
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
From microemulsion phase diagrams to hydrophilicity and hydration controlled adsorption: a dissipative particle dynamics modelling study of phospholipid assembly in bio oils†
Maisa
Vuorte
 ab and
Maria
Sammalkorpi
*abc
ab and
Maria
Sammalkorpi
*abc
aDepartment of Chemistry and Materials Science, School of Chemical Engineering, Aalto University, P.O. Box 16100, FI-00076 Aalto, Finland
bAcademy of Finland Center of Excellence in Life-Inspired Hybrid Materials (LIBER), Aalto University, P.O. Box 16100, FI-00076 Aalto, Finland
cDepartment of Bioproducts and Biosystems, School of Chemical Engineering, Aalto University, P.O. Box 16100, FI-00076 Aalto, Finland. E-mail: maria.sammalkorpi@aalto.fi
First published on 22nd June 2023
Abstract
We explore here the assembly and adsorption response of a ternary bio oil–phospholipid–water system via dissipative particle dynamics (DPD) simulations. The mesoscale, particle-based modelling approach allows the examination of large-scale self-assembly response of dipalmitoylphosphatidylcholine (DPPC) phospholipids in a model bio oil solvent (modelled by triglycerides) in the presence of varying amounts of water. We report the reverse micellar and microemulsion assembly phase diagrams of the ternary mixture, verifying the model against literature data. The results show water content vs phospholipid concentration dependent transitions in reverse micellar to network-like and various lamellar phases in bulk assembly. Examination of the DPPC adsorption to smooth, homogeneous adsorbate surfaces of differing polarity reveals that phospholipid adsorption response transitions between discrete assemblies on polyethylene-like hydrophobic to continuous coating on mica-like hydrophilic substrates as the function of phospholipid and water concentrations. The significance of the work is that the presented model for phospholipid assembly in apolar solvents is capable of predicting accurately large scale assembly response and morphology changes including adsorption response in terms of system variables. The presented parametrization and verification information of the model enable readily extending the approach to other systems. The work provides computational access for tuning lipid-based microemulsion systems and their adsorption.
1 Introduction
Biobased microemulsions exist in key roles in biological systems but also have high technological relevance in pharmaceutic materials, cosmetics, food additives, and drug delivery systems. In biology, for example lipid droplets that have a central role in cellular lipid storage and homeostasis, correspond to the emulsion phase.1,2 The technological benefits of biobased emulsions rise from biocompatibility, aqueous green chemistry processing, and high tunability of the materials response by additives and conditions.3,4One of the most studied biobased microemulsion forming systems are ternary mixtures of phospholipids, water, and a low polarity organic solvent.5–7 In such phospholipid based microemulsions, the hydrophobic tails of the phospholipid molecules reside in the low polarity solvent phase and the hydrophilic head groups face the water phase.6 For water-in-oil (w/o) microemulsions, this results in reverse micellar structures with assembly morphologies highly sensitive to water and other polar additives in the system8–11 but also to the apolar solvent species.12–15
The high water sensitivity means that the water/surfactant molar ratio w is a key structural parameter to control the characteristics of microemulsions and reverse micellar systems.16 Presence of water typically promotes surfactant aggregation, causes swelling of the micelles, and, at high enough w, results in phase separation.17 Actually, a debate exists, whether large reverse micelles can form in the absence of water or other polar additives in apolar solvents.18–21 In biobased systems, trace amounts of water readily persist due to the hygroscopic nature of many bio-based surfactants and solvents.22 Water binds to the polar surfactant head group and, at a threshold water content, reverse micelles with a water pool at the micelle core form. Further water addition leads to anisotropic swelling and elongation of the reverse micelles, and eventually a transformation into cylindrical or worm-like assemblies.23,24 At elevated w, typically large lamellar precipitates or hexagonal phases emerge.25,26 The sensitivity of the surfactant assembly to water content, and the eventually induced phase separation, have technological uses in, e.g. bio oil purification processes, but also tuning rheological and chemical properties of microemulsions.27
The sensitivity of phospholipid reverse micellar assemblies to apolar solvent species is also well demonstrated with the assembly response extensively studied in various organic solvents,8 including cyclohexane,28–30 benzene31 and toluene.32 The choice of solvent, particularly in terms of solvent molecular weight and possible polar moieties, has a strong impact on the aggregation behaviour.33–35 The existing works on the effect of solvent leave a gap over assembly response in bio oils, or triglycerides, as the nonpolar media. Works on phospholipid reverse micelles or emulsification in such solvents remain sparse,17,27,36,37 and focus on specific conditions. Interesting for the assembly response, bio oils can solubilize large amounts of water compared to most common organic solvents, and this is reflected as curious variations in their ternary system phase response.13,38 To address this gap, we focus here on the assembly response of phospholipids in bio oils at varying water contents, i.e. the microemulsion assembly response dependencies on the system composition.
These dependencies, assembly phase diagrams, are important in designing systems with desirable colligative, rheological, and spectroscopic properties, but also in engineering microenvironments, e.g., for chemical reactions control and catalysis.14 However, detailed characterization of microemulsions phase behaviour and their microscale structural features is challenging.39,40 Particularly challenging to capture are the smooth transitions between different types of microemulsions, characteristic to reverse micellar systems, see e.g. ref. 3 and 6. Often several different experimental techniques, including NMR,41 conductometry, electron microscopy,42 or scattering experiments,41,43,44 are used in tandem. Also labelling techniques provide information on assembly.12 More refined description of the microemulsion structure can be obtained by the combination of various experimental techniques with appropriate theoretical models.37,40,44–46
Besides bulk assembly response, also the adsorption response of ternary bio oil – phospholipid – water mixtures is broadly interesting. Phospholipids at air–water and oil–water interfaces have been widely studied, see ref. 6 for a comprehensive review. The adsorption to solid substrates is relevant, for example, in bio oils processing, where phospholipids and other impurities can be removed from the oil through adsorption onto substrates such as silica,47–49 or adsorbent clays.50,51 Surface adsorption is also key to interfacial lubrication properties,52 but also to charge transfer at electrified emulsion interfaces.53,54 It is notable, that most phospholipid adsorption studies focus on real bio oil systems including poorly characterised or unknown interfering compounds, and measuring adsorption indirectly through total phosphorus adsorption, or at phospholipid concentrations below the experimentally reported critical micellization concentration (CMC) values to disregard the effect of bulk self-assembly.50,55–57
Molecular modelling methods offer a versatile tool for accessing surfactant aggregation and dynamics on the molecular scale in well-defined molecular conditions. Particularly, atomistic molecular dynamics approaches have been used to examine water dynamics and phospholipid hydration in reverse micelles11,58,59 and the effect of cosurfactants, such as fatty acids, on reverse micelle morphology and dynamics.37 Due to time and length scales involved, most molecular modelling studies of reverse micelles and microemulsions focus on pre-built aggregates11,37,58 or planar oil–surfactant–water interfaces.60,61 Studies focusing on the initial aggregation process and or pre-aggregates forming from an initially random surfactant distribution in solution also exist.12,62–64 Additional challenges to molecular level modelling of surfactants in apolar solvents are posed by the low dielectric solvent medium and typically high viscosity of the oils as these lead to heterogeneous screening of electrostatics and slow structural relaxation (in comparison to water solutions), as well as, the poor transferability of parameters from aqueous systems to nonpolar oils.10,63,65,66 At equilibrium state level, thermodynamic modelling and theory approaches for the aggregation and adsorption response can be formulated, see e.g. ref. 67, but such approaches loose molecular level structural and assembly information. An interesting modelling solution in between thermodynamic state models and molecular level modelling are coarse-grained and mesoscale modelling approaches, especially dissipative particle dynamics (DPD).68–70 While the approach allows modelling assembly and dynamics at time scales in the microsecond to millisecond range and describing system sizes in the micrometer range which brings access to large scale assembly morphologies and their transitions71–75 and allows also mapping system dependencies on adsorption phases,76–79 the trade-off is the abstraction of chemical features and molecular scale interactions, e.g. hydrogen bonding and solvent shell formation. However, for surfactant systems, well argumented DPD parametrization schemes result in micellization response with good match to experimental observables80–83 and established scaling laws.84
Encouraged by the success of the mesoscale modelling approach to colloidal materials modelling and extracting predictions for large scale assembly in them, we develop here a DPD model for a ternary bio oil–phospholipid–water system to examine the assembly response of a phospholipid (dipalmitoylphosphatidylcholine, DPPC) in bio oil (triglyceride solvent) in the presence of varying amounts of water. The modelled system parameters correspond to a range in which the system is expected to form (w/o) microemulsions and reverse micellar systems. The model is verified against expected literature response of the assembly, obtaining a good match. Finally, adsorption response based on the developed model on smooth, homogeneous surfaces of differing polarity is mapped. The examined polarity changes correspond to changes in adsorbent hydrophilicity, or alternatively, chemical modification of the surface. Importantly, the results highlight a difference in phospholipid adsorption regime with the polarity of the surface as well as the role of water in determining the packing of surfactant on the adsorbent and modifying aggregation behaviour.
2 Computational methods
DPD is a particle-based, mesocale coarse-grained (CG) simulations method that relies on describing the molecular system by beads that capture the effective characteristics of regions of the system.85 The time evolution of the beads are obtained via integration of Newton's equations of motion. Additionally, the setup conserves momentum and preserves hydrodynamics. DPD is computationally efficient because of dissipative forces, a short cut-off distance, and pair-wise additive calculation between the DPD beads that interact via a soft-core potential.The DPD beads i interact via a total force68,86
 | (1) |
The DPD conservative force
 | (2) |
![[r with combining circumflex]](https://www.rsc.org/images/entities/b_char_0072_0302.gif) ij = rij/rij determines the force direction. The random FRij and dissipative FDij forces are
ij = rij/rij determines the force direction. The random FRij and dissipative FDij forces are| FDij = −γωD(rij)(ij·vij)ij | (3) |
| FRij = σωR(rij)ξijΔt−1/2ij, | (4) |
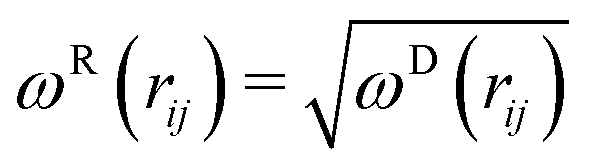 | (5) |
 | (6) |
To construct a DPD model for the phospholipid/oil/water ternary system, we focus on tuning the DPD conservative force FCij repulsion parameters aij, which relate to the chemical nature of the modelled system. We consider DPPC as the model phospholipid to be parametrized, the oil solvent to be bio oil, modelled by triglycerides, and water as the polar additive. For the DPD parametrization and bead coarse-graining, these components of the phospholipid/oil/water ternary system are considered to be composed of molecule regions corresponding chemically to four different DPD bead types, CHOL, GLYS, TAIL, and Water, see Fig. 1. The chosen degree of coarse-graining NCG = 8 is to allow construction of a model that can predict equilibrium assembly responses at length and time scales corresponding to large scale assembly morphology changes. For the adsorbent surface modelling, a DPD bead SURF corresponding to varying hydrophilicity to model chemically specific adsorbent surfaces and another DPD bead species BARRIER are introduced. The bead BARRIER is for the inner parts of the adsorbate and has a high repulsion to all other bead components in the system (aBARRIER,j = 160, for all bead species j other than the BARRIER beads). This provides a penetration barrier, needed to prevent species diffusion into and through the adsorbent, otherwise allowed by the soft core repulsion of DPD particles.
 | ||
| Fig. 1 Coarse-graining scheme for the DPD model of DPPC. An additional TAIL bead was added to the phospholipid hydrocarbon tails to capture better the molecular asymmetry. Model for triglyceride solvent was constructed from the hydrocarbon TAIL beads and a central GLYS bead. The water bead corresponds to ∼8 H2O molecules. | ||
The self-repulsion parameter aii for GLYS, TAIL, Water and the SURF beads is set to 25kBT, derived from water isothermal compressibility at DPD particle density ρ = 3, and a common choice also in DPD modelling of other solvent species.80,87,88 For CHOL beads, aii = 30 is used to better reflect the charged nature and increased self-attraction resulting from the zwitterionic DPPC head group. Notably, no electrostatic interactions are explicitly present in the model and the modification to CHOL self-repulsion is purely an effective modification. DPD electrostatics approaches based on a smeared charge do exist,84,89–91 and would be an option here. However, the molecular self-assembly response is dependent on the smearing.84 Additionally, charge smearing implies a uniform dielectric screening environment and here the solvent oil phase, the (hydrated) reverse micelle cores, and the adsorbent oil interface provide heterogenous, directionally varying screening. In considering the results of the current model, the lack of direct charge–charge interactions should be kept in mind, as e.g., electric double layer and electric dipole interactions do not rise from the model setup.
The DPD conservative force repulsion parameters aij between different bead types i and j connect with the Flory–Huggins mixing parameter χij as
 | (7) |
In this, α = 0.101 ± 0.001 for DPD particle density ρ ≥ 3. The parameter α is a constant related to the radial distribution function between the beads in a single component system. The Flory–Huggins mixing parameter was obtained from Hildebrand solubility parameters δi and δj92 for the components i and j as
 | (8) |
Here, Vbead is the DPD bead volume. The Hildebrand solubility parameter δ for each of the components corresponding to the DPD beads is defined based on cohesive energy Ecoh determined here via all-atom molecular dynamics (MD) simulations via the energy difference between the condensed phase and the gas phase of each bead component. Details of the parametrization MD simulations including the non-bonded energies and the resulting solubility parameters (Table S1) are provided in the ESI.†
The Hildebrand solution theory functions well for non-polar molecules, but fails to accurately describe molecules with high polarity, hydrogen bonding capability, or charged moieties. Other solubility theories, e.g. Hansen solubility theory, address this by identifying dispersion, dipole–dipole, and hydrogen bonding contributions in the cohesion energy. However, dividing the cohesion energy into such separate contributions lacks a physical basis. The parameters rise from an empirical estimate based on experimental data, group contribution methods, or from molecular modelling where explicit energy terms are typically associated for dispersion, dipole–dipole, and H-bonding interactions.80,93,94 In this work, the DPD interaction parameters derived from Hildebrand solubility theory were fine-tuned to match structural data of atomistic MD simulations of binary mixtures corresponding to molecules modelled by the different bead types. The protocol including the radial distribution function matching data are provided in ESI.† The obtained parametrization, i.e. the resulting set of DPD conservative force repulsion coefficients aij is presented in Table 1. Notably, also Hansen derived DPD interaction parameters require scaling to accurately replicate interfacial dynamics in real chemical systems.80
| CHOL | GLYS | TAIL | Water | |
|---|---|---|---|---|
| CHOL | 30 | |||
| GLYS | 40 | 25 | ||
| TAIL | 45 | 33 | 25 | |
| Water | 23 | 31 | 65 | 25 |
Additionally, to constrain the DPD beads into molecule-like compounds, see Fig. 1, harmonic potentials to bind two consecutive beads together by a spring and constraint angles between three consecutive beads are included in the DPD models corresponding to Fig. 1. These contribute to additional spring force contributions to the force terms of eqn (1)
| FS,bij = Kb(r − r0)2 | (9) |
| FS,aijk = Ka(θ − θ0)2. | (10) |
| Bond | r 0 [rc] | K b |
|---|---|---|
| CHOL–GLYS | 0.8 | 500 |
| GLYS–TAIL | 0.8 | 500 |
| TAIL–TAIL | 0.8 | 500 |
| Angle | θ 0 (°) | K a |
|---|---|---|
| CHOL–GLYS–TAIL | 120 | 5 |
| TAIL–TAIL–TAIL | 180 | 5 |
The DPD simulations employ a DPD particle density ρ = 3.0. The dissipative coefficient γ = 4.5. Following standard DPD formulation, all interacting beads, regardless of bead type or model chemical species, have the same mass mi and diameter rc. Reduced units, such that rc = mi = kBT = 1, are employed throughout the formulation and simulations. For the presented parametrization, the DPD cut-off distance rc = 0.891 nm when converted to real units based on the volume of 8 water molecules.
2.1 Simulated systems
Two types of simulation set-ups were examined: phospholipid/triglyceride/water bulk mixtures (no adsorbent surface), and bulk mixtures on a planar adsorbent surface. For bulk simulations, the DPD equivalent of 50 mM, 80 mM, 100 mM, 200 mM, or 500 mM DPPC was solvated in triglyceride solvent in a 50rc × 50rc × 50rc simulation box with 0, 0.5, 1, 2, or 4 water beads per DPPC molecule. Due to coarse-graining at 8 water molecules per DPD bead, this corresponds to 0–32 water molecules per DPPC molecule. All molecules were set initially randomly and uniformly distributed into the simulation box.The planar DPD surface was constructed by equilibrating a 40rc × 40rc × 5rc slab of identical DPD particles with density ρ = 3 and self-repulsion coefficient aii = 25.0. This slab was then centered perpendicular to the z-axis into a simulation box of size 40rc × 40rc × 100rc, resulting in the surface being periodic along the x,y-plane, and the z-axis being perpendicular to the plane. The remainder of the box was filled with DPD equivalent of 50 mM, 100 mM, or 200 mM DPPC solvated in triglyceride, with w = 0, 0.5, 1, or 2 water beads per DPPC molecule. Example visualizations of the simulated systems are presented in Fig. 2.
 | ||
| Fig. 2 Visualizations of example systems representing the simulated bulk self-assembly and surface adsorption systems. Coloring of DPD beads follows Fig. 1 for the beads corresponding to DPPC and water. Triglyceride solvent is omitted in visualization for clarity. The adsorbent surface consists of two types of beads with the chemically specific surface beads SURF in orange and the highly repulsive barrier beads BARRIER in white. | ||
For the adsorption study, to prevent unwanted solvent species diffusion into the surface as result of the soft interactions, the centermost slab of the relaxed surface slab was designated as highly repulsive DPD particles BARRIER that have a cross-repulsion coefficient of aij = 160.0 with all other DPD beads in the system. Sandwiching this, the outermost 1.2rc shell of the surface comprised of SURF beads that vary in their cross-repulsion coefficients aSURF,j according to the hydrophilicity variation captured by a hydrophilicity index λ. SURF beads have repulsive interactions
| aSURF,j = λaWater,j + (1 − λ)aTAIL,j | (11) |
All described DPD systems are simulated using a time step of Δτ = 0.01 for 5 × 106 time steps. Simulation trajectories were analyzed for the last 2 × 105 time steps. Simulations were performed by the Large-scale Atomic/Molecular Massively Parallel Simulator (LAMMPS) molecular dynamics program.98,99
3 Results and discussion
3.1 Phase behaviour of DPPC/water/triglyceride ternary systems
The phase diagram of the DPPC/triglyceride/water ternary system as a function of DPPC and water concentration, as predicted by the DPD modelling, is provided in Fig. 3. In the absence of water, the model predicts a CMC between 50 mM and 100 mM DPPC concentration, with concentrations exceeding the CMC resulting in small aggregates limited in size by the lack of water core. Notably, at 50 mM concentration, DPPC aggregation remains negligible and the surfactants form a nearly isotropic solution in the triglyceride. Literature reports the CMC of DPPC in soy-bean oil being ∼0.085 mM.100 The difference can be explained by the presence of trace water, but also impurities acting as nucleation centers in the experiments, as well as potential underestimation of the localized DPPC head group – head group interactions in the DPD mesoscale coarse-grained model. The introduction of water into the system even in the coarse-grained DPD model results in stronger assembly and more diverse phase behaviour. At low water concentrations, the model predicts DPPC to form elongated, ellipsoidal aggregates around a water core. Increase in hydration or DPPC concentration both promote the aggregates to elongate into worm-like reverse micelles that may form a network-like, connected structure. Finally, at sufficiently high water content, here w = 16, large lamellar aggregates and bicontinuous lamellar assemblies form.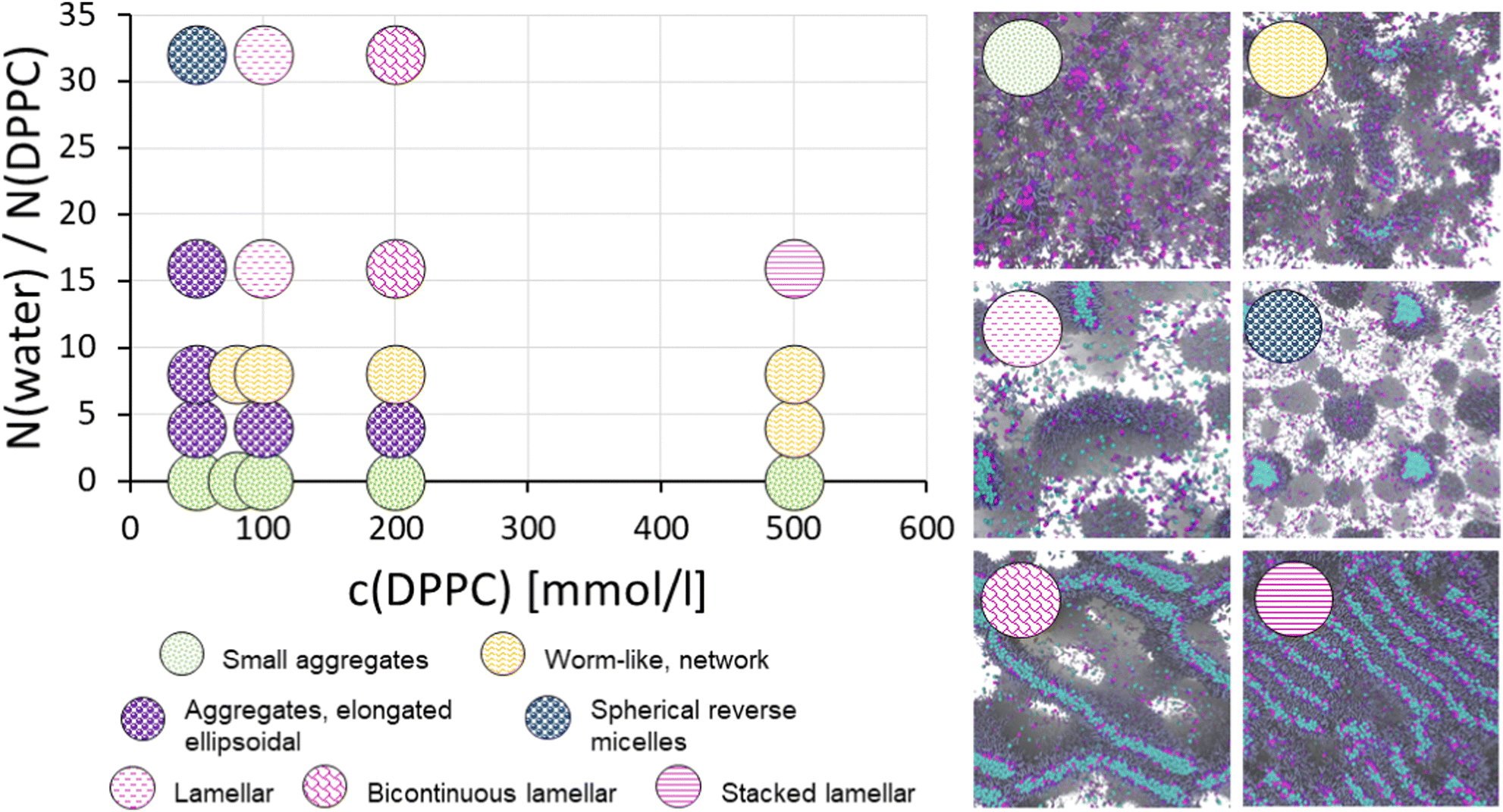 | ||
| Fig. 3 Assembly phase diagram for phospholipid (DPPC) in triglycerides solvent as a function of phospholipid concentration and water-to-DPPC ratio. The snapshots show representative visualizations from each assembly phase. The water-to-DPPC ratio w = N(water)/N(DPPC) has been converted from the DPD beads to number of molecules, i.e. one DPD water bead corresponds to 8 water molecules. | ||
The simulations indicate that DPPC forms aggregates in triglyceride even in the absence of water. The size distribution of the aggregates at different DPPC concentrations is presented in Fig. 4. The exponential shape of distribution indicates that aggregation follows step-wise, open association, meaning that the aggregates do not have a clearly preferential size. Such response is characteristic to step-wise surfactant aggregation in apolar solvents. Opposed to this would be the closed association leading to bias in preferred aggregate sizes and corresponding to the micellization peak characteristic to surfactant micellization aggregate size distributions in aqueous solutions. Notably, bias in aggregate size, indicating preference in aggregate size, may emerge even for surfactants in apolar solvents, assuming that sufficient attraction between the head groups exists: the bias has been observed even for non-ionic surfactants with sufficient hydrogen bonding capability at high concentrations.12,63 However, the effect is most relevant for strongly aggregating species, such as ionic surfactants, or in the presence of a gelating agent such as water, or other small polar molecule.
 | ||
| Fig. 4 DPPC aggregate size distributions for different DPPC concentrations in triglyceride in the DPD simulations. The data corresponds to water-to-DPPC ratio w = 0. | ||
In prior works, similar observations have been made for reverse micelles of other charged surfactants. For example, for AOT in isooctane, Urano et al.101 observed a rapid decrease in the free energy of surfactant insertion into a reverse micellar aggregate with increasing aggregation number for small aggregates. The free energy of surfactant insertion was reported to have a minimum near aggregate size of 20 AOT molecules and increase for aggregates larger than 30 AOTs.101 These findings support the existence of a biased step-wise aggregation regime (and consequently a preferential aggregate size, with likely also an aggregate geometry corresponding to the preferred size) for the strongly aggregating ionic surfactant, such as AOT, in an apolar solvent. Consistent with this, the data of Fig. 4 shows the aggregate size distribution deviates from the exponential distribution. Notably, at c(DPPC) ≥ 100 mM, instead of the exponential decay indicated by a straight line on the logarithmic scale, the distributions show a clear bend and favoring of the larger aggregates.
In the simulations, at c(DPPC) = 50 mM DPPC in the absence of water forms small, tightly packed, spherical aggregates. The structure of the formed aggregates was analysed by considering the radial distribution function (RDF) calculated between the CHOL beads of DPPC in the triglyceride solvent at different DPPC concentrations c(DPPC), see Fig. 5. Coinciding with the concentration induced bend in the distribution, associated with the bias in preferred size, the RDFs of Fig. 5 show the emergence of a second structural length scale. This is a telltale sign of the aggregates becoming elongated, i.e. instead of the RDF showing just the length scale corresponding to spherical aggregates, also the elongated dimension of the aggregate contributes to the RDF signal. The bend in the CHOL–CHOL RDF at r ≈ 1.5rc becomes more prominent with concentration. Additionally, the RDF data shows that the first peak in Fig. 5 shifts to larger r with increased phospholipid concentration, suggesting less dense packing in the reverse micelle core with increasing concentration. This connects directly with the increase in mean size indicated by the size distributions in Fig. 4.
 | ||
| Fig. 5 Radial distribution function RDF calculated between the CHOL beads of DPPC in triglyceride solvent in the absence of water (w = 0). The inset presents the RDF for cDPPC = 500 mM with the long range delocalized 2nd ordering peak marked with arrow. | ||
Based on literature, an increase in phospholipid concentration in apolar solvent such as the triglyceride here should lead to formation of an isotropic, viscoelastic, dense micellar phase.38,102 While this phase does not exhibit dominant lamellar, hexagonal, or cubic phase ordering, the presence of closed-packed structures for 60 wt% phospholipid in 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50 hexane:oil mixture has been observed.38
:50 hexane:oil mixture has been observed.38
In our simulations, at 500 mM (∼57 wt%) the DPPC aggregates start exhibiting some ordered packing, as indicated by the subtle long range ordering peak at r ≈ 5.8rc in the CHOL–CHOL RDF curve following the dip corresponding to the individual aggregates, see the inset of Fig. 5. The long-range ordering peak indicates not only correlations in the small aggregate positions but also depletion of solvent triglyceride as replaced by the aggregates. Notably, such long range order is absent for c(DPPC) < 500 mM, indicating the aggregates remain freely solvated in the triglyceride solvent. The location of the observed long range ordering peak r ≈ 5.8rc ≈ 5.1 nm is in excellent agreement with d-spacing of 5.45 nm from SAXS measurements of 60 wt% phospholipid in 50:50 hexane:oil mixture for the dense reverse micelle phase.38 Opposed, at low phospholipid concentrations, such long range order is not present.38,102,103 This means that the DPD model of this work captures the transition from a dilute micellar phase to a dense micellar phase with increased DPPC concentration. Notably, in the presented modelling, the extent of long-range ordering is limited by the heterogeneous size of the aggregates and transition from spherical to ellipsoidal micelles, as well as, the finite simulations box size. Despite the simulation box being extensive 50rc × 50rc × 50rc, the periodicity of the simulation box sets a constraint for the long-range assembly. This affects both capturing long range order and the periodic structures formed.
The transitions in reverse micellar shape and aggregation response in the presence of water in the system can be considered via changes in the surfactant packing parameter, that is, effective shape of the molecule determined by the head group size vs. size of the hydrophobic tails. Hydration increases the proportional size of the head group, which in this case promotes first elongation of the micelles and eventually leads to lamellar packing structures.
In oils, phospholipids can be expected to have a strongly bound water shell of 6–10 water molecules, hydrating the phospholipid head group and greatly limiting the growth of reverse micellar aggregates at small water concentrations.38 However, even trace amounts of water may trigger the elongation of phospholipid reverse micelles.8,28,104 Introduction of water in the phospholipid–oil system first leads to micelles transitioning from spherical to rod-like, and further addition of water promotes the growth of cylindrical micelles. Cylindrical micelles transition to worm-like micelles once their size exceeds their persistence length. Also branching may occur. The elongation and entanglement of the aggregates can cause an abrupt change in solution viscosity, potentially by several orders of magnitude.8,105 The amount of water needed for the formation of worm-like micelles depends strongly on the solvent; for phosphatidylcholine lipids in hydrocarbon solvents, transition of the solution into a gel occurs at w ≈ 7.8 Specifically for DPPC in triglyceride, a slightly higher transition water content is expected as the hydrophilic moiety of the triglyceride molecules also binds water, meaning that some of the water partitions into the solvent as well.
In the DPD simulations, at water-to-DPPC ratio w ≥ 16 the assembly morphologies correspond to large disk-like aggregates and lamellar phases. The formation of lamellar phases is also linked to changes in water dynamics, particularly greater mobility of the water DPD beads. Fig. 6 presents the scaled diffusion coefficients of the DPD water beads at different water-to-DPPC ratios. Notably, water diffusivity at DPPC concentrations ≤100 mM remains systematically low and largely independent of the degree of hydration (water-to-DPPC ratio w). This is expected as the assembly phases correspond to formation of either small rod-like or elongated and worm-like reverse micelles, in which water remains mostly bound tightly around the DPPC polar head groups and confined to the aggregate cores. The confinement limits the diffusion based movement. Conversely, for DPPC concentrations ≥200 mM, water mobility increases clearly as a function of hydration, with the water-to-DPPC ratio w controlling the diffusion. This matches expectations for network-like reverse micellar aggregate phases and lamellar structures as water has pathways for long-range propagation in such assembly morphologies. At elevated concentrations, in such assembly morphologies, water can be expected to have even free movement at the scale of the system. Previously, for similar microemulsion forming ternary systems, an increase in water mobility was linked to formation of bicontinuous inverse lamellar phases.106
 | ||
| Fig. 6 Top panel presents the scaled diffusion constant vs. water-to-DPPC ratio w = N(water)/N(DPPC) for different DPPC concentrations. Bottom panel presents the radial distribution function RDF calculated for DPD water beads around the DPPC head (CHOL + GLYS beads) for 200 mM DPPC in triglyceride solvent at varying water-to-DPPC ratios w = N(water)/N(DPPC). | ||
Water molecules in microemulsion droplets or reverse micelles can be classified into either two or three different types based on how tightly the water is bound. A common division is to consider water interacting with the micelle core–corona interface and with the surfactant polar head groups as bound water and the water deeper within the micelle core aqueous pool as water with bulk-like characteristics.107–109 The bound and the bulk-like water differ in their organization, mobility, and microviscosity; typically, tightly bound and bound (non-freezing and freezing) water are considered as different species.110–112 Similar bound and unbound water populations have been observed in e.g., polyelectrolyte multilayers112 but also in water adsorbed in doped carbon nanopores.113 The RDFs of water beads around the DPPC heads in Fig. 6 indicate that the increased water mobility results mainly from the amount of bulk-like water increasing, opposed to the bound water around the DPPC head group remaining at constant level. Analogous distinction between bound and bulk water has been observed in reverse micelles with sizeable water pools114 and also mapped in atomistic simulations.11 Molecular level interactions of water and the lipid head groups determine largely the binding strength and size of the head group water shell. For the DPD model here, and coarse-grained models in general, modelling the hydration number is mainly by the bead size and its polarity (conservative interaction parameter). Notably, the microemulsion droplet-like reverse micelles that have relatively large water cores at c(DPPC) = 50 mM and w = 32 have overall low diffusivity of the water. This is a direct consequence of the model being a coarse-grained mesoscale model and the diffusion coefficient capturing mobility only inside the water pool, now limited in size by the coarse-graining. The fine grained structure and detailed dynamics of water is lost. For comparison with atomistic detail simulations results, see ref. 11.
We conclude that the DPD model reproduces at qualitative level the DPPC assembly response in triglyceride in the absence of water, but also when water is added. Notably, the presence of CMC and small aggregate formation is predicted accurately, as is the emergence of dense micellar phase at higher DPPD concentrations. Furthermore, the transitions to elongated and wormlike aggregates, as expected based on literature, emerge upon addition of water, and further water addition transitions the assembly phases to various lamellar phases, as expected.
3.2 Adsorption of DPPC on adsorbents of differing polarity
As the DPD model reproduces bulk assembly response to good degree, we move to employing the model to map much less characterized phospholipid assembly response, that of surface adsorption from ternary DPPC/triglyceride/water system. First the adsorption of DPPC and water on planar surfaces of varying polarity was examined.Adsorption acts as a competing process for DPPC and water aggregation. Importantly, the results reveal a difference in adsorption regime on hydrophobic and hydrophilic surfaces, see Fig. 7 and 8. Fig. 7 presents the density of DPPC head groups on the adsorbent surface as a function of surface hydrophilicity parameter λ and the water-to-DPPC ratio w in the oil, modelling the oil–surfactant system hydration. Fig. 8 presents the corresponding adsorbed phospholipid and water quantifications for different DPPC concentrations. In the absence of water at w = 0, DPPC adsorption at the surface increases with DPPC concentration and surface hydrophilicity. On hydrophilic surfaces (λ = 0.8, comparable to a mica-like surface) a DPPC monolayer forms with coating saturation dependent on DPPC concentration, see Fig. 8. The formed monolayer coverages correspond with lipid surface areas of 0.85rc2 ≈ 0.68 nm2 for c(DPPC) = 50 mM, 0.57rc2 ≈ 0.45 nm2 for c(DPPC) = 100 mM, and 0.50rc2 ≈ 0.40 nm2 for c(DPPC) = 200 mM. The values compare well with prior experimental characterizations in which areas of 1.2–1.3 nm2 per DPPC lipid have been reported for adsorption from oil onto hydrophilic surfaces, including sepiolite, silica, silica–alumina and bleaching earths.50,57 The tighter packing in the simulations can be explained by the idealized setup, for example the flat, ideal surface. Notably, the resulting packing density predicted by the model at the hydrophilic substrate is much closer to what would be expected of phosphatidylcholine monolayers (0.6 nm2).61 Notably, the packing density is likely to be overestimated here due to insufficient repulsion between phosphatidylcholine head group DPD beads.
 | ||
| Fig. 7 Density maps of DPPC head group (CHOL + GLYS beads) distribution on different adsorbate surfaces as the function of surface hydrophilicity λ and water-to-DPPC ratio w = N(water)/N(DPPC) capturing the degree of hydration. Data corresponds to c(DPPC) = 100 mM. | ||
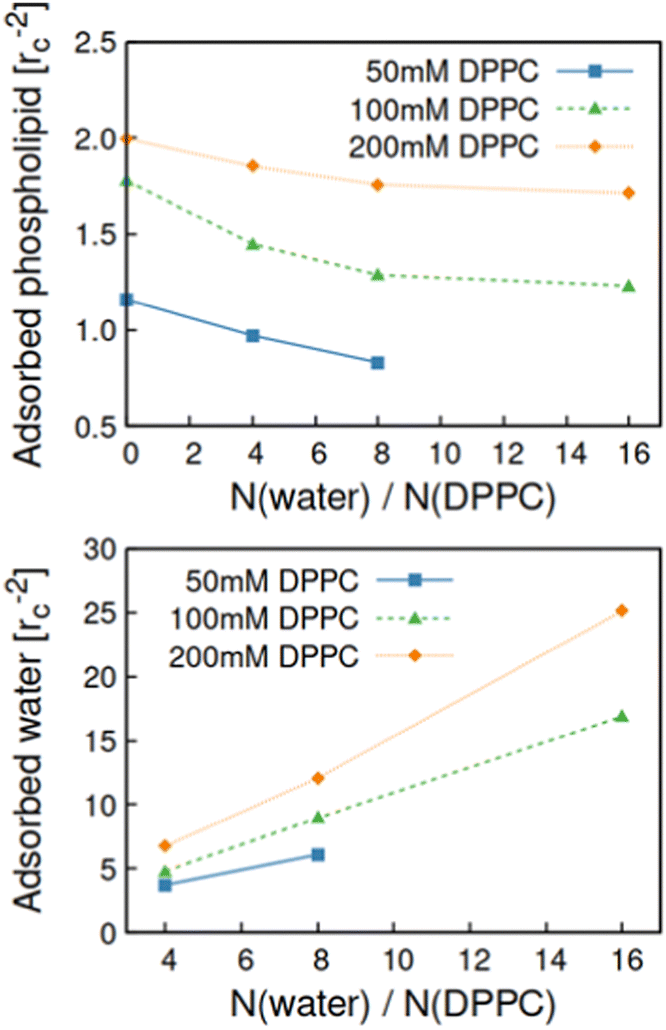 | ||
| Fig. 8 Top panel presents the surface density of adsorbed DPPC and the bottom panel the corresponding data for adsorbed water molecules on hydrophilic (λ = 0.8, mica-like substrate) adsorbent surface as a function of water-to-DPPC ratio in the system. | ||
In the ESI,† Fig. S1 summarizes the densities of the system constituents near the adsorbent. On hydrophilic surfaces (λ = 0.8, comparable to a mica-like surface), both DPPC head groups and water adsorb directly onto the surface. Here, water acts as a competing adsorbate, as shown by the decrease in DPPC adsorption in systems containing water. On hydrophobic surfaces (λ = 0.2, comparable to a polyethylene surface) both the water and DPPC head group density peaks shift away from the surface, while the DPPC tail peak shifts closer to the surface. This indicates either aggregate or lamellar like structures on the surface. On hydrophobic surfaces also the triglyceride solvent exhibits structuring at the interface, likely due to the interaction of sizable hydrophobic tails with the surface. Such ordering is absent on hydrophilic surfaces largely due to the limited steric availability of the hydrophilic central bead of the triglyceride molecule and formation of the phospholipid monolayer at on the surface.
Interestingly, the density mappings of Fig. 7 reveal the presence of clustered DPPC aggregates on hydrophobic surfaces (λ = 0.2, comparable to a polyethylene surface) at 100 mM DPPC concentration. These aggregates interact only weakly with the hydrophobic surface. The assembly at surface corresponds to the hydrophobic hydrocarbon tails facing both adsorbent surface and the solvent phase. Such reverse micellar aggregates on hydrophobic surfaces support previous experimental findings of minimal phospholipid adsorption on smooth hydrophobic polymer surfaces and increased adsorption on porous surfaces of similar chemistry.56 The findings could be explained by surface reverse micellar aggregates being trapped in the pore geometry of the adsorbate. Even step edges align and bind surfactants115 and any pore edges may have chemically different character, promoting aggregate pinning: on patterned and stepped surfaces, the length scale of the surface defects and the surfactant molecular size, as well, as chemical heterogeneity of the adsorbent surface leads to deviation from the planar adsorption response, see e.g., ref. 116–118. Notably, adsorbent surfaces decorated by adsorbed micelles have been previously reported for adsorption of both non-ionic and ionic surfactant onto silica in water and such assemblies may be described in literature as admicelles, bilayers, patchy bilayers, or interdigitated bilayers.119,120 In aqueous systems, an S-shaped adsorption isotherm response with initial hemimicelle adsorption at dilute surfactant concentration, followed by admicelle adsorption above CMC, when bulk aggregation occurs, can be expected.119 In the oil system, a more simple adsorption response is expected due to the lack of CMC.
The data of Fig. 9 shows that with increasing adsorbent hydrophilicity, surface coverage shifts from island like aggregate coating to the formation of an evenly distributed monolayer. This difference in adsorption regimes is indicated by the RDF curves for DPPC heads and tail with respect to the surface particles: the DPPC heads gradually shift away from direct contact with the adsorbent surface with increased adsorbent hydrophilicity parameter λ. An opposite trend is observed for DPPC tails, with direct adsorption of the tail groups with adsorbent on hydrophobic surfaces (λ = 0.2).
 | ||
| Fig. 9 Radial distribution function RDF calculated between DPPC heads and surface beads (top panel) and tails and surface beads (bottom panel) for adsorbent surfaces of differing hydrophobicity parameter λ. The presented data corresponds to DPPC concentration 100 mM and w = 0. Inset cartoons point out the transition from aggregate adsorption to monolayer coverage with increasing λ. | ||
The adsorption regimes observed in the absence of water persist also in the presence of water, see Fig. 7. However, water morphs the shape of the spherical surface aggregates into elongated ellipsoids. The ellipsoids undergo further growth with increasing water concentration. This results in increased DPPC adsorption on hydrophobic surfaces with increased water content, see Fig. 10. The aggregates present on the surface largely reflect those present in bulk solution. For example, at elevated water concentration, water-to-DPPC ratio w = 16, large inverse lamellar structures form on the surface, consistent with bulk phase behaviour. Here, due to adsorption being driven by diffusion, the time scale of the modelling may not accurately capture equilibrium state adsorption of larger aggregates, particularly slowly diffusing lamellar precipitates.
 | ||
| Fig. 10 Surface density of adsorbed DPPC on hydrophobic (λ = 0.2) adsorbent surface as a function of system water content. | ||
The competitive adsorbate character of water to DPPD on hydrophilic surfaces results in depletion of DPPC from the surface with increasing system water content, see Fig. 8. As most of the adsorbed water is bound to a phospholipid head group, the depletion of DPPC rises from the adsorbed hydrated DPPC head groups occupying a larger surface area than head groups with less water. The increase in head group radius can be expected to scale with w1/3 as it is a volumetric quantity. Experimentally, the effect of water on the adsorption of both hydratable and non-hydratable phospholipids has been shown to be minimal.37,57 For example, Lehtinen et al. showed negligible effect of up to 1.0 wt% water on DPPC adsorption from rapeseed oil onto acid activated sepiolite. However, the denser monolayer packing predicted by the model here is more sensitive to changes in the DPPC head group size with hydration. We hypothesise that the sparser packing of DPPC in experiments37 results from electrostatic repulsion between the lipid heads, which is largely unaffected by the formation of the first, strongly bound hydration layer around the head group. The employed DPD model considers the effect here as coarse-grained effective size change.
4 Conclusions
Here, we presented a DPD model for a DPPC/triglyceride/water ternary system and examined its bulk phase behaviour and adsorption properties. The bulk assembly response predicted by the model agreed very well with experimental assembly morphologies in large scale. We demonstrated that the system undergoes initial aggregation, with aggregate growth greatly limited in water-less systems. However, even for dry binary mixtures of DPPC and triglyceride solvent, onset of cooperative aggregation with increased phospholipid concentration was observed. Additionally, the model captures structural transition from a dilute micellar phase to a dense micellar phase. This coincides with elongation of the aggregate shape profile. When water is present, aggregates undergo growth and elongation from ellipsoidal to worm-like. Continued aggregate growth results in the formation of network like organogel phase and lamellar phase depending on oil water content. For the relatively high degree of coarse-graining (NCG = 8) used here to enable modelling large scale assembly and morphology changes in equilibrium, the agreement with experimental verification data is very good.For surface adsorption, we reported two different adsorption regimes depending on surface hydrophilicity: monolayer formation (hydrophilic adsorbent) and adsorption as aggregates (hydrophobic adsorbent). Water acted as a competing adsorbate on hydrophilic adsorbents but also promoted aggregate growth and adsorption of admicelles on hydrophobic surfaces. Similar admicelle adsorption regimes have been documented for surfactants in aqueous solvent.121–123 Here, we generalized this adsorption response for surfactants in oil. It is also interesting to speculate what response would other additives besides water cause. Small polar additives can be expected to behave like water, whereas e.g. hydrogen bonding ability will compete with both the solvent, surface and DPPC, see e.g. ref. 37. In prior work, we examined nitrogen compound adsorption from bio oil, resolving molecular mechanisms for related biospecies.65
We also reported that increasing hydration pushes some of the DPPC lipids from the surface. This depletion of phospholipids could largely be explained by the increased area of hydrated head groups. The adsorption of phospholipids is largely insensitive to water content in experiments mainly due to sparse packing of the lipid monolayer on the surface by electrostatic repulsion of the head groups.50 Here, the DPD model disregards electrostatic interactions, resulting in a denser packing structure of phospholipid monolayers despite the increase self-repulsion between DPPC head groups.
Overall, the work shows that DPD offers a versatile mesoscale modelling method for examining equilibrium structures and assembly phases for even complex, colloidal multicomponent systems. The obtained significant extension of the temporal and spatial resolution in comparison to, e.g. even coarse-grained classical atomistic MD approaches makes the approach powerful in terms of modelling reach. Additionally, the presented work provides guidelines for designing both microemulsion and reverse micellar bulk systems but also for predicting phospholipid adsorption response from bio oil solutions at interfaces of varying polarity.
Author contributions
Maisa Vuorte: conceptualization, data curation, formal analysis, investigation, visualization, writing – original draft, writing – review & editing; Maria Sammalkorpi: conceptualization, funding acquisition, project administration, resources, supervision, writing – review & editing.Data availability
Data for this paper are available at Etsin (https://etsin.fairdata.fi/) at https://doi.org/10.23729/c869a2b4-bc11-40c6-8063-448d84fe076b.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Academy of Finland through its Centres of Excellence Programme (2022–2029, LIBER) under project no. 346111 (M. S.) and Novo Nordisk Foundation under project no. NNF22OC0074060 (M. S.). The authors thank Dr. Susanna Kuitunen for useful discussions and feedback. We are grateful for the support by FinnCERES Materials Bioeconomy Ecosystem. Computational resources by CSC IT Centre for Finland, RAMI – RawMatters Finland Infrastructure, and Aalto Science-IT project are also gratefully acknowledged.Notes and references
- A. Thiam, R. J. Farese and T. Walther, Nat. Rev. Mol. Cell Biol., 2013, 14, 775–786 CrossRef CAS PubMed.
- J. Olzmann and P. Carvalho, Nat. Rev. Mol. Cell Biol., 2019, 20, 137–155 CrossRef CAS PubMed.
- B. Ozturk and D. J. McClements, Curr. Opin. Food Sci., 2016, 7, 1–6 CrossRef.
- C. Attaphong and D. A. Sabatini, Energy Fuels, 2013, 27, 6773–6780 CrossRef CAS.
- V. Klang and C. Valenta, J. Drug Delivery Sci. Technol., 2011, 21, 55–76 CrossRef CAS.
- R. Pichot, R. L. Watson and I. T. Norton, Int. J. Mol. Sci., 2013, 14, 11767–11794 CrossRef PubMed.
- M. Wang, W. Yan, Y. Zhou, L. Fan, Y. Liu and J. Li, Trends Food Sci. Technol., 2021, 118, 388–398 CrossRef CAS.
- R. Scartazzini and P. L. Luisi, J. Phys. Chem., 1988, 92, 829–833 CrossRef CAS.
- Y. A. Shchipunov and E. V. Shumilina, Mater. Sci. Eng., C, 1995, 3, 43–50 CrossRef.
- S. Vierros and M. Sammalkorpi, J. Chem. Phys., 2015, 142, 094902 CrossRef CAS PubMed.
- S. Vierros and M. Sammalkorpi, Phys. Chem. Chem. Phys., 2015, 17, 14951–14960 RSC.
- S. Vierros, M. Österberg and M. Sammalkorpi, Phys. Chem. Chem. Phys., 2018, 20, 27192–27204 RSC.
- E. Abuin, E. Lissi, R. Duarte, J. Silber and M. Biasutti, Langmuir, 2002, 18, 8340–8344 CrossRef CAS.
- J. P. Cason, M. E. Miller, J. B. Thompson and C. B. Roberts, J. Phys. Chem. B, 2001, 105, 2297–2302 CrossRef CAS.
- J. J. Silber, A. Biasutti, E. Abuin and E. Lissi, Adv. Colloid Interface Sci., 1999, 82, 189–252 CrossRef CAS.
- S. Mezzasalma, G. Koper and Y. A. Shchipunov, Langmuir, 2000, 16, 10564–10565 CrossRef CAS.
- R. Subramanian, S. Ichikawa, M. Nakajima, T. Kimura and T. Maekawa, Eur. J. Lipid Sci. Technol., 2001, 103, 93–97 CrossRef CAS.
- L. K. Shrestha, T. Sato, D. P. Acharya, T. Iwanaga, K. Aramaki and H. Kunieda, J. Phys. Chem. B, 2006, 110, 12266–12273 CrossRef CAS PubMed.
- L. K. Shrestha, O. Glatter and K. Aramaki, J. Phys. Chem. B, 2009, 113, 6290–6298 CrossRef CAS PubMed.
- L. K. Shrestha, G. R. Shrestha and K. Aramaki, J. Dispersion Sci. Technol., 2009, 30, 1525–1532 CrossRef CAS.
- D. J. Abdallah and R. G. Weiss, Adv. Mater., 2000, 12, 1237–1247 CrossRef CAS.
- Q. Guo, V. Singh and S. H. Behrens, Langmuir, 2010, 26, 3203–3207 CrossRef CAS PubMed.
- T. Koga and J. Terao, J. Agric. Food Chem., 1995, 43, 1450–1454 CrossRef CAS.
- L. K. Shrestha, R. G. Shrestha and K. Aramaki, Langmuir, 2011, 27, 5862–5873 CrossRef CAS PubMed.
- S. Zhang, Nat. Biotechnol., 2003, 21, 1171–1178 CrossRef CAS PubMed.
- B. Chen, A. Han, D. J. McClements and E. A. Decker, J. Agric. Food Chem., 2010, 58, 11993–11999 CrossRef CAS.
- L. Lei, Y. Ma, D. R. Kodali, J. Liang and H. T. Davis, J. Am. Oil Chem. Soc., 2003, 80, 383–388 CrossRef CAS.
- P. Schurtenberger and C. Cavaco, Langmuir, 1994, 10, 100–108 CrossRef CAS.
- D. Capitani, A. L. Segre, F. Dreher, P. Walde and P. L. Luisi, J. Phys. Chem., 1996, 100, 15211–15217 CrossRef CAS.
- I. Martiel, L. Sagalowicz and R. Mezzenga, Langmuir, 2013, 29, 15805–15812 CrossRef CAS.
- R. Costard, N. E. Levinger, E. T. Nibbering and T. Elsaesser, J. Phys. Chem. B, 2012, 116, 5752–5759 CrossRef CAS PubMed.
- R. Rodrguez, S. Vargas and D. Fernández-Velasco, J. Colloid Interface Sci., 1998, 197, 21–28 CrossRef PubMed.
- R. Angelico, A. Ceglie, U. Olsson and G. Palazzo, Langmuir, 2000, 16, 2124–2132 CrossRef CAS.
- R. Angelico, A. Ceglie, G. Colafemmina, F. Delfine, U. Olsson and G. Palazzo, Langmuir, 2004, 20, 619–631 CrossRef CAS PubMed.
- G. Palazzo, Soft Matter, 2013, 9, 10668–10677 RSC.
- S. Ichikawa, S. Sugiura, M. Nakajima, Y. Sano, M. Seki and S. Furusaki, Biochem. Eng. J., 2000, 6, 193–199 CrossRef CAS PubMed.
- O. P. Lehtinen, R. W. N. Nugroho, T. Lehtimaa, S. Vierros, P. Hiekkataipale, J. Ruokolainen, M. Sammalkorpi and M. Österberg, Colloids Surf., B, 2017, 160, 355–363 CrossRef CAS PubMed.
- R. Gupta, H. Muralidhara and H. Davis, Langmuir, 2001, 17, 5176–5183 CrossRef CAS.
- M. Gradzielski, Curr. Opin. Colloid Interface Sci., 2008, 13, 263–269 CrossRef CAS.
- M. Kahlweit, R. Strey, D. Haase, H. Kunieda, T. Schmeling, B. Faulhaber, M. Borkovec, H.-F. Eicke, G. Busse, F. Eggers, T. Funck, H. Richmann, L. Magid, O. Söderman, P. Stilbs, J. Winkler, A. Dittrich and W. Jahn, J. Colloid Interface Sci., 1987, 118, 436–453 CrossRef CAS.
- O. Regev, S. Ezrahi, A. Aserin, N. Garti, E. Wachtel, E. Kaler, A. Khan and Y. Talmon, Langmuir, 1996, 12, 668–674 CrossRef CAS.
- W. Jahn and R. Strey, J. Phys. Chem., 1988, 92, 2294–2301 CrossRef CAS.
- S.-H. Chen, Ann. Rev. Phys. Chem., 1986, 37, 351–399 CrossRef CAS.
- P. A. Penttila, S. Vierros, K. Utriainen, N. Carl, L. Rautkari, M. Sammalkorpi and M. Österberg, Langmuir, 2019, 35, 8373–8382 CAS.
- M. Cates, D. Andelman, S. Safran and D. Roux, Langmuir, 1988, 4, 802–806 CrossRef CAS.
- P. de Gennes and C. Taupin, J. Phys. Chem., 1982, 86, 2294–2304 CrossRef CAS.
- H. G. Brown and H. Snyder, J. Am. Oil Chem. Soc., 1989, 66, 353–355 CrossRef CAS.
- H. G. Brown and H. E. Snyder, J. Am. Oil Chem. Soc., 1985, 62, 753–756 CrossRef CAS.
- M. Catarino, E. Ferreira, A. P. S. Dias and J. Gomes, Chem. Eng. J., 2020, 386, 123930 CrossRef CAS.
- M. Laatikainen, W. Srithammavut, B. Toukoniitty, I. Turunen and T. Sainio, Adsorption, 2015, 21, 409–417 CrossRef CAS.
- W. E. Wallace Jr, L. C. Headley and K. C. Weber, J. Colloid Interface Sci., 1975, 51, 535–537 CrossRef.
- C. Murru, R. Badía-Laíño and M. E. Díaz-García, ACS Sustainable Chem. Eng., 2021, 9, 1459–1476 CrossRef CAS PubMed.
- P. Hazra, D. Chakrabarty and N. Sarkar, Langmuir, 2002, 18, 7872–7879 CrossRef CAS.
- M. Novaira, F. Moyano, M. A. Biasutti, J. J. Silber and N. M. Correa, Langmuir, 2008, 24, 4637–4646 CrossRef CAS PubMed.
- E. Sabah and M. Majdan, J. Food Eng., 2009, 91, 423–427 CrossRef CAS.
- M. Hancer, A. Patist, R. Kean and H. Muralidhara, Colloids Surf., A, 2002, 204, 31–41 CrossRef CAS.
- J. van Dalen and K. van Putte, Lipid/Fett, 1992, 94, 567–570 CrossRef.
- S. Abel, F. Sterpone, S. Bandyopadhyay and M. Marchi, J. Phys. Chem. B, 2004, 108, 19458–19466 CrossRef CAS.
- G. Folpini, T. Siebert, M. Woerner, S. Abel, D. Laage and T. Elsaesser, J. Phys. Chem. Lett., 2017, 8, 4492–4497 CrossRef CAS PubMed.
- G. Henneré, P. Prognon, F. Brion, V. Rosilio and I. Nicolis, THEOCHEM, 2009, 901, 174–185 CrossRef.
- G. Henneré, P. Prognon, F. Brion and I. Nicolis, Chem. Phys. Lipids, 2009, 157, 86–93 CrossRef.
- J. C. Shelley and M. Y. Shelley, Curr. Opin. Colloid Interface Sci., 2000, 5, 101–110 CrossRef CAS.
- M. Vuorte, S. Vierros, S. Kuitunen and M. Sammalkorpi, J. Colloid Interface Sci., 2020, 571, 55–65 CrossRef CAS PubMed.
- S. Vierros and M. Sammalkorpi, ACS Omega, 2019, 4, 15581–15592 CrossRef CAS PubMed.
- M. Vuorte, S. Kuitunen and M. Sammalkorpi, Phys. Chem. Chem. Phys., 2021, 23, 21840–21851 RSC.
- I. Leontyev and A. Stuchebrukhov, Phys. Chem. Chem. Phys., 2011, 13, 2613–2626 RSC.
- M. Vuorte, S. Kuitunen, P. R. Van Tassel and M. Sammalkorpi, J. Colloid Interface Sci., 2023, 630, 783–794 CrossRef CAS PubMed.
- P. Espanol and P. B. Warren, J. Chem. Phys., 2017, 146, 150901 CrossRef PubMed.
- P. Hoogerbrugge and J. Koelman, Europhys. Lett., 1992, 19, 155 CrossRef.
- J. Koelman and P. Hoogerbrugge, Europhys. Lett., 1993, 21, 363 CrossRef CAS.
- S. Javan Nikkhah and M. Sammalkorpi, J. Colloid Interface Sci., 2023, 635, 231–241 CrossRef CAS PubMed.
- S. Javan Nikkhah, E. Turunen, A. Lepo, T. Ala-Nissila and M. Sammalkorpi, Polymers, 2021, 13, 2193 CrossRef CAS.
- A. L. Harmat, S. Javan Nikkhah and M. Sammalkorpi, Polymer, 2021, 233, 124198 CrossRef CAS.
- Q. Zhu, T. R. Scott and D. R. Tree, Soft Matter, 2021, 17, 24–39 RSC.
- C. R. Iacovella, R. E. Rogers, S. C. Glotzer and M. J. Solomon, J. Chem. Phys., 2010, 133, 164903 CrossRef PubMed.
- A. Scacchi, K. Hasheminejad, S. Javan Nikkhah and M. Sammalkorpi, J. Colloid Interface Sci., 2023, 640, 809–819 CrossRef CAS PubMed.
- R. A. Gumerov, A. A. Rudov, W. Richtering, M. Möller and I. I. Potemkin, ACS Appl. Mater. Interfaces, 2017, 9, 31302–31316 CrossRef CAS.
- Z. Sumer and A. Striolo, Phys. Chem. Chem. Phys., 2018, 20, 30514–30524 RSC.
- P. Zhou, J. Hou, Y. Yan, J. Wang and W. Chen, Langmuir, 2019, 35, 8110–8120 CrossRef CAS PubMed.
- A. Khedr and A. Striolo, J. Chem. Theory Comput., 2018, 14, 6460–6471 CrossRef CAS PubMed.
- L. Zhen, K. Liu, D. Huang, X. Ren and R. Li, J. Dispers Sci. Technol., 2016, 37, 941–948 CrossRef CAS.
- D. Nivón-Ramrez, L. I. Reyes-Garca, R. Oviedo-Roa, R. Gómez-Balderas, C. Zuriaga-Monroy and J.-M. Martnez-Magadán, Colloids Surf., A, 2022, 128867 CrossRef.
- S. J. Gray, M. Walker, R. Hendrikse and M. R. Wilson, Soft Matter, 2023, 19, 3092–3103 RSC.
- R. Mao, M.-T. Lee, A. Vishnyakov and A. V. Neimark, J. Phys. Chem. B, 2015, 119, 11673–11683 CrossRef CAS PubMed.
- R. D. Groot and P. B. Warren, J. Chem. Phys., 1997, 107, 4423–4435 CrossRef CAS.
- C. Yang, X. Chen, H. Qiu, W. Zhuang, Y. Chai and J. Hao, J. Phys. Chem. B, 2006, 110, 21735–21740 CrossRef CAS.
- E. Mayoral, J. Arcos-Casarrubias and A. G. Goicochea, Colloids Surf., A, 2021, 615, 126244 CrossRef CAS.
- J. H. Velázquez, J. Santos-Santos and A. G. Goicochea, Mol. Sys. Design Eng., 2022, 7, 1650–1657 RSC.
- R. D. Groot, J. Chem. Phys., 2003, 118, 11265–11277 CrossRef CAS.
- H. Eslami, M. Khani and F. Muller-Plathe, J. Chem. Theory Comput., 2019, 15, 4197–4207 CrossRef CAS PubMed.
- H. Wang, X. Tang, D. M. Eike, R. G. Larson and P. H. Koenig, Langmuir, 2018, 34, 1564–1573 CrossRef CAS PubMed.
- A. Maiti and S. McGrother, J. Chem. Phys., 2004, 120, 1594–1601 CrossRef CAS.
- C. Hansen, Hansen Solubility Parameters: A User's Handbook, Second Edition, CRC Press, Boca Raton, Florida, USA, 2007 Search PubMed.
- T. Lindvig, M. L. Michelsen and G. M. Kontogeorgis, Fluid Phase Equilib., 2002, 203, 247–260 CrossRef CAS.
- M. Tadros, P. Hu and A. Adamson, J. Colloid Interface Sci., 1974, 49, 184–195 CrossRef CAS.
- C. Yang, T. Dabros, D. Li, J. Czarnecki and J. H. Masliyah, J. Colloid Interface Sci., 2001, 243, 128–135 CrossRef CAS.
- S. Nishimura, H. Tateyama, K. Tsunematsu and K. Jinnai, J. Colloid Interface Sci., 1992, 152, 359–367 CrossRef CAS.
- A. P. Thompson, H. M. Aktulga, R. Berger, D. S. Bolintineanu, W. M. Brown, P. S. Crozier, P. J. in't Veld, A. Kohlmeyer, S. G. Moore, T. D. Nguyen, R. Shan, M. J. Stevens, J. Tranchida, C. Trott and S. J. Plimpton, Comput. Phys. Commun., 2022, 271, 108171 CrossRef CAS.
- S. J. Plimpton, The LAMMPS molecular dynamics engine., Sandia National Lab.(SNL-NM), Albuquerque, NM (United States) technical report, 2017.
- R. Kanamoto, Y. Wada and G. Miyajima, J. Am. Oil Chem. Soc., 1981, 58, 1050–1053 CrossRef CAS.
- R. Urano, G. A. Pantelopulos and J. E. Straub, J. Phys. Chem. B, 2019, 123, 2546–2557 CrossRef CAS PubMed.
- G. Shearman, A. Tyler, N. Brooks, R. Templer, O. Ces, R. Law and J. Seddon, Liq. Cryst., 2010, 37, 679–694 CrossRef CAS.
- A. Tyler, G. Shearman, N. Brooks, H. Delacroix, R. Law, R. Templer, O. Ces and J. Seddon, Phys. Chem. Chem. Phys., 2011, 13, 3033–3038 RSC.
- S.-H. Tung, Y.-E. Huang and S. R. Raghavan, Langmuir, 2007, 23, 372–376 CrossRef CAS PubMed.
- L. S. K. Dassanayake, D. R. Kodali and S. Ueno, Curr. Opin. Colloid Interface Sci., 2011, 16, 432–439 CrossRef CAS.
- L. Chiappisi, L. Noirez and M. Gradzielski, J. Colloid Interface Sci., 2016, 473, 52–59 CrossRef CAS.
- Y. Ikushima, N. Saito and M. Arai, J. Colloid Interface Sci., 1997, 186, 254–263 CrossRef CAS PubMed.
- M. Wong, J. Thomas and M. Grätzel, J. Am. Chem. Soc., 1976, 98, 2391–2397 CrossRef CAS.
- A. D'Aprano, A. Lizzio, V. T. Liveri, F. Aliotta, C. Vasi and P. Migliardo, J. Phys. Chem., 1988, 92, 4436–4439 CrossRef.
- P. Batys, Y. Zhang, J. L. Lutkenhaus and M. Sammalkorpi, Macromolecules, 2018, 51, 8268–8277 CrossRef CAS.
- P. Batys, S. Kivistö, S. M. Lalwani, J. L. Lutkenhaus and M. Sammalkorpi, Soft Matter, 2019, 15, 7823–7831 RSC.
- C. I. Eneh, M. J. Bolen, P. C. Suarez-Martinez, A. L. Bachmann, T. J. Zimudzi, M. A. Hickner, P. Batys, M. Sammalkorpi and J. L. Lutkenhaus, Soft Matter, 2020, 16, 2291–2300 RSC.
- A. Striolo, A. A. Chialvo, P. T. Cummings and K. E. Gubbins, J. Chem. Phys., 2006, 124, 074710 CrossRef PubMed.
- T. K. Jain, M. Varshney and A. Maitra, J. Phys. Chem., 1989, 93, 7409–7416 CrossRef CAS.
- M. Sammalkorpi, A. Z. Panagiotopoulos and M. Haataja, J. Phys. Chem. B, 2008, 112, 12954–12961 CrossRef CAS PubMed.
- M. Suttipong, B. P. Grady and A. Striolo, Phys. Chem. Chem. Phys., 2014, 16, 16388–16398 RSC.
- M. Suttipong, B. P. Grady and A. Striolo, J. Phys. Chem. B, 2015, 119, 5467–5474 CrossRef CAS PubMed.
- M. Suttipong, B. P. Grady and A. Striolo, Soft Matter, 2017, 13, 862–874 RSC.
- R. Atkin, V. S. Craig and S. Biggs, Langmuir, 2000, 16, 9374–9380 CrossRef CAS.
- G. Despert and J. Oberdisse, Langmuir, 2003, 19, 7604–7610 CrossRef CAS.
- M. A. Yeskie and J. H. Harwell, J. Phys. Chem., 1988, 92, 2346–2352 CrossRef CAS.
- P. Somasundaran, T. W. Healy and D. Fuerstenau, J. Phys. Chem., 1964, 68, 3562–3566 CrossRef CAS.
- B.-Y. Zhu and T. Gu, Adv. Colloid Interface Sci., 1991, 37, 1–32 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Additional DPD simulations details, DPD parametrization based on atomistic MD simulations cohesion energy calculations, atomistic MD and DPD simulations RDF matching for binary mixtures, and density profiles for DPPC adsorption at planar surfaces. See DOI: https://doi.org/10.1039/d3sm00508a |
| This journal is © The Royal Society of Chemistry 2023 |