
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceQuantifying DNA-mediated liposome fusion kinetics with a fluidic trap†
Rodolphe
Marie
 *,
Martin K.
Rasmussen
and
Jonas N.
Pedersen
*,
Martin K.
Rasmussen
and
Jonas N.
Pedersen
Department of Health Technology, Technical University of Denmark, Ørsteds Plads Build. 345C, 2800 Kongens Lyngby, Denmark. E-mail: rcwm@dtu.dk
First published on 31st March 2023
Abstract
Self-assembly of synthetic lipid vesicles via lipid membrane fusion is a versatile tool for creating biomimetic nano- and micron-sized particles. These so-called liposomes are used in the development of biosensing platforms, design of drug delivery schemes, and for investigating protein-mediated fusion of biological membranes. This work demonstrates DNA-induced liposome fusion in a nanofluidic trap where the reaction occurs in a 15 femtoliter volume at homogeneous mixing. In contrast to current methods for fusion in bulk, we show that the fusion reaction follows second-order kinetics with a fusion rate of (170 ± 30)/(M−1s−1) times the square number of DNA molecules per liposome. The nanofluidic trapping gives a full characterization of the size and charge of the liposomes before and after fusion. The chip-based approach limits the amount of sample (down to 440 vesicles) and can be parallelized for systematic studies in synthetic biology, diagnostics, and drug delivery.
1 Introduction
Biological membrane fusion plays an important role in exocytosis and in certain paths of endocytosis, and it is the basis for cellular trafficking via extracellular vesicles.1 The fusion process can be triggered by certain proteins, where some are the members of the so-called SNARE protein family.2,3 Anchored in separate lipid membranes, these proteins force the membranes into close proximity (docking) that causes the amphiphilic lipid molecules to flip and rearrange the outer and inner leaflets of the lipid membrane (lipid mixing). This results in the formation of a fusion pore that brings fusion to completion (content mixing).1,4Artificial SNARE analogs mimic the SNARE complex mode of action by pulling two lipid membranes together. This has lead to the development of a number of artificial systems based on the direct interaction of the lipid membranes,5 covalent binding of boronic acid with diols,6 coiled-coil α-peptides,7,8 nucleic acids,9 and peptide nucleic acids.10 In particular, nucleic acids allow for systematic investigations of the membrane fusion kinetics through modifications of the base-pair sequence, the environment, and the anchoring of DNA strands to the lipid membranes.11 Recently, DNA-mediated fusion has also emerged as an engineering tool, where encoding DNA sequences give programmed sequential fusion in cascades.12 This provides a route to engineered exosomes with potential applications in diagnostics, drug delivery and synthetic biology.13
Despite the development of single-vesicle imaging assays with vesicles tethered to a surface,1,14 most experiments are still done in bulk as tethering may affect the fusion process. In bulk experiments, two homogeneous sub-populations of synthetic vesicles equipped with complementary biomolecules are prepared at high concentrations and characterized prior to fusion in a separate experiment. Then they are mixed to investigate, e.g., the reaction kinetics, the number of vesicles involved in the fusion process, and the nature of the fusion product. That is, if the fusion process stopped at the hemifused state (lipid mixing) where the outer leaflets have merged but the inner leaflets are separate,11 or if the product is a unilamellar vesicle, indicating complete fusion with content mixing.
Fusion is typically monitored with spectroscopy and fluorescence imaging as they provide the necessary sensitivity and time resolution to identify the different fusion steps (docking/lipid mixing/content mixing). Lipid mixing, which can occur without pore formation, is typically detected via fluorescence resonance energy transfer (FRET) assays,7,15,16 while pore formation is verified by loading one of the vesicle with a self-quenching single dye or a FRET pair.7,10,15,16 Most experiments in bulk report a detected signal from the fusion product that do not reach saturation, indicating an incomplete fusion reaction during the time of the experiment.9,11 This complicates the interpretation of the fusion kinetics and the extraction of reaction rates.
We use a nanofluidic trap as a femtoliter reactor to study the fusion of a few hundreds of liposomes (Fig. 1). The trap works by combining a diffusioosmotic flow in a funnel-shaped nanochannel with diffusiophoretic particle transport.17–20 Two samples of liposomes with complementary DNA probes and labelled with either FRET donors or acceptors are introduced sequentially in the nanaochannel and then trapped (Fig. 2). The increase in the detected FRET signal from the trapped liposomes shows successful lipid mixing due to DNA-mediated fusion. The time-dependent intensity fits a mathematical model based on irreversible, second-order kinetics, and the saturation of the intensity signal indicates that the reaction reaches completion. The fit provides the fusion rate [Fig. 3(a)–(e)] and quantifies how the fusion rate depends on the number of DNA strings per liposome [Fig. 3(f)]. A fit to the spatial distribution of the fluorescent acceptor signal in the trap returns the size and zeta potential of both the trapped vesicles prior to fusion and the fusion product (Fig. 4).
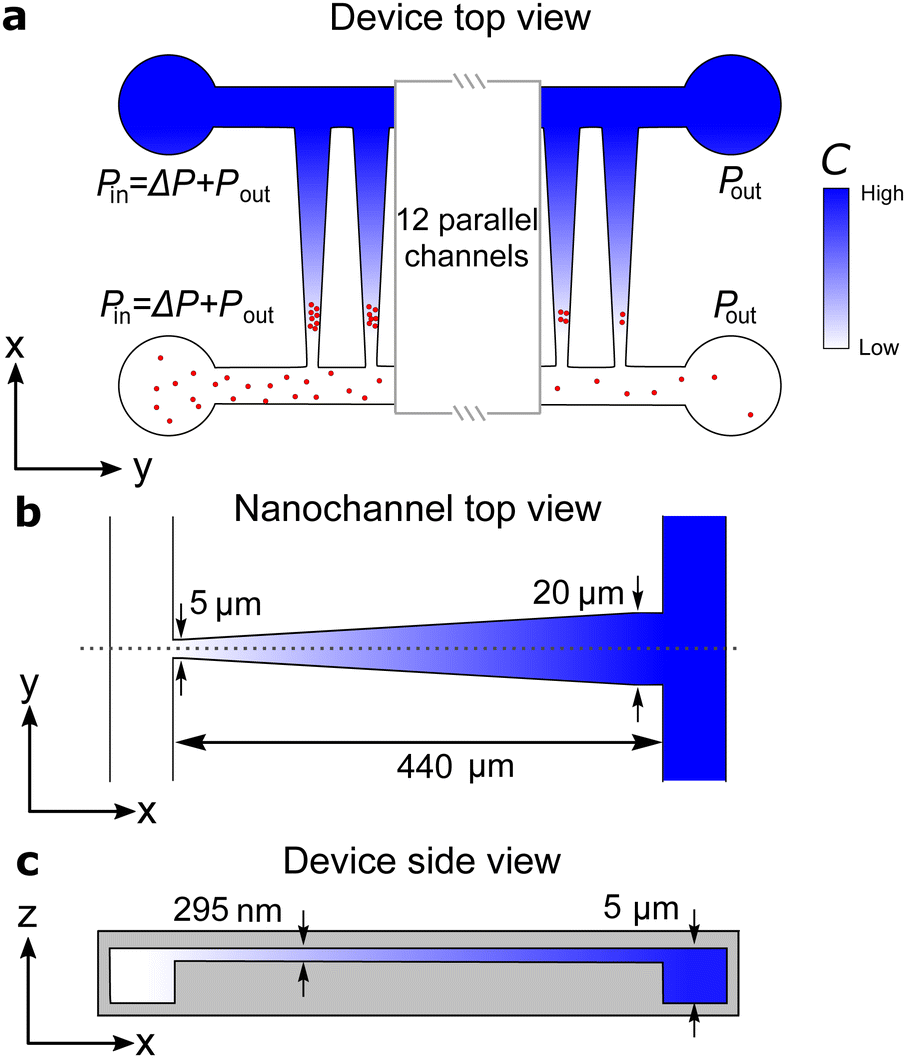 | ||
| Fig. 1 Dimensions of the nanofluidic device. (a) Top view of the device showing the two microchannels connected by an array of 16 funnel-shaped nanochannels. (b) Nanochanels are 440 μm long, 5 μm wide at the narrow end, and 20 μm wide at the other end. The wide end connects to the microchannel with the high salt solution. (c) The depths of the micro- and nanochannels are 5 μm and 295 nm, respectively. | ||
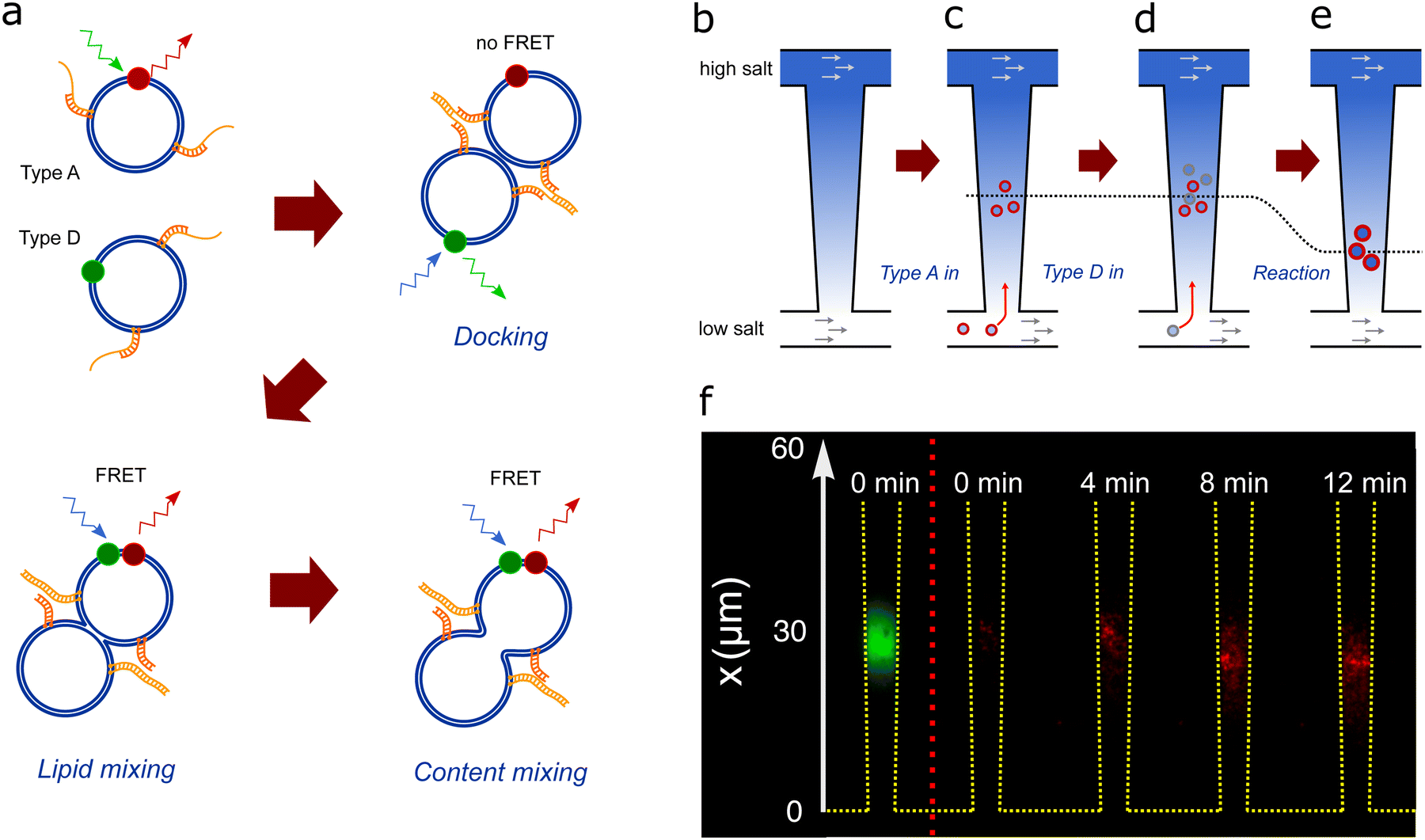 | ||
| Fig. 2 (a) Schematics of DNA-induced fusion of A- and D-liposomes. (b) Schematic of the nanofluidic trap consisting of two microchannels connected by a 440 μm -long funnel-shaped nanochannel, which is 20 μm and 5 μm wide at the narrow and wide ends, respectively (Fig. 1). Flows of solutions with different PBS concentrations in the two microchannels maintain a salinity gradient across the nanochannel. The gradient causes a diffusioosmotic flow inside the nanochannel. (c) A-liposomes are introduced in the lower microchannel and some of them migrate into the nanochannel and get trapped due to diffusiophoresis.22 (d) D-liposomes (grey) are introduced in the lower microchannel and some of these also migrate into the nanochannel, get trapped, and fuse with A-liposomes. (e) The trapping position (dotted line) depends on the size of the liposomes. Consequently, the trapping position shifts after fusion as the fusion products are larger than the original liposomes. (f) Composite microscope images of trapped liposomes. The leftmost image shows the acceptor dye signal from A-liposomes (green) trapped in the nanochannel before D-liposomes are introduced. The other images show the recorded FRET signal (red) after introduction of D-liposomes at time t = 0. Both types of liposomes have 50 DNA probes. | ||
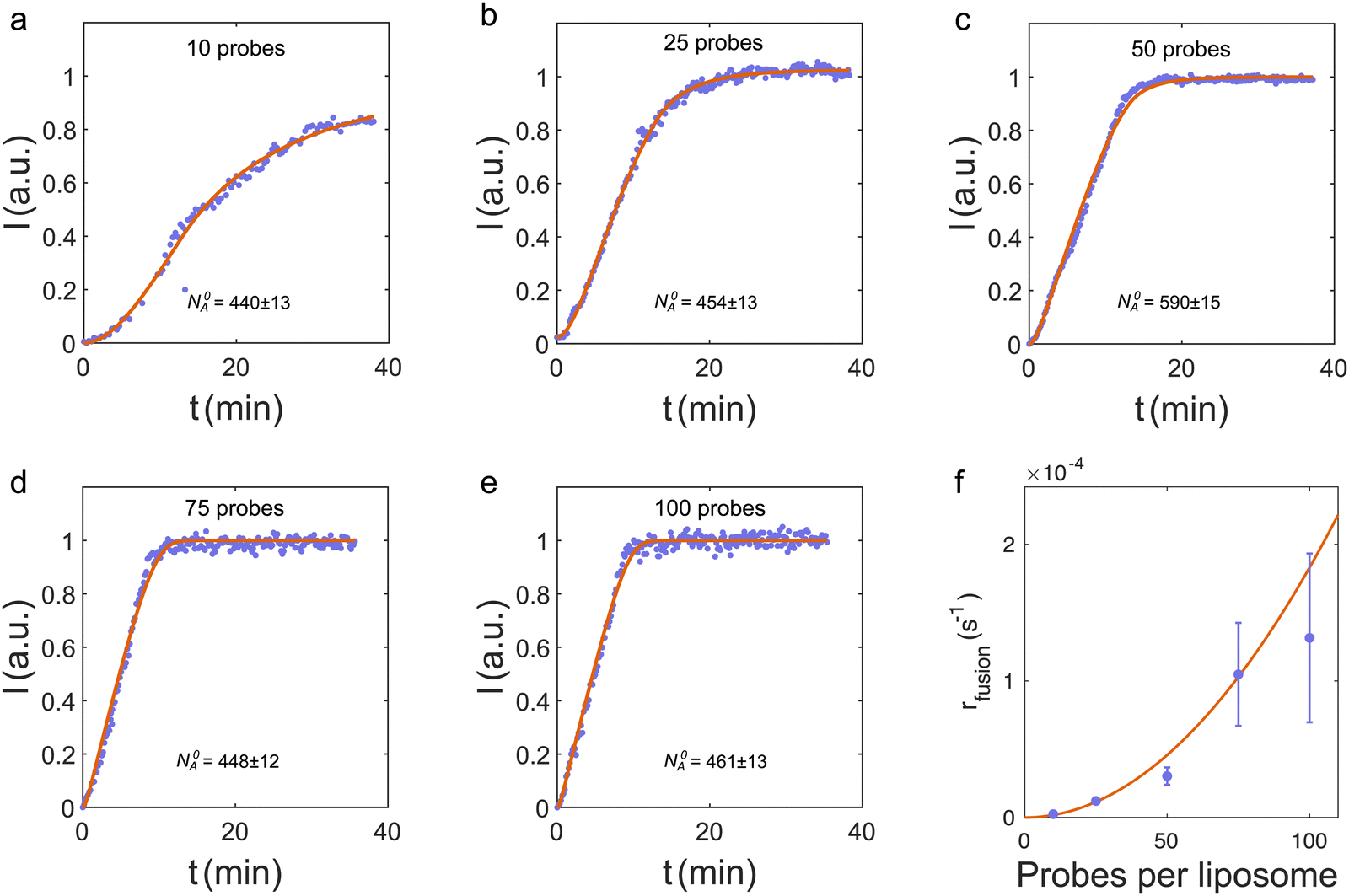 | ||
| Fig. 3 (a–e) Total FRET signal from the trap versus time for liposome fusion in the nanochannel (blue dots) and the corresponding fits to I(t) (red curves) for different number of DNA probes on the liposomes. Data in panel (c) are from the FRET signal in Fig. 2(e). (f) Fusion rates from fits to data in panels (a)–(e) (blue dots). Error bars are standard deviations obtained from simulations, where the input fusion rates are the values from the fits in panels (a)–(e) (see ESI† for details). The red curve is a weighted, least-squares fit to rfusion = kn2, where n is the number of probes on both donors and acceptors. | ||
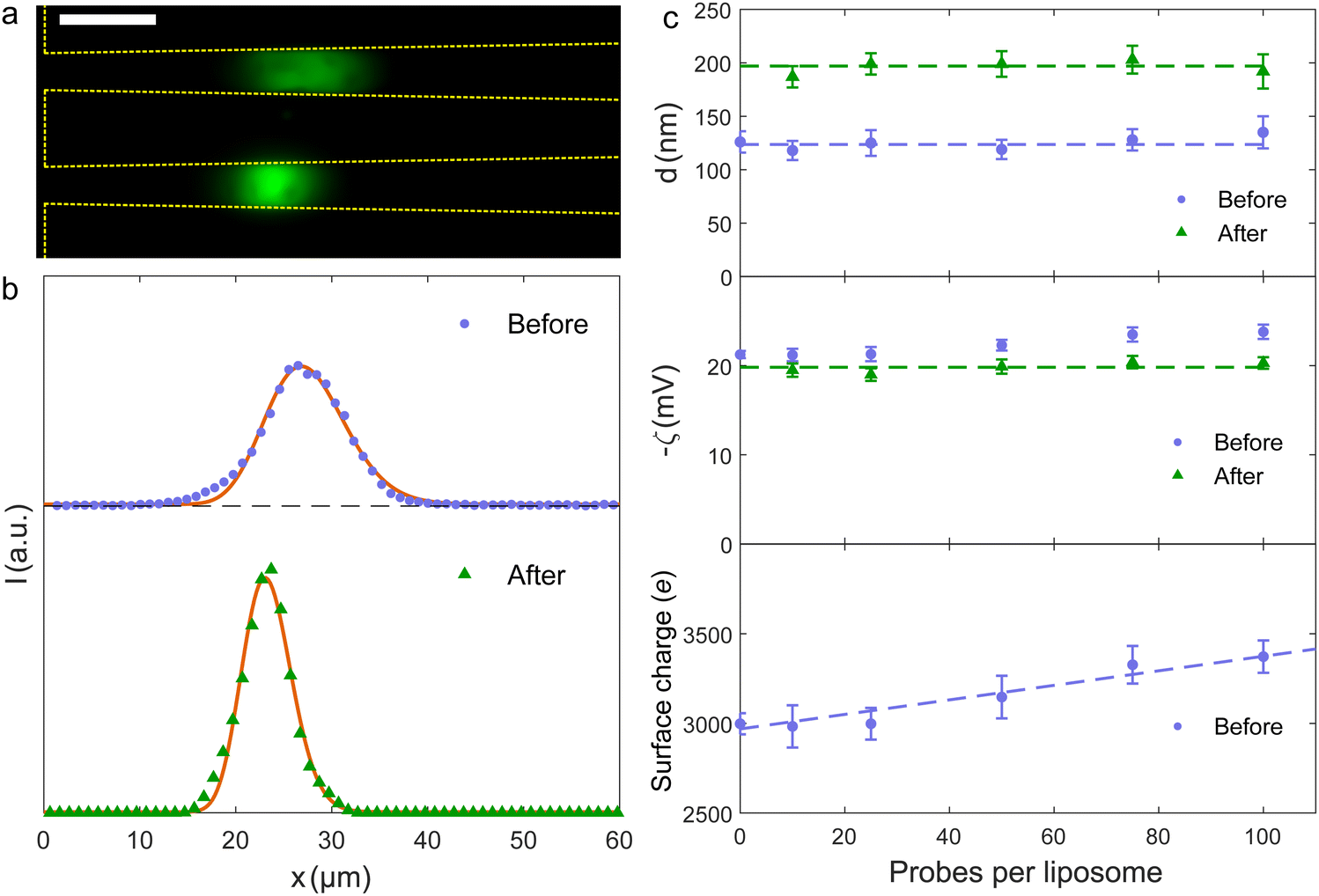 | ||
| Fig. 4 (a) Composite fluorescence microscopy image of liposomes with 50 probes per liposome in the trap before (upper) and after (lower) fusion. The fluorescence signal before fusion is the acceptor dye alone, the signal after is the FRET signal. Scale bar is 10 μm. (b) Fluorescence intensity along the nanochannel before and after liposome fusion for samples with 50 probes per liposome. Full curves are fits to eqn (3). (c) Fitted values of liposome diameters (upper) and zeta potentials (middle) before and after the fusion process. Dashed lines are common average values. The lower panel shows the calculated surface charge on the liposomes prior to fusion. | ||
2 Experimental
2.1 Liposome synthesis
Phospholipids were purchased from Avanti Polar Lipids. Liposomes with DOPC (1,2-dioleoyl-sn-glycero-3-phosphocholine), DOPE (1,2-dioleoyl-sn-glycero-3-phosphoethanolamine) and CH (cholesterol) were prepared in the ratio DOPC![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :DOPE:CH 2:1:1 by lipid film hydration similarly to ref. 9 with few modifications for the fluorescence staining. First, lipids and cholesterol were dissolved in a 9:1 tertiary butanol to H2O solution and mixed with either the dye DiO used as FRET donor (D) or the dye DiI used as FRET acceptor (A) to obtain a DOPC:DOPE:CH 2:1:1 mixture. The liposomes are denoted as A- and D-type, respectively. The mixture was freeze dried for 24 hours, and the resulting lipid film was then rehydrated with PBS and kept at 64 °C while being vortexed every 15 minutes for an hour. Note that because of the way the liposomes are labelled, both leaflets are labelled. DiI and DiO are carbocyanines that do not flip-flop once inserted in the lipid membrane.21 So within the time of the experiment, a given dye molecule stays either in the outer or inner leaflet. The samples were finally extruded through polycarbonate membranes with 100 nm holes to create liposomes with a mean diameter of dDLS = (118 ± 5) nm, measured by Dynamic Light Scattering (DLS). Liposomes were functionalized with DNA molecules on the same day.
:DOPE:CH 2:1:1 by lipid film hydration similarly to ref. 9 with few modifications for the fluorescence staining. First, lipids and cholesterol were dissolved in a 9:1 tertiary butanol to H2O solution and mixed with either the dye DiO used as FRET donor (D) or the dye DiI used as FRET acceptor (A) to obtain a DOPC:DOPE:CH 2:1:1 mixture. The liposomes are denoted as A- and D-type, respectively. The mixture was freeze dried for 24 hours, and the resulting lipid film was then rehydrated with PBS and kept at 64 °C while being vortexed every 15 minutes for an hour. Note that because of the way the liposomes are labelled, both leaflets are labelled. DiI and DiO are carbocyanines that do not flip-flop once inserted in the lipid membrane.21 So within the time of the experiment, a given dye molecule stays either in the outer or inner leaflet. The samples were finally extruded through polycarbonate membranes with 100 nm holes to create liposomes with a mean diameter of dDLS = (118 ± 5) nm, measured by Dynamic Light Scattering (DLS). Liposomes were functionalized with DNA molecules on the same day.
2.2 DNA hybridization and immobilization on liposomes
Two kinds of double-stranded DNA probes were made according to ref. 9. Probes were hybridized by mixing two single-stranded DNA molecules with 12 and 27 base pairs, giving a single-stranded overhang of 15 base pairs at one end and two cholesterol anchors at the opposite end. Probe A was hybridized from the single strings 3′-CH-AGGCAGCACGGA-5′ and 5′-CH-TCCGTCGTGCCTTATTTCTGATGTCCA-3′, and probe D was hybridized from the single strings 5′-CH-TCCGTCGTGCCT-3′ and 3′-CH-AGGCAGCACGGAATAAAGACTACAGGT-5′ (purchased from Biomers). The mixtures were then incubated at 30 °C for an hour.The A- and D-type DNA probes were mixed with the corresponding liposomes in the ratios 10, 25, 50, 75 and 100 DNA probes per liposome. The probes were immobilized on the liposome surfaces by simple mixing. Due to the double cholesterol anchors, we expect that all probes bind to liposomes.9 The DNA-modified liposomes were kept at 4 °C and used within a day of their preparation.
2.3 FRET assay
The excitation and emission spectra for both the donor and acceptor fluorophores are shown in Fig. S2 (ESI†) together with the fluorescence filter cube arrangements used for imaging. Fluorescence imaging is performed with a LED light source (CoolLED pe-300 ultra SB) and two different filter cubes. In both cases, the acceptor dye emission is imaged using an emission filter at 605/55 nm. Using the first cube, the acceptor dye is excited directly using a green LED through a 540/25 nm filter and a 565 nm dichroic mirror. With this setup, both the A-liposomes prior to fusion and the fusion products are localized in the trap. The recorded emission signal is shown in Fig. 2(f) and 4(a). Using the second cube, the acceptor dye is excited through FRET by exciting the donor dye with a 450 nm LED through a 470/28 nm filter (blue excitation). Here the dichroic mirror cut-off wavelength is 505 nm. The FRET intensity is corrected by first subtracting the cross-talk signal from the A-liposomes that are trapped before introducing D-liposomes [Fig. 2(f) at t = 0], then subtracting the cross-talk signal from the introduced D-liposomes. The latter is measured in a negative control experiment where none of the liposomes carried DNA-probes (Fig. S4, ESI†). No detectable increase of FRET signal was observed, indicating no membrane fusion.2.4 Nanofluidic device fabrication
The nanofluidic device consists of 16 parallel funnel-shaped nanochannels bridging two microchannels [Fig. 1(a)].22 Each nanochannel constitutes a nanofluidic trap with a height of 295 nm (so it easily accommodates the ∼120 nm-sized liposomes), a length L = 440 μm, and with widths at the narrow and wide ends wN = 5 μm and wW = 20 μm, respectively. The height of the microchannels are 5 μm. The nanofluidic device was injection molded from cyclic olefin polymer (TOPAS 5013) using a nickel master produced in a two-step UV-lithography and reactive ion etching process.23 The injection molded part was sealed with a 150 μm thick COC foil (TOPAS 5013) by UV-assisted thermal bonding using a mercury lamp (Dymax) and a manual hydraulic press (PO Weber).22The inner surfaces of the nanofluidic device were passivated by coating for 30 minutes with a 3:1 mixture of 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) and 1-palmitoyl-2-oleoyl-sn-glycero-3-[phospho-rac-(1-glycerol)] (POPG) dissolved in 70% ethanol.
2.5 Nanofluidic trapping and imaging
The salinity gradient used in the experiments was maintained by a continuous flow of PBS in the microchannels driven by a 5 mbar pressure difference using a pressure controller (Fluigent, MFCS-EX) that also ensures that there is no significant pressure drop across the nanochannel.22 The PBS concentrations in the microchannels at the narrow and wide ends of the nanochannels are CN = 0.01× PBS and CW = 10× PBS, respectively. Inside the nanochannel, the PBS concentration is given by eqn (S9) (ESI†). At the trapping position x = 20–35 μm [see Fig. 2(f) and 4(b)], the concentration is in the range of 0.9–1.6× PBS. So trapping and vesicle fusion occur close to physiological conditions (∼1× PBS). A-liposomes are introduced in the CN = 0.01 × PBS solution and some are trapped in the nanochannel. The supply of liposomes is stopped when ∼450 liposomes are trapped in the nanochannel by changing to a pure CN = 0.01× PBS solution. D-liposomes are then introduced while recording the FRET signal. The change of buffers alters the trapping position temporarily due to pressure fluctuations, but it does not influence the quantitative results.Liposomes are imaged using a 20×/0.50 objective on an inverted epifluorescence microscope (Nikon, Eclipse Ti2) equipped with an electron-multiplying charge-coupled device camera (Photometrics, Evolve 512). Images are averaged over 10 s to reduce noise.
3 Results & discussion
3.1 Trapping and liposome fusion
Continuous flows of PBS solutions with different concentrations in the two microchannels maintain a salt gradient across the nanochannels [Fig. 2(b)]. This causes a diffusioosmotic flow from high to low salt concentration inside the nanochannels, i.e. from the wide to the narrow end.18,22 Notice that there is no significant pressure-driven flow inside the nanochannels as there is no pressure drop across them.18,22The A-liposomes are introduced in the microchannel at the narrow end of the nanochannels, and some migrate from the microchannel into the nanochannels due to diffusiophoresis caused by the salt gradient.22,24 They are trapped inside the nanochannel where the diffusioosmotic and diffusiophoretic velocities are equal [Fig. 2(c)].19,20,22 The A-liposomes are removed from the microchannel when a few hundreds of liposomes are trapped. The trapped liposomes are imaged by a direct excitation of the acceptor dye [Fig. 2(f), left panel]. The number of trapped A-liposomes, N0A, is measured before introducing the D-liposomes by dividing the total fluorescence intensity in the trap by the intensity of a single liposome (separate experiment). No FRET signal is observed with only A-liposomes in the trap [Fig. 2(f), t = 0].
Fusion starts when D-liposomes are introduced in the microchannel from time t = 0 to t = Δtadd,D [Fig. 2(d)]. Some of the D-liposomes enter the nanochannels, get trapped, and dock with A-liposomes due to the complementary ssDNA overhangs [Fig. 2(e)]. After docking, fusion occurs and causes lipid mixing [Fig. 2(e)]. The donor and acceptor dyes diffuse in the membrane, get in close proximity, and cause a detectable FRET signal [Fig. 2(f), t > 0].
Fig. 3(a)–(e) shows the total FRET intensity from the trap versus time for different number of probes per liposome, but with an equal number of probes on both types of liposomes. For more than 25 probes per liposome, the FRET signal saturates within the ∼35 min measurement time [Fig. 3(b)–(e)]. Hence all the initially trapped A-liposomes fuse with a D-liposome. This is consistent with the observation that a surplus of D-liposomes are added to the trap (see below).
3.2 Reaction kinetics and modeling of the FRET signal
At the experimental conditions, approximately one D-liposome is added per second to a population of trapped A-liposomes. As the two types of liposomes have identical sizes and zeta potentials, they are trapped at the same position and within the same volume. Both types of liposomes diffuse in the trap, so we assume that fusion occurs in a fixed volume under well-mixed conditions. We model the fusion reaction as a second-order irreversible reaction, A + D → rfusionAD with the dynamics described by the coupled kinetic equations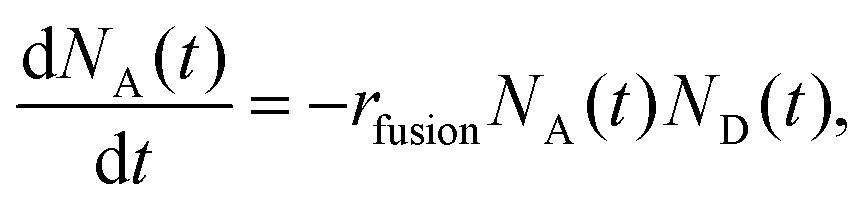 | (1) |
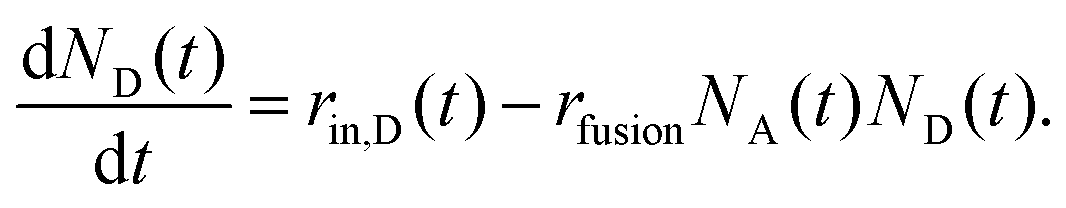 | (2) |
In the experiments, D-liposomes are only present in the microchannel for a finite time interval Δtadd,D = 14 min. So it is assumed that rin,D(t) = rin,D for 0 < t < Δtadd,D, and zero otherwise. Here rin,D = (0.9 ± 0.1) s−1 and the initial value of N0A is in the range of 400 to 600 (measured in separate experiments). The total number of D-liposomes added to the trap is ND,total = Δtadd,Drin,D ∼ 750, larger than the initial number of A-liposomes N0A.
The coupled equations, eqn (1) and (2), are solved numerically and fitted to the experimental FRET intensities with I(t) = I0NAD(t), where the scale factor I0 relates the FRET intensity to the concentration of fused liposomes. So, the only free parameters in the fit are the fusion rate rfusion and the scale factor I0. The full, red curves in Fig. 3(a)–(d) are fits to the recorded FRET signal, and the fitted values of rfusionversus the number of probes per liposome are shown in Fig. 3(e). Error bars are standard deviations obtained from simulations (ESI†). The simulations also show that the standard deviations can be significantly reduced by increasing N0A (ESI†).
Data suggest a quadratic relation between the fusion rate and the number of probes per liposome, similar to the docking probability of tethered vesicles with up to 50 DNA molecules per liposome.25 So the fusion rate is modeled as rfusion = kn2, where n is the number of probes per liposome on both the acceptors and the donors, and k is a constant. A weighted fit to the fusion rates in Fig. 3(f) gives k = (1.8 ± 0.3) × 10−8 s−1.
The reaction rate is converted to molar units by using that approximately 500 A-liposomes (N0A = 440–590) are trapped before fusion [ Fig. 3(a)–(e)]. The trapping volume is estimated as Vtrap = hw(x0)Ltrap, with the channel height h = 295 nm, and the width of the channel at the trapping position x0, w(x0) = 5.9 μm. The length of the trap is Ltrap = 8.7 μm, i.e., approximately the full width at half maximum of the distribution in the upper panel in Fig. 4(b) (see the ESI† for details). This gives a trapping volume Vtrap = 15 fL and an initial concentration ∼55 nM. So the fitted fusion rate is rMfusion = kMn2 in molar units with kM = kVtrapNAvogadro = (170 ± 30) M−1 s−1. With 10 to 100 probes per liposomes, the fusion rate rMfusion is in the range of 0.017 to 1.7 μm−1 s−1.
Previous experiments in bulk with similar double-anchored-DNA-coated liposomes did not show fusion results consistent with second-order kinetics, but an increase in FRET intensity without saturation.9,11 The cause of the qualitative difference between bulk experiments and the present trapping experiments is unclear, but with the fitted value for rMfusion we can estimate the time it takes to fuse 50% of the vesicles in bulk experiments.11 These show a time-scale of ∼2.5 minutes,11 in reasonable agreement with the estimated ∼3 min based on rMfusion with n = 100 probes per liposome (ESI†)
3.3 Fusion detection from the trapping profile
The spatial distribution of the trapped liposomes is detected from a direct excitation of the acceptor dye. The change in the distribution during the experiment reflects the fusion reaction, as the distribution depends on the size and charge of the liposomes.22 The upper panel in Fig. 4(a) shows the fluorescence emission intensity of A-liposomes with 50 DNA molecules per liposome before D-liposomes are introduced (t = 0). The lower panel shows the distribution after D-liposomes are introduced and fusion has occurred (t = 40 min).From these two images, the fluorescence intensity profile along the nanochannel before and after liposome fusion is extracted [Fig. 4(b)]. Fusion shifts the trapping position towards the narrower end of the nanochannel and decreases the width of the intensity profile. The decrease in width after the reaction clearly indicates that no unfused A-liposomes exists after 40 minutes because unfused liposomes would still be trapped at their initial position. This also agrees with Fig. 3(b)–(e), where saturation of the FRET signal indicates a completion of the fusion reaction, and with the observation that an excess of D-liposomes are added to the trap, ND,total > N0A.
The intensity profile is fitted with a mathematical expression for the concentration of liposomes in the nanochannel22
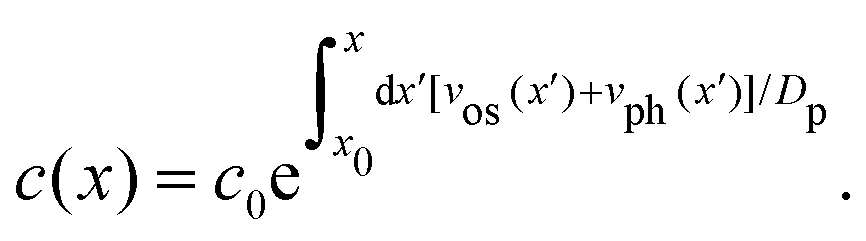 | (3) |
The fit of the fluorescence data in Fig. 4(b) to Ifluor(x) gives that the liposome size and zeta potential are dbefore = (128 ± 10) nm and ζbefore = (−22.3 ± 0.6) mV before fusion, and dafter = (207 ± 13) nm and ζafter = (−19.9 ± 0.8) mV after fusion. The results for samples with different numbers of DNA probes are summarized in Fig. 4(c). The fitted diameters before fusion are consistent with a common average diameter ![[d with combining macron]](https://www.rsc.org/images/entities/i_char_0064_0304.gif) before = (124 ± 4) nm (p = 0.91), in agreement with Dynamic Light Scattering (DLS) measurements (Section 2.1). After fusion, the common average value has increased to after = (197 ± 5) nm (p = 0.34).
before = (124 ± 4) nm (p = 0.91), in agreement with Dynamic Light Scattering (DLS) measurements (Section 2.1). After fusion, the common average value has increased to after = (197 ± 5) nm (p = 0.34).
Liposomes are assumed spherical before fusion as they are produced with extrusion.26 So the surface area and volume of an average liposome are Abefore = π2before and Vbefore = π3before/6, respectively. Pairwise fusion doubles the surface area, Aafter = 2Abefore, while different scenario exist for the volume. If fluid enters the lumen of the vesicles during fusion, the fusion product can eventually become spherical with a diameter 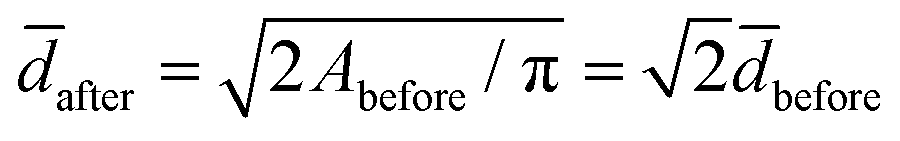 and a volume
and a volume 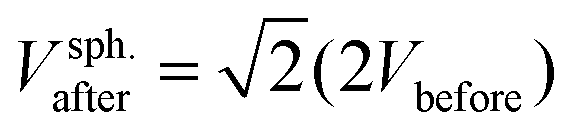 . If volume is conserved, the fusion product has a volume
. If volume is conserved, the fusion product has a volume 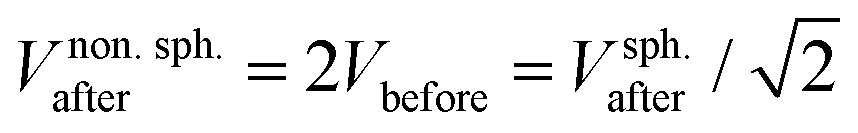 , and a nonspherical, elongated form.27
, and a nonspherical, elongated form.27
The experimental value for the average diameter of the fusion product is (13 ± 4)% larger than for a sphere with twice the initial surface area surface area, 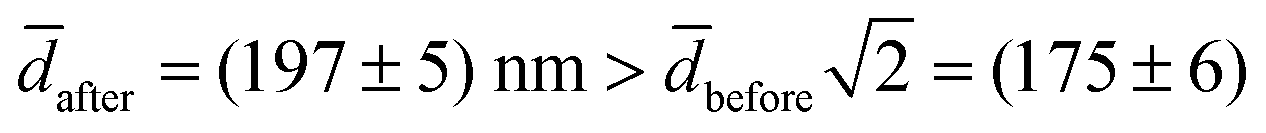 nm. We speculate that this could indicate a volume-conserving fusion process with a nonspherical fusion product. If the volume of the fusion product is conserved,
nm. We speculate that this could indicate a volume-conserving fusion process with a nonspherical fusion product. If the volume of the fusion product is conserved, 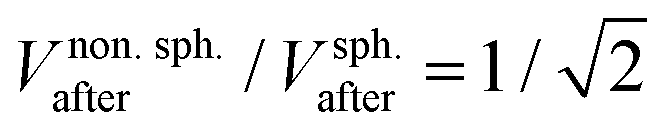 , the spontaneous-curvature model for zero spontaneous curvature predicts a prolate equilibrium shape, which we approximate with an ellipsoid with identical surface area and volume (see Fig. S3, ESI†).27 For an ellipsoid and a sphere with the same surface area, but where the ellipsoid has a volume which is
, the spontaneous-curvature model for zero spontaneous curvature predicts a prolate equilibrium shape, which we approximate with an ellipsoid with identical surface area and volume (see Fig. S3, ESI†).27 For an ellipsoid and a sphere with the same surface area, but where the ellipsoid has a volume which is 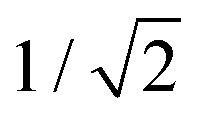 times the volume of the sphere, the ellipsoid's hydrodynamic radius is in bulk 4% larger than the sphere's.28 Confinement in a nanochannel could potentially enhance this difference due to the hydrodynamic effects of the nearby walls (ESI†).29 The present experimental data can, however, not rule out three-vesicle fusion as a spherical fusion product in this case would have an expected diameter of
times the volume of the sphere, the ellipsoid's hydrodynamic radius is in bulk 4% larger than the sphere's.28 Confinement in a nanochannel could potentially enhance this difference due to the hydrodynamic effects of the nearby walls (ESI†).29 The present experimental data can, however, not rule out three-vesicle fusion as a spherical fusion product in this case would have an expected diameter of 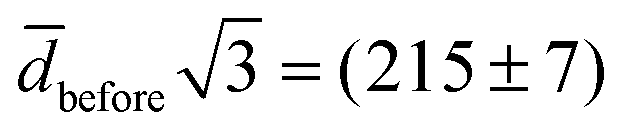 nm, comparable to after. Three-vesicle fusion is, however, unlikely on the time scale of the experiment.25
nm, comparable to after. Three-vesicle fusion is, however, unlikely on the time scale of the experiment.25
The fitted values for the zeta potential before fusion depend on the number of negatively charged DNA probes on the vesicles [Fig. 4(c), middle panel]. Using eqn (2.5.5) in ref. 30, the zeta potentials for the different number of DNA probes are converted to surface charges with before as the vesicle diameter [Fig. 4(c), lower panel]. A linear fit gives that each DNA probe adds a charge of (4.0 ± 0.7)e (p = 0.93). The zeta potentials after fusion are consistent with a common average value ![[small zeta, Greek, macron]](https://www.rsc.org/images/entities/i_char_e0c7.gif) after = (−19.8 ± 0.3) mV (p = 0.59). If charges were preserved during fusion, the surface charge density and, consequently, the zeta potential would, except for a weak size dependence, remain unchanged since both types of liposomes have the same number of DNA molecules. In contrast, Fig. 4(c) (middle panel) clearly shows that fusion alters the zeta potentials. The change increases with the number of DNA molecules. The origin of this change in zeta potential is unclear, but experiments show that some of the negatively charged DNA probes are ripped off during membrane mixing and that changes the surface charge of the fusion product (ESI†).
after = (−19.8 ± 0.3) mV (p = 0.59). If charges were preserved during fusion, the surface charge density and, consequently, the zeta potential would, except for a weak size dependence, remain unchanged since both types of liposomes have the same number of DNA molecules. In contrast, Fig. 4(c) (middle panel) clearly shows that fusion alters the zeta potentials. The change increases with the number of DNA molecules. The origin of this change in zeta potential is unclear, but experiments show that some of the negatively charged DNA probes are ripped off during membrane mixing and that changes the surface charge of the fusion product (ESI†).
3.4 Perspectives
In the present experiments, the fluorescence of the acceptor dye is only measured before and after membrane fusion to detect a fusion-induced size change (Fig. 4). Monitoring the spatial distribution during the entire experiment would provide real-time detection of the fusion reaction and a view on the fusion process that is complementary to the FRET detection scheme (Fig. 2). A bimodal liposome distribution is expected at intermediate time steps when liposome fusion gradually shifts the trapping position.While the present work focuses on the overall fusion kinetics through the detection of lipid mixing with a FRET assay, a possible extension is to add a fluorescence-based content-mixing assay.7,10,15,16 Here a fluorescent dye is added to the lumen of the liposomes, but the dye only gives a signal after fusion with content mixing. One of the drawbacks of such content-mixing assays when performed in bulk is that leakage creates false positives.31 In the nanofluidic trap, content escaping the vesicles would rapidly diffuse away from the trap and not contribute to the fluorescence signal.
Current approaches in bulk for the detection of lipid membrane fusion are not easily implemented for biological membranes, which has driven the development of single-particle assays with vesicles tethered to a surface for physical32–34 or chemical characterization.35 These assays monitor individual fusion events and can thus detect inhomogeneities in the fusion process. A similar analysis can be performed in the diffusiophoretic trap with only a few vesicles present and with the additional advantage of continuously monitoring the same particles without immobilization. The nanofluidic chip also allows handling of small volumes of dilute sample and controlling the rate at which reactants are introduced.36 The kinetics of other DNA-coated nanoparticles systems could also be characterized in the trap, e.g., crystallization of DNA-coated colloids.37–39
4 Conclusions
A nanofluidic device and a four-step protocol were used to collect a small number of liposomes in a nanofluidic trap, characterize the liposomes before fusion, fuse them with other liposomes, and characterize the size and charge of the fusion product. The method provides accurate measurements of the DNA-mediated lipid membrane fusion kinetics by combining nanofluidic trapping with a FRET-based assay for the detection of lipid mixing. In contrast to similar experiments performed in bulk, the fusion reaction is well-described by second-order kinetics with a reaction rate that depends quadratically on the number of DNA-probes covering the liposomes. As fusion occurs in a well-defined volume at homogeneous mixing, it is simpler to extract the fusion rates than for vesicles tethered to a surface, where the vesicle distribution is inhomogeneous across the surface and changes during the experiment.25The chip-based platform could also prove useful for studying self-assembly reactions where samples are sparse, e.g., with biological particles isolated from patients. The trapping increases the concentration and hence the reaction rate. Parts of or all the preliminary purification steps used, e.g., for exosomes, would also be circumvented. Combined with the chip format this has potential applications in the development of engineered exosomes for drug delivery schemes, where exosomes are fused with liposomes.13,40
Author contributions
Rodolphe Marie: conceptualization, methodology, formal analysis, writing – original draft, writing – review & editing, visualization, supervision. Martin K. Rasmussen: conceptualization, methodology, software, formal analysis, investigation, writing – original draft, visualization. Jonas N. Pedersen: conceptualization, software, formal analysis, writing – original draft, writing – review & editing, supervision.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Research reported in this publication is supported by the Novo Nordisk Foundation New Exploratory Research and Discovery programme, under grant number NNF21OC0068622.Notes and references
- H. Pick, A. C. Alves and H. Vogel, Chem. Rev., 2018, 118, 8598–8654 CrossRef CAS PubMed.
- R. Jahn, T. Lang and T. C. Südhof, Cell, 2003, 112, 519–533 CrossRef CAS PubMed.
- Y. A. Chen and R. H. Scheller, Nat. Rev. Mol. Cell Biol., 2001, 2, 98–106 CrossRef CAS PubMed.
- Y.-H. M. Chan, B. van Lengerich and S. G. Boxer, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 979–984 CrossRef CAS PubMed.
- S. M. Christensen, P. Y. Bolinger, N. S. Hatzakis, M. W. Mortensen and D. Stamou, Nat. Nanotechnol., 2012, 7, 51–55 CrossRef CAS PubMed.
- A. Kashiwada, M. Tsuboi and K. Matsuda, Chem. Commun., 2009, 695–697 RSC.
- Y. Gong, Y. Luo and D. Bong, J. Am. Chem. Soc., 2006, 128, 14430–14431 CrossRef CAS PubMed.
- N. L. Mora, A. L. Boyle, B. J. V. Kolck, A. Rossen, Š. Pokorná, A. Koukalová, R. Šachl, M. Hof and A. Kros, Sci. Rep., 2020, 10, 3087 CrossRef CAS PubMed.
- G. Stengel, R. Zahn and F. Höök, J. Am. Chem. Soc., 2007, 129, 9584–9585 CrossRef CAS PubMed.
- M. Sadek, D. Berndt, D. Milovanovic, R. Jahn and U. Diederichsen, ChemBioChem, 2016, 17, 479–485 CrossRef CAS PubMed.
- G. Stengel, L. Simonsson, R. A. Campbell and F. Hook, J. Phys. Chem. B, 2008, 112, 8264–8274 CrossRef CAS PubMed.
- P. M. Löffler, O. Ries, A. Rabe, A. H. Okholm, R. P. Thomsen, J. Kjems and S. Vogel, Angew. Chem., Int. Ed., 2017, 56, 13228–13231 CrossRef PubMed.
- M. Xie, F. Wang, J. Yang, Y. Guo, F. Ding, X. Lu, Y. Huang, Y. Li, X. Zhu and C. Zhang, Anal. Chem., 2022, 94, 13043–13051 CrossRef CAS PubMed.
- Z. Tian, J. Gong, M. Crowe, M. Lei, D. Li, B. Ji and J. Diao, Prog. Lipid Res., 2019, 73, 92–100 CrossRef CAS PubMed.
- W. Xu, J. Wang, J. E. Rothman and F. Pincet, Angew. Chem., 2015, 127, 14596–14600 CrossRef.
- B. L. Scott, J. S. Van Komen, S. Liu, T. Weber, T. J. Melia and J. A. McNew, Liposome fusion assay to monitor intracellular membrane fusion machines, Methods Enzymol., 2003, 372, 274–300 CAS.
- D. C. Prieve, J. L. Anderson, J. P. Ebel and M. E. Lowell, J. Fluid Mech., 1984, 148, 247–269 CrossRef CAS.
- C. Lee, C. Cottin-Bizonne, A.-L. Biance, P. Joseph, L. Bocquet and C. Ybert, Phys. Rev. Lett., 2014, 112, 244501 CrossRef CAS PubMed.
- S. Shin, J. T. Ault, P. B. Warren and H. A. Stone, Phys. Rev. X, 2017, 7, 041038 Search PubMed.
- S. Shin, Phys. Fluids, 2020, 32, 101302 CrossRef CAS.
- D. Wolf, Biochemistry, 1985, 24, 582–586 CrossRef CAS PubMed.
- M. K. Rasmussen, J. N. Pedersen and R. Marie, Nat. Commun., 2020, 11, 1–8 CrossRef.
- P. Utko, F. Persson, A. Kristensen and N. B. Larsen, Lab Chip, 2011, 11, 303–308 RSC.
- J. L. Anderson, Annu. Rev. Fluid Mech., 1989, 21, 61–99 CrossRef.
- Y.-H. M. Chan, P. Lenz and S. G. Boxer, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 18913–18918 CrossRef CAS PubMed.
- K. I. Mortensen, C. Tassone, N. Ehrlich, T. L. Andresen and H. Flyvbjerg, Nano Lett., 2018, 18, 2844–2851 CrossRef CAS PubMed.
- U. Seifert, K. Berndl and R. Lipowsky, Phys. Rev. A: At., Mol., Opt. Phys., 1991, 44, 1182–1202 CrossRef CAS PubMed.
- J. Pencer, G. F. White and F. R. Hallett, Biophys. J., 2001, 81, 2716–2728 CrossRef CAS PubMed.
- J. Happel and H. Brenner, Low Reynolds Number Hydrodynamics, Prentice Hall, 1965 Search PubMed.
- R. Hunter, Zeta potential in colloid science. Principles and Applications, Academic Press, 1981 Search PubMed.
- M. Kyoung, Y. Zhang, J. Diao, S. Chu and A. T. Brunger, Nat. Protoc., 2013, 8, 1–16 CrossRef CAS PubMed.
- S. Block, B. J. Fast, A. Lundgren, V. P. Zhdanov and F. Höök, Nat. Commun., 2016, 7, 12956 CrossRef CAS.
- S. Jõemetsa, P. Joyce, Q. Lubart, M. Mapar, E. Celauro, B. Agnarsson, S. Block, M. Bally, E. K. Esbjörner, G. D. Jeffries and F. Höök, Langmuir, 2020, 36, 9693–9700 CrossRef PubMed.
- R. Friedrich, S. Block, M. Alizadehheidari, S. Heider, J. Fritzsche, E. K. Esbjörner, F. Westerlund and M. Bally, Lab Chip, 2017, 17, 830–841 RSC.
- J. E. Sorrells, E. M. Martin, E. Aksamitiene, P. Mukherjee, A. Alex, E. J. Chaney, M. Marjanovic and S. A. Boppart, Sci. Rep., 2021, 11, 3308 CrossRef CAS PubMed.
- M. K. Rasmussen, J. N. Pedersen and R. Marie, ACS Sens., 2020, 5, 4057–4063 CrossRef CAS PubMed.
- Y. Wang, Y. Wang, X. Zheng, E. Ducrot, J. S. Yodh, M. Weck and D. J. Pine, Nat. Commun., 2015, 6, 7253 CrossRef CAS PubMed.
- C. A. Mirkin, R. L. Letsinger, R. C. Mucic and J. J. Storhoff, Nature, 1996, 382, 607–609 CrossRef CAS PubMed.
- A. P. Alivisatos, K. P. Johnsson, X. Peng, T. E. Wilson, C. J. Loweth, M. P. Bruchez and P. G. Schultz, Nature, 1996, 382, 609–611 CrossRef CAS PubMed.
- M. Piffoux, A. K. A. Silva, C. Wilhelm, F. Gazeau and D. Tareste, ACS Nano, 2018, 12, 6830–6842 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Theoretical background for the trapping mechanism including a description of the diffusio-osmotic and -phoretic velocities. In addition, experimental results showing the loss of charge during fusion. See DOI: https://doi.org/10.1039/d2sm01658c |
| This journal is © The Royal Society of Chemistry 2023 |