
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Elastoplastic behavior of anisotropic, physically crosslinked hydrogel networks comprising stiff, charged fibrils in an electrolyte†
Rebecca
Östmans‡
 ab,
Maria F.
Cortes Ruiz‡
ab,
Jowan
Rostami
a,
Farhiya Alex
Sellman
ab,
Lars
Wågberg
ab,
Stefan B.
Lindström
c and
Tobias
Benselfelt
*ad
ab,
Maria F.
Cortes Ruiz‡
ab,
Jowan
Rostami
a,
Farhiya Alex
Sellman
ab,
Lars
Wågberg
ab,
Stefan B.
Lindström
c and
Tobias
Benselfelt
*ad
aDepartment of Fibre and Polymer Technology, Division of Fibre Technology, KTH Royal Institute of Technology, 100 44 Stockholm, Sweden. E-mail: bense@kth.se
bWallenberg Wood Science Center, 100 44 Stockholm, Sweden
cFSCN Research Center, Mid Sweden University, 851 70 Sundsvall, Sweden
dSchool of Materials Science and Engineering, Nanyang Technological University, 639798 Singapore, Singapore. E-mail: gustaftobias.b@ntu.edu.sg
First published on 20th March 2023
Abstract
Fibrillar hydrogels are remarkably stiff, low-density networks that can hold vast amounts of water. These hydrogels can easily be made anisotropic by orienting the fibrils using different methods. Unlike the detailed and established descriptions of polymer gels, there is no coherent theoretical framework describing the elastoplastic behavior of fibrillar gels, especially concerning anisotropy. In this work, the swelling pressures of anisotropic fibrillar hydrogels made from cellulose nanofibrils were measured in the direction perpendicular to the fibril alignment. This experimental data was used to develop a model comprising three mechanical elements representing the network and the osmotic pressure due to non-ionic and ionic surface groups on the fibrils. At low solidity, the stiffness of the hydrogels was dominated by the ionic swelling pressure governed by the osmotic ingress of water. Fibrils with different functionality show the influence of aspect ratio, chemical functionality, and the remaining amount of hemicelluloses. This general model describes physically crosslinked hydrogels comprising fibrils with high flexural rigidity – that is, with a persistence length larger than the mesh size. The experimental technique is a framework to study and understand the importance of fibrillar networks for the evolution of multicellular organisms, like plants, and the influence of different components in plant cell walls.
Introduction
Hydrogels are crosslinked networks of polymers or nanoparticles holding considerable amounts of water, typically 70–90%. Biological tissues are hydrogels, and synthetic hydrogels have been successfully used as biocompatible materials for different applications.1–5 Anisotropic hydrogels formed by oriented elongated nanoparticles are a recent trend in the hydrogel community.6–10 They can mimic the extracellular matrix (ECM) of plants and animals to facilitate directed cellular growth and proliferation.9 They also enable the manufacturing of anisotropic shape-morphing of stimuli-responsive hydrogels towards more controlled sensing and actuation.11,12 Their anisotropy leads to anisotropic conduction of electrons, ions, and water in soft electronics to optimize the performance in a preferred direction.4,13,14Fibrillar hydrogels are easily made anisotropic by a bottom-up assembly of water-dispersed fibrils, for example, by vacuum filtration.6,9 Despite the promising properties and demonstrated applications of anisotropic fibrillar hydrogels; there is still no theoretical framework to describe their elastoplastic behavior under stress. Formulating such a framework was the aim of this work.
In the classical theory of polymeric hydrogels, the elastic behavior is described by an entropy elastic Gaussian chain model according to Flory and Rhener,15 while the Flory–Huggins theory describes the thermodynamics of mixing the components and the solvent.16,17 For fibrillar hydrogels, the thermodynamics of mixing can still be adapted, and the elastoplastic behavior can be qualitatively described by enthalpy elasticity – that is, bending or stretching of covalent bonds – and plastic friction.
Fibrillar hydrogels can be formed without crosslinking chemistry by volumetric restrictions known as colloidal glasses.18,19 Physical contact points induced by van der Waals interactions can further provide strength in the form of weak, temporary crosslinks or increased friction, while strong covalent crosslinking leads to a molecularly interconnected network. Regardless of whether the crosslinks are weak or strong, the mechanics of the networks should plausibly fit into one theoretical framework. However, the microstructural complexity of anisotropic nanoparticle networks is challenging to describe, and unlike polymer networks, a variety of models is likely needed to describe networks of particles with different shapes – that is, spheres, rods, or sheets.
Previously, the polymer gel model has been qualitatively used for anisotropic fibrillar networks,20,21 and their in-plane mechanical properties have been described.7 However, a quantitative description of the elastoplasticity of these networks is still incomplete. A proposed model for actin filaments is based on the relation between mesh size and shear modulus.9 This model is unsuitable for colloidal glasses, for which the influence of the charges on the particles considerably impacts the pressure balance. A simple model encompassing all fibrillar hydrogels with few mechanical elements, i.e., springs, dashpots, etc., is desirable.
This work moves toward a general elastoplastic model for anisotropic networks of charged fibrils based on network theory and measured swelling pressures of anisotropic fibrillar hydrogels made from charged cellulose nanofibrils (CNFs) as model fibrils. Note that this theoretical framework was not explicitly designed for cellulose fibrils and should be suitable for any network of stiff, charged fibrils. Hydrogels were prepared from different types of CNFs (Table 1) as model fibrillar hydrogels with different network microstructures to compare the theoretical description with experimental data. By changing the aspect ratio, the surface chemistry of the fibrils, or the ionic strength of the swelling medium, the mechanical behavior of the networks was described under compression perpendicular to the orientation of fibrils in the gel.
| CM-long | CM-short | Cationic-long | Holo | |
|---|---|---|---|---|
| Charge density (mmol g−1) | 0.52 | 0.36 | 0.46 | 0.13 |
| Width, d (nm) | 2.2 (0.7) | 2.6 (0.7) | 2.2 (0.6) | 4.0 (2.0) |
| Length, L (μm) | 0.8 (0.3) | 0.4 (0.2) | 1.0 (0.4) | 3.0 (2.0) |
| Aspect ratio | 360 | 150 | 450 | 750 |
The results show that the elastoplastic behavior of anisotropic hydrogels of stiff, charged fibrils can be described by a mechanical model of two springs, one representing the network and one the osmotic pressure, and one yielding element representing plastic sliding with friction at fibril contacts. For attractive gels, such as actin gels, the osmotic pressure in this model is negligible and can be removed. Conversely, for repulsive gels or glasses at low solidity, the compression stiffness of the gel is almost entirely determined by the ionic swelling pressure. A second-degree polynomial could describe the relationship between the network pressure and the solidity of the gel as it starts to dominate over the osmotic pressure when reaching critical solidity during compression. Different dimensions and chemistry of the fibrils also provided insight into the importance of the fibril characteristics.
This model provides a better understanding and utilization of anisotropic fibrillar hydrogels, such as their mechanical behavior for large deformations, the influence of anisotropy, and how they can be optimized in applications. It also demonstrates the robustness of cellulose networks in wet environments with water as an essential material component,22 providing insight into the evolutionary origin of cellulose as a turgor envelope for aquatic cells or into the basic understanding of the mechanical properties of the plant cell wall.23
Results and discussion
In this work, CNFs were used as model fibrils for experimental inspiration and support for a general theoretical model for anisotropic networks of stiff, charged fibrils. CNFs, consisting of a semicrystalline ordered structure of glucan molecules, are the structural building blocks of trees and plants that form strong and self-supporting hydrogel networks at diminishing concentrations containing as much as 99.99% water. These low-density networks are enabled by the fibrils having a high affinity for water,22 a high stiffness of ∼100 GPa,24 and a high aspect ratio of up to 800.25 These properties result in volume-spanning arrested states, also known as colloidal glasses or gels, if the concentration of these materials increases above a certain threshold or decreases the repulsive interactions between the materials.18 Upon water removal by filtration and subsequent drying, nanocellulose networks self-assemble into layered structures. When placed in water, these networks swell uniaxially, out-of-plane, into anisotropic hydrogels.8,21High and low aspect ratio carboxymethylated CNFs (CM-long and CM-short), high-aspect-ratio cationic CNFs (cationic-long), and CNFs with a high amount of remaining hemicellulose as a hydrophilic coating (Holo) were used to manufacture hydrogels in this work, and the fibril properties are shown in Table 1. The fibrils are easily charged by chemical modification to prepare CNFs with excellent colloidal stability.26,27 This work aimed for a moderate charge density of the fibrils around 0.5 mmol g−1. The charge of the Holo CNFs is determined by the specific chemistry used in the delignification process, which is aimed only at removing lignin and preserving both charged and uncharged hemicellulose.28,29
The swelling of isotropic polymer or fibrillar networks can be described by a balance of applied external pressure (P) and contributing material pressure components according to a classical polyelectrolyte gel model:
| P = Pchem + Pnet = Pmix + Pion + Pnet | (1) |
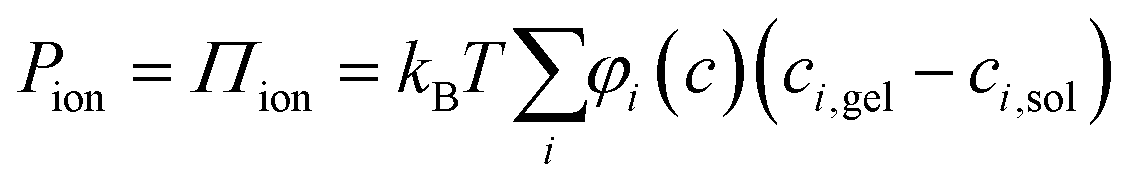 | (2) |
It has been shown that by measuring the dimension of a polymer hydrogel placed in solutions of controlled chemical potential in combination with the measurement of swelling pressure, it is possible separate Pchem and Pnet to better understand the dynamic properties of the hydrogel network.31,32 In the present work, the swelling pressure of anisotropic fibrillar hydrogels was directly measured as a function of solidity and surrounding electrolyte concentration to separate the influence of Pion from Pmix and Pnet. These measurements are simplified because these anisotropic hydrogels swell uniaxially, out-of-plane, under such conditions so that the strain is essentially unidirectional.6 Thus, compressing the hydrogel from its unrestricted equilibrium swelling (Fig. 1a and b), a new restricted equilibrium is reached where the measured normal force represents the swelling pressure at the given solidity.
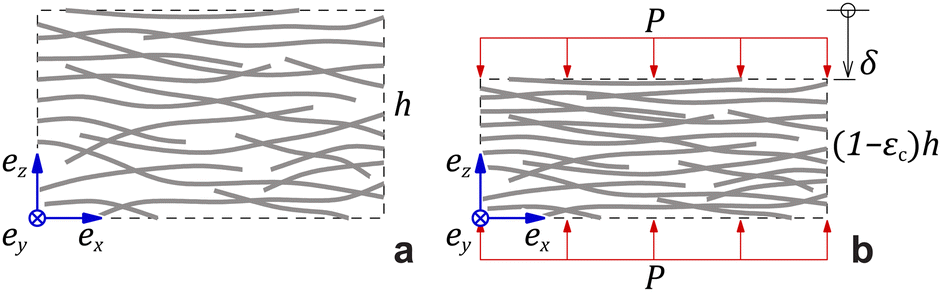 | ||
| Fig. 1 (a) Undeformed geometry with height h. (b) Deformed geometry at an out-of-plane compressive strain εc due to an applied pressure P. | ||
As noted, the anisotropy of the hydrogels results in anisotropic network stress and strain. The fibrillar hydrogel contains an electrolyte medium and a solid network phase of solidity ϕ. The volume-average stress tensor in the hydrogel material is:
| σ = −(Pmix + Pion)I + σnet, | (3) |
| σzz = −P, σxx = σyy = σxy = σxz = σxz ≈ 0. | (4) |
| Pnet = P − Pmix − Pion. | (5) |
| εzz = −εc, εxx = εyy = εIP, εxy = εxz = εyz ≈ 0, | (6) |
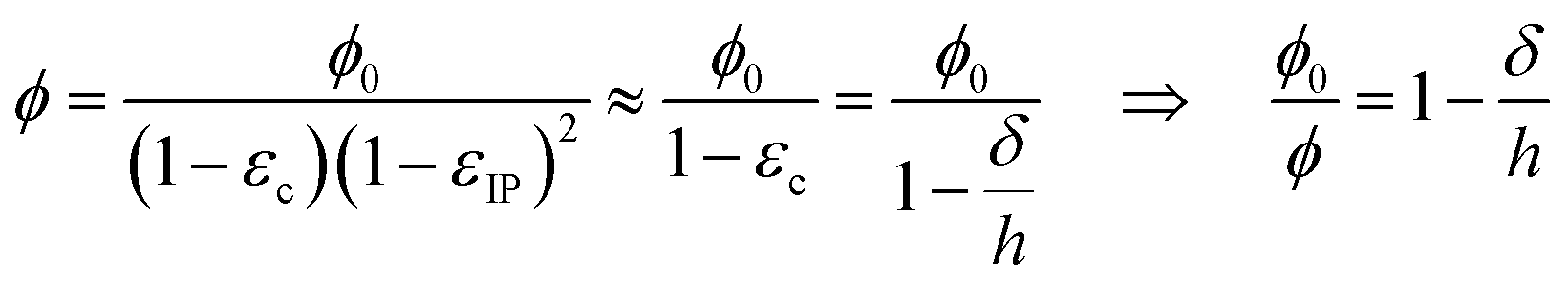 | (7) |
As illustrated in Fig. 2a, the out-of-plane swelling pressure of the hydrogel was measured as a function of the gap (h) between two parallel plates. The compression was changed stepwise to allow the hydrogel to approach equilibrium for each incremental compression. The pressures under dynamic compression are outside the current scope but are higher due to the long relaxation time for fibrillar networks. Fig. 2b–e shows the measured pressure in the hydrogels as a function of solidity. In the case of hydrogels made from carboxymethylated and cationic CNFs (Fig. 2b–d), the electrolyte concentration (ci,soleqn (2)) had a considerable influence on the degree of swelling, that is, on the unrestricted equilibrium solidity (ϕ0). With a reduced gap, the pressure increases as the ionic groups of the CNFs are forced into a smaller volume, increasing ci,gel in eqn (2). The slope of the increasing pressure thus depends on the surrounding electrolyte concentration up to a high-enough solidity where the repulsive network pressure (Pnet) dominates. The slopes at different electrolyte concentrations were the same in the network-dominated regime. In theory, the curves should have the same final pressure as ϕ approaches 1, corresponding to the compression of a solid sheet.
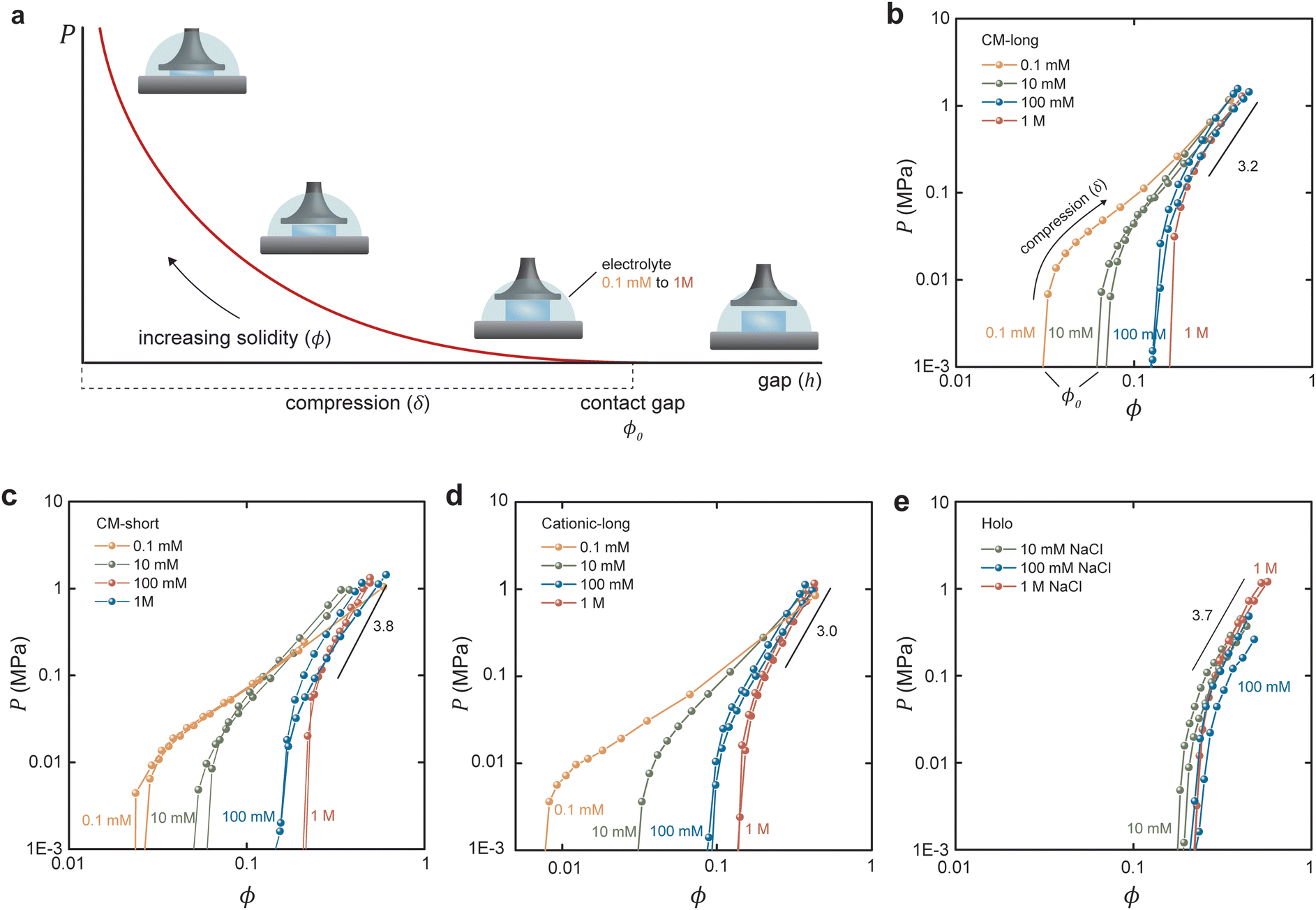 | ||
| Fig. 2 Equilibrium swelling pressures under compression. (a) Schematic illustration of the compression measurement. Equilibrium swelling pressures for (b) CM-long, (c) CM-short, (d) cationic-long, and (e) holo CNFs. Note that the x-axis in (d) has a different scale to fit the data of the highly swelling cationic CNFs. | ||
In contrast, the swelling of Holo CNFs (Fig. 2e) was not as influenced by the electrolyte concentration, which is interesting since these fibrils still carry a significant charge in the order of 0.2 mmol g−1.28,29 The hypothesis is that the hemicelluloses shell results in Pnet and Pmix that are much greater than Pion so that the ionic swelling pressure is not distinguishable. The most probable explanation is that the hemicelluloses increase Pnet by forming physical crosslinks due to polymer entanglement and interdiffusion during the initial drying of the sheets. Another possibility is that repulsive ionic pressure is dissipated by changing the conformation of the dynamic hemicellulose shell compared to the static cellulose fibril contact zone (Fig. S3, ESI†). This data adds to the discussion about the role of hemicelluloses in the mechanical properties of the plant cell wall under different conditions.23
The proposed one-dimensional model explaining the data in Fig. 2 is schematically depicted in Fig. 3a and comprises three elements: (i) elastic modulus Enet representing the enthalpy elastic part of the network, (ii) elastic modulus Echem representing mainly the ionic contribution but also the thermodynamics of mixing, and (iii) a yielding element Pyield representing a limiting force before sliding with friction ensues, manifested as plastic deformation of the network. The network spring and the yielding element lead to stick or slip depending on the compression (δ or εc); below the yield compressive strain (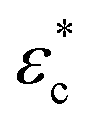 ), the network is elastic, while above
), the network is elastic, while above 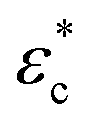 it deforms plastically. The yield is the reason for the unique stick-slip-stick behavior of fibril networks,33 meaning that, unlike crosslinked polymer networks, weakly associated fibril networks do not build elastic pressure as they are subjected to large deformation and thus have no elastic recoil. A relaxed fibril network has a similar Pnet in both extension and compression.
it deforms plastically. The yield is the reason for the unique stick-slip-stick behavior of fibril networks,33 meaning that, unlike crosslinked polymer networks, weakly associated fibril networks do not build elastic pressure as they are subjected to large deformation and thus have no elastic recoil. A relaxed fibril network has a similar Pnet in both extension and compression.
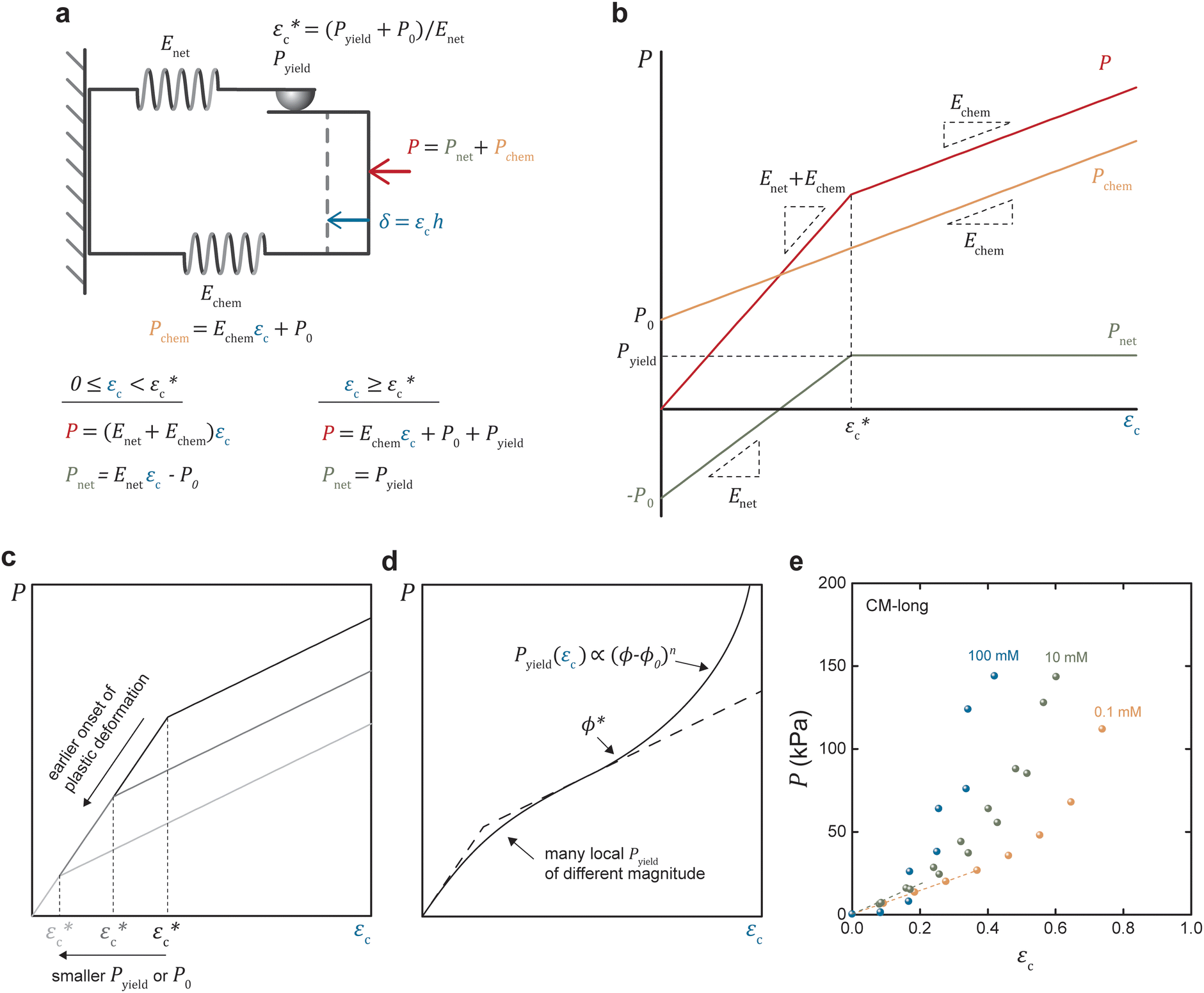 | ||
| Fig. 3 Elastoplastic model. (a) Schematic of the mechanical representation of the network, including equations. (b) Qualitative relationship between the plate pressure and absolute compression: outlining the different contributing pressures, with (c) showing the influence of the yielding pressure, and (d) the smoothing of the curve due to many local yielding pressures of different magnitude as well as the nonlinearity of the network at higher compression. (e) Relationship between plate pressure and compressive strain for CM-long hydrogels in different NaCl concentrations. | ||
The equations describing the elastoplastic behavior in different compression regimes are included in Fig. 3a. Fig. 3b shows a qualitative drawing of how the different pressures contribute to the total plate pressure (P) required to keep the compressed state. At zero strain (ϕ0), Pnet = Pchem which means that there is a swelling pressure (P0), which is canceled by an equal opposing, negative pressure from the network – that is, the same pressure balance that keeps a balloon under equilibrium. Thus, 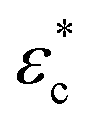 depends on both Pyield and P0, and determines the normal pressure required for yielding with subsequent plastic deformation. Yielding is observed as a change of the slope at a position depending on the magnitude of Pyield and P0, as illustrated in Fig. 3c. Immediate yielding from the start would result in a single straight line.
depends on both Pyield and P0, and determines the normal pressure required for yielding with subsequent plastic deformation. Yielding is observed as a change of the slope at a position depending on the magnitude of Pyield and P0, as illustrated in Fig. 3c. Immediate yielding from the start would result in a single straight line.
The model parameters will have different values for different types of fibrils other than cellulose. The model is general and not filled with constants for specific fibrils. The aim was to show the general behavior of anisotropic fibrillar networks.
The model assumes homogeneous properties. However, different structures in the network lead to many local yielding positions of varying Pyield, and the transition between the elastic and plastic deformation would be gradual, as demonstrated in Fig. 3d. Fig. 3d also illustrates that the model is limited to moderate compression before densification of the network dominates, seen as a nonlinear increase of the pressure.
The experimental data were replotted against compressive strain in Fig. 3e for the CM long at different ionic strengths to test how this model agrees with experiments (other samples in Fig. S4, ESI†). The initial response was linear, at least in 0.1 and 10 mM NaCl, indicating an immediate yielding (Pyield and 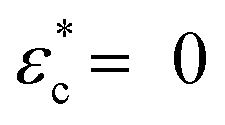 ) so that the initial resistance to compression comes almost exclusively from Pion. At 0.1 mM NaCl, compression of up to 50% was needed before Pnet was significant enough to induce nonlinearity. At 10 mM NaCl the linear part was up to about 25%, and at 100 mM NaCl, the response was nonlinear from the start. Increasing electrolyte concentration led to a diminishing Pion as ci,gel − ci,sol in eqn (2) approaches 0, and the network pressure dominates throughout the compression.
) so that the initial resistance to compression comes almost exclusively from Pion. At 0.1 mM NaCl, compression of up to 50% was needed before Pnet was significant enough to induce nonlinearity. At 10 mM NaCl the linear part was up to about 25%, and at 100 mM NaCl, the response was nonlinear from the start. Increasing electrolyte concentration led to a diminishing Pion as ci,gel − ci,sol in eqn (2) approaches 0, and the network pressure dominates throughout the compression.
In 1 M NaCl Pion and Pmix are expected to be so small in comparison to Pnet and can be neglected to estimate Pnet. A model for fiber networks under uniaxial compression was used to estimate the elastic modulus of the networks under these conditions. Compression of fiber networks can be described using a Taylor expansion:
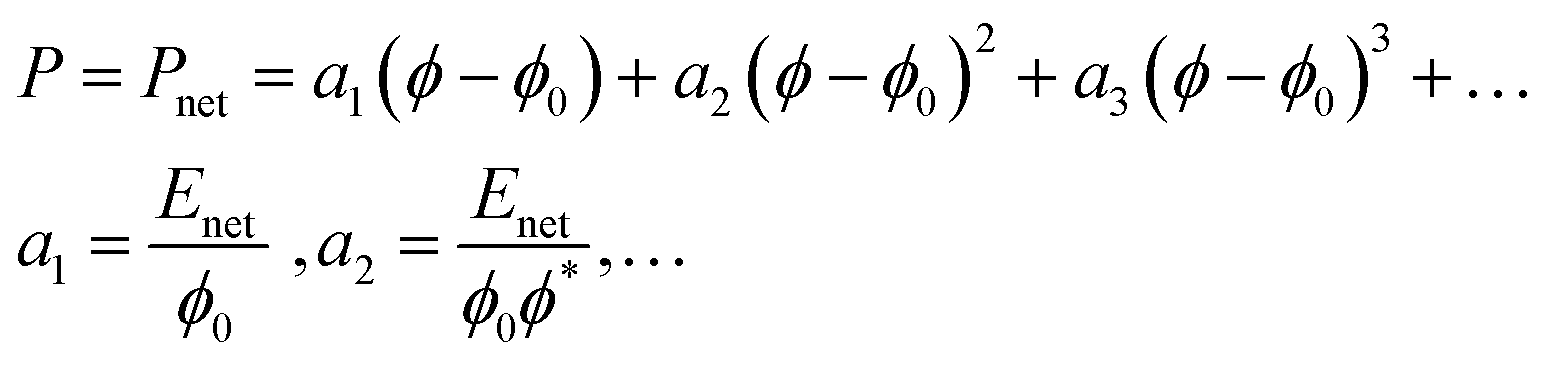 | (8) |
The nonlinearity exponent (n) in Fig. 3d was determined to n = 2 by polynomial fitting of the data at 1 M NaCl – that is, when the repulsion is switched off – meaning that only the first two terms in eqn (8) were needed to describe these anisotropic fibrillar networks under compression. The approximate fitted values of the Enet are ∼300 kPa for CM-long, ∼200 kPa for CM-short, ∼100 kPa for cationic-long, and ∼200 kPa for Holo (Table 2). These moduli are indeed low, considering the relatively high solidity (ϕ*) and highlight the anisotropy of these networks. As a reference, the in-plane (dynamic) tensile modulus of CNF networks at these concentrations is ∼1 GPa.7 However, the dynamic in-plane modulus is measured without time for relaxation, whereas these compression moduli are estimated from static equilibrium, which is probably much lower than the dynamic compression modulus. The uncertainty of the elastic modulus from this curve fitting would be reduced by more measurement points, which would also require consideration of the resolution in the measurement. However, the relative magnitudes are reasonable based on the aspect ratio and previous swelling experiments.20,26
| E net (kPa) | ϕ 0 | ϕ* | |
|---|---|---|---|
| CM-long | 330 ± 50 | 0.16 ± 0.00 | 0.41 ± 0.03 |
| CM-short | 180 ± 190 | 0.20 ± 0.01 | 0.3 ± 0.1 |
| 280 ± 240 | 0.20 ± 0.02 | 0.4 ± 0.2 | |
| Cationic-long | 70 ± 50 | 0.13 ± 0.01 | 0.24 ± 0.08 |
| 140 ± 80 | 0.13 ± 0.01 | 0.31 ± 0.09 | |
| Holo | 190 ± 220 | 0.22 ± 0.03 | 0.3 ± 0.2 |
| 170 ± 200 | 0.22 ± 0.03 | 0.3 ± 0.2 |
The equilibrium solidity (ϕ0) is plotted in Fig. 4 as a function of the aspect ratio of the different CNFs. Besides being dependent on the electrolyte concentration – that is, a higher equilibrium solidity at higher NaCl concentration according to eqn (2) – the aspect ratio of the fibrils also significantly impacted the network. A lower aspect ratio CM-short networks had a higher electrolyte sensitivity than CM-long, since the restrictive Pnet is smaller in comparison to Pion. At higher electrolyte concentrations, CM-short had a higher equilibrium solidity, probably because shorter fibrils can pack into a denser network during the initial drying resulting in a lower volume fraction of pores that could open when the network swelled in water at high electrolyte concentration. The CM-short network also expanded more at low electrolyte concentrations due to fewer load-bearing contacts per fibril. The slightly different charge densities of CM-short and CM-long probably also contributed to this behavior.
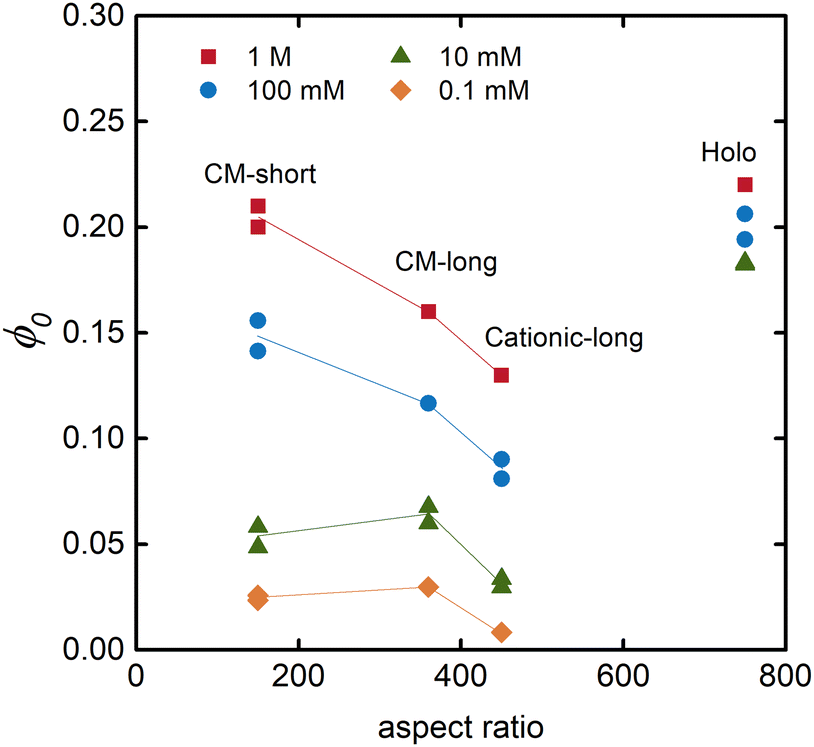 | ||
| Fig. 4 Relationship between the equilibrium solidity and the aspect ratio of the fibrils for hydrogels swollen in solutions with different concentrations of NaCl. | ||
Fig. 4 also shows that cationic CNFs had a lower equilibrium solidity, as also shown in other studies.20,26 The reason for this behavior of cationic CNFs is still not determined, but it is probably related to the bulkiness of quaternary amines, their complex hydration, and the fact that they are non-titrating. Thus, cationic CNFs are fully charged regardless of their concentration, which probably significantly impacts the network formation upon drying and the subsequent reswelling into hydrogels. Carboxymethyl groups induce a local acidic pH inside the hydrogel that can lead to protonation and a lower effective charge density, hence lower ci,gel in eqn (2).
Holo CNFs had a fairly electrolyte-independent equilibrium solidity, suggestively due to the entangling of hemicelluloses or their conformability to minimize the effect of Pion.
Conclusions
The results show that an elastoplastic model containing three elements can describe out-of-plane elastoplasticity of weakly associated anisotropic fibrillar hydrogels. At low solidity, the stiffness of these hydrogels is dominated by the ionic swelling pressure, but the hydrogels are still held together and are self-supporting due to the strong in-plane network. This structure can be utilized in designing actuators with impressive strength and direction of pressure that can beat polymer hydrogel actuators in speed and strain (work in progress).34,35 The same model can be used for in-plane properties with the difference that in this direction and under these swelling pressures, yielding can be neglected due to network connectivity, which is optimized to fully utilize the stiffness of the fibrils and maximize friction due to more contact area per fibril as they align with the plane.This simple model should, with slight modifications, be able to describe any fibrillar hydrogel with weak contact zones that can slide with friction, both net repulsive networks as in this work, or net attractive, which can be achieved by adding acid to protonate carboxyl groups on the CNFs. Covalent contacts between fibrils would need a modified model, and these are also not as anisotropic since the covalent crosslinks prevent the orientation of fibrils during drying. The model provides a framework to start understanding the high stiffness of fibrillar hydrogels at low solidity and their ability to hold vast amounts of water. It also provides insight into why nanofibrils were essential as a construction element in the evolution of complex multicellular ocean-based life forms and later defying gravity in land-based plants. Concerning plants, the influence of hemicellulose was considerable, demonstrating their role as a “mortar” between cellulose fibrils in the plant cell wall. The measurement technique demonstrated here provides a sensitive and utterly useful toolbox to investigate the influence of different hemicelluloses to map the roles of different chemical functionalities within the plant cell walls. The anisotropy of these hydrogels is reminiscent of the plant cell walls and should thus be an excellent model system for further investigations.
Experimental
Chemicals
Absolute ethanol and 2-propanol were purchased from VWR International AB (Sweden). Monochloroacetic acid ≥99.0%, glycidyl trimethylammonium chloride ≥90.0%, sodium chloride 99.999%, (3-aminopropyl)triethoxysilane (APTES) ≥98.0%, and sodium hydroxide (NaOH) were purchased from Sigma-Aldrich AB (Sweden). Hydrochloric acid (HCl) 37% and methanol were purchased from Thermo Fisher Scientific. Sodium bicarbonate ≥99.7% was purchased from Honeywell Fluka. Nordic paper Säffle AB, Sweden, kindly donated never-dried sulfite fibers (CM-long and cationic-long). All chemicals were used as received. Deionized (DI) water or ultrapure water (Merck Milli-Q 18.2 MΩ cm−1) was used throughout all experiments.Washing of cellulose fibers
Never-dried sulfite fibers (30 g, dry mass) were mechanically beaten (3000 revolutions) using a laboratory PFI mill (HAM-JERN, Norway). The fibers were soaked for 30 minutes in DI water (∼2.5 L) with pH adjusted to 2 using HCl (1 M). The fibers were then washed with DI water until the conductivity of the filtrate water was below 5 μS cm−1. The water was removed by filtration, and the fibers were placed for 30 minutes in sodium bicarbonate solution (0.001 M) at pH 9. Finally, the fibers were washed with DI water until the conductivity of the filtrate water was below 5 μS cm−1.Preparation of carboxymethylated cellulose nanofibrils
Anionic, low aspect ratio CNFs (CM-short) were purchased from RISE Bioeconomy (Stockholm, Sweden), and prepared according to the procedure from Wågberg et al.27 The CNF was further homogenized using a high-pressure microfluidizer (Microfluidizer M-110EH, Microfluidics Corp.) by three passages through 200/100 μm chambers (∼1650 bar).Anionic, high aspect ratio CNFs (CM-long) were prepared through carboxymethylation. Washed never dried sulfite fibers (30 g, dry mass) was solvent exchanged by soaking the fibers in ethanol (∼250 mL) for 15 minutes followed by filtering off excess ethanol, repeated three times. The fibers were then impregnated in a solution of chloroacetic acid (4.4 g) dissolved in isopropanol (153 mL) for 30 minutes. The impregnated fibers were then added to a mixture of NaOH (4 g) dissolved in methanol (114 mL) and isopropanol (455 mL) preheated to 82 °C. The carboxymethylation reaction was continued for 60 minutes. Following the reaction, the fibers were washed with DI water (∼5 L) followed by aqueous HCl solution (0.01 M, ∼5 L), and finally, DI water (∼5 L). After washing, the fibers were soaked in sodium bicarbonate solution (5 wt%, 2 L) for 60 minutes to convert the carboxyl groups to sodium form. A final washing with DI water was performed until the conductivity of the water filtrate was below 5 μS cm−1. The chemically modified fibers were finally homogenized using a high-pressure microfluidizer (Microfluidizer M-110EH, Microfluidics Corp.) with one passage through 400/200 μm chambers (∼1000 bar) and three passages through 200/100 μm chambers (∼1650 bar), to achieve fibrils in the hydrogel form (∼1 wt%).
Preparation of quaternized cellulose nanofibrils
Cationic, high aspect ratio CNFs (cationic-long) were prepared using quaternization reaction. The hydroxyl groups of the fibers were activated by soaking washed, never-dried sulfite fibers (28 g dry mass) in a NaOH solution (5 wt% NaOH, 7.5 wt% fibers) for 30 minutes before the reaction. The impregnated fibers were added to a low-density polyethylene plastic bag, followed by glycidyl trimethylammonium chloride (133.3 g). The plastic bag was placed in a water bath preheated to 65 °C, and the reaction time was set to 7 hours. After the reaction, the mixture was neutralized with a hydrochloric acid solution and washed with DI water until the conductivity of the water filtrate was below 5 μS cm−1. The chemically modified fibers were finally homogenized using a high-pressure microfluidizer (Microfluidizer M-110EH, Microfluidics Corp.), with one passage through 400/200 μm chambers (∼1000 bar) and three passages through 200/100 μm chambers (∼1650 bar), to achieve fibrils in the hydrogel form (∼1 wt%).Holo CNFs
Holo CNFs were kindly provided by Xuan Yang et al.28 and were prepared using peracetic acid delignification of matchstick-shaped wood pieces of Norway spruce followed by homogenization. Readers are referred to the original publication for the detailed protocol.Atomic force microscopy
The dimensions of the fibrils were characterized using AFM (Multimode 8, Bruker, Santa Barbara, USA) in the ScanAsyst mode using ScanAsyst air cantilevers. CNF dispersions (1.5 g L−1) for AFM measurements were prepared by ultrasonication for 10 minutes (∼80% amplitude) followed by centrifugation (1 h, 4000 rpm). Holo CNF dispersion was prepared by only centrifugation for 15 minutes. Anionic CNF and HOLO CNF surface was prepared by adsorbing APTES (1 g L−1) on a freshly cleaved mica surface, then rinsing the surface with Milli-Q water and adsorbing the anionic CNFs (0.01 g L−1). Finally, the surface was rinsed with Milli-Q water and dried with nitrogen gas. The cationic CNF surface was prepared by adsorbing cationic CNFs (0.005 g L−1) onto a freshly-cleaved mica surface, followed by drying with nitrogen gas. The average width was determined from 100 individual fibrils and the average length from 75 individual fibrils.Surface charge determination
Polyelectrolyte titration against either potassium polyvinylsulfonate or polydiallyldimethylammonium chloride using ParticleMetrix Stabino system (Germany) was used to determine the charge density of the fibrils. The charge density is reported as an average of three replicates.Preparation of anisotropic hydrogels
CNF in the hydrogel form (∼1 wt%) was diluted with ultrapure water to 0.2 wt%. The components were mixed using IKA Ultra-Turrax high-shear mixer (12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm, 10 minutes). CNF sheets (250 mg, dry mass) were prepared by overnight vacuum filtration of CNF dispersion through a Durapore membrane filter (PVDF, hydrophilic, 0.65 μm, Merck) in a microfiltration setup. The sheet was dried for 20 minutes in the sheet drier of a Rapid Köthen (Paper Testing Instruments GMBH, Austria), at 93 °C and at a reduced pressure of 95 kPa. The dry CNF sheets were cut into square pieces of 5 by 5 mm and placed in an electrolyte to equilibrate for 24 hours.
000 rpm, 10 minutes). CNF sheets (250 mg, dry mass) were prepared by overnight vacuum filtration of CNF dispersion through a Durapore membrane filter (PVDF, hydrophilic, 0.65 μm, Merck) in a microfiltration setup. The sheet was dried for 20 minutes in the sheet drier of a Rapid Köthen (Paper Testing Instruments GMBH, Austria), at 93 °C and at a reduced pressure of 95 kPa. The dry CNF sheets were cut into square pieces of 5 by 5 mm and placed in an electrolyte to equilibrate for 24 hours.
Compression measurement of anisotropic hydrogel
Compression measurements were performed using a DHR-2 rheometer (TA Instruments, New Castle, DE, USA) equipped with a Peltier plate system, an 8 mm diameter plate–plate geometry, and a solvent trap. Hydrogels equilibrated for 24 hours were placed between the compressing plates, and the electrolyte was added in excess. The gel was compressed by lowering the plate geometry in 10 steps. The normal force was recorded after equilibrium was reached for each step, typically after 20 to 60 min. The compression strain in each step was normalized for all samples to be the rounded down number of (swollen thickness − dry thickness)/10, so compression of roughly 6–9% in each step. The samples were protected from dehydration during the long measurement times using a solvent trap with electrolytes added to the perimeter to maintain a constant reservoir. The compression rate and relaxation time were optimized to provide enough time for the samples to equilibrate. The steps and conditions of each step are summarized in Table S1 (ESI†). In some instances, the system did not fully reach equilibrium after the final compression step, and the pressure was then estimated by extrapolation.Author contributions
Rebecca Östmans: data curation, formal analysis, investigation, methodology, resources, validation, writing – original draft. Maria F. Cortes Ruiz: data curation, formal analysis, investigation, methodology, validation, writing – original draft. Jowan Rostami: resources, writing – review and editing. Farhiya Alex Sellman: resources, writing – review and editing. Lars Wågberg: conceptualization, funding acquisition, supervision, writing – review and editing. Stefan B. Lindström: conceptualization, formal analysis, visualization, writing – original draft. Tobias Benselfelt: conceptualization, data curation, formal analysis, funding acquisition, investigation, methodology, project administration, supervision, visualization, writing – original draft.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Xuan (Justin) Yang is thanked for providing Holo-CNFs. The Knut and Alice Wallenberg Foundation are acknowledged for funding through the Wallenberg Wood Science Center (WWSC) and an individual fellowship for Tobias Benselfelt (KAW 2019.0564). Stefan B. Lindström works within the Neopulp research profile financed by the Knowledge Foundation, and also thanks SCA for financial support.References
- X. Liu, J. Liu, S. Lin and X. Zhao, Mater. Today, 2020, 36, 102–124 CrossRef.
- C.-Y. Lo, Y. Zhao, C. Kim, Y. Alsaid, R. Khodambashi, M. Peet, R. Fisher, H. Marvi, S. Berman, D. Aukes and X. He, Mater. Today, 2021, 50, 35–43 CrossRef CAS.
- G. Gerlach and K.-F. Arndt, Hydrogel sensors and actuators: engineering and technology, Springer Science & Business Media, 2009 Search PubMed.
- B. Lu, H. Yuk, S. Lin, N. Jian, K. Qu, J. Xu and X. Zhao, Nat. Commun., 2019, 10, 1043 CrossRef PubMed.
- H. Yuk, B. Lu and X. Zhao, Chem. Soc. Rev., 2019, 48, 1642–1667 RSC.
- T. Benselfelt and L. Wagberg, Biomacromolecules, 2019, 20, 2406–2412 CrossRef CAS PubMed.
- T. Benselfelt, M. Nordenström, S. B. Lindström and L. Wågberg, Adv. Mater. Interfaces, 2019, 6, 1900333 CrossRef.
- F. L. Hatton, J. Engstrom, J. Forsling, E. Malmstrom and A. Carlmark, RSC Adv., 2017, 7, 14947–14958 RSC.
- E. Prince and E. Kumacheva, Nat. Rev. Mater., 2019, 4, 99–115 CrossRef.
- K. Sano, Y. Ishida and T. Aida, Angew. Chem., Int. Ed., 2018, 57, 2532–2543 CrossRef CAS PubMed.
- X. Le, W. Lu, J. Zhang and T. Chen, Adv. Sci., 2019, 6, 1801584 CrossRef PubMed.
- Z. Sun, Y. Yamauchi, F. Araoka, Y. S. Kim, J. Bergueiro, Y. Ishida, Y. Ebina, T. Sasaki, T. Hikima and T. Aida, Angew. Chem., 2018, 130, 15998–16002 CrossRef.
- C. Qian, T. Higashigaki, T.-A. Asoh and H. Uyama, ACS Appl. Mater. Interfaces, 2020, 12, 27518–27525 CrossRef CAS PubMed.
- A. Malti, J. Edberg, H. Granberg, Z. U. Khan, J. W. Andreasen, X. Liu, D. Zhao, H. Zhang, Y. Yao, J. W. Brill, I. Engquist, M. Fahlman, L. Wågberg, X. Crispin and M. Berggren, Adv. Sci., 2015, 3, 1500305 CrossRef PubMed.
- P. Flory and J. Rehner, Chem. Phys., 1943, 11, 521–526 CAS.
- P. J. Flory, J. Chem. Phys., 1942, 10, 51–61 CrossRef CAS.
- M. L. Huggins, J. Chem. Phys., 1941, 9, 440 CrossRef CAS.
- M. Nordenström, T. Kaldéus, J. Erlandsson, T. Pettersson, E. Malmström and L. Wågberg, ACS Sustainable Chem. Eng., 2021, 9, 11003–11010 CrossRef.
- M. J. Solomon and P. T. Spicer, Soft Matter, 2010, 6, 1391–1400 RSC.
- T. Benselfelt, M. Nordenström, M. M. Hamedi and L. Wågberg, Nanoscale, 2019, 11, 3514–3520 RSC.
- T. Benselfelt, J. Engström and L. Wågberg, Green Chem., 2018, 20, 2558–2570 RSC.
- P. Chen, J. Wohlert, L. Berglund and I. Furó, J. Phys. Chem. Lett., 2022, 13, 5424–5430 CrossRef CAS PubMed.
- D. J. Cosgrove, Plant Physiol., 2022, 189, 1246–1277 CrossRef CAS PubMed.
- T. Nishino, K. Takano and K. Nakamae, J. Polym. Sci., Part B: Polym. Phys., 1995, 33, 1647–1651 CrossRef CAS.
- J. Rostami, T. Benselfelt, L. Maddalena, C. Avci, F. A. Sellman, G. C. Ciftci, P. A. Larsson, F. Carosio, F. Akhtar and W. Tian, Adv. Mater., 2022, 2204800 CrossRef CAS PubMed.
- A. Pei, N. Butchosa, L. A. Berglund and Q. Zhou, Soft Matter, 2013, 9, 2047–2055 RSC.
- L. Wågberg, G. Decher, M. Norgren, T. Lindström, M. Ankerfors and K. Axnäs, Langmuir, 2008, 24, 784–795 CrossRef PubMed.
- X. Yang, M. S. Reid, P. Olsén and L. A. Berglund, ACS Nano, 2020, 14, 724–735 CrossRef CAS PubMed.
- X. Yang and L. A. Berglund, Adv. Mater., 2021, 33, 2001118 CrossRef CAS PubMed.
- A. Katchalsky and I. Michaeli, J. Polym. Sci., 1955, 15, 69–86 CrossRef CAS.
- W. R. K. Illeperuma, J.-Y. Sun, Z. Suo and J. J. Vlassak, Soft Matter, 2013, 9, 8504–8511 RSC.
- R.-M. P. Karlsson, P. T. Larsson, S. Yu, S. A. Pendergraph, T. Pettersson, J. Hellwig and L. Wågberg, J. Colloid Interface Sci., 2018, 519, 119–129 CrossRef CAS PubMed.
- N. Mittal, F. Ansari, K. Gowda, C. Brouzet, P. Chen, P. T. Larsson, S. V. Roth, F. Lundell, L. Wågberg, N. A. Kotov and L. D. Söderberg, ACS Nano, 2018, 12, 6378–6388 CrossRef CAS PubMed.
- H. Na, Y.-W. Kang, C. S. Park, S. Jung, H.-Y. Kim and J.-Y. Sun, Science, 2022, 376, 301–307 CrossRef CAS PubMed.
- X. He, J. Zhu and C. Yang, Soft Matter, 2022, 18, 5177–5184 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2sm01571d |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |