Novel polymeric cobalt tetrabenzimidazole phthalocyanine for nanomolar detection of hydrogen peroxide†
Received
30th August 2022
, Accepted 1st November 2022
First published on 23rd November 2022
Abstract
An N4 macrocyclic complex of polymeric cobalt phthalocyanine substituted with a tetrabenzimidazole (poly(CoTBImPc)) moiety at the periphery was synthesized by a facile route. The redox behavior of poly(CoTBImPc) was confirmed by cyclic voltammetry and obtained redox peaks corresponding to the central metal (Co2+/Co1+) and phthalocyanine macrocycle (Pc2−/Pc3−). Furthermore, to enhance the conductivity, surface area, and sensitivity of the synthesized poly(CoTBImPc), carbon nanotubes (CNTs) were coated on a glassy carbon electrode (GCE/CNT/poly(CoTBmIPc)). Consequently, an amperometric sensor was fabricated for hydrogen peroxide (H2O2), and it manifested a linear response in the concentration range of 10 to 100 nM. The limit of detection (LOD) value observed for H2O2 was 2 nM with the configuration of GCE/CNT/poly(CoTBIPc) using the amperometry tool. The proposed sensor displayed outstanding reproducibility, repeatability, and high stability, without losing its main catalytic properties. Furthermore, the designed sensor has high selectivity for H2O2 even in the presence of interfering species in the solution.
Sustainability spotlight
The research and development of new phthalocyanines are very important for the electrochemical applications as it is utilized a lot in sensing applications and energy storage devices. However employing and use of cobalt (Co) metal were having limited reports for nanomolar detection of hydrogen peroxide (H2O2). So herein we describe the synthesis of polymeric cobalt tetrabenzimidazole phthalocyanine and the fabrication of an amperometric sensor with detailed characterization. A glassy carbon electrode was modified with the synthesized phthalocyanine and carbon nanotubes (CNTs) to determine the H2O2 content. The fabricated amperometric sensor manifests impeccable performances with high selectivity for H2O2 even in the presence of interfering species in solution. The present work aligns with affordable and clean energy, climate action for real UN SDG's.
|
1. Introduction
Metallophthalocyanines (MPcs) and their derivatives are well-known organic semiconducting materials belonging to the conjugated aromatic heterocyclic family.1–3 These N4 macrocycles exhibit intensive redox behavior, and their properties can be tuned by changing the appropriate peripheral substituents. They are primarily used for diverse applications like bio-active transducers, corrosion inhibition, sensing, catalysis, fuel cells, photoconducting, semiconducting, etc.4 Furthermore, Pcs have been widely studied for the functionalization of carbon nanotubes (CNTs) because they play an essential role in the enhancement of the performance of CNT-based devices as they demonstrate attractive electronic and photo-electronic properties.5,6
Furthermore, hydrogen peroxide (H2O2) is an antiseptic and can be applied to open wounds and cuts. Notably, H2O2 is used as a bleaching agent for teeth whitening. Even skin bleaching ingredients contain this substance. H2O2 detection and monitoring are significant because of their importance in recent years in various fields such as the food industry, electrochemical energy conversion, chemical synthesis, biotechnology, and environmental, clinical, and pharmaceutical applications.7–9 Among the various sensing techniques, electrochemical methods are simple, cheaper, and reproducible, and can be employed for on-site analysis because of their portability. Enzyme-based electrochemical biosensors for detecting H2O2 are extensively being researched but are expensive, complicated, and affected by temperature and pH due to the variation in the activity of an enzyme. Alternatively, non-enzymatic electrochemical sensors have attracted more attention because of their long-term stability, high sensitivity, pH-free, quick response time, and low cost.10–12
Chemically modified electrodes have enormous significance and advantages in real electrochemical sensing applications. The literature survey reports reveal that the reduction of H2O2 is accelerated at CNTs13–15 with an impressive lower over potential and enhanced reversibility in its voltammetric behavior. As a result of high conductivity, it potentially reciprocates fast electron transfer yielding the best electrochemical performance. Electrochemical sensors modified with a carbon nanomaterial composite film have displayed an excellent linear response range with a lower detection limit of 3 nmol L−1. Owing to their unique properties, we have fabricated a poly cobalt tetrabenzimidazole phthalocyanine–CNP organic composite hybrid and applied it as a non-enzyme electrochemical sensor to detect H2O2.
2. Experimental details
2.1. Materials
All the chemicals used for the synthesis of the phthalocyanine compound and electrochemical studies were purchased from Sigma Aldrich or Merck. The chemicals were utilized without any purification as obtained.
2.2. Synthesis of 2,2′-(butane-1,4-diyl)bis(1H-benzimidazole) compound (i)
Precursor (i) was prepared by using a mixture of o-phenylene diamine (OPDA) (3.23 g, 0.0290 mmol) and adipic acid (5.0 g, 0.0290 mmol) in a round bottom (RB) flask containing 50 mL polyphosphoric acid (PPA). Then, the reaction mixture was refluxed at 120–130 °C for eight hours, and the crude product was cooled to room temperature. Next, the crude product was thoroughly washed with water and dried. Then the targeted ones were obtained by recrystallization with the support of methanol.
Synthetic yield: 89%. Melting point: 104 °C. Mol. Wt. 290.36. Anal. for the compound (i), C18H18N4: calc. (%) C, 74.44; H, 6.10; N, 19.37. Found (%): C, 74.53; H, 5.99; N, 19.28. IR absorption bands (KBr (pellet), cm−1): 552, 703, 847, 902, 982, 1133, 1122, 1168, 1218, 1278, 1405, 1533, 1614, 1846, 2569, 3011, 3006 and 3401. Mass (m/z): 290 (M); 288 (M2−).
2.3. Synthesis of [2,2′]-(butyl)bis[4-(benzimidazole-1-yl)-phthalonitrile] ligand (ii)
Compound (ii) was prepared by combining compounds (i) (2.0 g, 0.0094 mmol), 4-nitrophthalonitrile (1.63 g, 0.0094 mmol) and potassium carbonate (1.55 g, 0.011 mmol) (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1.5) in a RB flask with dry dimethylformamide (DMF) (10 mL). The ground mixture was stirred at room temperature for one day in a N2 atmosphere. The progress and completion of the reaction were monitored by thin layer chromatography (TLC). After the reaction was completed, the crude product was transferred into a beaker containing ice water to obtain the residue. The precipitate was filtered and washed with water. Finally, compound (ii) was recrystallized from ethanol and dried in a vacuum.
:1:1.5) in a RB flask with dry dimethylformamide (DMF) (10 mL). The ground mixture was stirred at room temperature for one day in a N2 atmosphere. The progress and completion of the reaction were monitored by thin layer chromatography (TLC). After the reaction was completed, the crude product was transferred into a beaker containing ice water to obtain the residue. The precipitate was filtered and washed with water. Finally, compound (ii) was recrystallized from ethanol and dried in a vacuum.
Yield: 91%. Melting point: 215 °C. Mol. Wt. 542.55. Anal. for compound (ii) C34H22N8: calc. (%) C, 75.25; H, 4.02; N, 20.64. Found (%): C, 75.65; H, 3.99; N, 20.97. IR absorption bands (KBr (pellet), cm−1): 761, 821, 919, 955, 1086, 1123, 1266, 1317, 1585, 1543, 1685, 1782, 1898, 2244, 2890, 3021 and 3431. Mass (m/z): 542 (M); 543 (M1+).
2.4. Synthesis of cobalt(II)tetra[4[2,2′]-(butyl)bis[4-(benzimidazole-1-yl)]] phthalocyanine polymer, poly(CoTBImPc) (iii)
A ground mixture of compound (ii) (1.0 g, 0.0024 mmol), cobalt chloride (0.08 g, 0.00062 mmol), and dry 1-pentanol (25 mL), and a catalytic amount of 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) were taken in a RB flask and refluxed with continuous stirring at 140 °C for one day. Then, the dark blue-colored crude formed was cooled, filtered off, and washed successively with cold and hot water. Next, the blue-colored solid product was washed with hot ethanol, acetone, and hexane. Finally, the product was dried in a vacuum oven for one hour at 50 °C to obtain a bluish solid.
Yield: 92%. Mol. Wt. ∼ [2277.42]n. Anal. for compound poly(CoTBImPc) (iii) [C139H100N32Co]n: Calc. (%) C, 73.01; H, 4.35; N, 19.65; Co, 2.83. Found (%): C, 72.89; H, 4.66; N, 19.05; Co, 2.63. UV-VIS (DMSO, λmax(nm)): 324, 348, 612, 681. IR absorption bands (KBr (pellet), cm−1): 751, 761, 845, 954, 1096, 1104, 1245, 1328, 1385, 1426, 1407, 1621, 2701, 2848, 2921 and 3442.
2.5. Preparation of the analyte
0.1 M H2O2 stock solution was prepared by using double distilled water. The H2O2 working standard solution of 0.01 mM was prepared by dilution and was added in an increment of 10 μL into a cell containing 10 mL phosphate buffer solution pH 7 (PBS pH 7). The H2O2 concentration in the electrochemical cell after adding 10 μL H2O2 working standard solution was 10 nM.
2.6. Preparation of modified electrodes
The GC surface was cleaned by polishing with alumina 0.05 micron (Baikalox, Japan) on a polishing pad (Buehler, Ltd), washed with water, and subsequently rinsed with piranha solution for 10 min. Finally, the GCE was rinsed sequentially with a copious amount of double distilled water (DD water), acetone, and ethanol.
The electrode surface was modified by the drop coating method. The polymer ink was prepared by dispersing poly(CoTBImPc) (5 mg) in 1 mL isopropyl alcohol together with 10 μL of Nafion binder (5% wt) and was ultrasonicated for 30 min to yield a homogeneous solution. The polymeric electrode was constructed by drop casting 5 μL of the as-prepared poly(CoTBImPc) solution on the clean, glassy carbon electrode (GCE) and dried at room temperature and is represented as GCE/poly(CoTBImPc). Similarly, the hybrid composite electrode was constructed by dispersing 5 mg of a 1:4 ratio of CNT and CoTBImPc composite in 1 mL IPA with 10 μL of Nafion binder (5% wt). It was homogenized with the help of ultrasonication for 30 min. Then, 5 μL of the composite was drop cast on a GCE carefully to obtain a uniform film and dried at room temperature. The fabricated electrode is represented as GCE/CNP/poly(CoTBImPc). These two electrodes were utilized for the voltammetric and amperometric sensing of H2O2.
3. Characterization methods
The elemental compositions of all the new compounds were estimated by using a Vario EL III CHNS elemental analyzer. UV-Vis absorption spectroscopy was performed using an FLMSO4808 Ocean optics spectrometer in the wavelength range of 250–800 nm with 0.1 mM polymeric phthalocyanine dissolved in DMSO solvent. The KBr pellet technique was used to collect the FT-IR spectra from a FTIR PerkinElmer Spectrum-Two spectrophotometer in the region of 4000–500 cm−1. A GC/MS instrument of 6100 series from Agilent technologies was used for recording the mass data. Next, the powder X-ray diffraction (PXRD) profile was recorded from a Bruker D8 Advance X-ray diffraction machine. Thermograms were recorded using an STA6000 machine in the temperature region of 30 to 700 °C (heating rate of 10 °C min−1).
Electrochemical experiments were performed using a CHI6005E Electrochemical analyzer workstation with a three-electrode setup. The working electrode was a polymeric Pc immobilized glassy carbon electrode (GCE) with a surface area of 0.071 cm−2 (the diameter of the electrode is 3 mm). An Ag/AgCl electrode was employed as the reference electrode, and a Pt wire was used as the counter electrode. Before studying polymer immobilized electrodes, the electrochemical behavior of 0.1 mM poly(CoTBImPc) in DMSO containing TBAP as a supporting electrolyte was studied.16 Chronoamperometric and cyclic voltammetry experiments were conducted to detect and sense H2O2 in PBS at the polymeric Pc and composite electrodes. High purity N2 was used to remove dissolved O2 by purging for at least 20 minutes prior to each experiment and maintaining the nitrogen atmosphere over the solution during the measurements.
4. Results and discussion
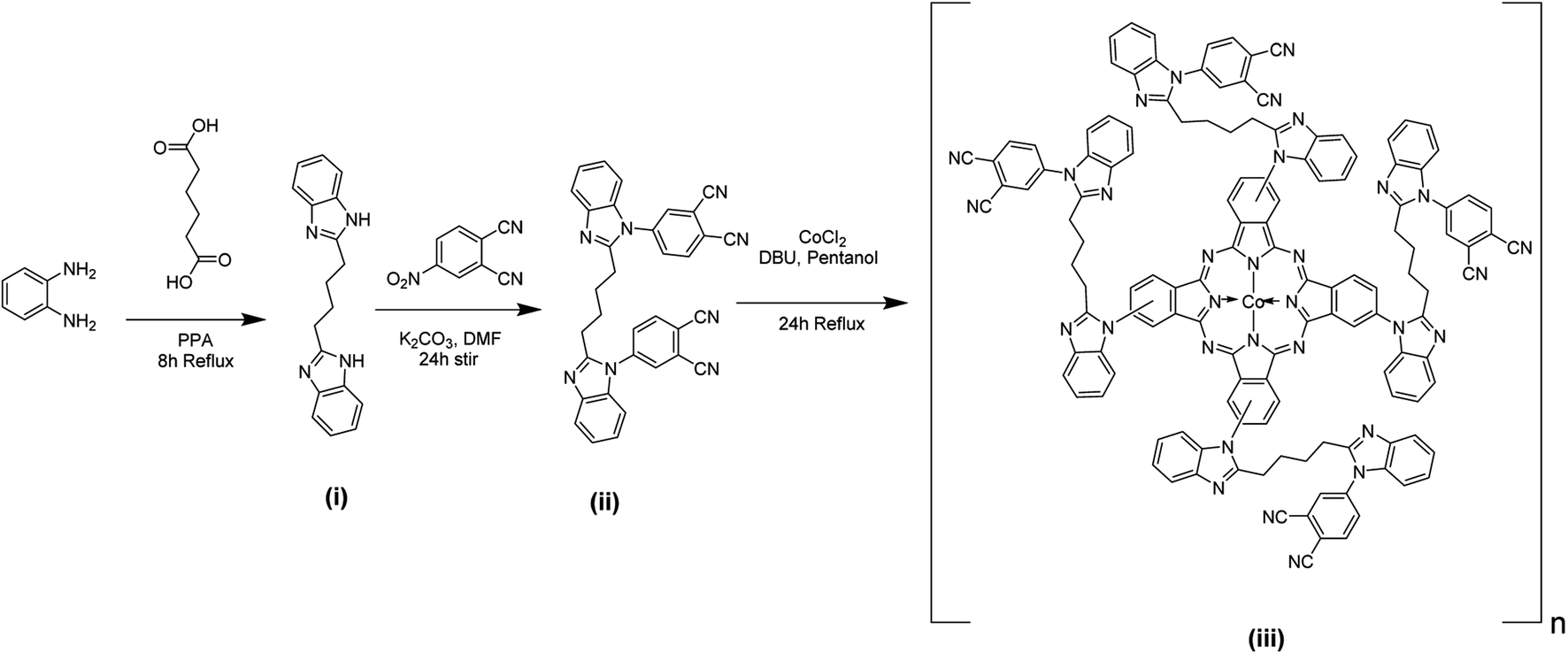
The schematic scheme for preparing phthalocyanine polymer is depicted in Scheme 1. OPDA reacted with a compound with a –COOH group to yield a benzimidazole-containing precursor (i). The benzimidazole precursor (i) undergoes a nucleophilic displacement reaction with nitrophthalonitrile to form the benzimidazole phthalonitrile ligand (ii). As shown in Scheme 1, ligand (ii) upon cyclization and condensation reactions with a metal salt in 1-pentanol forms polymeric phthalocyanine (iii). The ring closure and cyclotetramerization of the phthalonitrile ligand occur in the presence of organic “super-base” DBU yielding a macrocyclic structure. The ligand (ii) has two phthalonitrile groups, forming polymeric phthalocyanine from the monomeric phthalocyanines (iii).
 |
| | Scheme 1 Synthetic route for the preparation of polymeric cobalt phthalocyanine. | |
TLC monitored the progress and completion of each reaction step, and the synthesized compounds were purified and characterized using melting point, elemental analysis, FT-IR, and mass spectroscopy (mass spectral data are provided in the ESI†). The polymeric cobalt phthalocyanine complex was a blue to green product with solubility in DMSO, DMF, and concentrated H2SO4.
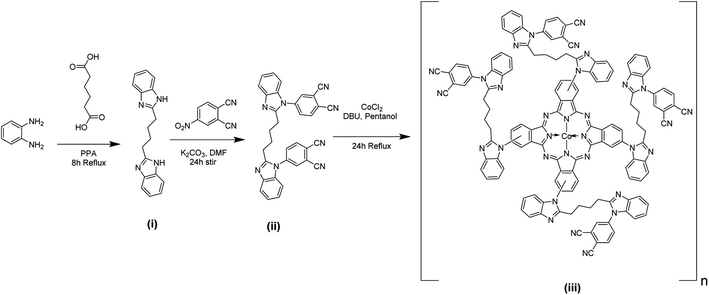
4.1. UV-vis spectroscopy
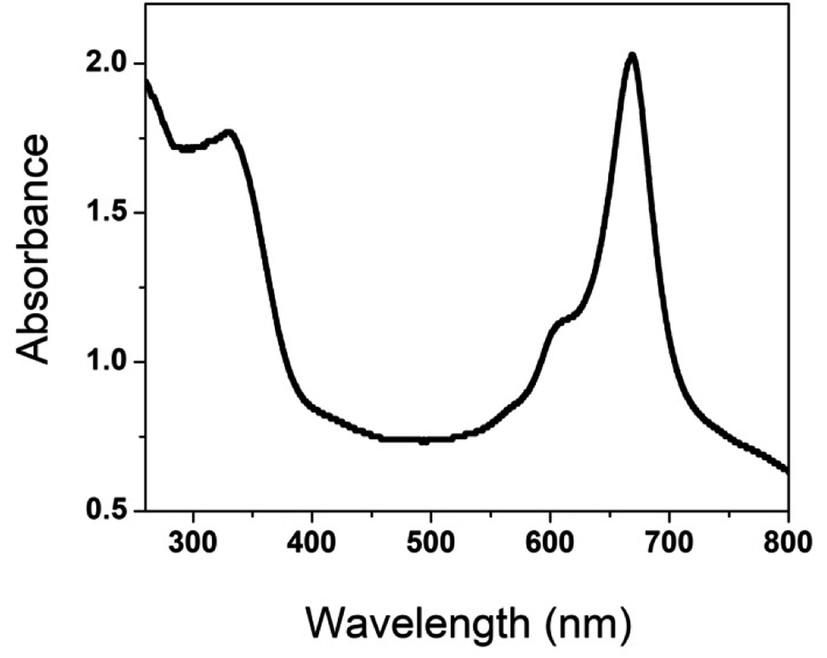
The UV-visible spectrum of poly(CoTBImPc) was recorded using 0.1 mM polymer in DMSO in the range of 250 to 800 nm, Fig. 1. The synthesized polymer showed sharp absorption peaks at 300–340, 590–610 (shoulder peak), and 650–705 nm. The peak observed in the UV region at 300–350 nm is called the B-band and is attributed to the n–π* transition. The intense Q-band was observed at 650–705 nm, and the phthalocyanine's deep, intense blue/green color can be ascribed to this peak, which arises because of the π–π* transition of the conjugated macrocycle.17,18 The peak observed at 590–610 nm can be accounted for by the Pc molecule's oligomer and dimeric forms and fine vibronic structure. The polymeric phthalocyanine showed a red shift in the absorption maximum of B and Q bands compared to the parent cobalt phthalocyanine due to extended conjugation, electron delocalization, and planarity of the molecule.18
 |
| | Fig. 1 UV-Visible spectrum of (iii). | |
4.2. FTIR characterization
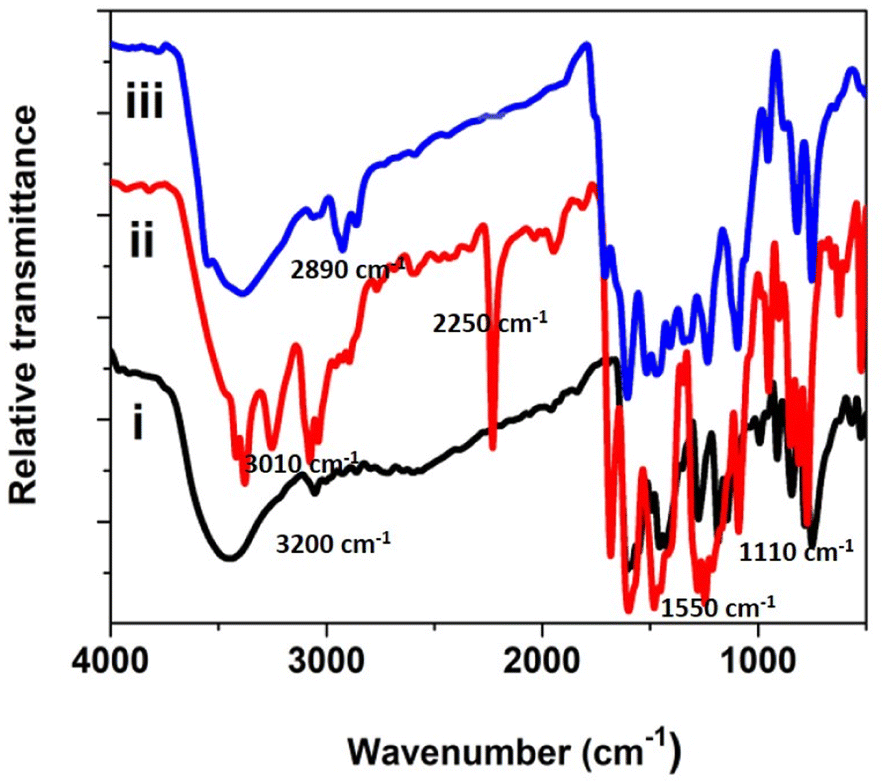
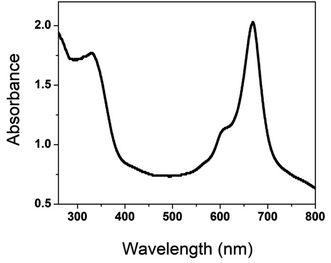
FT-IR spectra for the precursor, ligand, and phthalocyanine polymer were recorded in the region 500 to 4000 cm−1, Fig. 2. The FTIR spectrum of the precursor compound exhibited a vibrational band at 1540–1620 cm−1, which corresponds to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C stretching and 2870–3020 cm−1 to C–H stretching. In addition, a weak band was observed around 3200 cm−1, attributed to a secondary amine.
C stretching and 2870–3020 cm−1 to C–H stretching. In addition, a weak band was observed around 3200 cm−1, attributed to a secondary amine.
 |
| | Fig. 2 FT-IR spectra of the precursor, ligand and polymeric phthalocyanine. | |
A new sharp peak was noticed around 2250 cm−1 for the ligand corresponding to the –CN group. The –CN group's appearance in the ligand confirms the formation of the conjugated phthalonitrile ligand. In addition to TLC, IR was helpful in successfully monitoring the completion of the reaction to form cyano ligands from their respective imidazole precursors by carefully scrutinizing the evolution of the cyano group peak at ∼2250 cm−1. The synthesized phthalocyanine complex displayed peaks at 745–760, 875–895, 965–990, 1004–1110, and 1111–1135 cm−1, which can be related to the phthalocyanine skeletal vibrations.18–24 Additionally, the IR spectra of the polymeric Pc did not display a peak for the –CN group meaning that the cyano groups have been utilized to form a polymeric phthalocyanine structure. The less intense and broader nature of the bands in the IR spectrum confirms the polymeric nature of phthalocyanine.
4.3. Mass spectra
The mass spectra of the precursor and ligand were recorded to get information related to these compounds' molecular weight and fragmentation pattern. The mass spectra of the precursor and ligand are presented in Fig. S1 and S2.† The synthesized precursor and ligand showed a (M+ or M−) molecular ion peak in mass spectra, which coincided with the theoretical molecular weight of the compounds. For example, the molecular weight of the precursor is 290. Therefore, its mass spectrum displayed peaks at 290 and 288, corresponding to M. and M − 2 molecular ion peaks. In contrast, the ligand has a theoretical molecular weight of 542, and its mass spectrum displayed peaks at 542 and 543, which can be accounted for M. and M + 1 molecular ion peaks. This confirms the successful formation of the precursor and ligand.
4.4. X-ray diffraction studies
The powder X-ray diffraction pattern of the synthesized phthalocyanine polymer was recorded in the 2θ range of 5–70°, as shown in Fig. S3.† The pattern was found to be highly noisy with the peaks' broader nature, revealing that the synthesized Pc polymer is highly amorphous.
4.5. TGA studies
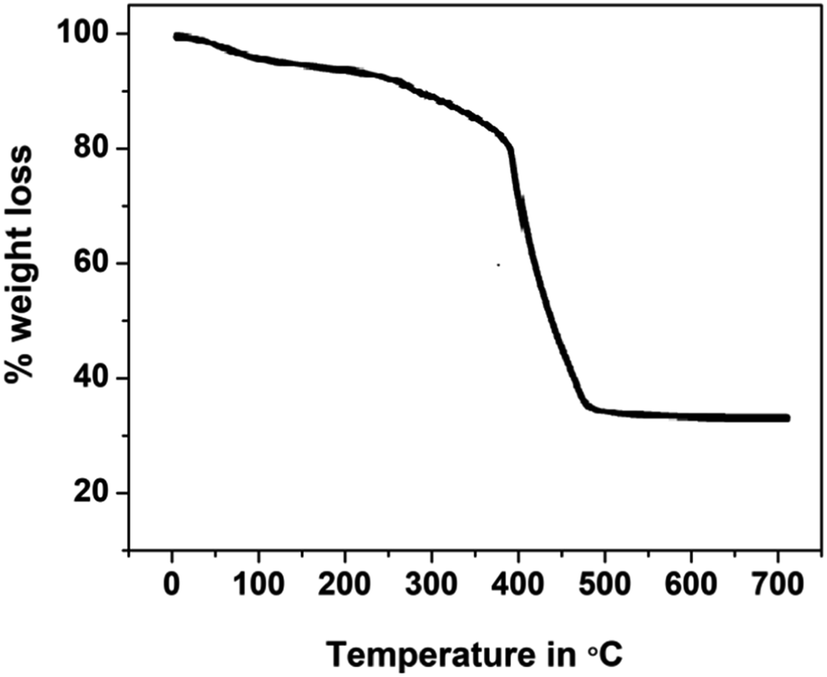
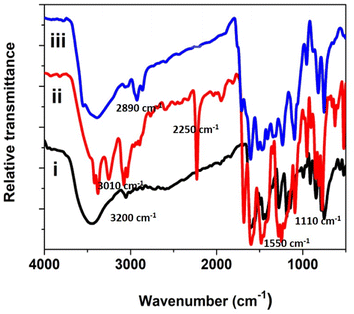
Thermogravimetric studies yield information related to the synthesized compounds' thermal stability and decomposition characteristics. The thermogram for the phthalocyanine polymer was recorded in the temperature range of 30 to 700 °C, Fig. 3. The thermogram indicates that the Pc polymer degrades mainly in three steps in an air atmosphere. The first mass loss in the region below 175 °C is due to the loss of volatile components, water, and moisture from the sample with an exothermic decomposition. In the second step, the terminal substituents detached in the temperature region of 300–400 °C. The final decomposition step occurs fast in an oxidizing atmosphere and results in the accelerated loss of mass corresponding to the degradation of the phthalocyanine structure after 400 °C. The stable and final product was formed after the decomposition of the polymer in the oxidizing atmosphere, and the amount of the stable product is equivalent to the theoretical mass of metal oxide present in the polymeric sample loaded for the analysis. Polymeric Pc showed better thermal stability than monomeric Pcs, which may account for the extended conjugation, delocalization of π-electrons, and coplanarity of the macromolecule.24–27
 |
| | Fig. 3 TGA analytical curve for polyCoTBImPc. | |
5. Electrochemical studies
5.1. Solution CV
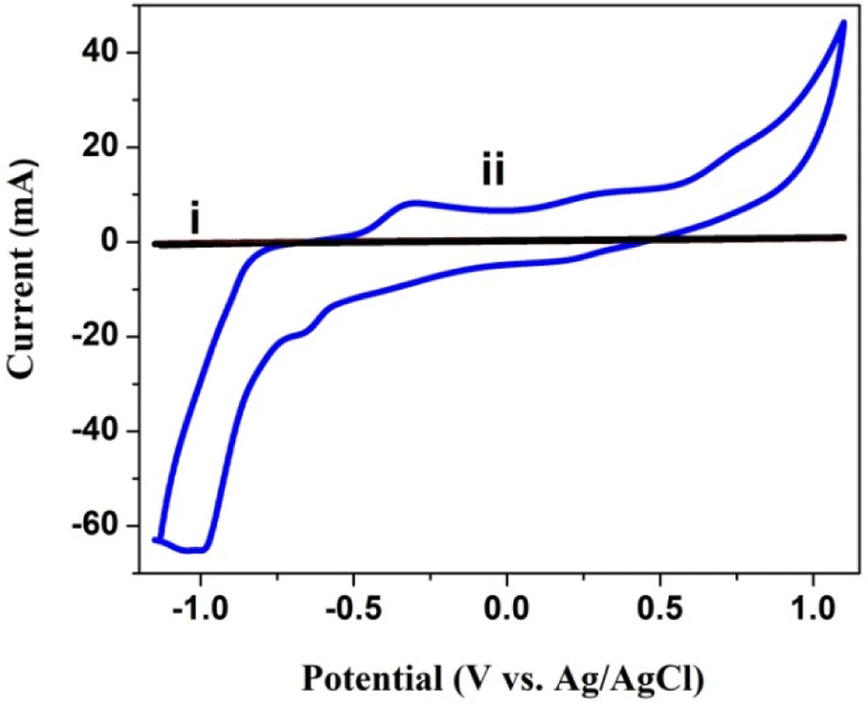
5.0 mg poly(CoTBImPc) c was used for the solution cyclic voltammetry study in DMSO containing 0.1 M TBAP as the supporting electrolyte, and the potential was cycled in 1.0 to −1.0 V at a scan rate of 50 mV s−1. Before the experiment, the DMSO solution was purged with a constant flow of N2 gas to remove the dissolved oxygen for 20 min. As a result, the solution CV of the bare GCE and poly(CoTBImPc) coated electrode is presented in Fig. 4. The CV of poly(CoTBImPc) showed two redox peaks around −0.4 V and 0.3 V, which can be assigned to the macrocycle (Pc2−/Pc3−) and the other redox peak for the redox activity of cobalt metal ions (Co2+/Co3+), respectively, as per literature reports.26,28
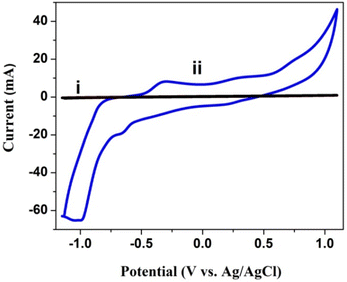 |
| | Fig. 4 CV of the bare GCE (i) and with (ii) polyCoTBImPc in DMSO under the current of N2 gas at 50 mV s−1 with 0.1 M TBAP. | |
5.2. Charge transfer studies
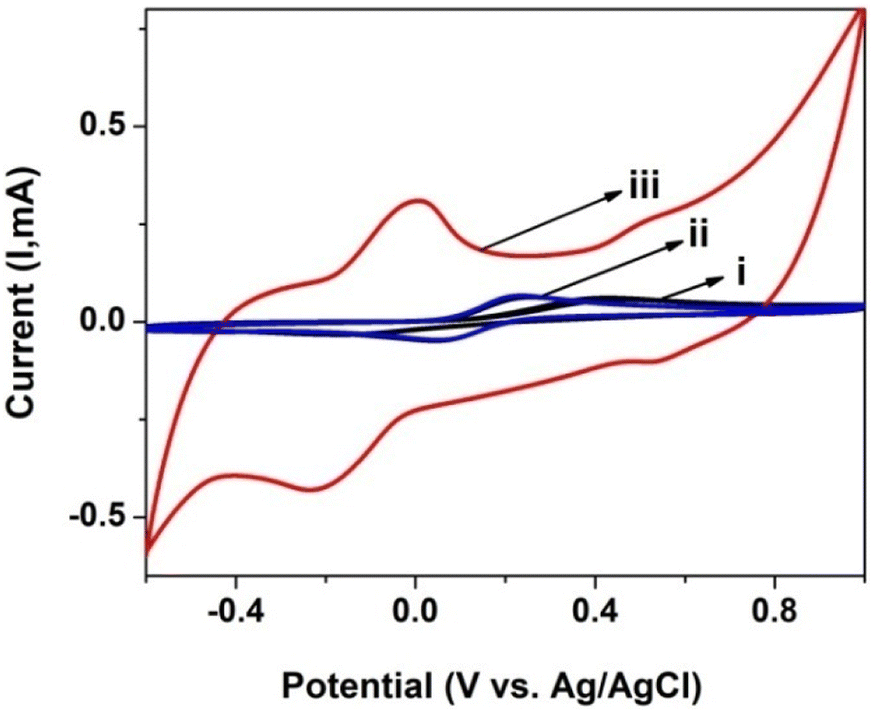
CV was also used to investigate the electrochemical response and charge transfer kinetics of the electrodes in PBS with a 0.1 mM [Fe(CN)6]3−/4− redox couple. Fig. 5 shows the CVs for the [Fe(CN)6]3−/4− redox pair on a bare GCE, GCE/poly(CoTBImPc) and GCE/CNP/poly(CoTBImPc) in PBS at a scan rate of 50 mV s−1. The separation of the anodic peak from the cathodic peak of the redox probe is slightly more for GCE/CNP/poly(CoTBImPc) than the bare GCE/GCE/poly(CoTBImPc) electrode. This indicates that the electron mediation is faster at the GCE/CNP/poly(CoTBImPc) electrode in comparison to bare GCE and GCE/poly(CoTBImPc) electrodes. The faster and rapid electron exchange kinetics for [Fe(CN)6]3−/4− on hybrid GCE/CNP/poly(CoTBImPc) compared to the Pc film is a result of a superior surface area and conductivity provided by the CNTs to the hybrid system.
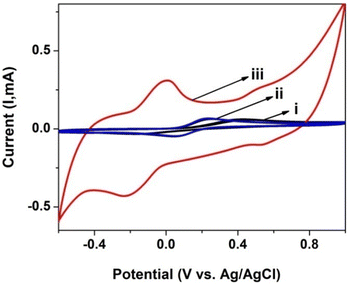 |
| | Fig. 5 CV profile for 0.1 mM [Fe(CN)6]3−/4− at (i) the bare GCE, (ii) GCE/poly(CoTBImPc) and (iii) GCE/CNT/poly(CoTBImPc) in PBS (pH 7). | |
5.3. Impedance studies
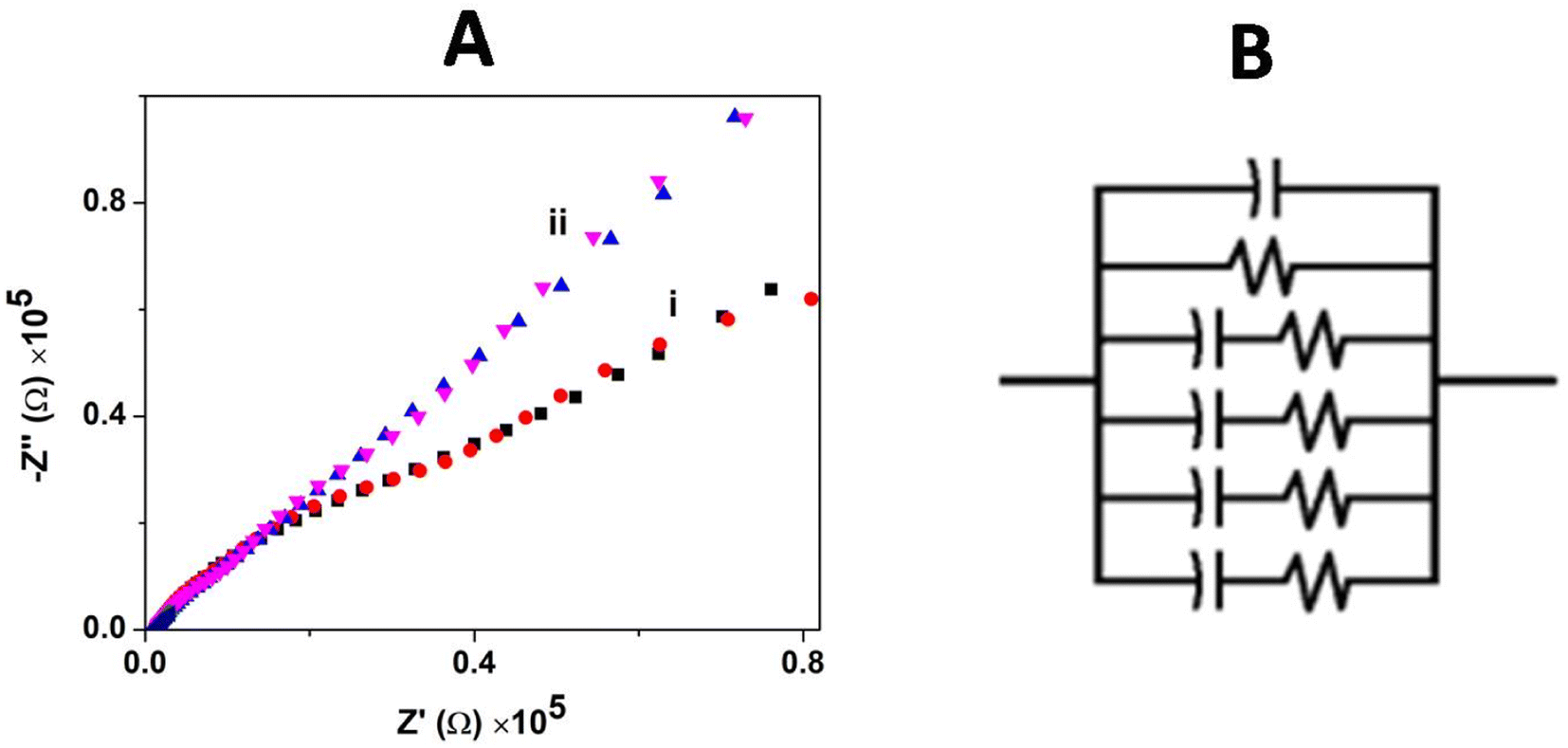
The charge transfer and interfacial behavior of the modified GC electrode surface were understood using electrochemical impedance spectroscopy (EIS). The Nyquist plot (Zimaginaryvs. Zreal) for the bare GCE and GCE/CNP/poly(CoTBImPc) hybrid composite electrode in [Fe(CN)6]3−/4− aqueous solution with an amplitude potential of 0.05 V in a wide range of 0.01–1000 kHz is shown in Fig. 6A.
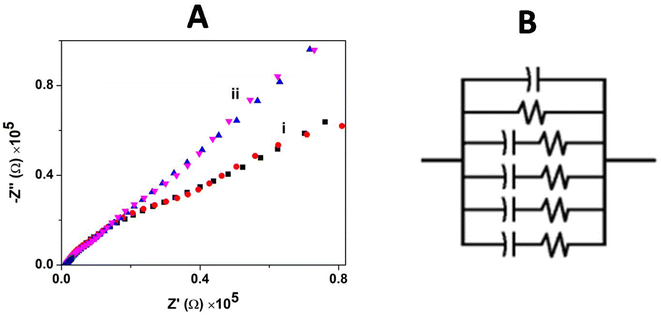 |
| | Fig. 6 (A) Impedance spectra for (i) the bare GCE and (ii) GCE/CNT/poly(CoTBImPc) in 0.1 mM [Fe(CN)6]3−/4− in PBS. (B) Equivalent circuit used to fit the Nyquist plot. | |
The impedance plot for the GCE and polymeric film electrode was fitted with a Randles circuit as shown in Fig. 6B. The equivalent circuit used to fit the experimental data is (CR(CR)(CR)(CR)(CR)), where R is the electrolyte resistance, and C is the capacitance arising at the electrode interface and its surrounding electrolyte. The values of the equivalent circuit data are summarized in Table 1.
Table 1 Equivalent circuit parameters for the electrodes
| Electrodes |
R
1 (Ω) |
C
1 (F) |
R
2 (Ω) |
C
2 (F) |
R
3 (Ω) |
C
3 (F) |
R
4 (Ω) |
C
4 (F) |
R
5 (Ω) |
C
5 (F) |
| Bare GCE |
2.049 × 10−9 |
1.647 × 104 |
8.384 × 10−7 |
465.9 |
3.783 × 10−7 |
226.3 |
1.353 × 10−6 |
3157 |
6.195 × 10−6 |
5341 |
| GCE/CNP/poly(CoTBImPc) |
2.026 × 10−9 |
1.060 × 104 |
2.627 × 10−6 |
824 |
5.063 × 10−8 |
2957 |
4.524 × 10−5 |
306.9 |
2.141 × 10−4 |
313.4 |
5.4. Application of the CNT/poly(CoTBImPc) electrode for the detection of H2O2
The polymeric Pc film on the GCE was used to detect H2O2 in PBS under a nitrogen atmosphere. Fig. S4† shows the cyclic voltammograms for with and without 100 nM H2O2 at bare, GCE/poly(CoTBImPc) and with and without 10 nM H2O2 at GCE/CNP/poly(CoTBImPc) for the electrocatalytic reduction of H2O2. The polymer Pc film on the electrode showed redox peaks, which are compatible with the peaks of polymeric benzimidazole Pc in solution, as shown in Fig. 7A. As can be noticed from the cyclic voltammograms, the current was less at the bare electrode for the reduction of H2O2. In contrast, the polymeric electrodes showed a larger current response with less overpotential. Furthermore, the current response was larger for the hybrid composite electrode than for the Pc electrode's polymerized film. The larger current response and lower overpotential for the composite electrode may be due to the larger surface area and conductivity of CNTs, which improves the charge transfer and distribution of the polymer film.
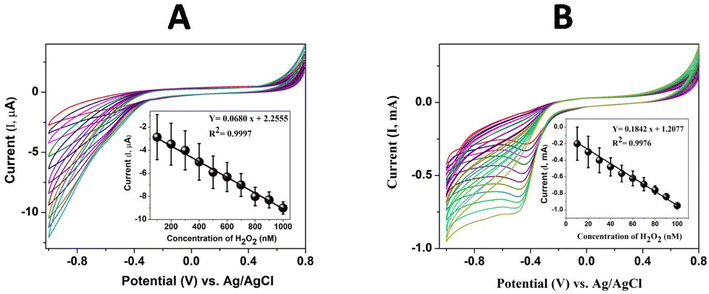 |
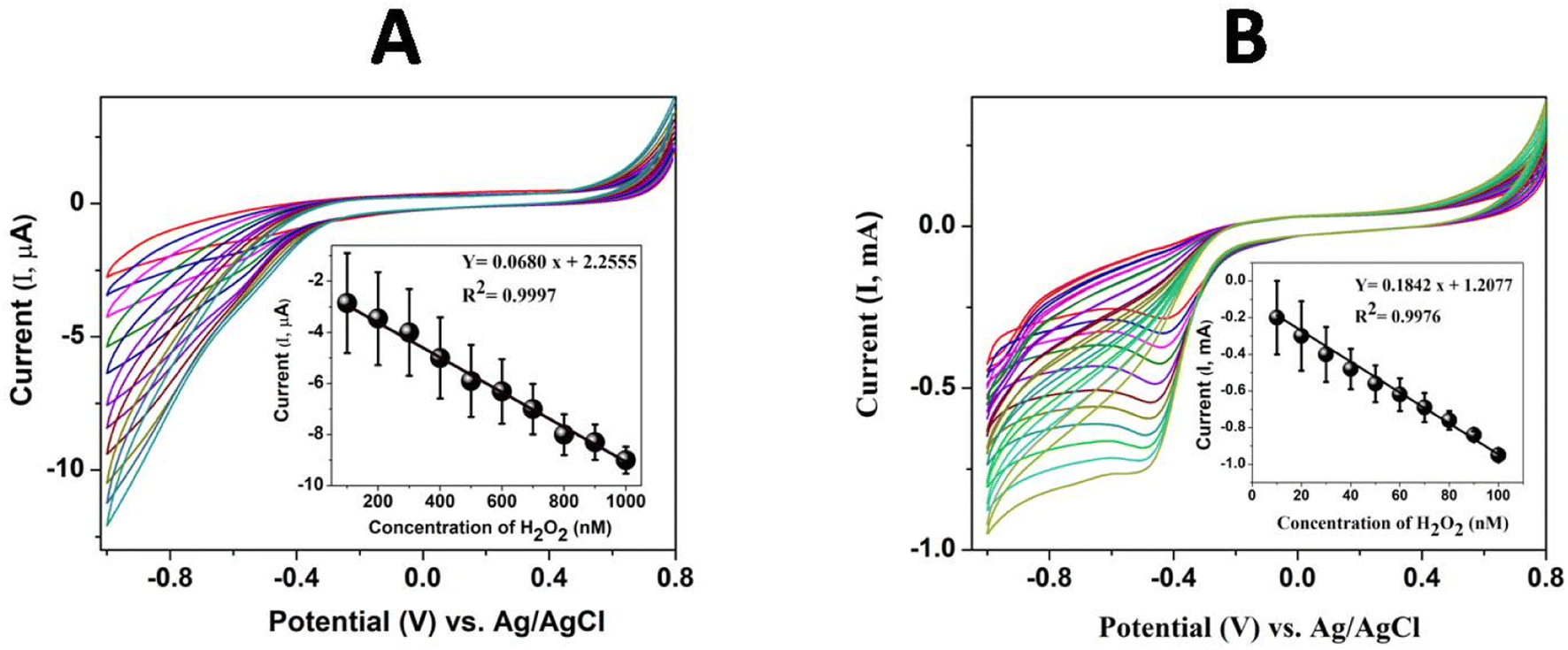
| | Fig. 7 Cyclic voltammograms at (A) GCE/poly(CoTBImPc) for different concentrations of H2O2 (100 to 1000 nM) and (B) GCE/CNT/poly(CoTBImPc) for different concentrations of H2O2 (10 to 100 nM) in PBS. Inset: plot of anodic peak current vs. H2O2 concentration at −0.45 V. | |
5.5. Electrocatalytic sensing of H2O2
For the sensing study, poly(CoTBImPc)/CNP was modified on a glassy carbon electrode (GCE) due to its easy fabrication, renewability, good reproducibility, and outstanding stability. The preliminary results of electrocatalytic reduction of H2O2 with higher reduction current and lower overpotential at the polymer Pc electrodes attracted us to study the current response for different concentrations of H2O2 in PBS (pH 7). The detection of H2O2 was examined using GCE/poly(CoTBImPc) and CNT/polyCoTBImPc at 50 mV s−1 in PBS. The electrocatalytic reduction of H2O2 was observed from −0.3 V to −0.8 V with an increased current in the cyclic voltammogram, but no clear or sharp peak was noticed. The plot of peak current vs. concentration of H2O2 at −0.45 V displayed a linear response in the range (100 to 1000 nM) with straight line characteristics depicted by Y = 0.0680 x + 2.2555 (R2 = 0.9997) with LOD = 30 nM and a sensitivity value of 0.9700 μA nM−1 cm−2 for GCE/poly(CoTBImPc).
Cyclic voltammograms at CNT/poly(CoTBImPc) were recorded for various concentrations of H2O2 (10 to 100 nM), Fig. 7B. Cyclic voltammograms showed a clear and sharp peak at −0.45 V for the reduction of H2O2 at CNT/poly(CoTBImPc). The cathodic current at −0.45 V increased with an increase in the concentration of H2O2, which can be accounted for by the reduction of H2O2. The increase in the reduction current for H2O2 was observed at lower overpotential on composite hybrid electrodes compared to GCE and polymeric Pc film electrodes. The enhanced current and lower overpotential displayed by CNT/poly(CoTBImPc) indicate the catalytic nature and synergic effect of the organic hybrid composite electrode in reducing H2O2.
The plot of cathodic peak current vs. concentration of H2O2 at −0.45 V was linear in the range 10 to 100 nM H2O2 at GCE/CNT/poly(CoTBImPc) with an insightful straight line characteristics Y = 0.1842x + 1.2077 (R2 = 0.9976) and LOD = 5 nM and a sensitivity of 2.6276 mA nM−1 cm−2. The GCE/CNT/poly(CoTBImPc) electrode exhibited better sensitivity, LOD and wide linearity scale for the detection of H2O2 compared to GCE/poly(CoTBImPc) as shown in Fig. 7B.
The linear concentration range and corresponding peak current obtained for H2O2 reduction at CNT/poly(CoTBImPc) were found to be higher than those of poly(CoTBImPc), which may account for the improved electrical conductivity, higher surface area, and also a larger number of active sites available at the composite due to the presence of CNTs.
5.6. Effect of scan rate
CVs were recorded for 50 nM H2O2 in PBS (pH 7) at different scan rates from 25 to 500 mV s−1 on the GCE/CNT/poly(CoTBImPc) composite electrode as shown in Fig S5.† The current increased with an increase in the scan rate for the reduction of H2O2. The plot of current response vs. square root of the scan rate at −0.45 V is shown in Fig S5,† which exhibited a linear plot indicating diffusion-controlled mass transfer phenomena between the electrode and electrolyte.18,29
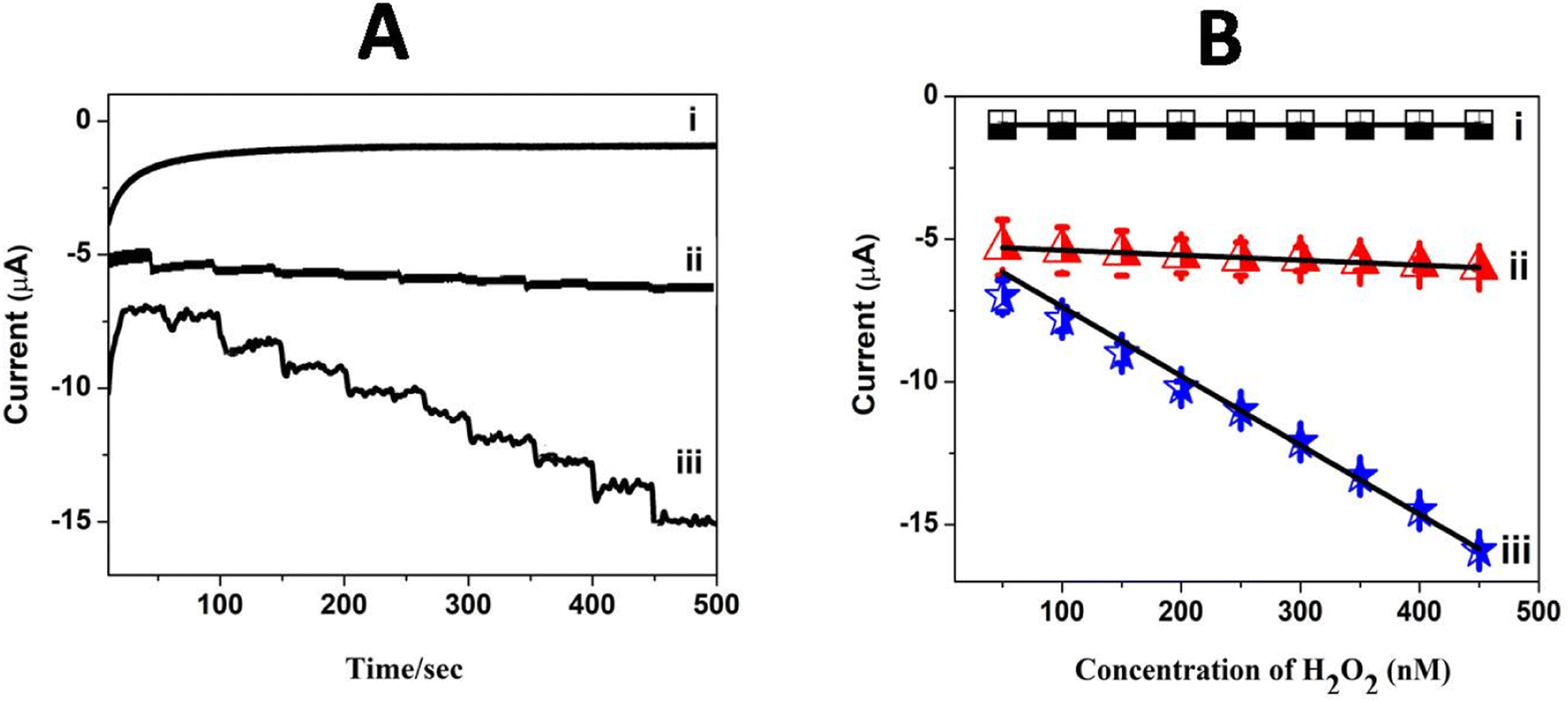
5.7. Amperometric detection of H2O2
The linear current (μA) response with a larger sensitivity value observed in cyclic voltammetry for H2O2 reduction encouraged the construction of an amperometric sensor for H2O2 using Pc polymer electrodes. Amperometric sensing of H2O2 was carried out at GCE/poly(CoTBImPc) and GCE/CNT/poly(CoTBImPc) in PBS with a constant potential of −0.45 V under stirring conditions. The current–time response was monitored with the repetitive addition of 10 nM H2O2 with a time interval of 50 seconds under constant stirring (500 rpm) at the bare and modified electrodes, as shown in Fig. 8A. Furthermore, the linear characteristic range for H2O2 reduction at poly(CoTBImPc) and CNT/poly(CoTBImPc) was in the concentration range of (10 to 100 nM). Both the polymeric and composite electrodes displayed a rapid increase in the amperometric response for successive addition of the H2O2 analyte, and the signal got stabilized in 2–3 s, which is an important parameter in the development of an efficient sensor. 90% of the steady state current was realized within 3 s after the addition of H2O2, which indicates the rapid detection and efficacy of the designed sensors for the analyte. In addition, the amperometric current decreased linearly with the H2O2 concentration. Fig. 8B demonstrates the plot for the amperometric current as a function of increase in concentration and the plot manifested a linear response in the 10 to 100 nM range with R2 = 0.9951 and 0.9888 for the poly(CoTBImPc) and CNT/poly(CoTBImPc) electrodes. The R2 value indicates better linear dependence between the amperometric current and concentration of H2O2 at the Pc and hybrid modified GC electrodes. The regression equation for the straight line characteristics was given by y = 0.0174x + 5.2151 and y = 0.2420x + 4.945 for GCE/poly(CoTBImPc) and GCE/CNP-poly(CoTBImPc) respectively. The LOD response observed at 3 nM and 2 nM for GCE/poly(CoTBImPc) and GCE/CNT/poly(CoTBImPc) electrode with sensitivity value 0.2482 μA μM−1 cm−2 and 3.4522 mA nM−1 cm−2 respectively. This reveals that the composite hybrid modified electrode provided good sensitivity towards the detection of H2O2. The composite hybrid electrode displayed an enhanced current response for H2O2 reduction compared to that of the polymeric Pc alone film.
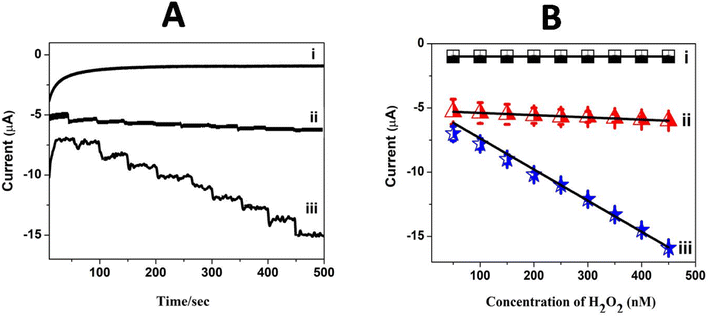 |
| | Fig. 8 (A) Chronoamperometric curves for the successive addition of H2O2 (10 to 100 nM) in pH 7 PBS at (i) the bare GCE, (ii) GCE/poly(CoTBImPc) and (iii) GCE/CNT/poly(CoTBImPc) at −0.45 V under stirring condition. (B) Plot of amperometric current vs. conc. of H2O2 on different electrodes. | |
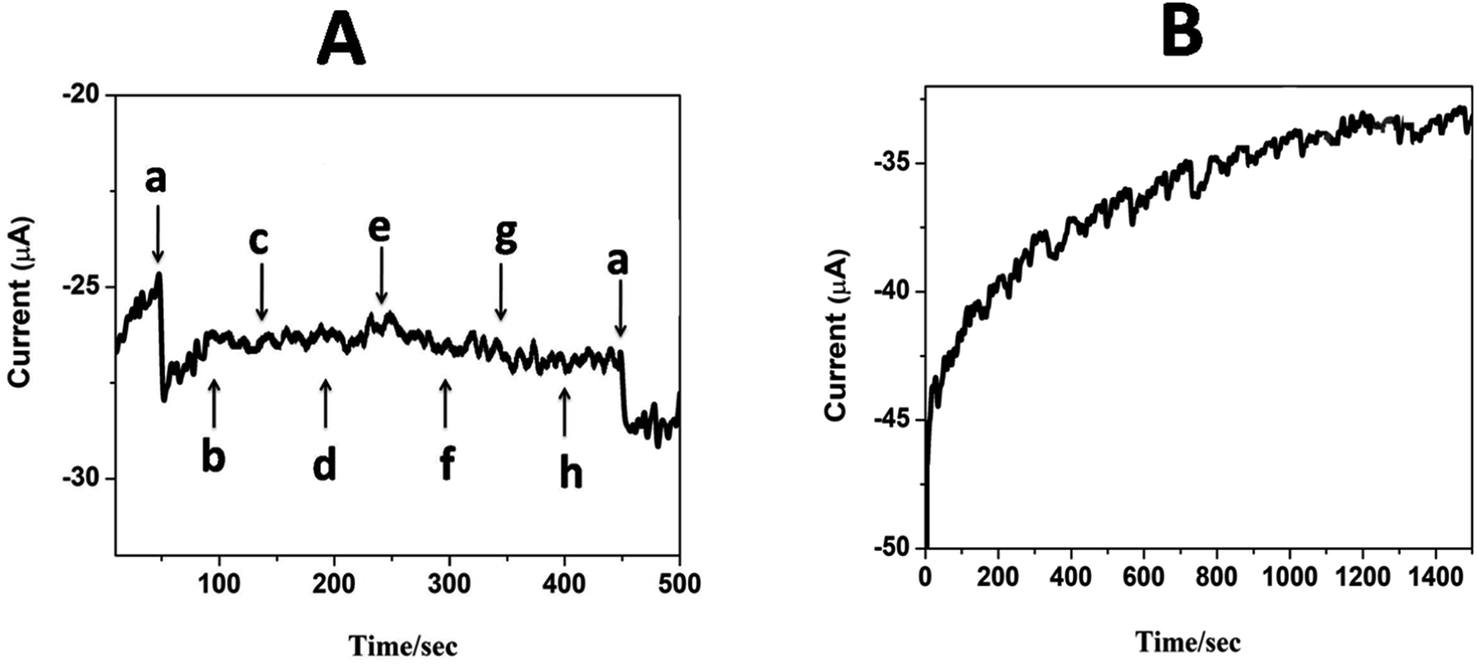
5.8. Sensitivity, selectivity and stability studies
The selectivity and specificity of the fabricated GCE/CNT/poly(CoTBImPc) sensor were investigated in a detailed way for the sensing of H2O2 in the existence of electroactive biological species and salts. Fig. 9A reveals the amperometric response for (a) 20 nM H2O2 and the successive addition of 30 mM of (b) glucose, (c) ascorbic acid, (d) dopamine, (e) NaNO2, (f) hydroquinone, (g) KCl, and (h) KNO3 at a regular interval of 50 s into PBS at a potential of −0.45 V on the CNT/poly(CoTBImPc) electrode. Amperometric response on the addition of H2O2 in the presence of different interfering compounds in PBS at CNT/poly(CoTBImPc) is depicted in Fig. 9A. The amperogram clearly showed a reductive growth in amperometric current on the addition of 20 nM H2O2. Furthermore, it was also noticed that even after the addition of a high concentration (30 mM) of the interfering compounds like glucose, ascorbic acid, dopamine, NaNO2, hydroquinone, KCl, and KNO3, the composite electrode did not have any significant response indicating that the interfering ions did not influence the H2O2 detection. Therefore, the non-interference of compounds like glucose, ascorbic acid, dopamine, NaNO2, hydroquinone, KCl, and KNO3 in phosphate buffer solution during the amperometric sensing of H2O2 indicates the excellent selectivity and superiority of the proposed electrode.
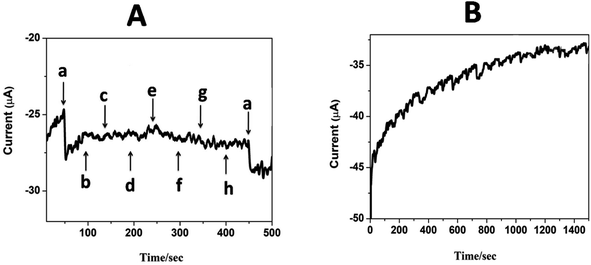 |
| | Fig. 9 (A) Amperometric i–t response for (a) 20 nM H2O2 and 1 mM (b) glucose, (c) ascorbic acid, (d) dopamine, (e) NaNO2, (f) hydroquinone, (g) KCl and (h) KNO3 in pH 7 PBS at GCE/CNP-poly(CoTBImPc). | |
The stability of the composite electrode was monitored by comparing the amperometric response of the newly prepared electrode and after storing the electrode for 14 days. The amperometric current response of the electrode for 14 days was in good agreement with that of the new electrode's initial response, indicating that the proposed sensor is stable even after 14 days. Furthermore, when the electrode was scanned for 100 cycles, it exhibited only a 4% loss in the peak current, which indicates the good electrochemical stability of the composite polymer electrodes. Fig. 9B shows the amperometric stability experiment of the CNT/poly(CoTBImPc) electrode up to a 1500 s time interval in PBS at a potential of −0.45 V. In this time interval, the electrode showed stable performance till the completion of the experiments. This revealed that the CNT/poly(CoTBImPc) electrode is stable and suitable for detecting H2O2 (Fig. 10). Finally, the essential analytical parameters observed in this work for detecting H2O2 were compared, and the results are documented in Table 2.
Table 2 Analytical parameters observed for the detection of H2O2
| Electrode |
Technique |
Sensitivity |
Linear range (nM) |
LOD (nM) |
| Poly(CoTBImPc) |
Cyclic voltammetry |
0.9700 μA nM−1 cm−2 |
100–1000 |
30 |
| CNT/poly (CoTBImPc) |
Cyclic voltammetry |
2.6276 mA nM−1 cm−2 |
10–100 |
5 |
| Poly(CoTBImPc) |
Amperometry |
0.2482 mA nM−1 cm−2 |
10–100 |
3 |
| CNT/poly (CoTBImPc) |
Amperometry |
3.4522 mA nM−1 cm−2 |
10–100 |
2 |
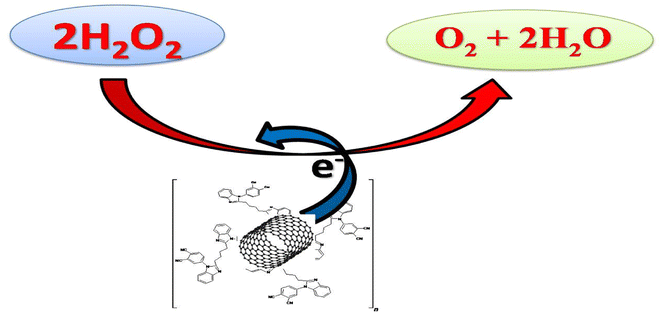 |
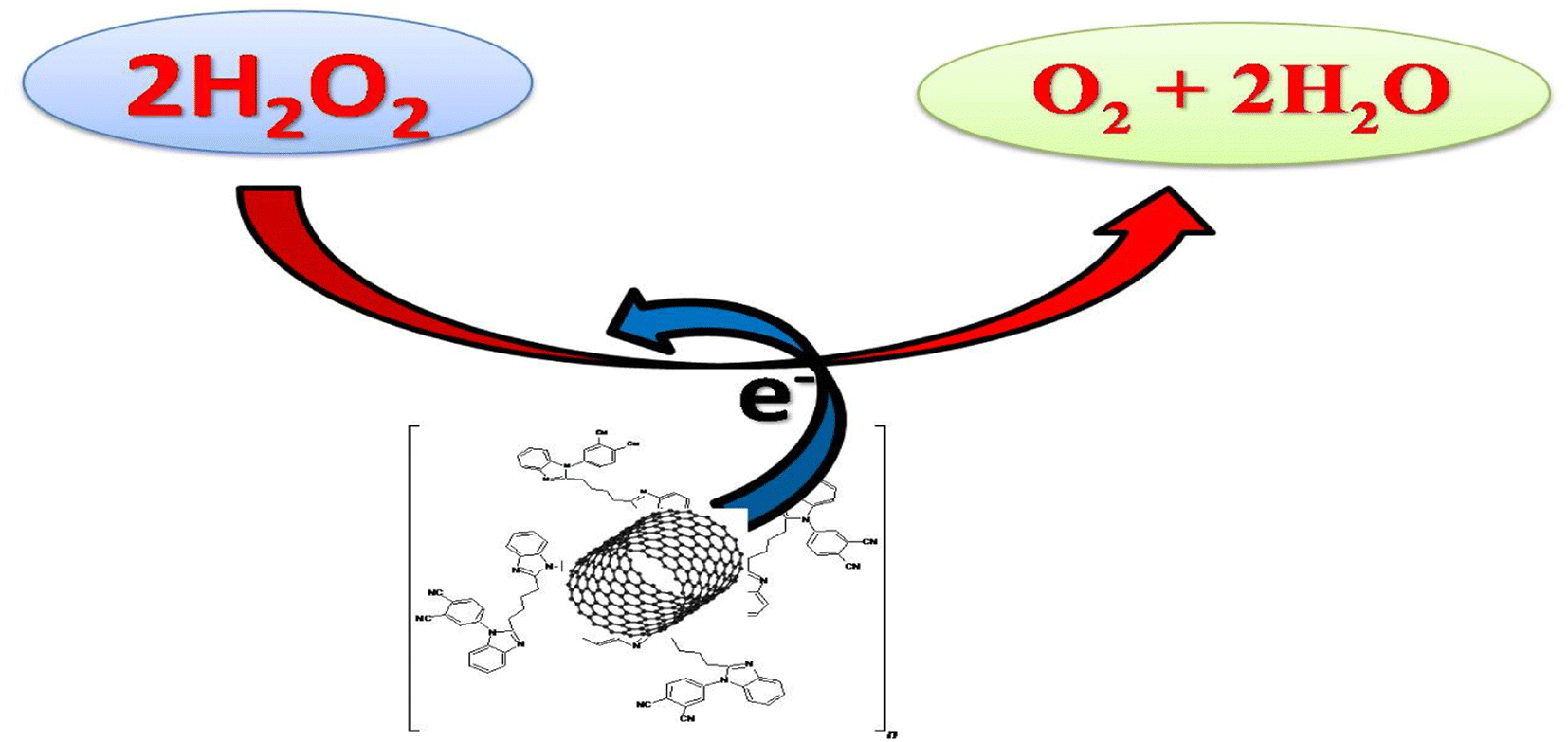
| | Fig. 10 Mechanism of H2O2 reduction at the CNT/poly(CoTBImPc) electrode. | |
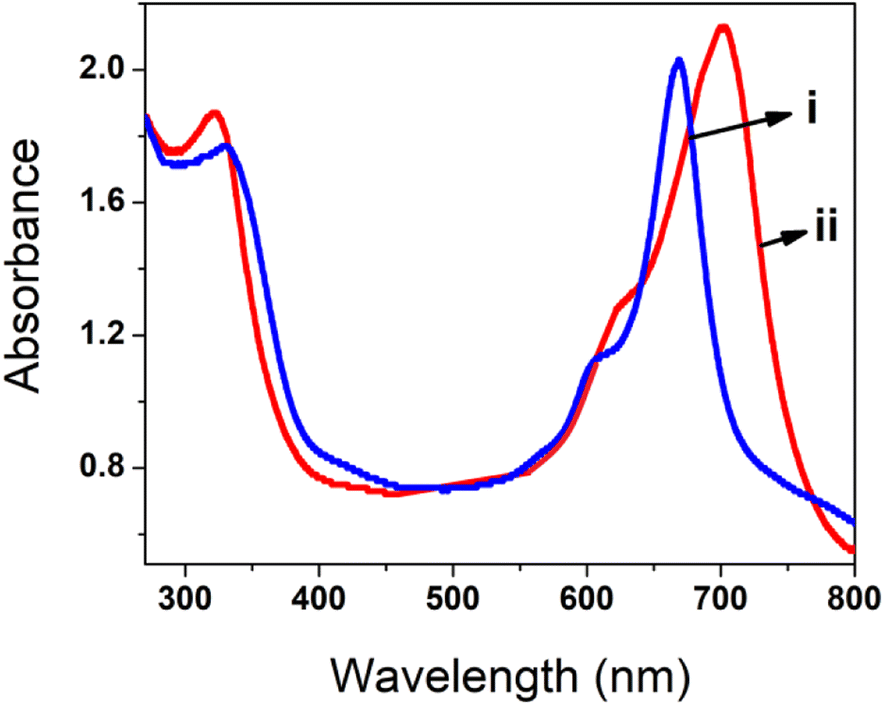
To understand the interaction of polyCoTBImPc with the H2O2 analyte, UV-vis spectrometry in DMSO was carried out (Fig. 11). The absorption spectrum of 10 mM poly(CoTBImPc) in DMSO demonstrated a narrow Q-band. In contrast, adding 100 nM H2O2 analyte leads to shifting of the Q-band towards a more extended wavelength region. This might result from adduct or axial complexation between the central metal of the phthalocyanine macrocycle and H2O2. The results obtained in this work for the detection of H2O2 were also compared with the literature reports as summarized in Table 3.
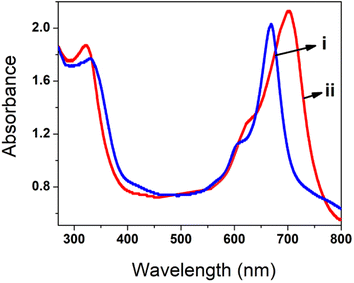 |
| | Fig. 11 Absorption spectra for (i) poly(CoTBImPc) and (ii) poly(CoTBImPc) and H2O2 in DMSO solvent. | |
Table 3 Comparison of GCE/CNT/poly(CoTBImPc) with different electrodes in the literature for the sensing of H2O2
| Electrode material |
Method |
Dynamic range (μM) |
LOD (μM) |
Ref. |
| GO/poly(CoTBIPc) |
CV |
2–200 |
0.6 |
16
|
| Ni(OH)2 nanoplates |
CV |
— |
1.0 |
30
|
|
n-Octylpyridinum hexafluorophosphate |
CV |
20–800 |
12 |
31
|
| [PFeW11O39]4− polyoxoanion |
CV |
10–200 |
7.4 |
32
|
| PtNPs/NPGE |
CV |
0.1–20.0 |
0.72 |
33
|
| Cu2O/N graphene/GCE |
CV |
5.0–357 |
0.80 |
34
|
| Poly(CoTBImPc) |
CV |
0.1–1 |
0.030 |
This work |
| CNT/poly (CoTBImPc) |
CV |
0.01–0.1 |
0.005 |
This work |
| Poly(CoTBImPc) |
CA |
0.01–0.1 |
0.003 |
This work |
| CNT/poly (CoTBImPc) |
CA |
0.01–0.1 |
0.002 |
This work |
5.9. Reproducibility and repeatability of modified electrodes
Reproducibility and repeatability play important roles in the design of the efficient sensor. To study the reproducibility of poly(CoTBImPc) and CNT/poly(CoTBImPc) electrodes, five modified electrodes were developed under similar conditions. The H2O2 detection was performed amperometrically for all the constructed electrodes using a 10 nM H2O2 solution. The relative standard deviation was found to be 2.1% for poly(CoTBImPc) and 1.8% for CNT/poly(CoTBImPc). The sensor's repeatability was evaluated for the detection of H2O2 (10 nM) by taking four duplicate amperometric experiments in PBS (pH 7) using poly(CoTBImPc) and CNT/poly(CoTBImPc) electrodes. The RSD was found to be 2.1% for poly(CoTBImPc) and 1.4% for CNT/poly(CoTBImPc). The RSD values are in the permissible range; therefore, the proposed amperometric sensor shows satisfactory reproducibility and repeatability.
5.10. Real sample analysis
Table 4 displays the analytical result obtained for determining hydrogen peroxide in commercially available 3% hydrogen peroxide using the CNT/poly(CoTBImPc) electrode. The well-defined CV responses were observed for each addition of an accurate sample in the PBS (pH 7) electrolyte. The response current reached 95% steady-state current within 5–7 s of the sample injection, and the resulting amperogram was consistent with lab sample results. The concentration-dependent linear calibration plot showed good linearity. A cheap and fast responsive electrochemical device and an instantaneous analytical device were proposed for the detection of H2O2 present in commercially available 3% hydrogen peroxide samples.
Table 4 Analytical data obtained for commercial 3% H2O2 analysis
| Sample |
Theoretical value (nM) |
Detected (nM) |
Recovery (%) |
RSD (%) |
| Commercially available 3% hydrogen peroxide |
20 |
21 |
105 |
2.50 |
| 40 |
41 |
102 |
1.85 |
| 70 |
72 |
103 |
1.55 |
5.11. Interaction between the phthalocyanine and carbon nanotubes
Finally, the designed poly(CoTBImPc) supported on carbon nanotubes was submitted to different treatments to modify the interaction between the active phase and the support. The prepared materials have been studied primarily as electrochemical sensors for detecting H2O2. Particular emphasis has been placed on determining the importance of the strength of the interaction between the phthalocyanine and the support; using CNT modifications, the electrochemical behavior and surface chemistry of the resulting materials have been improved and have led to a better electron transfer. Moreover, these nanocomposites provide a synergic effect that enhances the response properties of the modified electrodes so that GCE/CNT/poly(CoTBImPc) showed better analytical parameters with a greater sensitivity (3.4522 mA nM−1 cm−2), LOD (2 nM), and linear range (10–100 nM).
6. Conclusion
The present work demonstrates the successful synthesis of poly(CoTBImPc) and its application for the nanomolar detection of H2O2. On successive addition of H2O2, polyCoTBImPc-CNT/GCE shows clear separation and consistent growth in peak current for the reduction of H2O2 compared to Pc alone and the bare GCE. The superior performance of the composite electrode may be ascribed to the presence of CNTs, which have a high conductivity and surface area. GCE/CNT/poly(CoTBImPc) showed better analytical parameters with a greater sensitivity (3.4522 mA nM−1 cm−2), LOD (2 nM), and linear range (10–100 nM). Furthermore, the electrode exhibits greater reproducibility, repeatability, and high stability and also shows good selectivity for H2O2 even in the presence of interfering compounds like electroactive biological species and inorganic salts.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This research work was supported by the Department of Science and Technology (DST) grant No. SERB/F/9388/2016-17, and the support from VGST K-FIST (GRD No. 555) and DST-FIST grant SR/FST/CSI-274/2016 is also acknowledged. CPK thanks VSK University for the fellowship.
References
- S. Ambily and C. S. Menon, Determination of the thermal activation energy and optical band gap of cobalt phthalocyanine thin films, Mater. Lett., 1998, 34, 124–127 CrossRef CAS.
- A. Zanguina, M. Bayo-Bangoura, K. Bayo and G. V. Ouedraogo, IR and UV-Visible Spectra of Iron(ii) Phthalocyanine Complexes With Phosphine or Phosphite, Bull. Chem. Soc. Ethiop., 2002, 16, 73–79 CrossRef CAS.
- C. J. Schramm, Chemical, Spectral, Structural, and Charge Transport Properties of the “Molecular Metals” Produced by Iodination of Nickel Phthalocyanine, Chem. Soc., 1980, 102, 6702–6713 CrossRef CAS.
- D. Zhang, J. Zhang, H. Shi, X. Guo, Y. Guo and R. Zhang,
et al., Redox-active microsized metal-organic framework for efficient nonenzymatic H2O2sensing, Sens. Actuators, B, 2015, 221, 224–229 CrossRef CAS.
- B. Wang, Y. Wu, X. Wang, Z. Chen and C. He, Copper phthalocyanine non covalent functionalized single-walled carbon nanotubes with enhanced NH3 sensing performance, Sens. Actuators, B, 2013, 190, 157–164 CrossRef.
- X. Zhou, B. Wang, Y. Wu, Z. Chena and C. He, Lead phthalocyanine modified carbon nanotubes with enhanced NH3 sensing performance, Sens. Actuators, B, 2012, 171–172, 398–404 Search PubMed.
- H. Hosseini, S. J. T. Rezaei, P. Rahmani, R. Sharifi, M. R. Nabid and A. Bagheri, Nonenzymatic glucose and hydrogen peroxide sensors based on catalyticproperties of palladium nanoparticles/poly (3, 4-ethylenedioxythiophene)nanofibers, Sens. Actuators, B, 2014, 195, 85–91 CrossRef CAS.
- L. Yang, C. Xu, W. Ye and W. Liu, An electrochemical sensor for H2O2based on anew co-metal-organic framework modified electrode, Sens. Actuators, B, 2015, 215, 489–496 CrossRef CAS.
- M.-Q. Wang, Y. Zhang, S.-J. Bao, Y.-N. Yu and C. Ye, Ni (II)-based metal-organicframework anchored on carbon nanotubes for highly sensitive non-enzymatichydrogen peroxide sensing, Electrochim. Acta, 2016, 190, 365–370 CrossRef CAS.
- B. Zhao, Z. Liu, Z. Liu, G. Liu, Z. Li and J. Wang,
et al., Silver microspheres for application as hydrogen peroxide sensor, Electrochem. Commun., 2009, 11, 1707–1710 CrossRef CAS.
- X. Cai, B. Ogorevc, G. Tavcar and J. Wang, Indium–tin oxide film electrode ascatalytic amperometric sensor for hydrogen peroxide, Analyst, 1995, 120, 2579–2583 RSC.
- L. Shang and S. Dong, Design of fluorescent assays for cyanide and hydrogenperoxide based on the inner filter effect of metal nanoparticles, Anal. Chem., 2009, 81, 1465–1470 CrossRef CAS PubMed.
- M. D. Rubianes and G. A. Rivas, Carbon nanotubes paste electrode, Electrochem. Commun., 2003, 5, 689–694 CrossRef CAS; M. D. Rubianes and G. A. Rivas, Enzymatic Biosensors Based on Carbon Nanotubes Paste Electrodes, Electroanalysis, 2005, 17, 73–78 CrossRef.
- J. Wang, M. Musameh and Y. Lin, Solubilization of Carbon Nanotubes by Nafion toward the Preparation of Amperometric Biosensors, J. Am. Chem. Soc., 2003, 125, 2408 CrossRef CAS PubMed.
- N. S. Lawrence, R. P. Deo and J. Wang, Comparison of the Electrochemical Reactivity of Electrodes Modified with Carbon Nanotubes from Different Sources, Electroanalysis, 2005, 17, 64 CrossRef.
- A. S. Veeresh, M. Imadadulla, N. Manjunatha, A. Shambulinga, K. R. Hemantha Kumar, S. Swamy and K. S. Lokesh, Synthesis and electropolymerization of cobalt tetraamine benzamidephthalocyanine macro cycle for the amperometric sensing of dopamine, J. Electroanal. Chem., 2019, 838, 33–40 CrossRef.
- C. P. Keshavananda Prabhu, N. Manjunatha, A. Shambhulinga, M. Imadadulla, K. H. Shivaprasad, M. K. Amshumali and K. S. Lokesh, Synthesis and characterization of novel imine substituted phthalocyanine for sensing of L-cysteine, J. Electroanal. Chem., 2019, 834, 130–137 CrossRef CAS.
- X. Li, X.-H. Peng, B.-D. Zheng, J. Tang, Y. Zhao, Bi-Y. Zheng, M.-R. Ke and J.-D. Huang, New application of phthalocyanine molecules: from photodynamic therapy to photothermal therapy by means of structural regulation rather than formation of aggregates, Chem. Sci., 2018, 9, 2098–2104 RSC.
- E. Dube, D. O. Oluwole, E. Prinsloo and T. Nyokong, A gold-chitosan composite with low symmetry zinc phthalocyanine for enhanced singlet oxygen generation and improved photodynamic therapy activity, New J. Chem., 2018, 42, 10214–10225 RSC.
- T. Nyokong and S. Vilakazi, Phthalocyanines and related complexes as electrocatalysts for the detection of nitric oxide, Talanta, 2003, 61, 27–35 CrossRef CAS PubMed.
- N. Manjunatha, A. Shambhulinga, M. Imadadulla, S. Swamy and K. S. Lokesh, Electro polymerized octabenzimidazole phthalocyanine as an amperometric sensor for hydrazine, J. Electroanal. Chem., 2019, 839, 238–246 CrossRef.
- C. P. Keshavananda Prabhu, N. Manjunatha, A. Shambhulinga, I. Mohammed, M. Palanna, A. S. Veeresh, D. Akshitha and L. K. Sannegowda, A comparative study of carboxylic acid and benzimidazole phthalocyanines and their surface modification for dopamine sensing, J. Electroanal. Chem., 2019, 847, 113262 CrossRef CAS.
- M. Durmus and V. Ahsen, Water-soluble cationic gallium(III) and indium(III) phthalocyanines for photodynamic therapy, J. Inorg. Biochem., 2010, 104, 297–309 CrossRef CAS PubMed.
- J. Oni and T. Nyokong, Simultaneous voltammetric determination of dopamine and serotonin on carbon paste electrodes modified with iron (II) phthalocyanine complexes, Anal. Chim. Acta, 2001, 434, 9–21 CrossRef CAS.
- N. Manjunatha, A. Shambhulinga, M. Imadadulla, C. P. K. Prabhu and K. S. Lokesh, Chemisorbed palladium phthalocyanine for simultaneous determination of biomolecules, Microchem. J., 2018, 143, 82–91 CrossRef.
- V. Mani, S.-T. Huang, R. Devasenathipathy and T. C. K. Yang, Electropolymerization of cobalt tetraamino-phthalocyanine at reduced graphene oxide for electrochemical determination of cysteine and hydrazine, RSC Adv., 2016, 44, 38463–38469 RSC.
- A. L. Verma, S. Saxena, G. S. S. Saini, V. Gaur and V. K. Jain, Hydrogen peroxide vaporsensor using metal-phthalocyanine functionalized carbon nanotubes, ThinSolid Films, 2011, 519, 8144–8148 CrossRef CAS.
- Y. Zhao and X. C. Song, Synthesis and electrocatalytic property of Ni(OH)2 nanoplates for H2O2 reduction, Micro Nano Lett., 2011, 6, 995–997 CrossRef CAS.
- B. Haghighi, H. Hamidi and L. Gorton, Formation of arobustandstable film comprising ionic liquid and polyoxometalate on glassy carbon electrode modified with multiwalled carbon nanotubes: toward sensitive and fast detection of hydrogen peroxide and iodate, Electrochim. Acta, 2010, 55, 4750–4757 CrossRef CAS.
- K. Thenmozhi and S. S. Narayanan, Electrochemical sensor for H2O2 based on thionin immobilized 3-aminopropyltrimethoxy silane derived sol-gel thin film electrode, Sens. Actuators, B, 2007, 125, 195–201 CrossRef CAS.
- G. Yin, L. Xing, X.-J. Ma and J. Wan, Non-enzymatic hydrogenperoxide sensor based on a nanoporous gold electrode modified with platinum nanoparticles, Chem. Pap., 2014, 68, 435–441 CAS.
- B. BinJiang, X. W. Wei, F. H. Wu, K. L. Wu, L. Chen, G. Z. Yuan, C. Dong and Y. Ye, A non enzymatic hydrogen peroxide sensor based on a glassy carbon electrode modified with cuprous oxide and nitrogen-doped graphene in a nafion matrix, Microchim. Acta, 2014, 181, 1463–1470 CrossRef.
- K. Dhara and D. R. Mahapatra, Recent advances in electrochemical nonenzymatic hydrogen peroxide sensors based on nanomaterials: a review, J. Mater. Sci., 2019, 54, 12319–12357 CrossRef CAS.
- B. B. Jiang, X. W. Wei and F. H. Wu,
et al., A non-enzymatic hydrogen peroxide sensor based on a glassy carbon electrode modified with cuprous oxide and nitrogen-doped graphene in a nafion matrix, Microchim. Acta, 2014, 181, 1463–1470 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a,
Kenkera Rayappa
Naveen
*a,
Kenkera Rayappa
Naveen