
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Compression of colloidal monolayers at liquid interfaces: in situ vs. ex situ investigation†
Keumkyung
Kuk
 a,
Vahan
Abgarjan
a,
Lukas
Gregel
a,
Yichu
Zhou
a,
Virginia
Carrasco Fadanelli
b,
Ivo
Buttinoni
b and
Matthias
Karg
*a
a,
Vahan
Abgarjan
a,
Lukas
Gregel
a,
Yichu
Zhou
a,
Virginia
Carrasco Fadanelli
b,
Ivo
Buttinoni
b and
Matthias
Karg
*a
aInstitut für Physikalische Chemie I: Kolloide und Nanooptik, Heinrich-Heine-Universität Düsseldorf, Universitätsstr. 1, 40225, Düsseldorf, Germany. E-mail: karg@hhu.de
bInstitut für Experimentelle Physik der kondensierten Materie, Heinrich-Heine-Universität Düsseldorf, Universitätsstr. 1, 40225, Düsseldorf, Germany
First published on 18th November 2022
Abstract
The assembly of colloidal particles at liquid/liquid or air/liquid interfaces is a versatile procedure to create microstructured monolayers and study their behavior under compression. When combined with soft and deformable particles such as microgels, compression is used to tune not only the interparticle distance but also the underlying microstructure of the monolayer. So far, the great majority of studies on microgel-laden interfaces are conducted ex situ after transfer to solid substrates, for example, via Langmuir–Blodgett deposition. This type of analysis relies on the stringent assumption that the microstructure is conserved during transfer and subsequent drying. In this work, we couple a Langmuir trough to a custom-built small-angle light scattering setup to monitor colloidal monolayers in situ during compression. By comparing the results with ex situ and in situ microscopy measurements, we conclude that Langmuir–Blodgett deposition can alter the structural properties of the colloidal monolayers significantly.
Inroduction
Colloidal monolayers at liquid interfaces, namely, micro- and nanoparticle-laden liquid interfaces, are widely used in fundamental and applied studies. Colloidal particles can self-assemble, in fact, in two-dimensional materials with properties (e.g. photonic or electronic) similar to those of atomic structures. However, unlike atomic counterparts, the colloidal building blocks can be engineered in terms of the chemical composition,1–4 shape,4–7 and morphology,8,9 in order to tailor the assembly behavior and spatial arrangement. One of the methods for preparing colloidal monolayers is via confining particles at the flat interfacial plane between two immiscible fluids (e.g. an air/water or oil/water interface).10 This approach offers great advantages not only for studies of gas–liquid–solid phase transitions as the particle concentration (i.e., the number of particles per unit area) can be tuned in situ by means of lateral barriers,11,12 but also for scalable fabrications for both planar and curved surfaces with areas ranging from cm2 up to m2 scales.13–15 In the latter approach, the microstructures at the liquid interface are transferred and deposited on solid surfaces (Langmuir–Blodgett deposition) to obtain dried colloidal films, e.g. for coating or photonic applications.16–20 In contrast to assemblies of rigid spheres, soft colloidal objects like microgels and nanogels21 can be deformed, for example, under external compression giving access to richer phase diagrams and complex superstructures.22,23The structural properties of colloid-laden interfaces are typically extracted from microscopy images by detecting the centers of mass of the colloidal units.24–28 This “particle-tracking” method quickly becomes time-consuming and computationally demanding in the presence of many particles. Even more importantly, it can be only applied above the Abbe diffraction limit. For example, due to the mostly small sizes synthesized to date, assemblies of coreless and core–shell microgels have been mainly characterized ex situ – by looking at dried samples with atomic force or electron microscopes – under the assumption that the structure is unaltered during Langmuir–Blodgett deposition.8,11,12,29–33 Only recently, in situ observation of local regions of the particle-laden interface was achieved via atomic force microscopy.34
In this study, we propose an in situ method – a Langmuir trough combined with small-angle light scattering (LT-SALS) – to characterize colloidal monolayers at the air/water interface. To demonstrate its versatility, colloids of different morphologies were monitored during compression: silica particles (rigid spheres), poly-N-isopropylacrylamide (PNIPAM) microgels (soft spheres) and silica-PNIPAM core–shell microgels (hard core–soft shell spheres). The focus of our study, however, lies on the assembly of the core–shell (CS) microgel system. We first present the results from an ex situ structural analysis using Langmuir–Blodgett deposition. Then, we compare these results to an in situ analysis performed using LT-SALS as well as fluorescence microscopy. Our results indicate that there are severe structural differences between the microstructures of CS microgels at air/water interfaces and after transfer to a solid substrate. These drying effects are in stark contrast with the widely accepted assumption that the interfacial structure is replicated during Langmuir–Blodgett deposition for microgel type building blocks. We discuss analogies and differences with existing works as well as possible reasons for the observed structural changes during drying.
Results
Core–shell microgels
We prepared monolayers of CS microgels at an air/water interface in a Langmuir trough and studied their structure under compression using ex situ light microscopy (Method 1), in situ fluorescence microscopy (Method 2) and in situ small-angle light scattering (Method 3).CS microgels possess two relevant length scales: the diameter of the incompressible core (here, silica) and the thickness of the soft, deformable shell (here, PNIPAM). In bulk, these length scales simply define the boundaries of the interparticle interactions. When the microgels are confined and spread at the air/water (or oil/water) interface, the situation becomes more complex because the shells laterally deform at the interface leading to changes in shell morphology and shape, and consequently the total diameter, Di (interfacial diameter), which is larger than the bulk hydrodynamic diameter, Dh.12,23,35,36 Generally, there are three different scenarios for the spatial arrangement of CS microgels at air/water or oil/water interfaces: at very low number of particles per unit area (nP/A), i.e., for near-zero surface pressures, the CS microgels mostly stay apart in an unordered, fluid-like state. In the second regime, as the nP/A increases, the microgel shells start to touch (shell–shell contact) more frequently. Finally, in the third regime, the shells are squeezed and/or interpenetrated (core–core contact) until the critical point, where the monolayer buckles, breaks and/or is pushed into the subphase (water). Theoretically, if the energy difference between the partially and fully overlapped shells is small enough, energy minimization is achieved by the overlap of shells in some directions at the cost of other neighboring shells,37–39 leading to a change in the symmetry of the monolayer. In experimental studies, however, such a symmetry change of the microgel monolayer with increasing nP/A has only been partially observed.40 In most cases, core–shell structured microgels12,29,30 and coreless microgels31–33,41–43 seem to undergo an “isostructural solid–solid phase transition” characterized by microgels in hexagonal arrangement with “shell–shell” contact versus a denser phase, also of hexagonal order, where the microgels are in “core–core” contact.11,44 Note that the interparticle distance in “core–core” contact includes the diameter of the core, Dc, as well as the compressed microgel shell. The discrepancies between numerical and experimental studies concerning the phase behavior at interfaces have been ascribed to capillary forces and a highly nonlinear mechanical response of the polymer chains (i.e. the PNIPAM corona) under compression11,30,33,40
In the following, we report the same “isostructural solid–solid phase transition” only during ex situ characterization (Method 1) of monolayers of micron-sized CS microgels. Remarkably, this phenomenon is not observed during in situ experiments (Methods 2 and 3).
Method 1 (ex situ microscopy)
The ex situ microstructural analysis relies on the microscopic investigation of the colloidal monolayer upon transfer from the liquid interface to a solid substrate followed by drying. This leads to dry, substrate-supported colloidal monolayers. When the transfer to the solid substrate is done continuously while the monolayer is compressed in the Langmuir trough, the monolayer position on the substrate can be linked to the corresponding surface pressure at the liquid interface.11,44 In this study, the CS microgel system used for the in situ and ex situ comparison has a core diameter Dc = 340 ± 20 nm and a total hydrodynamic diameter Dh = 920 ± 18 nm (see Synthesis Section for more details). Fig. 1A shows the measured compression isotherm during the Langmuir–Blodgett deposition along with the corresponding microscopy images. Note that we use a linear color coding from light blue to black linked with low to high surface pressure (Π) throughout this article. From the mid Π regime (16.3 mN m−1 and higher), the images were taken in dark field mode to facilitate image analysis. The monolayer images at lower Π were recorded in bright field mode. With increasing compression, i.e., decreasing available area, A, the surface pressure increases continuously. In the low Π regime, the CS microgels are not homogeneously distributed over the accessible area (see microscopy images) but rather show hexagonal arrangements with shell–shell contacts and some voids among numerous crystalline domains. This indicates the presence of attractive interparticle interactions despite the large interparticle distances, in agreement with previously reported results from in situ and ex situ analysis of CS microgel monolayers.8,12,44 As Π increases, the crystalline domains grow while the voids close. In the high Π regime, we observe the formation of CS microgel clusters in “core–core” contact. The critical surface pressure for the start of this “isostructural solid–solid phase transition” is around 16–18 mN m−1 for the presented CS microgel, which can be also identified both in the splitting of the first peak of the radial distribution functions (RDFs, Fig. 1B) and in the diffraction patterns of the dried monolayers (Fig. 2). Although the ex situ “core–core” distance should lie within the detection limit of our current SALS setup, the microstructures produce diffuse scattering patterns (Fig. 2C and D), instead of revealing two distinctive length scales. This is due to the fact that the “isostructural phase transitions” is only locally isostructural, i.e. the monolayers, on mm2 scale, do not show a defined symmetry. The transition is more pronounced for higher values of Π, i.e. the diffuse contribution to the scattering patterns increases with increasing Π. For low to medium values of Π, the RDFs are characterized by the first peak corresponding to the interparticle distance, i.e. center-to-center distance from ex situ image (Dim,exc–c), whereas for high Π above the critical value (e.g. 23.2 mN m−1), the peak splits near Dh. Fig. 1C reflects the appearance of these two distinct interparticle distances, as the value of Dim,exc–c approaches Dh.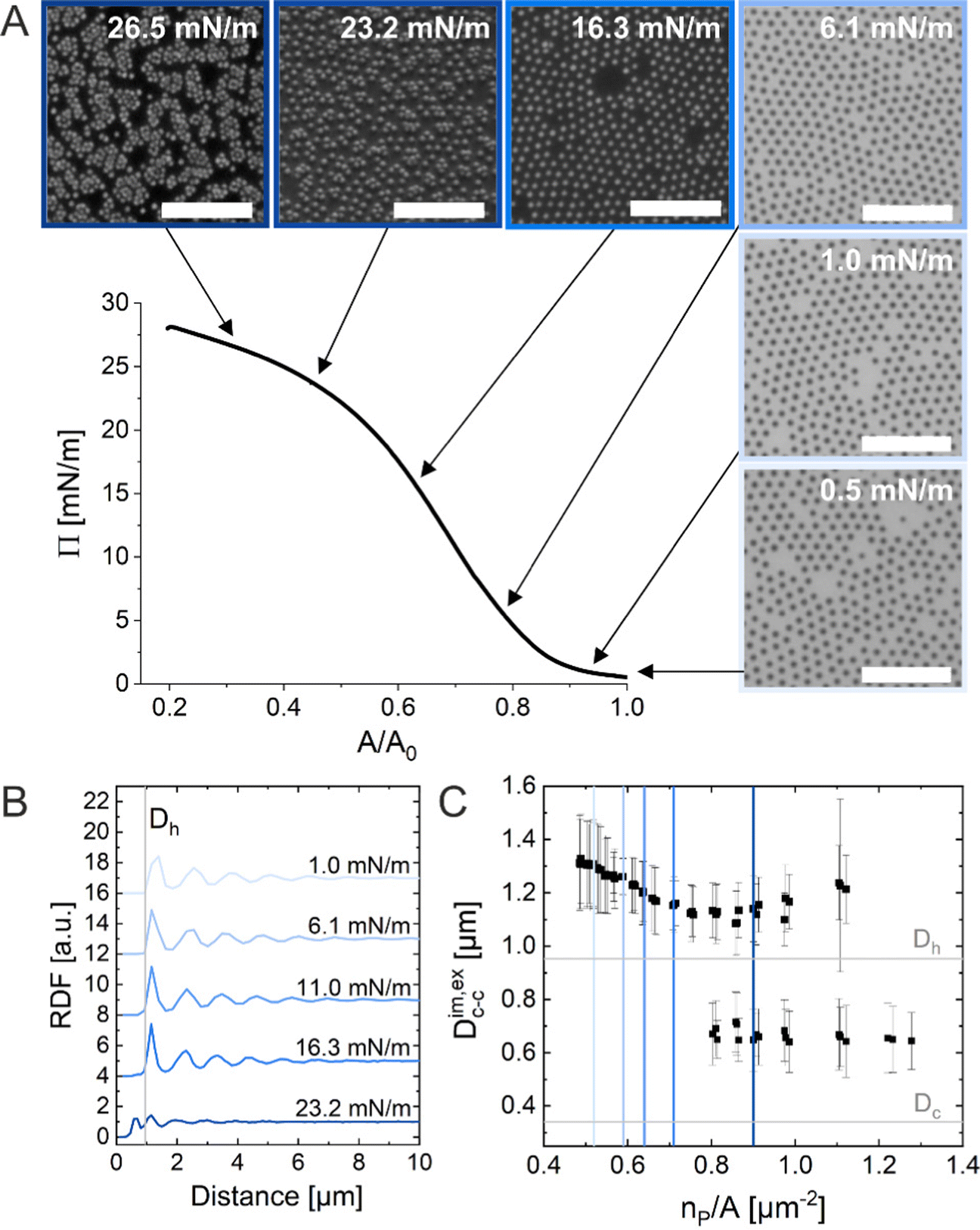 | ||
| Fig. 1 (A) Compression isotherm of the CS microgels at the air/water interface: surface pressure (Π) as a function of normalized area (A/A0). The inserted images correspond to optical microscopy images taken ex situ using the substrate-supported monolayers obtained from simultaneous Langmuir–Blodgett deposition (dried monolayer). The black arrows indicate the corresponding Π for each microscopy image. The scale bars correspond to 10 μm. (B) Radial distribution functions (RDF) for different Π. (C) Interparticle distance Dim,exc–c as a function of the number of particles per unit area (np/A). The colored vertical reference lines indicate the corresponding Π from (B). | ||
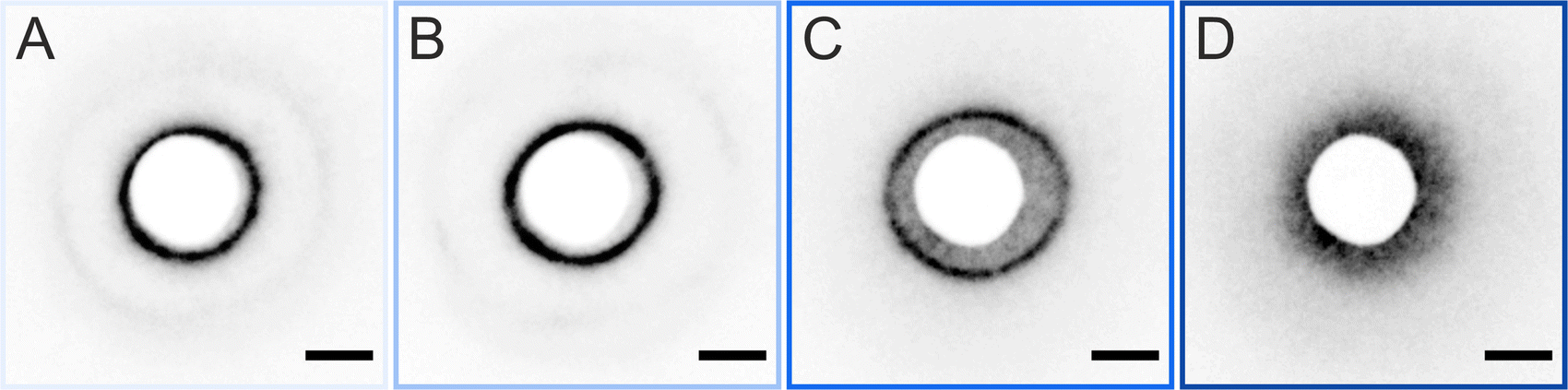 | ||
| Fig. 2 Scattering patterns of the dried CS microgel monolayer. The corresponding surface pressures (Π) are (A) 0.5 mM m−1, (B) 6 mM m−1, (C) 19 mM m−1, and (D) 24 mM m−1. The scale bars correspond to 10 mm. | ||
In summary, the ex situ analysis reveals that the CS microgel monolayers undergo an “isostructural solid–solid phase transition” upon compression, in agreement with previous studies.29,40
Method 2 (in situ microscopy)
In situ analysis of the monolayers of CS microgels at the air/water interface under compression was done by combining fluorescence microscopy with a microscopy trough, i.e., a trough equipped with an optical window. Fig. 3A shows representative microscopy images taken at various values of Π during compression. At near zero Π, we observe clusters of CS microgels due to attractive (capillary) interparticle interactions (see Fig. S1 in ESI†), as also reported for other large (Dh > 700 nm) coreless and CS microgels.8,44–46 In the regime of low Π, we observe similar microstructures as for the ex situ analysis after transfer to a substrate. However, the degree of order appears to be lower at the air/water interface. For medium-to-high values of Π, the comparison with ex situ results reveals striking differences: unlike Dim,exc–c, the in situ interparticle distances, Dim,exc–c, continuously decrease and the degrees of order increase with increasing Π. An “isostructural solid–solid phase transition” is not observed, in contrast to the assembly behavior reported for other similarly-sized coreless and CS microgels.29,44 This becomes even more evident when looking at selected RDFs as presented in Fig. 3B. The first double peak in the RDFs for higher Π is not present. Furthermore, the higher degree of order is reflected by the large number of distinct peaks in the RDF computed for the highest Π. In contrast to the high Π regime studied in the ex situ analysis, the monolayer possesses pronounced long-range order when studied at the air/water interface. Fig. 3C shows the evolution of Dim,exc–c with nP/A in direct comparison with the values obtained from ex situ analysis (Dim,exc–c, shadowed area). The data clearly shows a continuous decrease in Dim,exc–c with increasing nP/A, indicating a continuous compression of the soft colloidal monolayer. Starting from approximately 1.5 μm, Dim,exc–c decreases linearly with increasing nP/A until a pronounced deviation from the ex situ results appears when approaching distances that are close to Dh. The final values, at high compression, are slightly larger than half the initial Dim,exc–c and lie – until the monolayer buckles – in between the two distinct distances (shell–shell and “core–core”) determined by the ex situ analysis.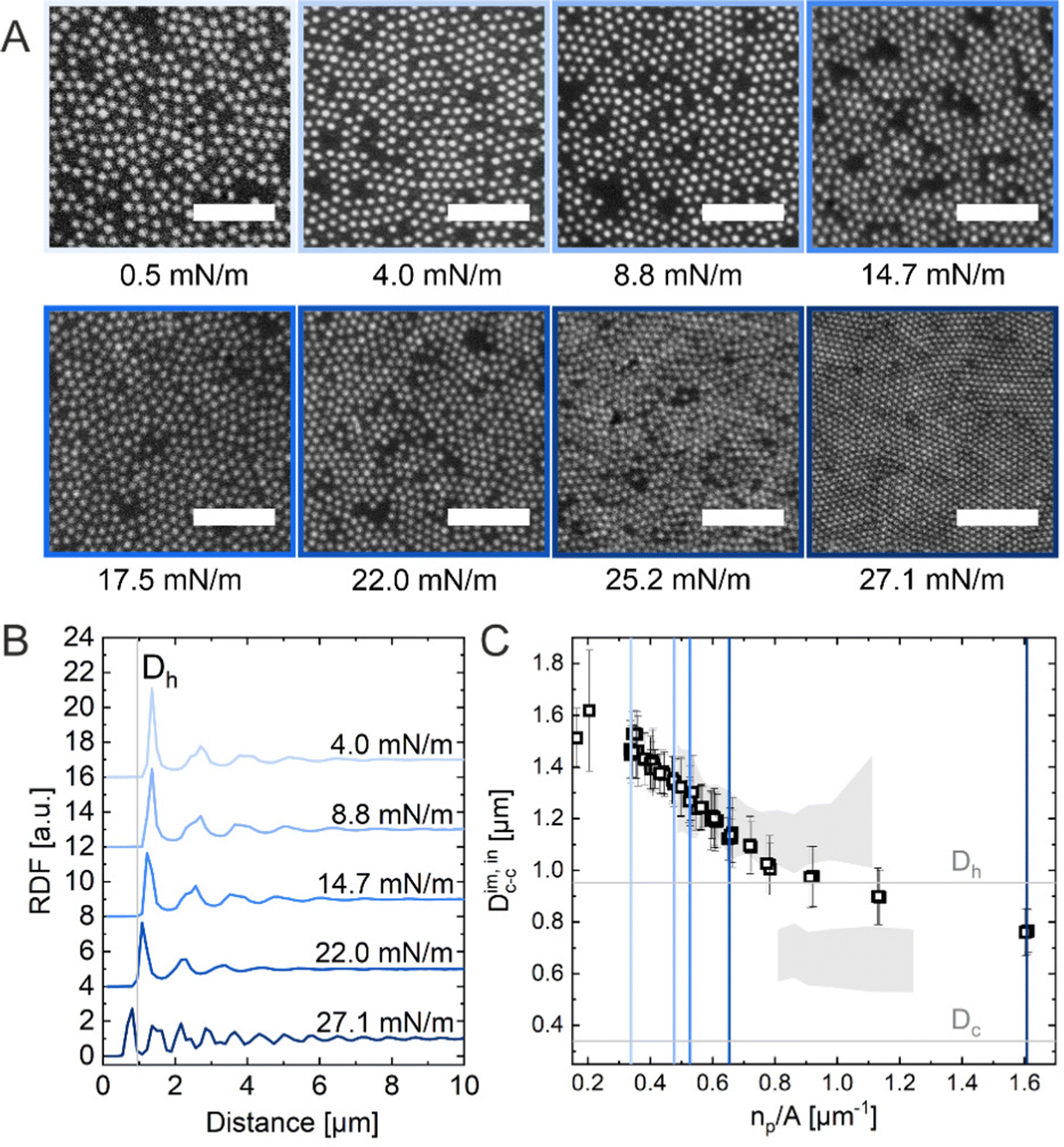 | ||
| Fig. 3 (A) Fluorescence microscopy images of the CS microgel monolayer at the air/water interface at various surface pressures (Π). The scale bars correspond to 10 μm. (B) Radial distribution functions (RDF) for different Π. (C) Interparticle distance Dim,exc–c as a function of the number of particles per unit area (np/A). The colored vertical reference lines indicate the corresponding Π from (B) and the shadowed area represents the data points from the ex situ measurements (Fig. 1C). | ||
To summarize, the in situ measurements using fluorescence microscopy revealed significant differences not only in the microstructure of the monolayer but also in terms of the evolution of the interparticle distance and a noticeable shift in np/A for corresponding Π (see Fig. S2A and B in ESI† for more details). For the studied CS microgels, these findings point towards a pronounced drying and/or substrate effect upon transfer by Langmuir–Blodgett deposition, as typically performed for such ex situ microstructure analysis. We address this further when discussing the LT-SALS experiments in the next section.
Method 3 (in situ LT-SALS)
We realized a custom-built setup that combines a Langmuir trough featuring a transparent glass window in the trough bottom (microscopy trough) with a custom-built SALS setup that allows to measure diffraction patterns at high frame rates (up to 30 frames per second, in our case). The details of this setup are provided in the Experimental Section and in the ESI.† Furthermore, the ESI† addresses the achievable q-range for various laser wavelength highlighting the versatility of the presented LT-SALS method. Fig. 4 shows the compression isotherm along with six selected frames recorded by LT-SALS. The selected frames are correlated to the respective values of Π in the isotherm as indicated by the black arrows. The full video recorded during the compression can be found in the ESI.† The diffraction patterns evolve from a small to a larger ring in a continuous manner in the low to mid Π regime, indicating a continuous decrease in Dc–c in real space. This continuous evolution of the diffraction ring goes on well beyond the critical value of Π where the structural transition was observed in the ex situ analysis (Method 1 and Fig. 2). This is in agreement with the results from in situ analysis using fluorescence microscopy (Method 2). As the compression proceeds approaching the high Π regime, the diffraction pattern moves more rapidly away from the center revealing diffraction peaks with six-fold symmetry. After the maximum Π is passed at approximately 34 mN m−1, the scattering intensity around the beam stop increases abruptly, indicating the collapse of the monolayer (data are not shown; the collapse can be seen in the Video, ESI†).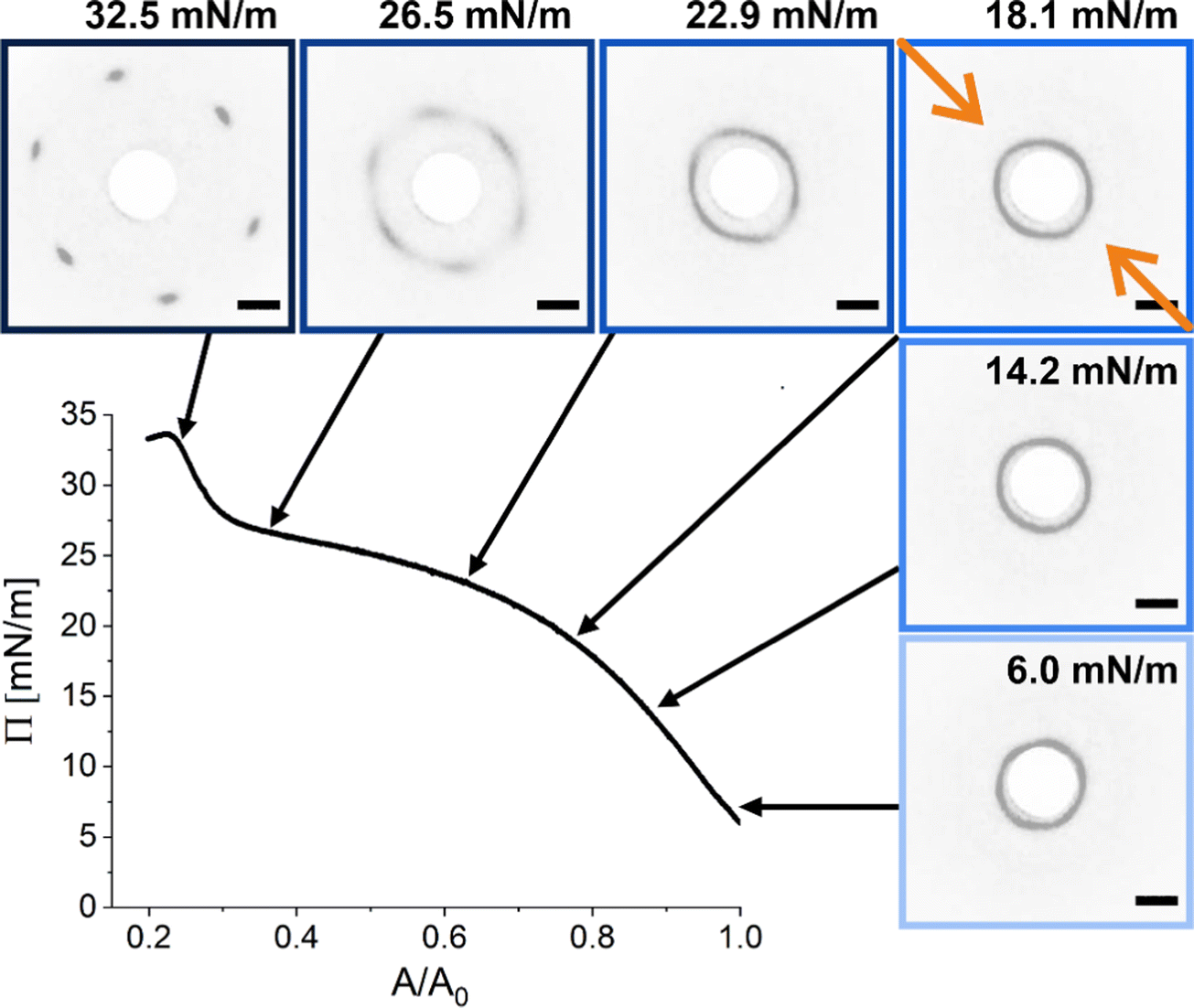 | ||
| Fig. 4 Compression isotherm of the monolayer of CS microgels at the air/water interface: surface pressure (Π) as a function of the normalized area (A/A0). The images are diffraction patterns of selected frames from a video recorded during the compression (see also ESI†). The black arrows indicate the corresponding Π at time of measurement. The greyscale of the images was inverted for better visibility. The orange arrows indicate the compression direction and the white circle at the center is the beam stop. The scale bars correspond to 10 mm (real space dimensions on the detection screen). | ||
Fig. 5A shows the radial averaging of the intensity of the SALS images as a function of the magnitude of the scattering vector q. The resulting scattering profiles show single peaks that correspond to the structure factor of the monolayer. With increasing compression, the position of the structure factor maximum shifts to larger q, i.e., smaller real space distances, and its intensity drops significantly in the high Π regime. The oval shape of the diffraction pattern in the mid Π regime and the increasing full width at half maxima (FWHM) in radial averaging provide interesting insight into the order of the uniaxially compressed monolayer. This, however, is out of the scope of this work and thus will not be discussed here. The calculated real space interparticle distance from the position of the structure factor maximum, DSALSc–c (see ESI† for more details), is plotted against Π in Fig. 5B, which also contains the values of Dim,exc–c from the in situ fluorescence microscopy (Method 2) as shadowed area for direct comparison. The two data sets overlap and demonstrate the continuous decrease in interparticle distance with increasing Π. There is no indication of the “isostructural solid–solid phase transition” as observed in the ex situ analysis (Method 1). The increase in degree of order during the compression can be monitored by azimuthal averaging at the respective structure factor maximum as shown in Fig. 5C. With increasing Π, the azimuthal profiles show a transition from a rather isotropic signal to pronounced Bragg peaks at 60° intervals indicating the hexagonal arrangement of the CS microgels in the monolayer. The FWHMs of all six peaks were averaged (FWHMavg) and plotted against DSALSc–c in Fig. 5D. The FWHMavg notably lowers before the DSALSc–c reaches values similar to Dh, which supports the previous observation of increasing order.
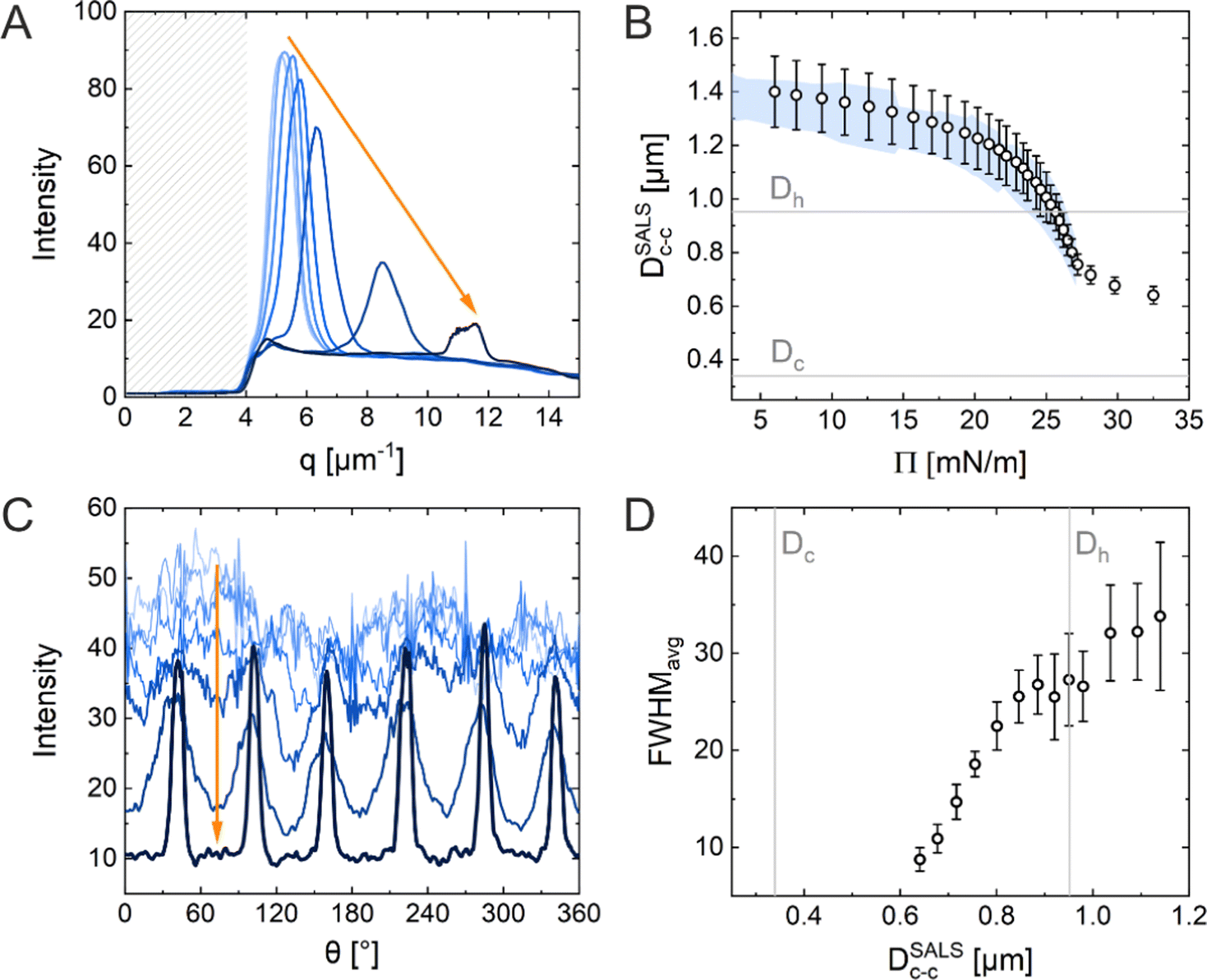 | ||
Fig. 5 (A) Normalized integrated scattering intensity as a function of the magnitude of the scattering vector ![[q with combining right harpoon above (vector)]](https://www.rsc.org/images/entities/i_char_0071_20d1.gif) obtained from radial averaging of the diffraction patterns. The shadowed area indicates the area covered by the beam stop. (B) Calculated interparticle distances obtained from structure factor analysis of the radially averaged data shown in (A), DSALSc–c as a function of surface pressure (Π). The blue-colored area represents the data set from the in situ fluorescence microscopy. (C) Normalized integrated intensity as a function of the azimuthal angle (θ) as analysis results of azimuthal averaging. (D) Width of the Bragg peaks FWHMavg averaged for all six peaks as a function of DSALSc–c. The vertical grey lines highlight the core and total CS microgel diameter. obtained from radial averaging of the diffraction patterns. The shadowed area indicates the area covered by the beam stop. (B) Calculated interparticle distances obtained from structure factor analysis of the radially averaged data shown in (A), DSALSc–c as a function of surface pressure (Π). The blue-colored area represents the data set from the in situ fluorescence microscopy. (C) Normalized integrated intensity as a function of the azimuthal angle (θ) as analysis results of azimuthal averaging. (D) Width of the Bragg peaks FWHMavg averaged for all six peaks as a function of DSALSc–c. The vertical grey lines highlight the core and total CS microgel diameter. | ||
In conclusion, the results obtained using the three Methods (ex situ microscopy, in situ microscopy and in situ LT-SALS) are compared in Fig. 6A. The grey and blue shadowed areas illustrate the calculated interparticle distances from LT-SALS and in situ fluorescence microscopy, respectively, whereas the filled squares correspond to the ex situ measurements. The graph highlights the conflict between the ex situ and in situ analysis of our CS microgel monolayers. The continuous evolution of interparticle distance in monolayers at the air/water interface during the continuous compression was also observed for other CS microgels, as illustrated in Fig. 6B and C, where the interparticle distance (normalized by the core diameter) is plotted as a function of Π for CS microgels with different shell thickness (Fig. 6B) and overall hydrodynamic diameters ranging from 770 to 1170 nm (Fig. 6C).
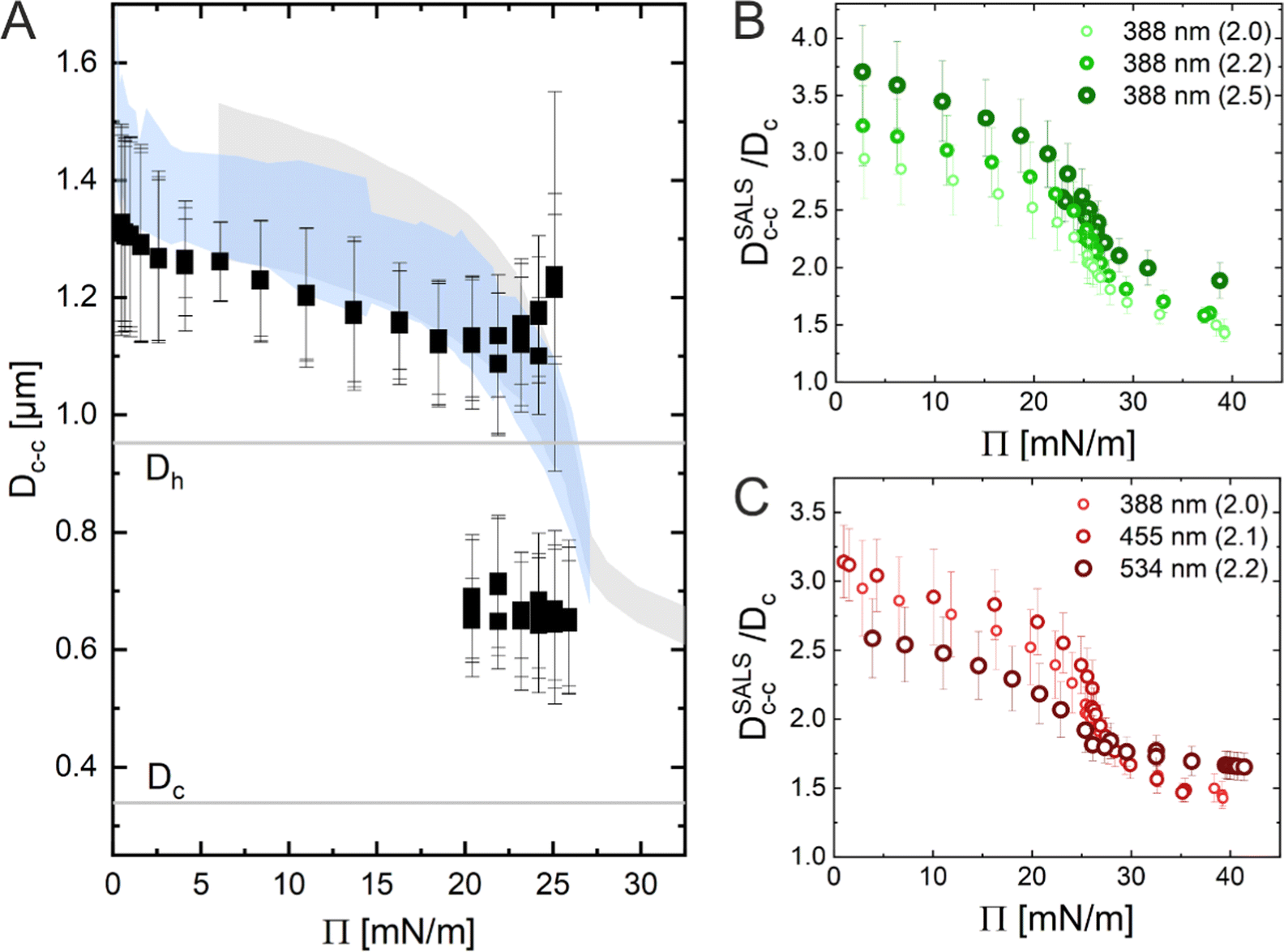 | ||
| Fig. 6 (A) Dc–cversus surface pressure (Π) plot illustrating the difference between ex situ (black squares) and in situ (blue and grey shadows) measurements. (B) LT-SALS analysis results from core–shell microgels with various shell thickness and (C) different overall sizes. The numbers outside of the parentheses in the legend denote the diameter of the silica core (i.e. 388 nm) and the number inside of the parentheses their shell-to-core size ratio (i.e. 2.0). | ||
Application of LT-SALS to other colloidal systems
In this section, we would like to briefly emphasize the versatility of LT-SALS by showing data for two additional representative colloidal systems, i.e., silica particles (as an example of rigid spheres) and PNIPAM microgels without rigid cores (as an example of soft spheres). In Fig. 7, the compression isotherms are shown along with the diffraction patterns for selected values of Π.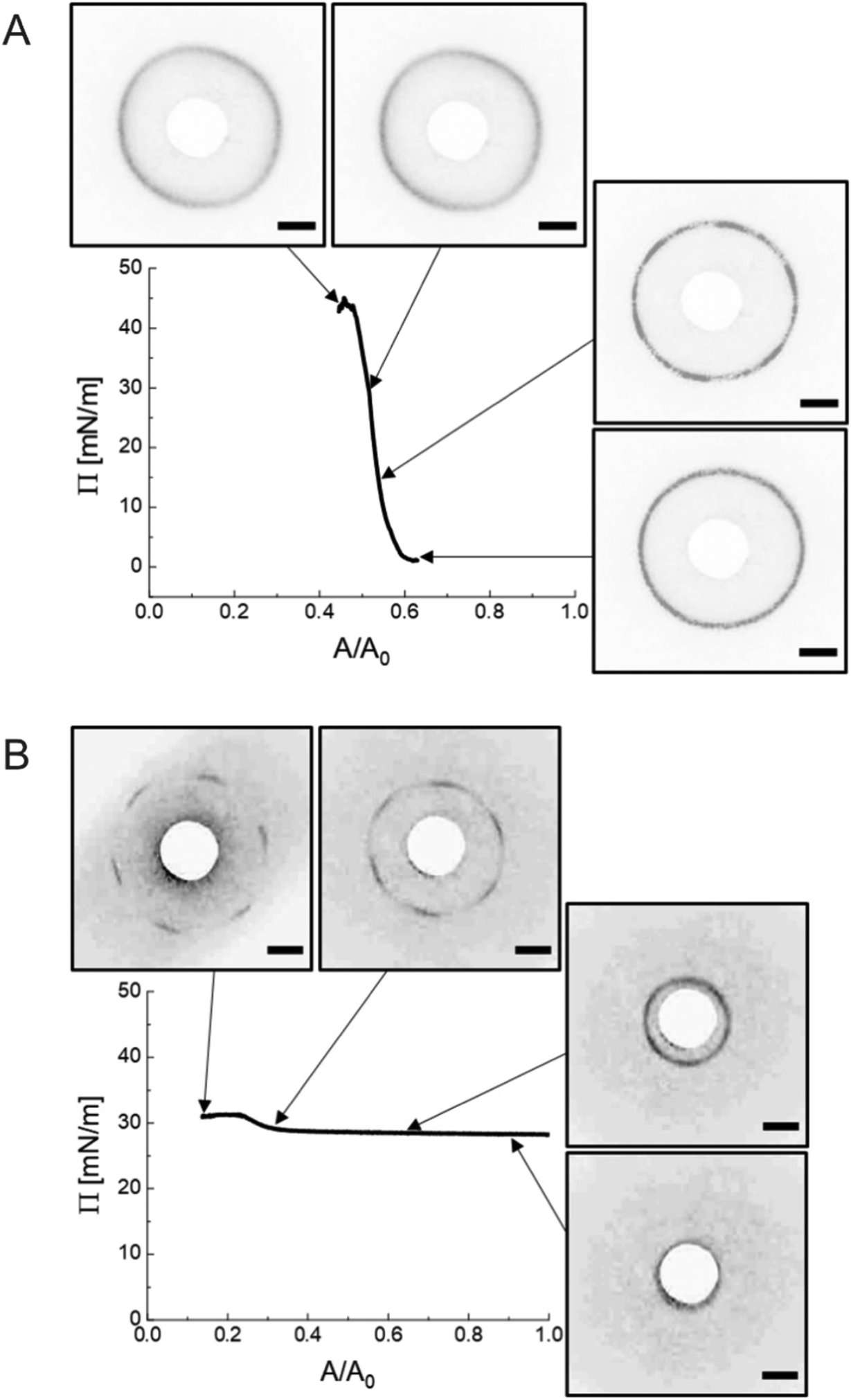 | ||
| Fig. 7 (A) Π against A/A0, compression isotherm for silica particles along with the diffraction patterns at corresponding Π. (B) Π against A/A0, compression isotherm for PNIPAM microgels along with the diffraction patterns at corresponding Π. The contrast of the images was adjusted for better visibility. The scale bars correspond to 10 mm. | ||
The incompressible nature of the silica particles (diameter measured by transmission electron microscopy, DTEM = 695 ± 22 nm) with a nearly hard-spheres interaction potential is well depicted by the steeply increasing Π over relatively small changes in accessible area A (Fig. 7A). The diffraction images corresponding to different values of Π reveal diffraction rings indicative of polycrystalline microstructures without any preferred domain orientation, as reported for other hard sphere and hard sphere-like systems.47–49 As such, there are no distinguishable Bragg peaks. During compression, the position of the diffraction ring remains nearly unchanged (DSALSc–c = 719 ± 15 nm). Therefore, the system is characterized by only one length scale from the beginning to the end of the compression as expected for hard spheres in contact. This characteristic length scale, i.e. in situ interparticle spacing, of the silica particle monolayer is also present in the ex situ microscopic images and the diffraction patterns of the dried silica monolayer as shown in Fig. S3 (ESI†).
The diffraction pattern obtained from the monolayers of the PNIPAM microgels (Dh = 858 ± 41 nm) goes through a transition from a diffraction ring (unordered state) to six distinct Bragg peaks (hexagonally ordered state) near the maximum Π. The monolayers showed rather small changes in Π per area reduced over the course of compression. The experiment was conducted in a highly compressed state not only because the high Π regime is where the structural change is most visible but also because the interparticle distance in the low Π regime is far too large to be resolved by diffraction analysis with our current setup. Fig. 7B illustrates LT-SALS measurement from the final stage of the compression. The position of the diffraction patterns changes from the edge of the beam stop (DSALSc–c ≈ 1690 nm) and moves away from the center to the furthest peak position, i.e., the smallest possible interparticle distance (DSALSc–c = 762 ± 128 nm), although there is only a slight change in Π. The in situ fluorescence microscopy (Method 2) at lower Π (0–29.2 mN m−1) in Fig. S4 (ESI†) confirms that the interparticle distance at the air/water interface evolves in a continuous manner throughout the compression also for these coreless microgels. Additionally, the contrast in the spatial arrangements between the in situ monolayer and the dried microgels (Fig. S5, ESI†), which resembles reported dried monolayer of similarly sized CS microgels,40 further supports our conclusion that the drying process accompanies structural changes.
Discussion
“Isostructural phase transitions” in microgel-laden monolayers under compression have been ascribed to the combination of attractive capillary forces and local failures of the polymer shells, which would otherwise prevent “core–core” contact. Both contributions depend on various parameters including the degree of deformability of the shells (which is mainly determined by the crosslinker density50), the size of the core and the shell, the materials, and the overall synthesis protocol. Ex situ measurements suggest that microgels with low crosslinker density show a more continuous evolution of the interparticle distance, whereas higher crosslinker densities give rise to “isostructural phase transitions”.29,31–33 However, we want to note that small microgels with low crosslinker density tend to self-assemble into less ordered structures.51 Phase transitions seem also to be more likely when large microgels (e.g., Dh = 1450 nm44) are used or when the polymer shells are thicker in the case of CS microgels.29 Nonetheless, there exist still several controversial results; for example, Vogel et al.52 and Rauh et al.12 studied CS microgels of similar size, but “isostructural phase transitions” were only observed in ref. 12. Importantly, all these results are based on ex situ measurements and only recent works started to provide in situ data.34,53 In particular, acquiring structural information on statistically relevant areas remains challenging.In this manuscript, we used in situ methods (Method 2 and Method 3) to investigate monolayers of CS microgels of size and crosslinker density similar to references29,33,40,46 and, to a smaller extent, monolayers made of coreless microgels similar to ref. 44 and 45. In all cases, our results strongly point towards a continuous evolution of the interparticle distance, i.e. no “isostructural phase transition”. The direct comparison with ex situ measurements (Method 1) suggests that the “structural transitions” are an artifact of the transfer and/or drying process. As such, the conflicting literature can be partly explained by taking into account the further complexity introduced by the ex situ measurement protocol. For example, in contrast to the often applied synchronized Langmuir–Blodgett deposition during compression also depositions at fixed surface pressures were performed.31 What happens when a microgel-laden monolayer is transferred onto a solid substrate? Fig. 8A illustrates a sketch of a typical CS microgel, while Fig. 8B1 and C1 depict CS microgels with shells of different deformability at the air/water interface. As water evaporates, the microgels approach the substrate and the bottom part of the microgels will start to touch the substrate, most likely causing further deformations as illustrated in Fig. 8B2 and C2. The contact area between the microgels and the substrate and the resulting adhesion depends on (1) the properties of the microgels (e.g., their morphology), the ones of the underlying surface (e.g., its wettability) and the transfer protocol (e.g., deposition speed).54,55 As the level of subphase lowers further, the microgels protrude more and more from the liquid film as shown in Fig. 8B3 and C3, leading to a deformation of the meniscus and attractive immersion capillary forces.56–58 Although these forces have not been measured experimentally for CS microgels, they qualitatively explain the formation of clusters as monolayers are transferred to the substrate.
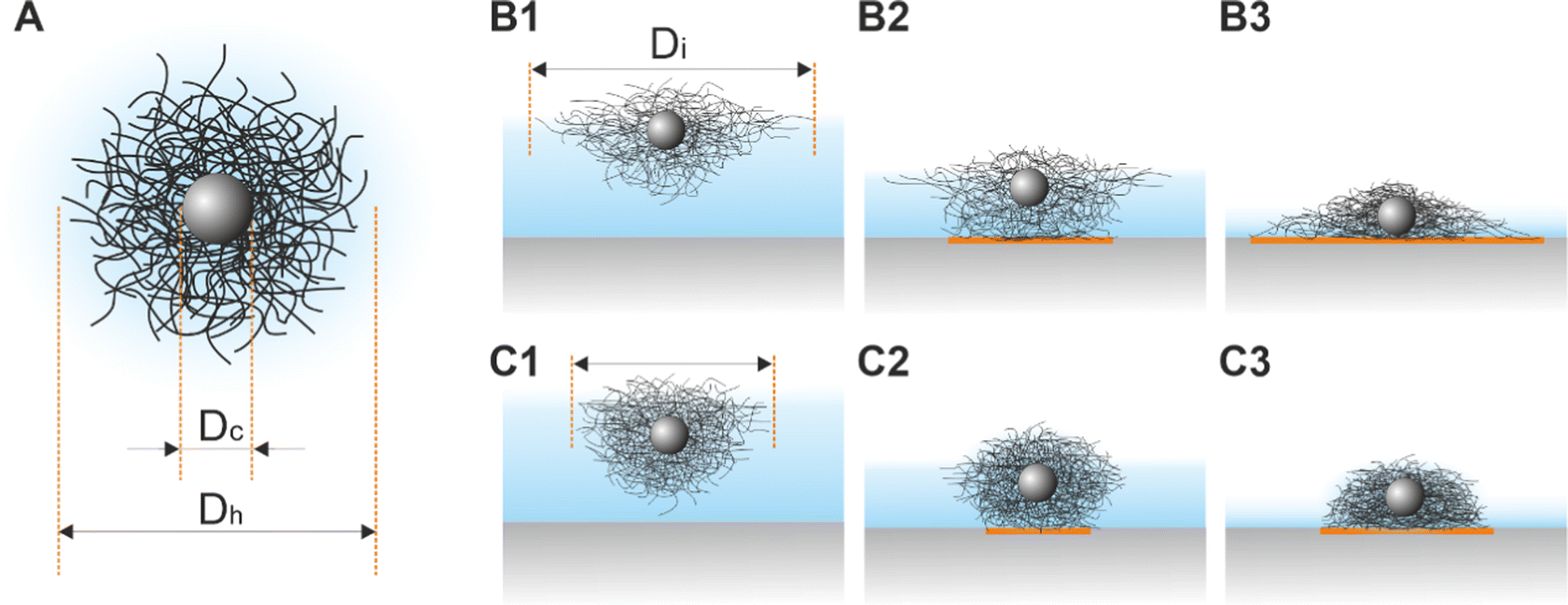 | ||
| Fig. 8 (A) Schematic illustration of the structure of a CS microgel. Dc denotes the diameter of the core and Dh the hydrodynamic diameter. (B1–B3) CS microgels with high deformability adsorbed at the air/water interface at three different drying stages. Di denotes the diameter at the interface and the orange area depicts the contact area between the microgel and the substrate. (C1–C3) the same set of illustrations for CS microgels with low deformability. | ||
We briefly verified that the transfer protocol affects the ex situ assemblies by drying CS microgel monolayers with overall hydrodynamic diameters ranging from approximately 500 to 1000 nm59 (5 mol% crosslinker density) with two different drying conditions; ‘slow’ drying at ambient conditions against open air, and ‘fast’ drying using a heat gun. Fig. 9 shows that structural changes – consistent with an “isostructural phase transition” at the interface – appear only after slow evaporation (blue panels). This observation implies that the microgels have enough time to rearrange when the monolayer is dried slowly under ambient conditions. This is in line with the experimental and theoretical findings of Volk et al.60 It is noteworthy, however, that the “freezing” of monolayers by fast drying has its limits and will depend on the core dimension, the shell-to-core size ratio, and the crosslinker density. A similar conclusion was drawn by Vasudevan et al.46 Furthermore, as the temperature influences the microgel fraction in the water subphase and mostly along the vertical direction,53 differences in the adhesion and contact area with the substrate during drying are expected. The AFM images of Fig. S6 (ESI†) reveal such a structural difference between slow and fast-dried monolayers. In particular, the phase images show the difference in contact area on the substrate. Similarly, Bochenek et al. have also reported that the drying conditions have a direct influence on the resulting microstructure.41,42
 | ||
| Fig. 9 Influence of drying conditions of CS microgel monolayers transferred to solid substrates at different surface pressures of 10 mN m−1 (A), 20 mN m−1 (B), and 30 mN m−1 (C). Left panel: Fast drying using a heat gun. Right panel: Slow drying against air at ambient conditions. Shown are results from CS microgels with different core sizes (105, 245, and 388 nm, from top to bottom) and shell-to-core size ratios. The scale bars correspond to 5 μm. The inset in C2 is an AFM image of the corresponding monolayer. The scale bar corresponds to 2 μm. The microgels are labeled with core diameter along with their shell-to-core size ratio in parentheses. | ||
Conclusion
In this work, we have investigated the isothermal compression of different colloidal monolayers assembled at air/water interfaces using a Langmuir trough in combination with a self-built setup for small-angle light scattering measurements (LT-SALS). This setup allowed us to measure the interparticle distances and characterize the structural order of the monolayers in situ, while the total available surface area was continuously reduced by the barriers of the Langmuir trough. When using core–shell microgels with rigid cores and soft and deformable shells, we found stark differences between microstructures analyzed ex situ (i.e., monolayers that were transferred to solid substrates) in comparison with the in situ structural analysis based on optical diffraction. The ex situ analysis revealed an “isostructural phase transition” from core–shell microgels in shell–shell contact to “core–core” contact during compression. In contrast, the in situ analysis revealed a continuous decrease of interparticle distance as the monolayer is compressed. No phase transition was observed. This key result was also confirmed by in situ real space analysis of the monolayer using fluorescence microscopy. As a proof of concept, we also demonstrated that the in situ investigation using small-angle light scattering can be also applied to monolayers of rigid particles as well as low optical contrast PNIPAM microgels.LT-SALS is fast, non-destructive and relatively easy to set up from low cost components. Compared to in situ optical microscopy, it has several important advantages: (1) very large monolayer areas >1 mm2 can be probed. Such large areas correspond to, for example, >40 × 104 core–shell microgels that are simultaneously probed. (2) It is not necessary to have markers or strong refractive index contrast in colloid systems under investigation. (3) The measurement is less sensitive to external interferences such as vibrations. (4) Microstructural phase transitions become evident immediately due to changes of the diffraction pattern, i.e., a transition from a diffraction ring to Bragg peaks revealing the transition from a disordered to an ordered state. (5) The processing and analysis (e.g., radial averaging, peak position and width) of the diffraction patterns is much less prone to errors and less time-consuming when compared to real space analysis of microscopy images for which the centers of mass of all imaged particles have to be identified.
We believe that the presented methodology will stimulate further research on colloidal monolayers at liquid/liquid or air/liquid interfaces, in particular when softness and deformability of objects are studied.50,61 Furthermore, the fact that phase transitions can be directly monitored in situ at the respective interface will allow systematic studies required to achieve a more comprehensive understanding of colloidal assembly at interfaces22 and enable on-demand tailoring of colloidal microstructures on solid substrates – provided that the transfer protocol to the substrate is suitable to maintain the microstructure. The next important steps in this line of research are further investigation on the role of the transfer protocol as well as the surface chemistry of the substrate during the drying procedure.
Materials
Ethanol (Sigma-Aldrich, 99.8%), ethanol (Heinrich-Heine-University, central chemical storage, p.a.), tetraethyl orthosilicate (TEOS, Sigma-Aldrich, 98%), chloroform (Fischer Scientific, 99.8%), ammonium hydroxide solution (NH3 (aq.), AppliChem, 32%), rhodamine B isothiocyanate (RITC, Sigma-Aldrich, mixed isomers), methacryloxyethyl thiocarbamoyl rhodamine B (MRB, Polysciences, Inc.), 3-aminopropyltrimethoxysilane (APS, Sigma-Aldrich, 97%), 3-(trimethoxysilyl)propyl methacrylate (MPS, Sigma-Aldrich, 98%), N,N′-methylenebisacrylamide (BIS, Sigma-Aldrich, 98%), potassium peroxodisulfate (KPS, Sigma-Aldrich, 99%), and sodium chloride (NaCl, Sigma-Aldrich, 99.5%) were used as received. Water was purified by Milli-Q system (18.2 MΩ cm−1) and N-isopropylacrylamide (NIPAM, TCI, 97%) by recrystallization from cyclohexane (Fisher Scientific, 99.8%).Synthesis
Silica particles and silica-PNIPAM CS microgels
The detailed synthesis protocol for both silica nanoparticles and micron-sized silica-PNIPAM microgels can be found in.59 In short, silica particles were synthesized via the well-known Stöber procedure. RITC dye was incorporated in the particles that were used for fluorescence microscopy experiments. The PNIPAM shell encapsulation was done via seeded precipitation polymerization.The silica particles used to create monolayers at the air/water interface were measured to be 695 ± 22 nm (126 particles counted) in diameter by TEM. Its ethanolic dispersion was mixed with chloroform with 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4 volume ratio to assist the spreading of the silica particles at the air/water interface. Surface charges were screened by adding 100 mM NaCl in the aqueous subphase of the Langmuir trough in order to achieve rigid sphere-like interactions.
:4 volume ratio to assist the spreading of the silica particles at the air/water interface. Surface charges were screened by adding 100 mM NaCl in the aqueous subphase of the Langmuir trough in order to achieve rigid sphere-like interactions.
The main CS microgels used for the in situ and ex situ comparison had a core with a diameter of 340 ± 20 nm. The Dh of the total CS microgel was measured to be 920 ± 18 nm at 20 °C using dynamic light scattering (DLS). The purified dispersion was freeze-dried, re-dispersed in ethanol with 5 w/v% and stored on a 3D shaker overnight prior to the monolayer deposition at the air/water interface. Ethanol was used as spreading agent.
PNIPAM microgel synthesis
The synthesis protocol for the PNIPAM microgels was adopted from a previously published work.62 5 g of PNIPAM and 50 mg of BIS were dissolved in 50 mL of water in a three-neck round-bottom flask equipped with a reflux condenser and a magnetic stirrer. 1 mg of MRB dye was dissolved in 1 mL of water and added to the flask. The mixture was heated to 40 °C and purged with nitrogen while stirring. 20 mL of the mixture was transferred to another flask, where 10 mL of additional water was added. The rest was kept in a syringe with a needle and placed on a syringe pump for the continuous feeding of the monomers. The mixture in the flask was heated to 80 °C and equilibrated. 10.4 mg of KPS was dissolved in 2 mL water and added to the flask. Once the dispersion started to become turbid, indicating that the polymerization was initiated, the syringe pump was started with the speed of 1 mL min−1. 5 minutes after the feeding process, the polymerization was quenched by dipping the flask in an ice bath and the dispersion was filtered through glass wool. The synthesized PNIPAM microgels were dialyzed against water for two weeks, freeze-dried and re-dispersed in ethanol (1 w/v%) as for the CS microgels. The size of the PNIPAM microgels at 20 °C was determined by DLS (Dh = 858 nm ± 41 nm).Experimental
Ex situ investigation after Langmuir–Blodgett deposition (Method 1)
For the ex situ analysis, we followed the well-established protocol to study the phase behavior of CS microgel monolayers at the air/water interface.12,29,30 According to the protocol, the microgel monolayer at the air/water interface is simultaneously transferred to a substrate during the compression, dried as the substrate is pulled out and examined under a microscope, hence referred to as an ex situ approach. The total duration of the substrate pulled out is often matched with the total duration of the compression, consequently enabling the position of the substrate to the corresponding Π tracing. This link between the substrate position to Π was established under the assumption that the number of particles transferred from the air/water interface per time is negligible thus does not influence the measured value of Π.The transfer of the monolayer was carried out using a Langmuir–Blodgett deposition trough (Microtrough G2, Kibron Inc.) equipped with a film balance, two Delrin barriers, a dip coater and an acrylic cover box. A standard microscope glass slide was treated in an ultrasonic bath sequentially with Hellmanex aqueous solution (2 vol%), water (2 ×) and in ethanol (2 ×) for 15 minutes each. The cleaned glass slide was then cut in half along its length and the position markings were carved on its back to trace the corresponding Π at the moment of monolayer transfer (see Fig. 10A for a schematic illustration of the procedure). Before the deposition of the particle monolayer, the trough and the barriers were thoroughly cleaned with water, ethanol and again rinsed with water. The trough was then filled with water with the barriers closed. The glass slide was thoroughly rinsed with water before being mounted to the dip coater (parallel to the barriers and perpendicular to the air/water interface), positioned at the center of the trough and lowered 55 mm below the interface. An aspirator with a narrow tip was used to remove any residual floating substances at the interface between the two barriers as well as to flatten the interface by lowering its level to the height of the trough wall. A Wilhelmy plate was rinsed with water and ethanol and held over a flame to remove any impurities and cooled before it was mounted to the film balance. The barriers were then opened to the maximum area. Only when the fluctuation of the surface tension value was below 0.3 mN m−1 while opening the barriers, the particles were deposited at the air/water interface using a 10 or 20 μL micropipette. The colloidal dispersion was treated alternating between vortex mixing and sonication for 2–4 minutes prior to the deposition. The injection was done slowly at a shallow angle with the tip of the micropipette gently touching the interface. The compression was started with the speed of 150 mm2 min−1 after at least 15 minutes of equilibration time after deposition. The glass substrate was pulled out simultaneously at the speed of 84 mm min−1 over a time that matched the total duration of the compression. The glass substrate was left hanging until it was completely dried. Three images were taken every 2.5 mm using an upright microscope (Eclipse LV150N, Nikon). The acquired images were processed and analyzed by ImageJ (1.53k, National Institutes of Health, USA) alongside the recorded compression isotherm. Additional information regarding image processing can be found in ESI.† All experiments were done at room temperature. The interparticle distances were determined from the first peak of the radial distribution functions (RDFs) and are denoted as Dim,exc–c, where D stands for “distance”, im for “image”, ex for “ex situ” and c–c for “center-to-center”.
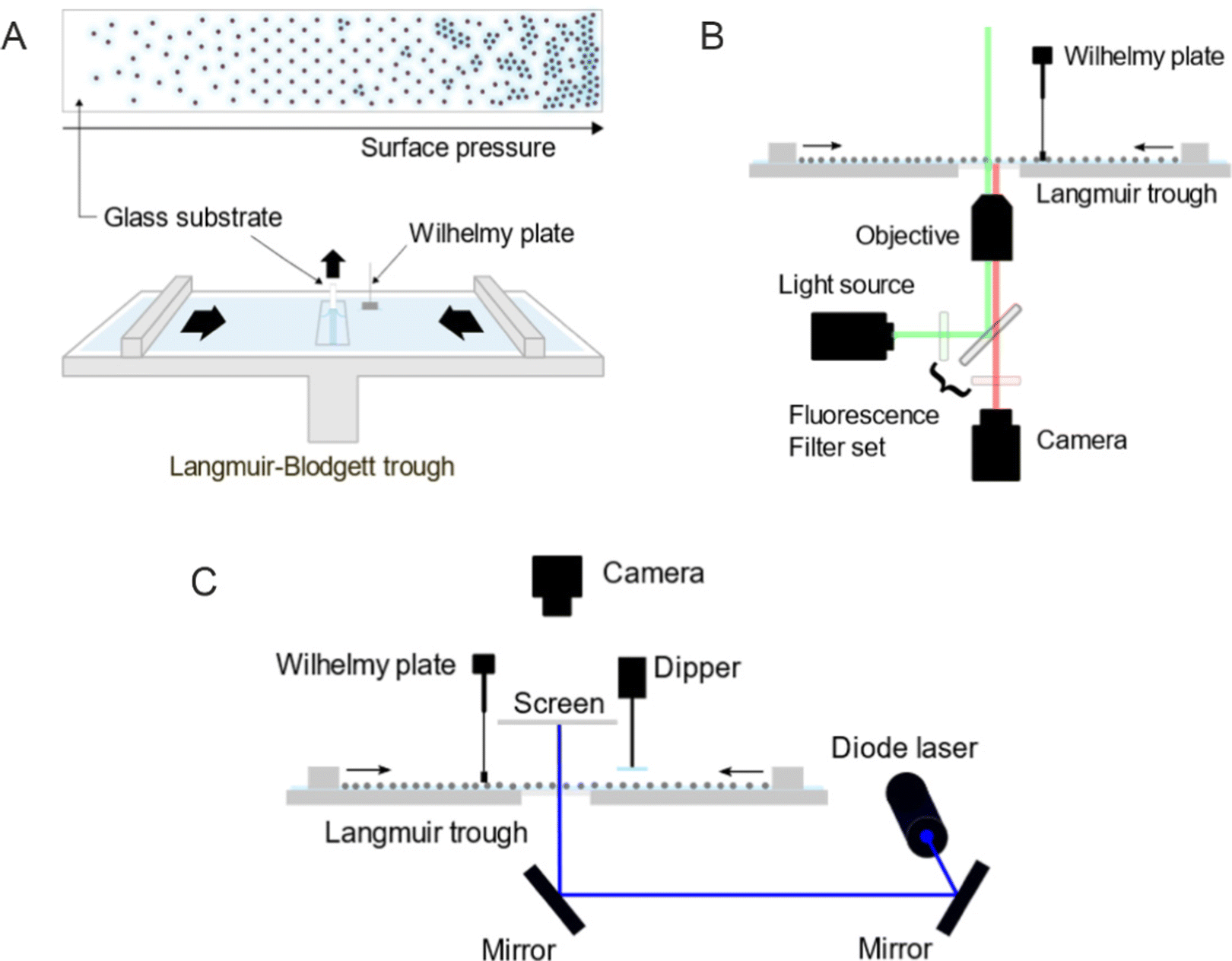 | ||
| Fig. 10 Schematic illustration of (A) the transfer of a colloidal monolayer to a glass substrate using a Langmuir–Blodgett trough, (B) the Langmuir trough combined with a fluorescence microscope, and (C) the Langmuir trough combined with a small-angle light scattering (LT-SALS) setup. | ||
In situ investigation by fluorescence microscopy combined with a Langmuir trough (Method 2)
The in situ measurements by fluorescence microscopy were conducted in another Langmuir trough (KSV NIMA inverted, Biolin Scientific) equipped with two Delrin barriers, an inverted microscopy trough and a black acrylic cabinet. A microscope (Olympus IX73) equipped with a mercury lamp, a fluorescence filter set, a CMOS camera and a 60× objective was used to probe the colloid-laden interface at various values of Π. The setup was placed on an optical table combined with pneumatic vibration isolation (Nexus, Thorlabs Inc.) and is illustrated in Fig. 10B. The microscopy trough was cleaned and prepared in the same way as for the ex situ experiments previously described. Once the air/water interface was clean enough (surface pressure fluctuation below 0.3 mN m−1, measured by a Wilhelmy plate), the microgels were deposited at the interface. The compression was done stepwise with the compression speed of 10 mm min−1. Images were taken after at least 15 minutes of equilibration time at each step. Three different volumes of the microgel dispersion were used and consequently three compression isotherms were measured to address the full range of Π. Three images were acquired from different positions for each measured Π and processed by ImageJ alongside the Π measured at the moment of image acquisition. All experiments were done at room temperature.In situ investigation by LT-SALS (Method 3)
The in situ measurements by LT-SALS were performed with the same Langmuir trough (KSV NIMA) used for the in situ fluorescence microscopy experiments. Along with the Langmuir trough, the cabinet was placed on an optical table, on which a blue diode laser (MediaLas, LDM-20-405, 20 mW, 405 nm) and two mirrors (Thorlabs, BB1-E02) were set up. A customized metal frames was installed around the Langmuir trough to mount a paper screen and a CCD camera (Thorlabs, DCU223C-MVL6WA) above the trough. The schematic of the setup is depicted in Fig. 10C (see also the ESI† for more details regarding the setup and laser alignment). The monolayer deposition at the air/water interface was done as for the in situ fluorescence microscopy study. The compression was started after at least 15 minutes of equilibration time with the speed of 150 mm2 min−1. The laser intensity was adjusted with a neutral density filter (Thorlabs, NDC-50C-4M). Diffraction patterns were recorded during the entire compression at 0.2 frames per second. The resulting video was analyzed alongside the recorded compression isotherm using ImageJ (one data point every five seconds). All experiments were done at room temperature.Dynamic light scattering (DLS)
DLS measurements were performed with a 3D LS spectrometer (LS Instruments) at a constant temperature of 20 °C. The measurements were repeated three times with 40 seconds of acquisition time. The device was equipped with a HeNe laser (632.8 nm), a decalin bath and two avalanche photodiodes in pseudo cross-correlation mode as detectors. The dilute samples (volume fraction ≪ 0.001) were measured in borosilicate cuvettes with an outside diameter of 10 mm. The obtained intensity-time autocorrelation functions were analyzed using cumulant analysis.Transmission electron microscopy (TEM)
TEM measurements were performed using a JEOL JEM-2100 Plus microscope operated in bright-field mode at 80 kV acceleration voltage. The sample preparation was done by applying a drop of the respective particle dispersion on a carbon-coated copper grid (200 mesh, Science Services) and drying at room temperature. The captured images were then processed with ImageJ for the size analysis.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
The authors acknowledge the German Research Foundation (DFG) and the state of NRW for funding the cryo-TEM (INST 208/749-1 FUGG). M. K. acknowledges the DFG for funding under grant KA3880/6-1. The authors also would like to thank Sonja Schiller (HHU Düsseldorf) for assistance in building the setup, Dr. Kiran Kaithakkal Jathavedan for providing the PNIPAM microgels, Marius Otten (HHU Düsseldorf) for TEM imaging and Jonathan Garthe (HHU Düsseldorf) for Langmuir–Blodgett deposition of many samples. K. K. would like to express special thanks to Prof. Michel Cloitre, Prof. Igor Potemkin, Prof. Jérôme J. Crassous, Prof. Janne-Mieke Meijer, Prof. Walter Richtering and others for inspiring lectures and fruitful discussions during the 3rd International Summer School on functional microgels and microgel systems. The authors acknowledge the reviewers for their constructive feedback and helpful suggestions.References
- R. Ghosh Chaudhuri and S. Paria, Chem. Rev., 2012, 112, 2373–2433 CrossRef CAS.
- Z. Gong, T. Hueckel, G.-R. Yi and S. Sacanna, Nature, 2017, 550, 234–238 CrossRef PubMed.
- M. He, J. P. Gales, É. Ducrot, Z. Gong, G.-R. Yi, S. Sacanna and D. J. Pine, Nature, 2020, 585, 524–529 CrossRef CAS.
- J. Zhang, B. A. Grzybowski and S. Granick, Langmuir, 2017, 33, 6964–6977 CrossRef CAS.
- P. F. Damasceno, M. Engel and S. C. Glotzer, Science, 2012, 337, 453–457 CrossRef CAS.
- H. R. Vutukuri, A. Imhof and A. Van Blaaderen, Angew. Chem., Int. Ed., 2014, 53, 13830–13834 CrossRef CAS PubMed.
- S. Ni, J. Leemann, I. Buttinoni, L. Isa and H. Wolf, Sci. Adv., 2016, 2, e1501779 CrossRef.
- S. A. Vasudevan, A. Rauh, L. Barbera, M. Karg and L. Isa, Langmuir, 2018, 34, 886–895 CrossRef CAS.
- J. Harrer, S. Ciarella, M. Rey, H. Löwen, L. M. C. Janssen and N. Vogel, Soft Matter, 2021, 17, 4504–4516 RSC.
- B. P. Binks and T. S. Horozov, Colloidal particles at liquid interfaces, Cambridge University Press, 2006 Search PubMed.
- M. Rey, M. Á. Fernández-Rodríguez, M. Steinacher, L. Scheidegger, K. Geisel, W. Richtering, T. M. Squires and L. Isa, Soft Matter, 2016, 12, 3545–3557 RSC.
- A. Rauh, M. Rey, L. Barbera, M. Zanini, M. Karg and L. Isa, Soft Matter, 2017, 13, 158–169 RSC.
- T. Kohoutek, M. Parchine, M. Bardosova and M. E. Pemble, Colloids Surf., A, 2020, 593, 124625 CrossRef CAS.
- I. T. Chen, E. Schappell, X. Zhang and C.-H. Chang, Microsyst. Nanoeng., 2020, 6, 22 CrossRef CAS.
- M. Parchine, J. McGrath, M. Bardosova and M. E. Pemble, Langmuir, 2016, 32, 5862–5869 CrossRef CAS PubMed.
- J. D. Joannopoulos, S. G. Johnson, J. N. Winn and R. D. Meade, Photonic Crystals: Molding the Flow of Light, Princeton Univ. Press, Princeton, NJ, 2008 Search PubMed.
- J. Ge and Y. Yin, Angew. Chem., Int. Ed., 2011, 50, 1492–1522 CrossRef CAS PubMed.
- M. Bardosova, M. E. Pemble, I. M. Povey and R. H. Tredgold, Adv. Mater., 2010, 22, 3104–3124 CrossRef CAS PubMed.
- S. Jiang, A. Van Dyk, A. Maurice, J. Bohling, D. Fasano and S. Brownell, Chem. Soc. Rev., 2017, 46, 3792–3807 RSC.
- G. G. Roberts, Adv. Phys., 1985, 34, 475–512 CrossRef CAS.
- M. Karg, A. Pich, T. Hellweg, T. Hoare, L. A. Lyon, J. J. Crassous, D. Suzuki, R. A. Gumerov, S. Schneider, I. I. Potemkin and W. Richtering, Langmuir, 2019, 35, 6231–6255 CrossRef CAS.
- D. Feller and M. Karg, Soft Matter, 2022, 18, 6301–6312 RSC.
- M. Rey, M. A. Fernandez-Rodriguez, M. Karg, L. Isa and N. Vogel, Acc. Chem. Res., 2020, 53, 414–424 CrossRef CAS PubMed.
- I. Buttinoni, M. Steinacher, H. T. Spanke, J. Pokki, S. Bahmann, B. Nelson, G. Foffi and L. Isa, Phys. Rev. E, 2017, 95, 012610 CrossRef PubMed.
- I. Buttinoni and R. P. A. Dullens, J. Phys.: Mater., 2021, 4, 025001 CAS.
- K. Dietrich, G. Volpe, M. N. Sulaiman, D. Renggli, I. Buttinoni and L. Isa, Phys. Rev. Lett., 2018, 120, 268004 CrossRef CAS PubMed.
- A. D. Law, D. M. A. Buzza and T. S. Horozov, Phys. Rev. Lett., 2011, 106, 128302 CrossRef PubMed.
- A. Yethiraj and A. Van Blaaderen, Nature, 2003, 421, 513–517 CrossRef CAS PubMed.
- J. S. J. Tang, R. S. Bader, E. S. A. Goerlitzer, J. F. Wendisch, G. R. Bourret, M. Rey and N. Vogel, ACS Omega, 2018, 3, 12089–12098 CrossRef CAS.
- J. Menath, J. Eatson, R. Brilmayer, A. Andrieu-Brunsen, D. M. A. Buzza and N. Vogel, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2113394118 CrossRef CAS PubMed.
- C. Picard, P. Garrigue, M.-C. Tatry, V. Lapeyre, S. Ravaine, V. Schmitt and V. Ravaine, Langmuir, 2017, 33, 7968–7981 CrossRef CAS PubMed.
- M. Rey, X. Hou, J. S. J. Tang and N. Vogel, Soft Matter, 2017, 13, 8717–8727 RSC.
- S. Ciarella, M. Rey, J. Harrer, N. Holstein, M. Ickler, H. Löwen, N. Vogel and L. M. C. Janssen, Langmuir, 2021, 37, 5364–5375 CrossRef CAS PubMed.
- J. Vialetto, S. N. Ramakrishna and L. Isa, arXiv, 2022, preprint, arXiv:2204.09324.
- T. Honold, K. Volk, A. Rauh, J. P. S. Fitzgerald and M. Karg, J. Mater. Chem. C, 2015, 3, 11449–11457 RSC.
- K. Volk, J. P. S. Fitzgerald, M. Retsch and M. Karg, Adv. Mater., 2015, 27, 7332 CrossRef CAS PubMed.
- E. A. Jagla, Phys. Rev. E, 1998, 58, 1478–1486 CrossRef CAS.
- T. Dotera, T. Oshiro and P. Ziherl, Nature, 2014, 506, 208–211 CrossRef CAS.
- W. R. C. Somerville, A. D. Law, M. Rey, N. Vogel, A. J. Archer and D. M. A. Buzza, Soft Matter, 2020, 16, 3564–3573 RSC.
- M. Ickler, J. Menath, L. Holstein, M. Rey, D. M. A. Buzza and N. Vogel, Soft Matter, 2022, 18, 5585–5597 RSC.
- S. Bochenek, A. Scotti, W. Ogieglo, M. Á. Fernández-Rodríguez, M. F. Schulte, R. A. Gumerov, N. V. Bushuev, I. I. Potemkin, M. Wessling, L. Isa and W. Richtering, Langmuir, 2019, 35, 16780–16792 CrossRef CAS.
- S. Bochenek, A. Scotti and W. Richtering, Soft Matter, 2021, 17, 976–988 RSC.
- J. Vialetto, N. Nussbaum, J. Bergfreund, P. Fischer and L. Isa, J. Colloid Interface Sci., 2022, 608, 2584–2592 CrossRef CAS.
- L. Scheidegger, M. Á. Fernández-Rodríguez, K. Geisel, M. Zanini, R. Elnathan, W. Richtering and L. Isa, Phys. Chem. Chem. Phys., 2017, 19, 8671–8680 RSC.
- S. Huang, K. Gawlitza, R. Von Klitzing, L. Gilson, J. Nowak, S. Odenbach, W. Steffen and G. K. Auernhammer, Langmuir, 2016, 32, 712–722 CrossRef CAS PubMed.
- S. A. Vasudevan, A. Rauh, M. Kröger, M. Karg and L. Isa, Langmuir, 2018, 34, 15370–15382 CrossRef CAS PubMed.
- N. L. Smith, A. Coukouma, S. Dubnik and S. A. Asher, Phys. Chem. Chem. Phys., 2017, 19, 31813–31822 RSC.
- X. Qin, W. Liu, G. Liu, C. Ren, C. Liu, H. Li and Y. Cao, J. Appl. Polym. Sci., 2020, 137, 49299 CrossRef CAS.
- Y. Cao, G. Liu, B. Zheng, X. Wang, H. Li, G. Wang, L. Zhao and Y. Wang, Soft Matter, 2021, 17, 4969–4978 RSC.
- A. Scotti, M. F. Schulte, C. G. Lopez, J. J. Crassous, S. Bochenek and W. Richtering, Chem. Rev., 2022, 122(13), 11675–11700 CrossRef CAS.
- A. Scotti, S. Bochenek, M. Brugnoni, M. A. Fernandez-Rodriguez, M. F. Schulte, J. E. Houston, A. P. H. Gelissen, I. I. Potemkin, L. Isa and W. Richtering, Nat. Commun., 2019, 10, 1418 CrossRef CAS.
- N. Vogel, C. Fernández-López, J. Pérez-Juste, L. M. Liz-Marzán, K. Landfester and C. K. Weiss, Langmuir, 2012, 28, 8985–8993 CrossRef CAS.
- S. Bochenek, F. Camerin, E. Zaccarelli, A. Maestro, M. M. Schmidt, W. Richtering and A. Scotti, Nat. Commun., 2022, 13, 3744 CrossRef CAS PubMed.
- L. Hoppe Alvarez, S. Eisold, R. A. Gumerov, M. Strauch, A. A. Rudov, P. Lenssen, D. Merhof, I. I. Potemkin, U. Simon and D. Wöll, Nano Lett., 2019, 19, 8862–8867 CrossRef CAS.
- L. Hoppe Alvarez, A. A. Rudov, R. A. Gumerov, P. Lenssen, U. Simon, I. I. Potemkin and D. Wöll, Phys. Chem. Chem. Phys., 2021, 23, 4927–4934 RSC.
- N. Denkov, O. Velev, P. Kralchevski, I. Ivanov, H. Yoshimura and K. Nagayama, Langmuir, 1992, 8, 3183–3190 CrossRef CAS.
- P. A. Kralchevsky and K. Nagayama, Adv. Colloid Interface Sci., 2000, 85, 145–192 CrossRef CAS.
- N. Vogel, C. K. Weiss and K. Landfester, Soft Matter, 2012, 8, 4044–4061 RSC.
- K. Kuk, L. Gregel, V. Abgarjan, C. Croonenbrock, S. Hänsch and M. Karg, Gels, 2022, 8, 516 CrossRef CAS.
- K. Volk, F. Deißenbeck, S. Mandal, H. Löwen and M. Karg, Phys. Chem. Chem. Phys., 2019, 21, 19153–19162 RSC.
- F. Pinaud, K. Geisel, P. Massé, B. Catargi, L. Isa, W. Richtering, V. Ravaine and V. Schmitt, Soft Matter, 2014, 10, 6963–6974 RSC.
- T. Still, K. Chen, A. M. Alsayed, K. B. Aptowicz and A. G. Yodh, J. Colloid Interface Sci., 2013, 405, 96–102 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Additional comparison of ex and in situ measurements, interparticle distance calculation from diffraction images, LT-SALS measurement with different wavelength lasers, LT-SALS setup and monolayer preparation for LT-SALS, image processing steps and other details (PDF), a video of LT-SALS measurement (AVI). See DOI: https://doi.org/10.1039/d2sm01125e |
| This journal is © The Royal Society of Chemistry 2023 |