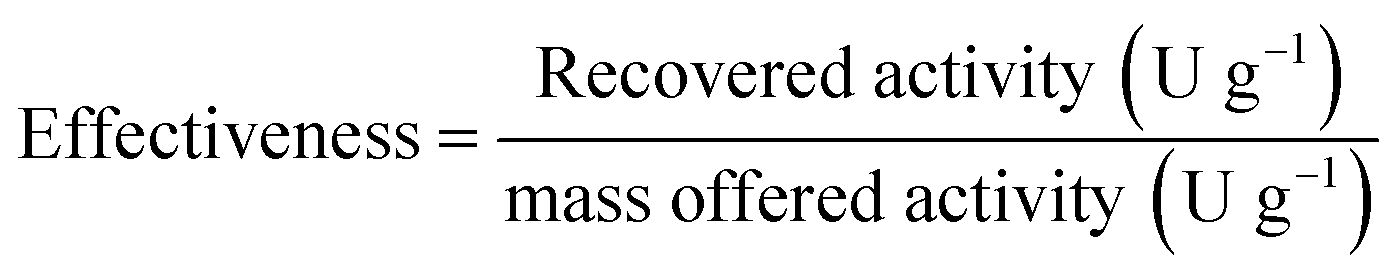
| Conversion = ([BHMF]n × [HMF]0−1) × 100 |
| Selectivity = ([BHMF]n/([HMF]0 − [HMF]n)) × 100 |
| Vol. productivity (g L−1 h−1) = [BHMF] (g L−1)/reaction time (h) |
| E-Factorreagents = total reagents mass (g)/product mass (g) |
| E-Factorwater = total water mass (g)/product mass (g) |
| E-Factorcatalyst = total catalyst mass (g)/product mass (g) |
Author contributions
The manuscript was written through the contribution of all authors. Particularly, J. F. K. carried out the experimental work. J. F. K. and F. L. G. performed data curation, formal analysis and writing of the original draft. A. P. and F. L. G. conducted project administration and provided resources. All authors have discussed the results, reviewed the manuscript and given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
European Union's Horizon 2020 funding from the research and innovation program under the Marie Skłodowska-Curie grant agreement no. 860414 (Interfaces) is gratefully acknowledged. F. L. G. acknowledges support from Ikerbasque. This work was performed under the Maria de Maeztu Units of Excellence Program from the Spanish State Research Agency; MDM-2017-0720 (CIC biomaGUNE).Notes and references
- S. Kang, J. Fu and G. Zhang, Renewable Sustainable Energy Rev., 2018, 94, 340–362 CrossRef CAS.
- J. B. Binder and R. T. Raines, J. Am. Chem. Soc., 2009, 131, 1979–1985 CrossRef CAS PubMed.
- X. Li, R. Xu, J. Yang, S. Nie, D. Liu, Y. Liu and C. Si, Ind. Crops Prod., 2019, 130, 184–197 CrossRef CAS.
- S. Baraldi, G. Fantin, G. Di Carmine, D. Ragno, A. Brandolese, A. Massi, O. Bortolini, N. Marchetti and P. P. Giovannini, RSC Adv., 2019, 9, 29044–29050 RSC.
- Y. Jiang, A. J. Woortman, G. O. Alberda van Ekenstein, D. M. Petrovic and K. Loos, Biomacromolecules, 2014, 15, 2482–2493 CrossRef CAS PubMed.
- J. Ohyama, A. Esaki, Y. Yamamoto, S. Arai and A. Satsuma, RSC Adv., 2013, 3, 1033–1036 RSC.
- C. Post, D. Maniar, V. S. Voet, R. Folkersma and K. Loos, ACS Omega, 2023, 8, 8991–9003 CrossRef CAS PubMed.
- B. Wozniak, S. Tin and J. G. de Vries, Chem. Sci., 2019, 10, 6024–6034 RSC.
- M. Chatterjee, T. Ishizaka and H. Kawanami, Green Chem., 2014, 16, 4734–4739 RSC.
- L. Hu, A. He, X. Liu, J. Xia, J. Xu, S. Zhou and J. Xu, ACS Sustain. Chem. Eng., 2018, 6, 15915–15935 CrossRef CAS.
- T. Mizugaki, Y. Nagatsu, K. Togo, Z. Maeno, T. Mitsudome, K. Jitsukawa and K. Kaneda, Green Chem., 2015, 17, 5136–5139 RSC.
- K. Vikanova, E. Redina, G. Kapustin, M. Chernova, O. Tkachenko, V. Nissenbaum and L. Kustov, ACS Sustain. Chem. Eng., 2021, 9, 1161–1171 CrossRef CAS.
- B. Zhu, C. Chen, L. Huai, Z. Zhou, L. Wang and J. Zhang, Appl. Catal., B, 2021, 297, 120396 CrossRef CAS.
- P. Domínguez de María and N. Guajardo, ChemSusChem, 2017, 10, 4123–4134 CrossRef PubMed.
- Y. M. Li, X. Y. Zhang, N. Li, P. Xu, W. Y. Lou and M. H. Zong, ChemSusChem, 2017, 10, 372–378 CrossRef CAS PubMed.
- A. Petri, G. Masia and O. Piccolo, Catal. Commun., 2018, 114, 15–18 CrossRef CAS.
- S. Chang, X. He, B. Li and X. Pan, Front. Chem., 2021, 9, 635191 CrossRef CAS PubMed.
- R. C. Rodrigues, C. Ortiz, Á. Berenguer-Murcia, R. Torres and R. Fernández-Lafuente, Chem. Soc. Rev., 2013, 42, 6290–6307 RSC.
- M. C. Franssen, P. Steunenberg, E. L. Scott, H. Zuilhof and J. P. Sanders, Chem. Soc. Rev., 2013, 42, 6491–6533 RSC.
- A. Liese and L. Hilterhaus, Chem. Soc. Rev., 2013, 42, 6236–6249 RSC.
- C. Garcia-Galan, Á. Berenguer-Murcia, R. Fernandez-Lafuente and R. C. Rodrigues, Adv. Synth. Catal., 2011, 353, 2885–2904 CrossRef CAS.
- A. I. Benítez-Mateos, M. L. Contente, D. Roura Padrosa and F. Paradisi, React. Chem. Eng., 2021, 6, 599–611 RSC.
- P. De Santis, L.-E. Meyer and S. Kara, React. Chem. Eng., 2020, 5, 2155–2184 RSC.
- J. Rocha-Martín, B. d. l. Rivas, R. Muñoz, J. M. Guisán and F. López-Gallego, ChemCatChem, 2012, 4, 1279–1288 CrossRef.
- S. Velasco-Lozano, J. Santiago-Arcos, J. A. Mayoral and F. López-Gallego, ChemCatChem, 2020, 12, 3030–3041 CrossRef CAS.
- A. Pick, B. Rühmann, J. Schmid and V. Sieber, Appl. Microbiol. Biotechnol., 2013, 97, 5815–5824 CrossRef CAS PubMed.
- A. Pick, W. Ott, T. Howe, J. Schmid and V. Sieber, J. Biotechnol., 2014, 189, 157–165 CrossRef CAS PubMed.
- E. Vázquez-Figueroa, J. Chaparro-Riggers and A. S. Bommarius, ChemBioChem, 2007, 8, 2295–2301 CrossRef PubMed.
- Q. Wu, M.-H. Zong and N. Li, ACS Catal., 2023, 13, 9404–9414 CrossRef CAS.
- A. H. Orrego, D. Andrés-Sanz, S. Velasco-Lozano, M. Sanchez-Costa, J. Berenguer, J. M. Guisan, J. Rocha-Martin and F. López-Gallego, Catal. Sci. Technol., 2021, 11, 3217–3230 RSC.
- A. Barzegar, A. A. Moosavi-Movahedi, J. Z. Pedersen and M. Miroliaei, Enzyme Microb. Technol., 2009, 45, 73–79 CrossRef CAS.
- A. Bastida, P. Sabuquillo, P. Armisen, R. Fernández-Lafuente, J. Huguet and J. M. Guisan, Biotechnol. Bioeng., 1998, 58, 486–493 CrossRef CAS PubMed.
- J. M. Bolivar, C. Mateo, V. Grazu, A. V. Carrascosa, B. C. Pessela and J. M. Guisan, Process Biochem., 2010, 45, 1692–1698 CrossRef CAS.
- C. Mateo, G. Fernández-Lorente, E. Cortés, J. L. Garcia, R. Fernández-Lafuente and J. M. Guisan, Biotechnol. Bioeng., 2001, 76, 269–276 CrossRef CAS PubMed.
- C. Mateo, V. Grazu, J. M. Palomo, F. Lopez-Gallego, R. Fernandez-Lafuente and J. M. Guisan, Nat. Protoc., 2007, 2, 1022–1033 CrossRef CAS PubMed.
- V. Grazu, L. Betancor, T. Montes, F. Lopez-Gallego, J. M. Guisan and R. Fernandez-Lafuente, Enzyme Microb. Technol., 2006, 38, 960–966 CrossRef CAS.
- A. H. Orrego, M. Romero-Fernández, M. D. C. Millán-Linares, M. D. M. Yust, J. M. Guisán and J. Rocha-Martin, Catalysts, 2018, 8, 333 CrossRef.
- C. Mateo, O. Abian, G. Fernández-Lorente, J. Pedroche, R. Fernández-Lafuente and J. M. Guisan, Biotechnol. Prog., 2002, 18, 629–634 CrossRef CAS PubMed.
- P. M. P. Souza, D. Carballares, N. Lopez-Carrobles, L. R. Gonçalves, F. Lopez-Gallego, S. Rodrigues and R. Fernandez-Lafuente, Int. J. Biol. Macromol., 2021, 191, 79–91 CrossRef CAS PubMed.
- J. Xia, S. Jiang, J. Liu, W. Yang, Z. Qiu, X. Liu, A. He, D. Li and J. Xu, Mol. Catal., 2023, 542, 113122 CrossRef CAS.
- P. D. de María, Curr. Opin. Green Sustainable Chem., 2021, 31, 100514 CrossRef.
- P. D. de María, Green Chem., 2022, 24, 9620–9628 RSC.
- F. W. Studier, Protein Expression Purif., 2005, 41, 207–234 CrossRef CAS PubMed.
- M. M. Bradford, Anal. Biochem., 1976, 72, 248–254 CrossRef CAS PubMed.
- C. Mateo, V. Grazú, B. Pessela, T. Montes, J. Palomo, R. Torres, F. López-Gallego, R. Fernández-Lafuente and J. Guisán, Biochem. Soc. Trans., 2007, 35, 1593–1601 CrossRef CAS PubMed.
- C. Mateo, R. Torres, G. Fernández-Lorente, C. Ortiz, M. Fuentes, A. Hidalgo, F. López-Gallego, O. Abian, J. M. Palomo and L. Betancor, Biomacromolecules, 2003, 4, 772–777 CrossRef CAS PubMed.
- C. Mateo, J. M. Palomo, M. Fuentes, L. Betancor, V. Grazu, F. López-Gallego, B. C. Pessela, A. Hidalgo, G. Fernández-Lorente and R. Fernández-Lafuente, Enzyme Microb. Technol., 2006, 39, 274–280 CrossRef CAS.
- S. Martins de Oliveira, S. Velasco-Lozano, A. H. Orrego, J. Rocha-Martin, S. Moreno-Perez, J. M. Fraile, F. Lopez-Gallego and J. M. Guisán, Biomacromolecules, 2021, 22, 927–937 CrossRef CAS PubMed.
- T. Matsuo, T. Kobayashi, Y. Kimura, M. Tsuchiyama, T. Oh, T. Sakamoto and S. Adachi, Biotechnol. Lett., 2008, 30, 2151–2156 CrossRef CAS PubMed.
- J. Santiago-Arcos, S. Velasco-Lozano and F. López-Gallego, Biomacromolecules, 2023, 24, 929–942 CrossRef CAS PubMed.
- R. Fernandez-Lafuente, C. Rosell, V. Rodríguez, C. Santana, G. Soler, A. Bastida and J. M. Guisán, Enzyme Microb. Technol., 1993, 15, 546–550 CrossRef CAS PubMed.
- F. López-Gallego, L. Betancor, C. Mateo, A. Hidalgo, N. Alonso-Morales, G. Dellamora-Ortiz, J. M. Guisán and R. Fernández-Lafuente, J. Biotechnol., 2005, 119, 70–75 CrossRef PubMed.
- L. Betancor, F. López-Gallego, A. Hidalgo, N. Alonso-Morales, G. D.-O. C. Mateo, R. Fernández-Lafuente and J. M. Guisán, Enzyme Microb. Technol., 2006, 39, 877–882 CrossRef CAS.
- A. J. Cooper, M. Conway and S. M. Hutson, Anal. Biochem., 2002, 308, 100–105 CrossRef CAS PubMed.
- A. H. Orrego, M. Romero-Fernández, M. Millán-Linares, M. Yust, J. M. Guisán and J. Rocha-Martin, Catalysts, 2018, 8, 333 CrossRef.
- R. Fernández-Lafuente, V. Rodríguez, C. Mateo, G. Penzol, O. Hernández-Justiz, G. Irazoqui, A. Villarino, K. Ovsejevi, F. Batista and J. M. Guisán, J. Mol. Catal. B: Enzym., 1999, 7, 181–189 CrossRef.
- O. Barbosa, C. Ortiz, Á. Berenguer-Murcia, R. Torres, R. C. Rodrigues and R. Fernandez-Lafuente, RSC Adv., 2014, 4, 1583–1600 RSC.
- R. Morellon-Sterling, D. Carballares, S. Arana-Peña, E.-H. Siar, S. A. Braham and R. Fernandez-Lafuente, ACS Sustain. Chem. Eng., 2021, 9, 7508–7518 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3su00178d |
| This journal is © The Royal Society of Chemistry 2023 |