
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBio-based vitrimers from divanillic acid and epoxidized soybean oil†
Yunfan
Zhang
 ,
Enomoto
Yukiko
and
Iwata
Tadahisa
*
,
Enomoto
Yukiko
and
Iwata
Tadahisa
*
Department of Biomaterial Sciences, Graduate School of Agricultural and Life Sciences, The University of Tokyo, 1-1-1 Yayoi, Bunkyo-ku, Tokyo 113-8657, Japan. E-mail: atiwata@g.ecc.u-tokyo.ac.jp; Fax: +81-3-5841-1304; Tel: +81-3-58415266
First published on 8th March 2023
Abstract
Divanillic acid (DVA), a symmetrical dimer of vanillic acid, is a novel aromatic building block for bio-based polymers. In the present study, we prepared DVA-derived crosslinked vitrimers, which constitute a new class of thermosetting polymers formed via dynamic covalent bonds. We cured two series of bio-based vitrimers, OHESO-x and BuESO-x (x represents the COOH/epoxy ratio, x = 1.5, 1.0, 0.7) derived from DVA and butoxylated DVA, respectively, with epoxidized soybean oil (ESO) in the presence of a transesterification catalyst. Depending on the side chain structures and x values, the glass transition temperatures (Tgs) and Young's moduli of the vitrimer films varied from 20 to 62 °C and from 3 to 286 MPa, respectively. The OHESO-x vitrimers had higher Tgs and Young's moduli than the BuESO-x vitrimers with the same x values. This was due to the hydrogen bonds formed by the free phenolic hydroxyl groups and unreacted carboxylic acid groups. We confirmed dynamic transesterification by stress relaxation testing, and characterized the reprocessability and self-healing properties of the vitrimers.
Sustainability spotlightBio-based polymers have received considerable attention in both academia and industry owing to their outstanding environmental benignity. However, most commercialized bio-based polymers are aliphatic polyesters, such as poly(lactic acid) and poly(butylene succinate), which fail to satisfy all market demands because of their limited thermal and mechanical properties. As a result, there has been growing interest in using bio-based aromatics to develop bio-based aromatic polymers with enhanced material properties. This work focused on developing high-performance and recyclable polymeric materials using renewable natural feedstocks by applying the concept of vitrimer, which is a class of polymers crosslinked via dynamic covalent bonds. It aligns with the UN's Sustainable Development Goals 12 (responsible consumption and production), 13 (climate action) and 15 (life on land). |
1 Introduction
The world is currently facing some tough challenges, such as global energy supply and environmental issues. Therefore, polymeric materials made from renewable natural feedstocks (i.e., bio-based polymers) are attracting burgeoning attention worldwide owing to their huge potential as alternatives to petroleum-based polymers.1,2 Representative bio-based polymers include aliphatic bio-based polyesters derived from the fermentation products of carbohydrates, such as poly(lactic acid) (PLA) and poly(butylene succinate) (PBS), which have been extensively studied and industrialized.3,4 Recently, researchers have focused on biomass-derived aromatics with the aim of obtaining high-performance aromatic polymers with versatile applicability. For example, poly(ethylene furandicarboxylate) has received considerable interest owing to its outstanding thermal and gas barrier properties.5,6 The use of lignin fragments or other plant-derived aromatics, such as vanillic acid7,8 and gallic acid,9 to prepare polymers is also a current trend.We recently systematically synthesized and characterized aromatic bio-based polymers derived from divanillic acid (DVA), which is a symmetrical dimer of vanillic acid, such as polyesters,10–13 polyamides,14,15 and polyketones.16 DVA and divanillin (DV) are seen as promising bio-based building blocks because they can be synthesized using enzymes as green catalysts,17,18 and their biphenyl structures increase the rigidity of the polymer chains, thereby endowing the material with favorable thermal and mechanical properties. Nevertheless, according to previous works by Cramail et al.19–22 and our own group, it is challenging to use DVA or DV to prepare polymers directly. The free phenolic hydroxyl groups are usually alkoxylated to enhance the miscibility of DVA/DV with other comonomers. Therefore, the preparation of DVA/DV-derived polymers is not sufficiently cost-effective. Furthermore, unlike some conventional aromatic polymers such as poly(ethylene terephthalate), linear DVA/DV-derived polymers, especially DVA-derived polyesters, are virtually impossible to crystallize. This significantly limits their practical use. Cramail et al. proposed a new approach to preparing DV-derived polymers: the formation of a thermosetting resin with a network topology cured from easy-to-prepare DV-based epoxy monomers.23 Likewise, based on the material design strategies used for polyesters and epoxy resins,24,25 we considered using DVA to prepare recyclable crosslinked polymers with favorable material performances by applying the concept of the vitrimer.
Vitrimers are a class of polymeric materials that are crosslinked by dynamic covalent bonds.26,27 Under certain conditions (usually high temperature), bond exchange reactions occur and the molecular chains become mobile. The materials then become malleable and processable (i.e., exhibit thermoplasticity). Because vitrimers undergo various types of bond exchange reactions, including transesterification,26,28–31 transamination of vinylogous urethanes32,33 and diketoenamines,34 dioxaborolane metathesis,35,36 silyl ether transalkoxylation37 and metathesis38etc., vitrimers provide more possibilities for the design and construction of polymer networks. Furthermore, vitrimers can be recycled by physical or chemical methods, and some of them are biodegradable.35 Therefore, they are potential candidates for a new generation of eco-friendly crosslinked polymeric materials.
Bio-based vitrimers have received growing attention in recent years because they provide a facile means of utilizing biomass-derived compounds with multiple functional groups or complicated chemical structures.39,40 For example, several research groups have prepared vitrimers from compounds or macromolecules such as rosin derivatives,41,42 tannic acid,43 and modified lignin;44,45 they have little or no polymerizability (i.e., are not suitable for traditional polymerization). Among the biomass-derived monomers, vanillin and epoxidized soybean oil (ESO) have been extensively studied as building blocks for bio-based vitrimers or other dynamically crosslinked polymers.28,46–54 This is because vanillin has a relatively simple chemical structure among naturally occurring aromatics, and ESO is a low-cost, industrially produced epoxy. In many previous works, some petrochemicals or multiple reaction steps were still necessary to introduce exchangeable moieties (e.g., vinylogous urethane and imine bonds) into the monomers. This fact made the process less environmentally friendly. In addition, although bio-based aliphatic acids, such as citric acid and sebacic acid, have been used to obtain transesterification-based vitrimers, there is a lack of research focusing on the direct use of bio-based aromatic acids as vitrimer monomers. Therefore, as we introduced earlier, DVA may offer a promising solution as a bio-based aromatic acid for the preparation of bio-based vitrimers with enhanced material properties.
In this present work, we prepared and characterized the fully bio-based vitrimers based on the dynamic transesterification of DVA or butoxylated DVA (BuDVA) and ESO. We cured DVA/BuDVA–ESO mixtures in the presence of zinc acetylacetonate hydrate (a transesterification catalyst), and subsequently fabricated vitrimer films. Next, we investigated the effects of the side chain (i.e., the phenolic hydroxyl and butoxy groups of DVA and BuDVA, respectively) and the stoichiometric ratios of the carboxylic acid/epoxy groups on the thermal and mechanical properties of the vitrimers. At high temperatures, we confirmed transesterification by stress relaxation testing. We also determined the reprocessability, self-healing, and shape memory properties of the obtained vitrimers.
2 Experimental
2.1 Materials
Zinc acetylacetonate hydrate [Zn(acac)2, 98%] and potassium hydroxide (KOH, >85%) were purchased from Wako Pure Chemical Industries, Ltd. The ESO (epoxide equivalent weight = 233.2 g eq.−1) was generously donated by New Japan Chemical Co., Ltd. All other reagents were obtained commercially and used as received.2.2 Synthesis of DVA and BuDVA
DVA and BuDVA were synthesized by the hydrolysis of dimethyl divanillate (Me-DVA) and dimethyl dibutoxy divanillate (Me-BuDVA) (Scheme 1); Me-DVA and Me-BuDVA were prepared according to the method we have used previously.12 | ||
| Scheme 1 Synthesis of divanillic acid (DVA) and dibutoxydivanillic acid (BuDVA). | ||
DVA.55 Me-DVA (10.00 g, 27.6 mmol, 1 equiv.) and KOH (12.38 g, 220.8 mmol, 8 equiv.) were dissolved in a tetrahydrofuran (THF)/water solution (350 mL, 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v). After stirring the mixture at 70 °C overnight, the red aqueous layer was acidified with hydrochloric acid to induce precipitation. The precipitate was filtered, washed several times with deionized water, and dried in vacuo at. DVA was obtained as a pale orange solid (8.86 g, 96% yield).
:1, v/v). After stirring the mixture at 70 °C overnight, the red aqueous layer was acidified with hydrochloric acid to induce precipitation. The precipitate was filtered, washed several times with deionized water, and dried in vacuo at. DVA was obtained as a pale orange solid (8.86 g, 96% yield).
BuDVA. Me-BuDVA (10.00 g, 21.1 mmol, 1 equiv.) and KOH (9.46 g, 168.6 mmol, 8 equiv.) were dissolved in 30 mL of methanol. After stirring the mixture at 40 °C overnight, we washed it with ethyl acetate, and acidified the colorless (or light yellow) aqueous layer with hydrochloric acid to induce precipitation. The precipitate was filtered, washed several times with deionized water, and dried in vacuo at 70 °C. BuDVA was obtained as a white powder (7.72 g, 82% yield).
2.3 Preparation of DVA/BuDVA–ESO vitrimers
As shown in Fig. 1, two series of vitrimers, named OHESO-x and BuESO-x, were prepared in the present study. OH and Bu represent the DVA (R = H) and BuDVA (R = Bu) monomers, respectively, and x represents the ratio of carboxylic acid (COOH) groups to epoxy groups (i.e., x = COOH/epoxy, n/n). The formulation of the vitrimers is summarized in Table 1. We used 5 mol% (relative to the amount of COOH groups) of Zn(acac)2 to catalyze the transesterification.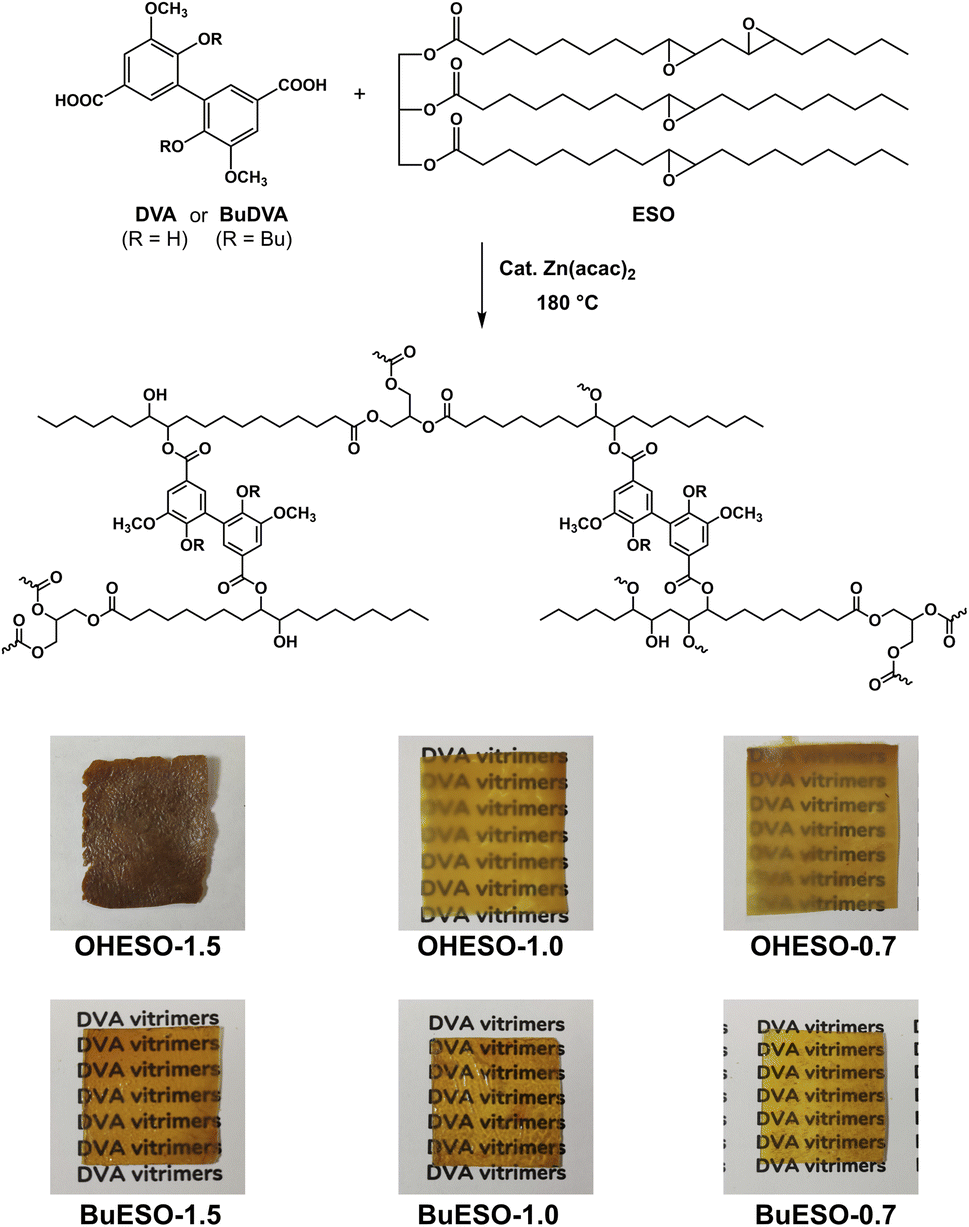 | ||
| Fig. 1 Preparation and images of the DVA/BuDVA–ESO vitrimer films. | ||
| Codes | ESO (g) | Epoxy group (mmol) | Diacid | Diacid (g) | COOH group (mmol) | x |
|---|---|---|---|---|---|---|
| a x = n(COOH group)/n(epoxy group). | ||||||
| OHESO-0.7 | 2.00 | 8.57 | DVA | 1.00 | 6.00 | 0.7 |
| OHESO-1.0 | 1.43 | 8.57 | 1.0 | |||
| OHESO-1.5 | 2.15 | 12.86 | 1.5 | |||
| BuESO-0.7 | 2.00 | 8.57 | BuDVA | 1.34 | 6.00 | 0.7 |
| BuESO-1.0 | 1.91 | 8.57 | 1.0 | |||
| BuESO-1.5 | 2.87 | 12.86 | 1.5 | |||
2.4 Characterization
δ (tanδ = E′/Eʺ) values were recorded.
The shape memory behavior of OHESO-1.0 was investigated in a series of heating-and-reheating processes. A strip of OHESO-1.0 film was reshaped and recovered at T = 100 °C (above its Tg). Memory erasing and rewriting of the original specimen were also conducted at T = 180 °C. The details are discussed in the ESI.†
3 Results and discussion
3.1 Monomers and materials preparation
The diacid monomers DVA and BuDVA were synthesized via the hydrolysis of their dimethyl esters (i.e., Me-DVA and Me-BuDVA, respectively). As shown in Fig. 2(a) and (b), the proton signals at δ ≈ 3.9 ppm, attributed to the methyl esters of Me-DVA or Me-BuDVA, have vanished compared with in the 1H NMR spectra of Me-DVA and Me-BuDVA in our previous work.12 Furthermore, the proton signals at δ >12 ppm (δ = 12.54 and 12.88 ppm for DVA and BuDVA, respectively) were assigned to COOH protons. All other signals can be assigned to the corresponding protons, the details of which are provided in Section S1.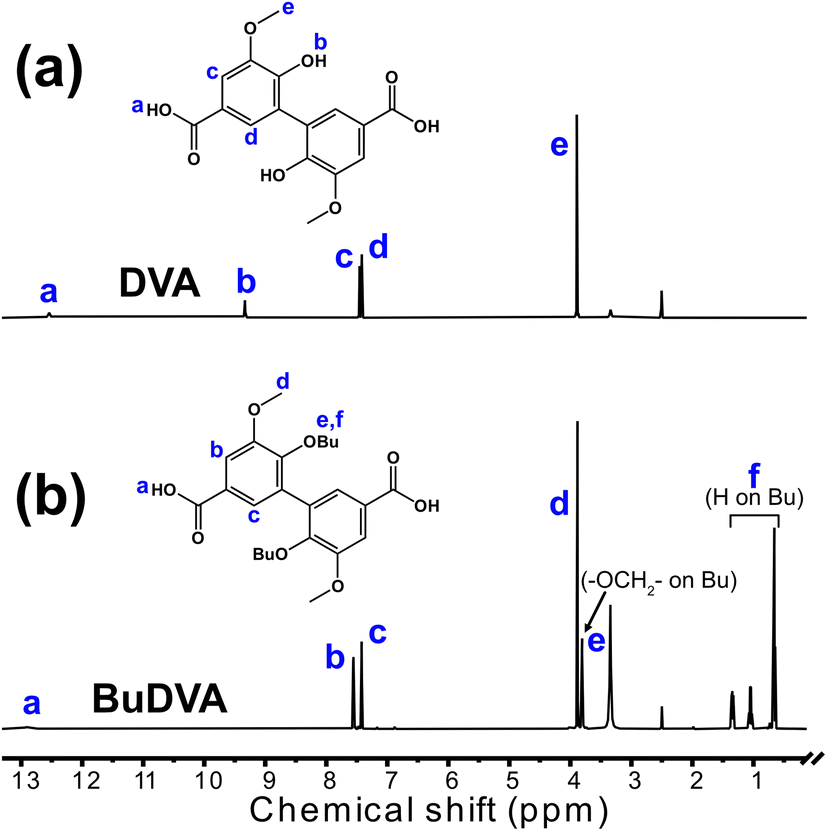 | ||
| Fig. 2 1H NMR spectra of (a) DVA and (b) BuDVA in DMSO-d6. The peaks are described in detail in the ESI.† | ||
OHESO-x and BuESO-x vitrimers with x = 0.7, 1.0, and 1.5 were prepared in the present study. As shown in Fig. 1, all the BuESO-x films were transparent and homogeneous in color, OHESO-0.7 and OHESO-1.0 were semi-transparent, and OHESO-1.5 was opaque. It was considered that the BuDVA–ESO mixtures were prepared homogeneously, as mentioned in Section 2.3. In contrast, in the DVA–ESO mixtures, the free phenolic hydroxyl groups of the DVA molecules can form hydrogen bonds with the surrounding phenolic OH and/or carbonyl groups, and readily aggregate together. Consequently, the OHESO-x vitrimers were fabricated from immiscible DVA–ESO mixtures and were heterogeneous in appearance. These cured vitrimer films only swelled (or partially dissolved) in conventional organic solvents, so we studied their structures by swelling experiments and FT-IR spectroscopy. The swell ratios and gel contents of the vitrimers are listed in Table S1.† All the cured vitrimers exhibited high gel content values (>80%), which suggests that crosslinked networks had formed. For both OHESO-x and BuESO-x, vitrimers with x = 1.0 exhibited relatively high gel content values, which corresponded to their higher crosslink densities compared with those with x = 0.7 and 1.5. We attributed this result to the fact that the diacids did not completely react with the epoxy or hydroxyl groups (especially for vitrimers with x = 1.5), and therefore some free COOH groups remained. The unreacted COOH groups were also confirmed by FT-IR. During the swelling process, some linear segments, including unreacted monomers and uncrosslinked oligomers, were extracted from the vitrimer matrix. However, the quantitative relationship between the extracts and the difference in crosslinking density remains unclear.
The IR spectra of the monomers and cured OHESO-1.0 and BuESO-1.0 vitrimers are shown in Fig. 3, and the spectra of other vitrimers with x = 0.7 and 1.5 are shown in Fig. S2 and S3.† In the present study, we focused on three regions of the IR spectra: (1) 3800–3200 cm−1, (2) 1780–1620 cm−1, and (3) 870–800 cm−1, which correspond to the absorption of hydroxyl groups, carbonyl groups, and epoxy groups, respectively.55,57,58 In the 3800–3200 cm−1 region, there was broad absorption attributable to the free aliphatic OH groups, which derived from the ring-opening of the epoxy groups, of all the BuESO-x and OHESO-0.7 vitrimers (Fig. S2 and S3†). For the OHESO-1.5 and OHESO-1.0 vitrimers, these broad peaks overlapped with the absorption of the free phenolic OH and COOH groups. As mentioned before, because DVA molecules tend to aggregate together owing to the interaction of hydrogen bonds, some COOH groups remained unreacted during the curing process. In the 1780–1620 cm−1 region, there were peaks close to 1710 cm−1, which were assigned to the carbonyl groups of the aromatic esters, between the peaks attributable to the carbonyl groups of ESO (1740 cm−1) and those of the original aromatic acid (1685 and 1674 cm−1 for DVA and BuDVA, respectively). This indicates that the aromatic acid groups reacted with the epoxy groups (or reacted with the OH groups generated from ring-opening), and formed new ester bonds during curing and transesterification. Moreover, for all the OHESO-x and BuESO-1.5 vitrimers, there were shoulder peaks corresponding to the COOH groups, which also suggests that some free COOH groups remained. In the 870–800 cm−1 region, the characteristic peaks attributable to epoxy groups (843 and 823 cm−1) of the ESO vanished after curing in both the OHESO-x and BuESO-x vitrimers. This demonstrates that all the oxirane rings had been opened. The peak near 820 cm−1 produced by OHESO-x—the intensity of which increased as x increased—may have derived from the unreacted COOH groups. According to the previous research,59,60 the homopolymerization of epoxy tends to occur at a relatively high level in a high epoxy/COOH environment (i.e., off-stoichiometry) and at high curing temperatures, and the absorption of the C–O–C ether bond would appear in the regions of 1200–1100 cm−1. In the present work, even in the cases of x = 0.7, we did not observe any distinguishable absorption of ether bonds. Overall, the results discussed above indicate that we had successfully prepared DVA/BuDVA–ESO vitrimers crosslinked by ester bonds. However, unreacted carboxylic acid groups remained in the matrices of the OHESO-x and BuESO-1.5 vitrimers.
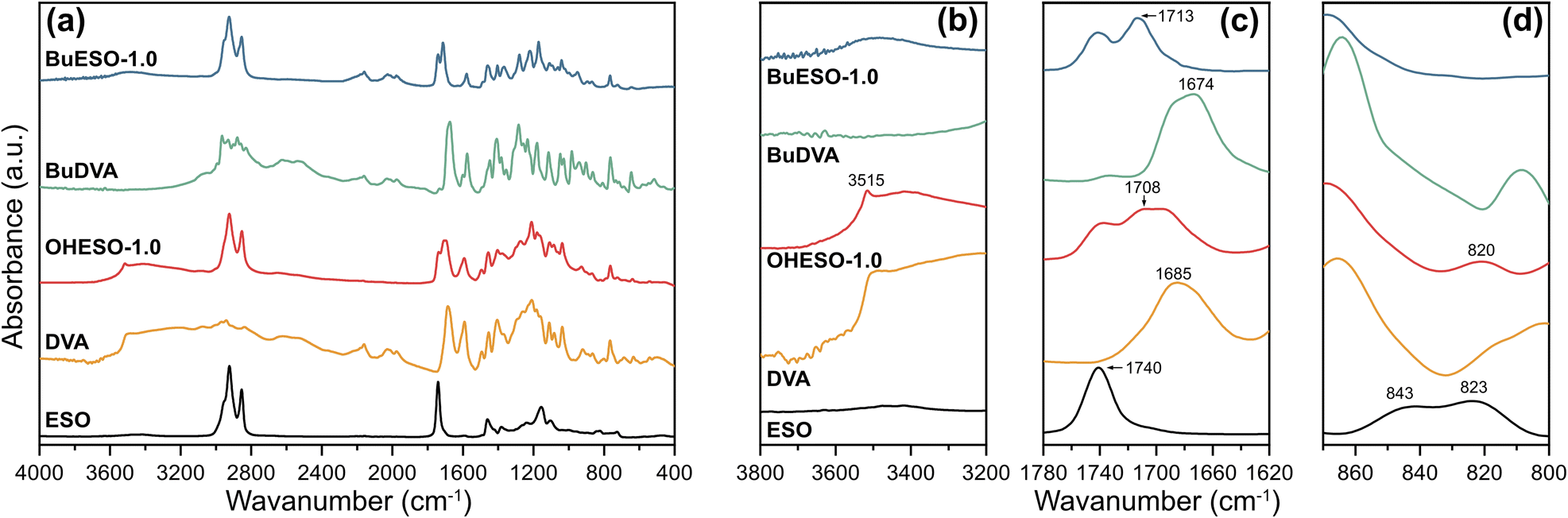 | ||
| Fig. 3 (a) FT-IR spectra of ESO, DVA, OHESO-1.0, BuDVA, and BuESO-1.0, and enlarged views of regions (b) 3800–3200 cm−1, (c) 1780–1620 cm−1, and (d) 870–800 cm−1. | ||
3.2 Thermal properties of the vitrimers
The thermal properties of the vitrimer films were assessed by TGA and DMA. Table 2 lists the Tg, the temperature at which the mass loss became 5% (Td5%), and the temperature at which the maximum degradation rate occurred (Tdmax) of each film. The TGA and derivative TG (DTG) curves of the vitrimers are shown in Fig. 4. The thermal degradation of each vitrimer began at approximately 240 °C, which is similar to the degradation temperature of an epoxy resin.61–63 The OHESO-x vitrimers had lower Td5% values than the BuESO-x vitrimers because they had more free phenolic OH groups and unreacted COOH groups. The OHESO-x and BuESO-x vitrimers had comparable Tdmax values, but had lower Td5% values than the aromatic bio-based polyesters, such as DVA-derived polyesters reported by us previously,12,13 and furandicarboxylic acid-derived polyesters5,64 (e.g., Td5% > 360 °C and Tdmax > 400 °C). These results may be explained as follows. At low temperatures, dehydration occurs much faster in OH-rich environments (i.e., when a large number of aliphatic OH groups are generated by ring-opening). This causes faster chain scission in vitrimers.63 At higher temperatures, vitrimers exhibit similar thermal degradation behaviors, such as β-hydrogen scission, to those of polyesters. Therefore, their Tdmax values occurred in the same temperature ranges.| Codes | T g (°C) | T d5% (°C) | T dmax (°C) | σ max (MPa) | ε max (MPa) | E (MPa) |
|---|---|---|---|---|---|---|
|
a By DMA (peak temperature of tanδ).
b The temperature at which the mass loss became 5%.
c The temperature at which the maximum degradation rate occurred.
d The maximum tensile strength.
e The elongation at break.
f The Young's modulus.
g The films were too brittle to conduct tensile tests.
|
||||||
| OHESO-1.5 | 62 | 324 | 393 | Not measuredg | ||
| OHESO-1.0 | 45 | 314 | 405 | 13.6 ± 1.1 | 13.6 ± 2 | 286 ± 49 |
| OHESO-0.7 | 43 | 332 | 413 | 12.9 ± 1.4 | 23.6 ± 13.7 | 276 ± 81 |
| BuESO-1.5 | 44 | 358 | 414 | Not measuredg | ||
| BuESO-1.0 | 36 | 362 | 416 | 4.8 ± 0.6 | 69 ± 12 | 57 ± 11 |
| BuESO-0.7 | 20 | 357 | 409 | 1.9 ± 0.3 | 116 ± 16 | 2.9 ± 0.5 |
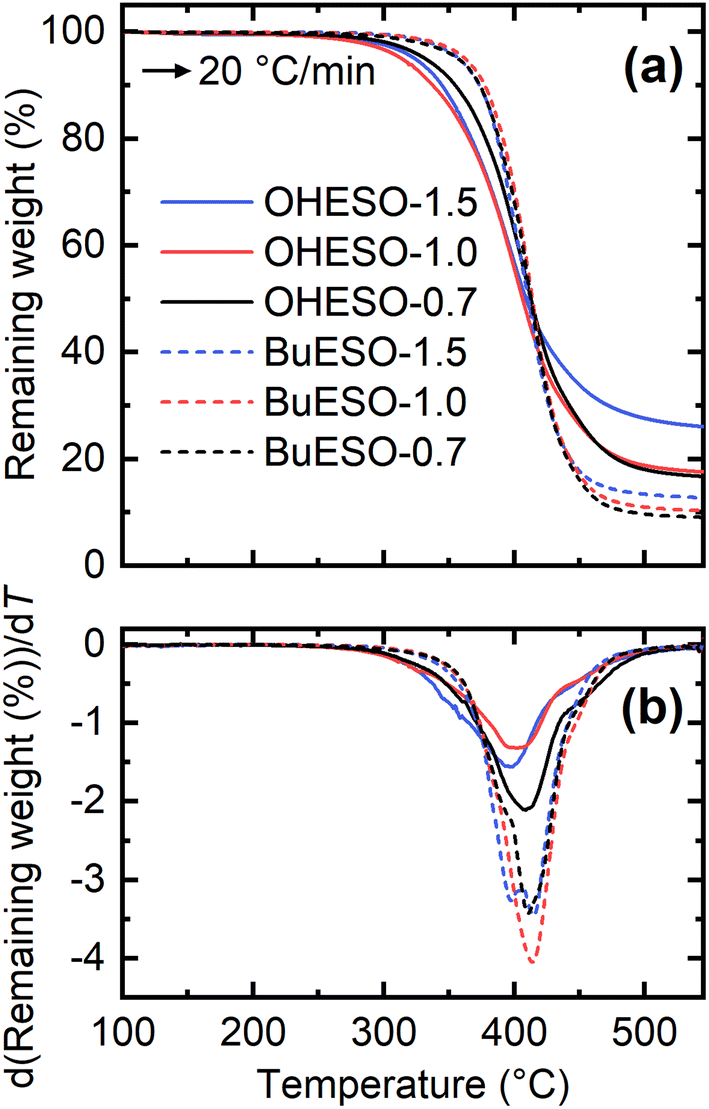 | ||
| Fig. 4 (a) TGA and (b) derivative TG (DTG) thermograms produced by OHESO-x and BuESO-x at a heating rate of 20 °C min−1. | ||
The Tg values of the vitrimers were determined by DMA according to the peak temperatures of tanδ (the loss or damping factor) in the present study (Fig. 5). The Tg values of the OHESO-x and BuESO-x vitrimers were in the ranges 43–62 °C and 20–44 °C, respectively. The Tg values of OHESO-1.0 and OHESO-0.7 were almost equal, and were lower than that of OHESO-1.5. The Tg of BuESO-x increased as x increased. Generally, a high gel content corresponds to a high crosslinking density and Tg value of the crosslinked polymer. It was noticed that the order of Tgs was different from that of gel contents. The heterogeneity in vitrimer networks possibly caused this discrepancy. The Tgs of the OHESO-x vitrimers were higher than those of the BuESO-x vitrimers of the same x values, even though the gel contents of the BuESO-x vitrimers were higher than those of the OHESO-x vitrimers. This may be explained by the large free volume generated by the long Bu side chains of BuDVA, which endowed the molecular chains of the BuESO-x vitrimers with greater mobility. Furthermore, the hydrogen bonds formed by the free phenolic OH groups acted as physical crosslinks. As mentioned earlier, bio-based carboxylic acids and epoxy compounds have been used to develop vitrimer materials before. For example, Altuna et al.28 prepared citric acid-ESO-based vitrimers (Tg < 30 °C), and Zhang et al.44 prepared ozonated-lignin-based vitrimers (Tg > 90 °C). Therefore, high-rigidity monomers (e.g., cyclic or aromatic monomers) with multiple functional groups (i.e., more crosslinking points) can be used to prepare vitrimers with high Tg values.
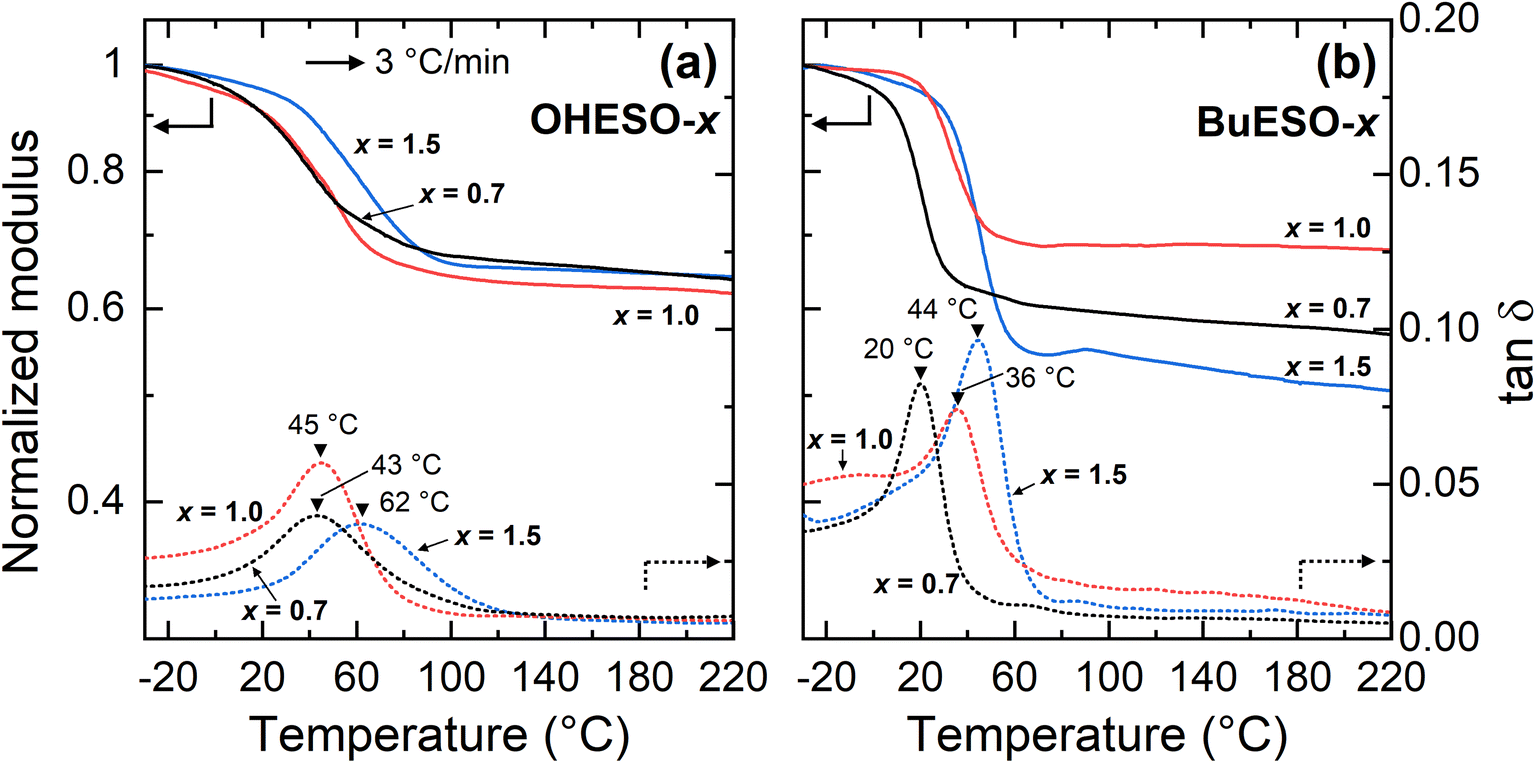 | ||
| Fig. 5 DMA thermograms of (a) OHESO-x and (b) BuESO-x obtained at a heating rate of 3 °C min−1. The Tg values are indicated by black triangles. | ||
3.3 Stress relaxation behavior
Stress relaxation is a common phenomenon of polymer materials. When a polymer is subjected to stress, its molecular chains tend to move to relax the stress, and relaxation becomes more rapid as the temperature increases.65 In vitrimers, stress relaxation behavior is closely related to the bond-exchange reaction.26 Therefore, we performed stress relaxation tests in the present work to confirm transesterification and evaluate the reaction rate. The stress relaxation curves of OHESO-1.0 and BuESO-1.0 at 170–200 °C, and those of the OHESO-x and BuESO-x vitrimers at 200 °C, are shown in Fig. 6.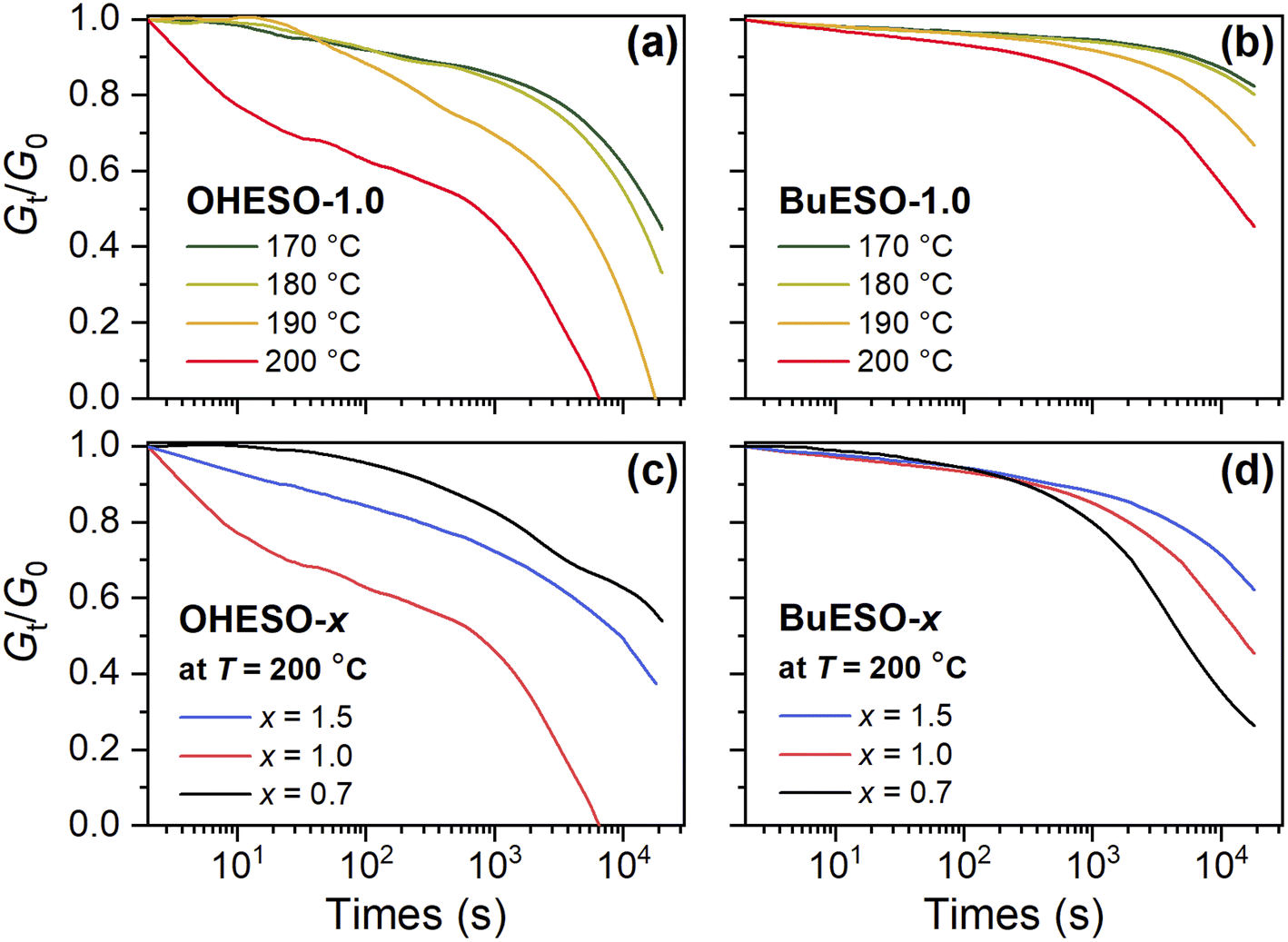 | ||
| Fig. 6 Normalized stress relaxation curves of (a) OHESO-1.0 and (b) BuESO-1.0 at T = 170–200 °C, and normalized stress relaxation curves of (c) OHESO-x and (d) BuESO-x at T = 200 °C. | ||
We measured the Gt/G0 change in the time range 0–20000 s, where Gt and G0 represent the relaxation moduli at times t and 0, respectively. Except for in the case of BuESO-1.0 at T = 170 and 180 °C, there was an apparent decrease in Gt/G0 in each stress relaxation curve. This indicates that transesterification did occur during heating. Moreover, as shown in Fig. 6(a) and (b), the Gt/G0 values decreased more rapidly as the temperature increased, corresponding to the increase in the transesterification rates. At T = 200 °C, BuESO-0.7 exhibited the most rapid decrease in Gt/G0 [Fig. 6(d)]. This may have been because the concentration of free aliphatic OH groups was higher in BuESO-0.7 than in BuESO-1.0 and BuESO-1.5. Therefore, BuESO-0.7 underwent more rapid transesterification.28 However, OHESO-0.7 and OHESO-1.0 had the lowest and highest relaxation rates of the OHESO-x vitrimers, respectively [Fig. 6(c)]. Unfortunately, we did not succeed in finding a plausible explanation for this irregular result, which may have been caused by the high heterogeneity of the OHESO-x matrix.
In a general Maxwell model, Gt/G0 can be expressed by the simple exponential function:
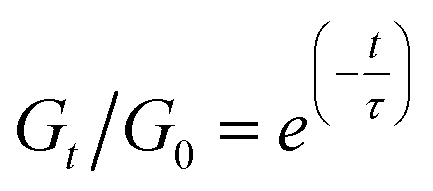
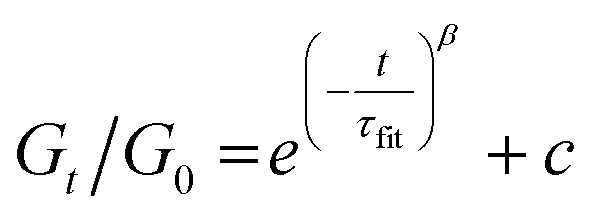
Although this function cannot accurately describe the relaxation behaviors of the OHESO-x vitrimers in the time ranges t < 102 s, the relaxation of the vitrimers in the present work was relatively slow (i.e., the Gt/G0 values decreased to less than 0.5 at t < 102 s). Therefore, this function can be used to describe the relaxation behaviors in the long time ranges to evaluate the fitted relaxation time (τfit). The experimental data fitted well using this function, with R2 values higher than 0.99. The best-fit values of τfit, and the β and c parameters are summarized in Table S2.†
The relaxation times of the vitrimers in the present study were obviously longer than those of other transesterification-based vitrimers, the τ values of which are typically in the range 102 to 103 s at T values close to 200 °C.29,41,44,69 In other words, the transesterification rates of the DVA vitrimers were lower than those reported in most previous studies. To the best of our knowledge, only Altuna et al. have reported comparable slow relaxation behavior (τ > 20000 s at T = 160 °C) in citric acid-ESO-based vitrimers.28 There are many factors that affect the rates of stress relaxation, such as the types and concentrations of catalysts,70 crosslinking densities,71,72 branch numbers on crosslinker,73 and polarity of the networks,74etc. We attributed the low relaxation rates of our vitrimers to the bulky and rigid chemical structures of the crosslinkers (i.e., DVA and BuDVA), which reduced the movability of molecular chains and thus slowed down the reorganization of networks.
We calculated the approximate activation energy (Ea) of the transesterification reaction using the Arrhenius equation:26,70
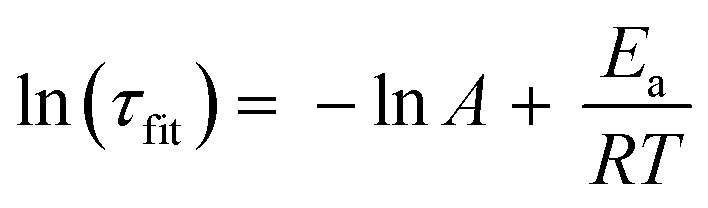
3.4 Mechanical properties of the pristine and reprocessed vitrimer films
The stress–strain curves of OHESO-x and BuESO-x (x = 0.7 and 1.0) are shown in Fig. 7, and their mechanical properties are summarized in Table 2. BuESO-1.0 had higher σmax and E values, but lower εmax values than BuESO-0.7, which agreed with the result that BuESO-1.0 had a higher crosslink density. However, OHESO-1.0 had σmax and E values that were only slightly higher than those of OHESO-0.7. This resembled their respective Tg relationships (the Tg of OHESO-1.0 was only slightly higher than that of OHESO-0.7). The increase in x had a more pronounced effect on the thermal and mechanical properties of BuESO-x compared with OHESO-x. Although the crosslink density of OHESO-1.0 was higher than that of OHESO-0.7, the unreacted and aggregated DVA moieties acted as defects, and caused a deterioration in the material properties. Therefore, OHESO-1.5 and BuESO-1.5 (noted that there were also unreacted COOH groups remained in BuESO-1.5) became too brittle for tensile tests.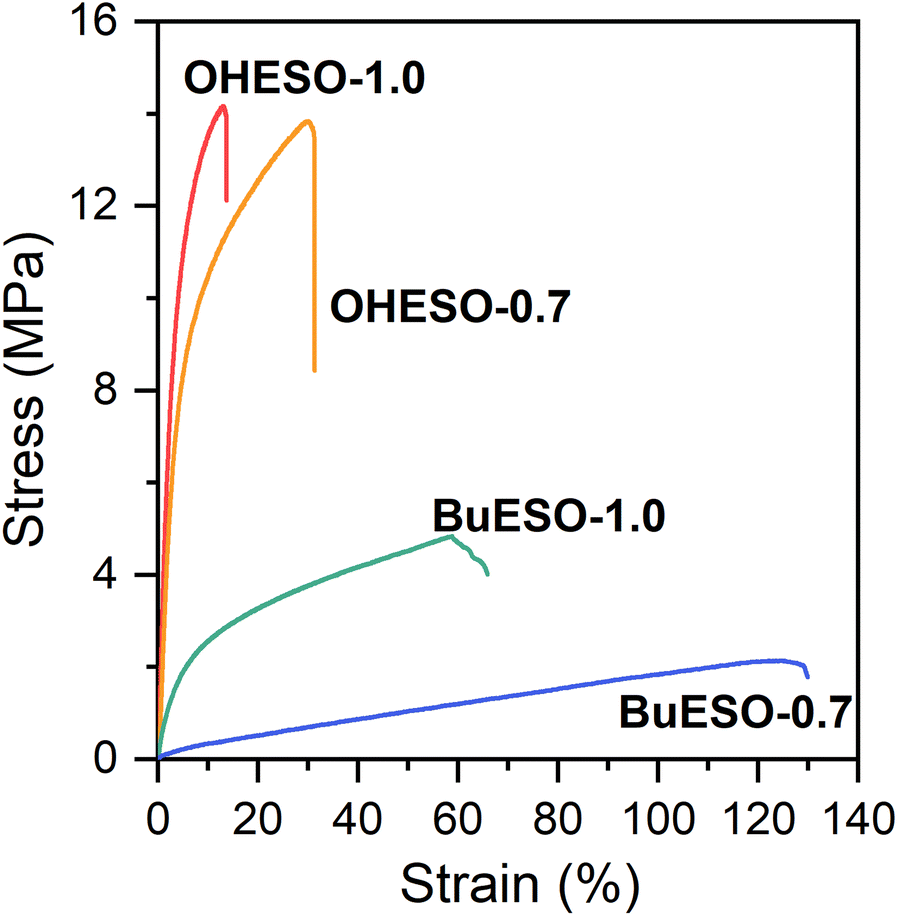 | ||
| Fig. 7 Stress–strain curves of OHESO-x and BuESO-x vitrimer films (x = 0.7 and 1.0). | ||
One of the attractive properties of vitrimers is that they can be reprocessed and remolded at the high-temperatures at which bond exchange occurs. Therefore, vitrimers can be used to develop recyclable thermosets. In the present study, we investigated the mechanical properties of reprocessed OHESO-1.0 and BuESO-1.0 vitrimer films over five reprocessing cycles using the method described in Section 2.4. Images and stress–strain curves of the reprocessed films, and the retention of their mechanical properties after reprocessing, are shown in Fig. 8. The σmax, εmax, and E values are summarized in Table S3.† After reprocessing, the films were still coherent and self-standing, but became softer (especially the OHESO-1.0 film), and darkened in color compared with the pristine films. Similar cases have been reported previously.41,49 The σmax and E values of OHESO-1.0 and BuESO-1.0 decreased to 40–50% of their original values during reprocessing. The εmax of the reprocessed BuESO-1.0 fluctuated between 70% and 110% of its original value, whereas that of the reprocessed OHESO-1.0 increased by more than 140% of its original value after three cycles of processing. This may be attributed to the decrease in crosslink density owing to thermal degradation at the high reprocessing temperature (230 °C), which was confirmed by reduced intensities of hydroxyl group absorption via FT-IR (Fig. S7 and S8†). This deterioration of mechanical properties can be improved by grinding the fragments into fine powders or prolonging the curing time, although it should be noted that thermal degradation should also be taken into account.75 Using chemical methods to recycle vitrimer materials is also a good way of avoiding thermal degradation.41,49,76 These results suggest that although the reprocessed DVA-derived vitrimers cannot be used for the same application as the pristine vitrimers, owing to their inferior properties, they had better reprocessability and recyclability compared to traditional thermosets.
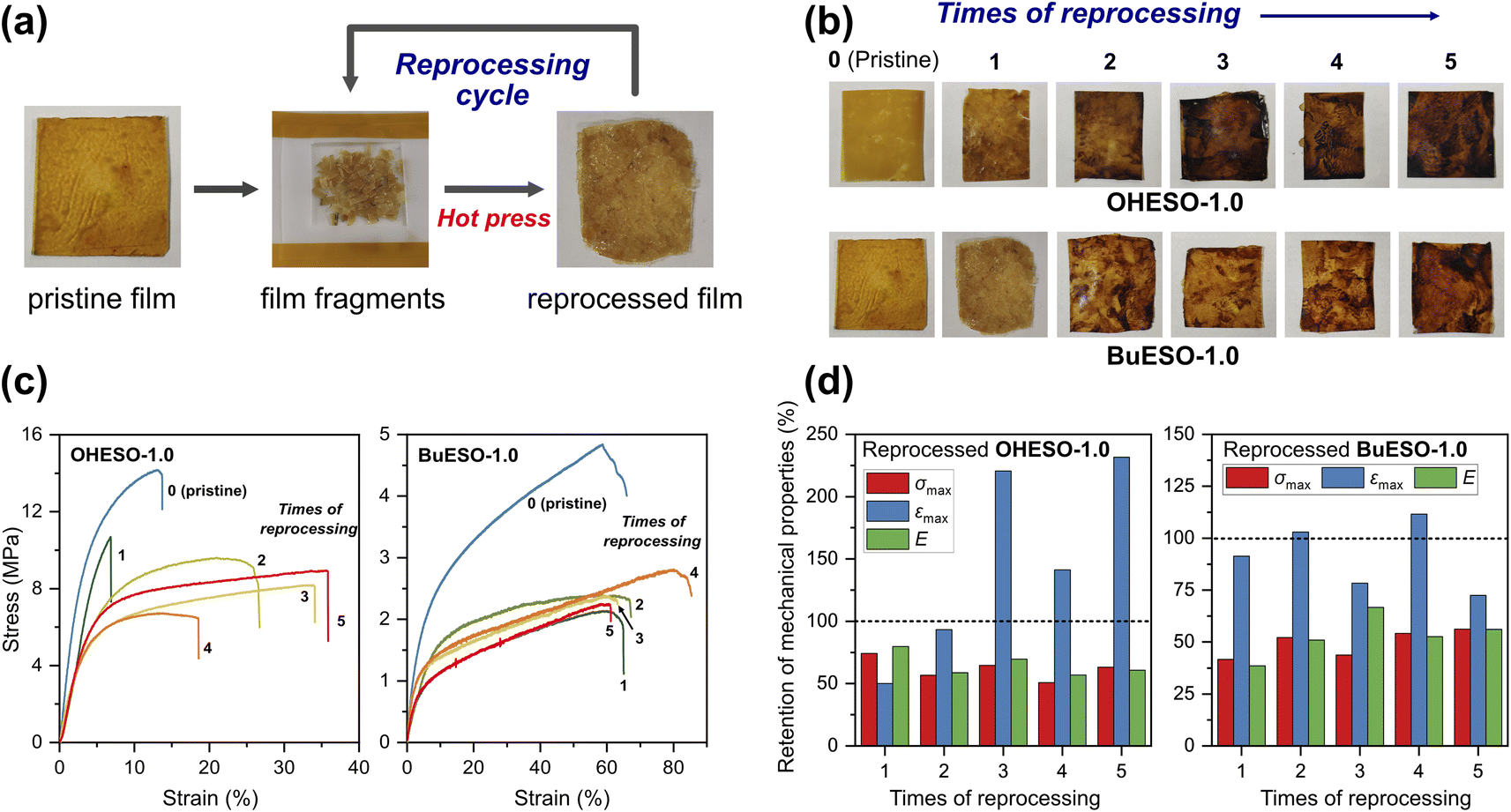 | ||
| Fig. 8 (a) Reprocessing the vitrimer films. (b) Images of the OHESO-1.0 and BuESO-1.0 films obtained whilst reprocessing them five times. (c) Stress–strain curves of the pristine and reprocessed OHESO-1.0 and BuESO-1.0 films. (d) Retention rates of the mechanical properties of the reprocessed vitrimer films. The retention rates at 100% are indicated by dashed lines. | ||
3.5 Self-healing property
Self-healing is a characteristic of dynamically crosslinked polymers. Bond exchange occurs during heating, which induces the rearrangement of molecular chains near the cracks. Consequently, the molecular chains refill the cracks, and the matrix material is healed. As shown in Fig. 9, we investigated the self-healing behaviors of OHESO-1.0 and BuESO-1.0 at T = 100 and 180 °C in the present work. | ||
| Fig. 9 Optical microscope images of (a–d) OHESO-1.0 and (e–h) BuESO-1.0 films healed at T = 100 and 180 °C. The scale bar represents 200 μm. | ||
At T = 100 °C [Fig. 9(a), (b), (e), and (f)], the crack in OHESO-1.0 was barely repaired after 2 h. In contrast, the crack width in BuESO-1.0 decreased from approximately 53 μm to approximately 7 μm in 1 h. Over 85% of the crack was healed (on the basis of crack width). OHESO-1.0 only exhibited self-healing at high temperatures. At T = 180 °C, [Fig. 9(c), (d), (g), and (h)] the crack width of OHESO-1.0 decreased slowly from approximately 20 to 15 μm over 2 h, whereas the crack in BuESO-1.0 was almost completely healed after 30 min. Obviously, BuESO-1.0 exhibited much higher repair rates at a wide range of temperatures compared with OHESO-1.0. This can be attributed to the hydrogen bonds formed by the free phenolic OH groups in OHESO-1.0. As mentioned previously, hydrogen bonds act as physical crosslinks, and hinder the movement of molecular chains, thereby slowing self-healing. In contrast to previous reports,77,78 we did not observe complete healing in either OHESO-1.0 or BuESO-1.0, confirming the slow transesterification rates of our vitrimers (i.e., their Ea values were relatively high).
Conclusions
In the present study, we prepared two series of bio-based vitrimers, i.e., OHESO-x and BuESO-x, by curing DVA or BuDVA with ESO in the presence of a zinc-containing catalyst. Each vitrimer exhibited satisfactory thermal stability (Td5% > 300 °C). Depending on their side chains and x values, the Tg values, Young's moduli, maximum elongation values, and tensile strengths of the vitrimers were in the ranges 20–62 °C, 3–286 MPa, 13–116%, and 2–14 MPa, respectively. As a result of the hydrogen bonds formed by the free phenolic hydroxyl groups and unreacted carboxylic acid groups remaining in the OHESO-x vitrimers, and the large free volumes generated by the butyl side chains in the BuESO-x vitrimers, the OHESO-x vitrimers had higher Tg, Young's modulus, and maximum tensile strength values than the BuESO-x vitrimers with the same x values. At temperatures approaching 200 °C, both series of vitrimers exhibited stress relaxation resulting from dynamic bond exchange reactions (i.e., transesterification). We also investigated the shape memory, self-healing, and reprocessability behaviors of OHESO-1.0 and BuESO-1.0. After reprocessing (up to five cycles), the mechanical properties of the reprocessed films recovered to approximately 50% of those of the pristine films.This present work demonstrates the feasibility of using symmetrical bio-based aromatic divanillic acid to prepare dynamically crosslinked vitrimers with favorable material properties and reprocessability. It also provides some new insights into the use of biomass-derived feedstocks to develop sustainable adhesives, coatings, and alternatives to other conventional thermosetting materials. Further research on the preparation of homogenous vitrimer films, and improvements to their thermal and mechanical properties are required before they will be suitable for practical applications.
Author contributions
Yunfan Zhang: Conceptualization, Investigation, Writing – original draft, Funding acquisition. Yukiko Enomoto: Conceptualization, Supervision, Writing – review & editing, Funding acquisition. Tadahisa Iwata: Writing – review & editing, Supervision, Funding acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the financial support of the JSPS KAKENHI [grant number JP 20K06159, to Y. E.] and the JST SPRING from the University of Tokyo [grant number JPMJSP2108, to Y. Z.]. We thank New Japan Chemical Co., Ltd for providing the epoxidized soybean oil, and Professor Tsuguyuki Saito and Associate Professor Tetsuo Yamaguchi (both from the University of Tokyo) for the use of the rheometer.References
- R. Mülhaupt, Macromol. Chem. Phys., 2013, 214, 159–174 CrossRef.
- S. Ramesh Kumar, P. Shaiju, K. E. O’Connor and R. B. Padamati, Curr. Opin. Green Sustainable Chem., 2020, 21, 75–81 CrossRef CAS.
- E. Castro-Aguirre, F. Iniguez-Franco, H. Samsudin, X. Fang and R. Auras, Adv. Drug Delivery Rev., 2016, 107, 333–366 CrossRef CAS PubMed.
- S. A. Rafiqah, A. Khalina, A. S. Harmaen, I. A. Tawakkal, K. Zaman, M. Asim, M. N. Nurrazi and C. H. Lee, Polymers, 2021, 13, 1436 CrossRef CAS PubMed.
- G. Z. Papageorgiou, D. G. Papageorgiou, Z. Terzopoulou and D. N. Bikiaris, Eur. Polym. J., 2016, 83, 202–229 CrossRef CAS.
- Z. Terzopoulou, L. Papadopoulos, A. Zamboulis, D. G. Papageorgiou, G. Z. Papageorgiou and D. N. Bikiaris, Polymers, 2020, 12, 1209 CrossRef CAS PubMed.
- S. Zhang, Z. Cheng, S. Zeng, G. Li, J. Xiong, L. Ding and M. Gauthier, J. Appl. Polym. Sci., 2020, 137, 49189 CrossRef CAS.
- E. Xanthopoulou, Z. Terzopoulou, A. Zamboulis, L. Papadopoulos, K. Tsongas, D. Tzetzis, G. Z. Papageorgiou and D. N. Bikiaris, ACS Sustainable Chem. Eng., 2021, 9, 1383–1397 CrossRef CAS.
- T. Goto, T. Iwata and H. Abe, Biomacromolecules, 2019, 20, 318–325 CrossRef CAS PubMed.
- Y. Enomoto and T. Iwata, Polymer, 2020, 193, 122330 CrossRef CAS.
- K. Fujieda, Y. Enomoto, Y. Zhang and T. Iwata, Polymer, 2022, 257, 125241 CrossRef CAS.
- Y. Zhang, Y. Enomoto and T. Iwata, Polymer, 2020, 203, 122751 CrossRef CAS.
- Y. Zhang, Y. Enomoto and T. Iwata, Polym. Degrad. Stab., 2021, 192, 109706 CrossRef CAS.
- K. Yagura, Y. Enomoto and T. Iwata, Polymer, 2022, 256, 125222 CrossRef CAS.
- K. Yagura, Y. Zhang, Y. Enomoto and T. Iwata, Polymer, 2021, 228, 123907 CrossRef CAS.
- Y. Enomoto and T. Iwata, Eur. Polym. J., 2021, 154, 110526 CrossRef CAS.
- R. T. Nishimura, C. H. Giammanco and D. A. Vosburg, J. Chem. Educ., 2010, 87, 526–527 CrossRef CAS.
- A. Llevot, E. Grau, S. Carlotti, S. Grelier and H. Cramail, J. Mol. Catal. B: Enzym., 2016, 125, 34–41 CrossRef CAS.
- S. S. Kuhire, A. B. Ichake, E. Grau, H. Cramail and P. P. Wadgaonkar, Eur. Polym. J., 2018, 109, 257–264 CrossRef CAS.
- A. Llevot, E. Grau, S. Carlotti, S. Grelier and H. Cramail, Polym. Chem., 2015, 6, 7693–7700 RSC.
- A. Llevot, E. Grau, S. Carlotti, S. Grelier and H. Cramail, Polym. Chem., 2015, 6, 6058–6066 RSC.
- E. Savonnet, C. Le Coz, E. Grau, S. Grelier, B. Defoort and H. Cramail, Front. Chem., 2019, 7, 606 CrossRef CAS PubMed.
- E. Savonnet, E. Grau, S. Grelier, B. Defoort and H. Cramail, ACS Sustainable Chem. Eng., 2018, 6, 11008–11017 CrossRef CAS.
- J. Liu, L. Zhang, W. Shun, J. Dai, Y. Peng and X. Liu, J. Polym. Sci., 2021, 59, 1474–1490 CrossRef CAS.
- Q. Zhang, M. Song, Y. Xu, W. Wang, Z. Wang and L. Zhang, Prog. Polym. Sci., 2021, 120, 101430 CrossRef CAS.
- D. Montarnal, M. Capelot, F. Tournilhac and L. Leibler, Science, 2011, 334, 965 CrossRef CAS PubMed.
- W. Denissen, J. M. Winne and F. E. Du Prez, Chem. Sci., 2016, 7, 30–38 RSC.
- F. I. Altuna, V. Pettarin and R. J. J. Williams, Green Chem., 2013, 15, 3360–3366 RSC.
- T. Liu, C. Hao, L. Wang, Y. Li, W. Liu, J. Xin and J. Zhang, Macromolecules, 2017, 50, 8588–8597 CrossRef CAS.
- T. Liu, C. Hao, S. Zhang, X. Yang, L. Wang, J. Han, Y. Li, J. Xin and J. Zhang, Macromolecules, 2018, 51, 5577–5585 CrossRef CAS.
- M. Capelot, D. Montarnal, F. Tournilhac and L. Leibler, J. Am. Chem. Soc., 2012, 134, 7664–7667 CrossRef CAS PubMed.
- J. J. Lessard, L. F. Garcia, C. P. Easterling, M. B. Sims, K. C. Bentz, S. Arencibia, D. A. Savin and B. S. Sumerlin, Macromolecules, 2019, 52, 2105–2111 CrossRef CAS.
- W. Denissen, G. Rivero, R. Nicolaÿ, L. Leibler, J. M. Winne and F. E. Du Prez, Adv. Funct. Mater., 2015, 25, 2451–2457 CrossRef CAS.
- P. R. Christensen, A. M. Scheuermann, K. E. Loeffler and B. A. Helms, Nat. Chem., 2019, 11, 442–448 CrossRef CAS PubMed.
- A. Zych, J. Tellers, L. Bertolacci, L. Ceseracciu, L. Marini, G. Mancini and A. Athanassiou, ACS Appl. Polym. Mater., 2021, 3, 1135–1144 CrossRef CAS.
- M. Röttger, T. Domenech, R. van der Weegen, A. Breuillac, R. Nicolaÿ and L. Leibler, Science, 2017, 356, 62–65 CrossRef PubMed.
- Y. Nishimura, J. Chung, H. Muradyan and Z. Guan, J. Am. Chem. Soc., 2017, 139, 14881–14884 CrossRef CAS PubMed.
- C. A. Tretbar, J. A. Neal and Z. Guan, J. Am. Chem. Soc., 2019, 141, 16595–16599 CrossRef CAS PubMed.
- B. Krishnakumar, A. Pucci, P. P. Wadgaonkar, I. Kumar, W. H. Binder and S. Rana, Chem. Eng. J., 2022, 433, 133261 CrossRef CAS.
- D. J. Fortman, J. P. Brutman, G. X. De Hoe, R. L. Snyder, W. R. Dichtel and M. A. Hillmyer, ACS Sustainable Chem. Eng., 2018, 6, 11145–11159 CrossRef CAS.
- X. Yang, L. Guo, X. Xu, S. Shang and H. Liu, Mater. Des., 2020, 186, 108248 CrossRef CAS.
- Y. Zeng, J. Li, S. Liu and B. Yang, Polymers, 2021, 13, 3386 CrossRef CAS PubMed.
- X. Feng, J. Fan, A. Li and G. Li, ACS Sustainable Chem. Eng., 2020, 8, 874–883 CrossRef CAS.
- S. Zhang, T. Liu, C. Hao, L. Wang, J. Han, H. Liu and J. Zhang, Green Chem., 2018, 20, 2995–3000 RSC.
- C. Hao, T. Liu, S. Zhang, L. Brown, R. Li, J. Xin, T. Zhong, L. Jiang and J. Zhang, ChemSusChem, 2019, 12, 1049–1058 CrossRef CAS PubMed.
- M. Qi, Y.-J. Xu, W.-H. Rao, X. Luo, L. Chen and Y.-Z. Wang, RSC Adv., 2018, 8, 26948–26958 RSC.
- Q. Yu, X. Peng, Y. Wang, H. Geng, A. Xu, X. Zhang, W. Xu and D. Ye, Eur. Polym. J., 2019, 117, 55–63 CrossRef CAS.
- Y.-Y. Liu, J. He, Y.-D. Li, X.-L. Zhao and J.-B. Zeng, Compos. Commun., 2020, 22, 100445 CrossRef.
- H. Memon, H. Y. Liu, M. A. Rashid, L. Chen, Q. R. Jiang, L. Y. Zhang, Y. Wei, W. S. Liu and Y. P. Qiu, Macromolecules, 2020, 53, 621–630 CrossRef CAS.
- X.-L. Zhao, Y.-Y. Liu, Y. Weng, Y.-D. Li and J.-B. Zeng, ACS Sustainable Chem. Eng., 2020, 8, 15020–15029 CrossRef CAS.
- K. P. Cortés-Guzmán, A. R. Parikh, M. L. Sparacin, A. K. Remy, L. Adegoke, C. Chitrakar, M. Ecker, W. E. Voit and R. A. Smaldone, ACS Sustainable Chem. Eng., 2022, 10, 13091–13099 CrossRef.
- S. Engelen, A. A. Wróblewska, K. De Bruycker, R. Aksakal, V. Ladmiral, S. Caillol and F. E. Du Prez, Polym. Chem., 2022, 13, 2665–2673 RSC.
- S. Grauzeliene, M. Kastanauskas, V. Talacka and J. Ostrauskaite, ACS Appl. Polym. Mater., 2022, 4, 6103–6110 CrossRef PubMed.
- A. Roig, P. Hidalgo, X. Ramis, S. De la Flor and À. Serra, ACS Appl. Polym. Mater., 2022, 4, 9341–9350 CrossRef CAS.
- J. F. Scholtes and O. Trapp, Angew. Chem., Int. Ed., 2019, 58, 6306–6310 CrossRef CAS PubMed.
- C. A. Schneider, W. S. Rasband and K. W. Eliceiri, Nat. Methods, 2012, 9, 671–675 CrossRef CAS PubMed.
- A. Adhvaryu and S. Z. Erhan, Ind. Crops Prod., 2002, 15, 247–254 CrossRef CAS.
- J. S. Lupoi, S. Singh, R. Parthasarathi, B. A. Simmons and R. J. Henry, Renew. Sustain. Energy Rev., 2015, 49, 871–906 CrossRef CAS.
- S. Chappuis, P. Edera, M. Cloitre and F. Tournilhac, Macromolecules, 2022, 55, 6982–6991 CrossRef CAS.
- Q.-A. Poutrel, J. J. Blaker, C. Soutis, F. Tournilhac and M. Gresil, Polym. Chem., 2020, 11, 5327–5338 RSC.
- L.-H. Lee, J. Polym. Sci., Part A: Gen. Pap., 1965, 3, 859–882 CrossRef CAS.
- M. Natarajan and S. C. Murugavel, Polym. Bull., 2017, 74, 3319–3340 CrossRef CAS.
- P.-Y. Kuo, L. de Assis Barros, Y.-C. Sheen, M. Sain, J. S. Y. Tjong and N. Yan, J. Anal. Appl. Pyrolysis, 2016, 117, 199–213 CrossRef CAS.
- Z. Terzopoulou, V. Tsanaktsis, M. Nerantzaki, D. S. Achilias, T. Vaimakis, G. Z. Papageorgiou and D. N. Bikiaris, J. Anal. Appl. Pyrolysis, 2016, 117, 162–175 CrossRef CAS.
- A. V. Tobolsky, J. Appl. Phys., 1956, 27, 673–685 CrossRef CAS.
- E. E. L. Maassen, J. P. A. Heuts and R. P. Sijbesma, Polym. Chem., 2021, 12, 3640–3649 RSC.
- Z. Song, Z. Wang and S. Cai, Mech. Mater., 2021, 153, 103687 CrossRef.
- K. S. Fancey, J. Mater. Sci., 2005, 40, 4827–4831 CrossRef CAS.
- J. P. Brutman, P. A. Delgado and M. A. Hillmyer, ACS Macro Lett., 2014, 3, 607–610 CrossRef CAS PubMed.
- M. Capelot, M. M. Unterlass, F. Tournilhac and L. Leibler, ACS Macro Lett., 2012, 1, 789–792 CrossRef CAS PubMed.
- M. Chen, H. Si, H. Zhang, L. Zhou, Y. Wu, L. Song, M. Kang and X.-L. Zhao, Macromolecules, 2021, 54, 10110–10117 CrossRef CAS.
- M. Hayashi and R. Yano, Macromolecules, 2020, 53, 182–189 CrossRef CAS.
- T. Isogai and M. Hayashi, Macromolecules, 2022, 55, 6661–6670 CrossRef CAS.
- S. K. Schoustra, T. Groeneveld and M. M. J. Smulders, Polym. Chem., 2021, 12, 1635–1642 RSC.
- K. Yu, P. Taynton, W. Zhang, M. L. Dunn and H. J. Qi, RSC Adv., 2014, 4, 10108–10117 RSC.
- W. Zhao, Z. Feng, Z. Liang, Y. Lv, F. Xiang, C. Xiong, C. Duan, L. Dai and Y. Ni, ACS Appl. Mater. Interfaces, 2019, 11, 36090–36099 CrossRef CAS PubMed.
- M. Hayashi, R. Yano and A. Takasu, Polym. Chem., 2019, 10, 2047–2056 RSC.
- W.-Q. Yuan, G.-L. Liu, C. Huang, Y.-D. Li and J.-B. Zeng, Macromolecules, 2020, 53(22), 9847–9858 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Product analysis, experimental details and supplementary figures. See DOI: https://doi.org/10.1039/d2su00140c |
| This journal is © The Royal Society of Chemistry 2023 |