
Homogeneous electrochemical water oxidation catalyzed by cobalt complexes with an amine–pyridine ligand†
Yanmei
Chen
*a,
Xiangyu
Meng
 b,
Xiaoli
Chen
a,
Xinyi
Li
a,
Hui
Ye
a,
Shanshan
Liu
a,
Zhijun
Ruan
a,
Xiangming
Liang
*c and
Junqi
Lin
*a
b,
Xiaoli
Chen
a,
Xinyi
Li
a,
Hui
Ye
a,
Shanshan
Liu
a,
Zhijun
Ruan
a,
Xiangming
Liang
*c and
Junqi
Lin
*a
aHubei Key Laboratory of Processing and Application of Catalytic Materials, College of Chemistry and Chemical Engineering, Huanggang Normal University, Huanggang, 438000, China. E-mail: cingym@163.com; linjunqi@hgnu.edu.cn
bHenan Provincial Key Laboratory of Nanocomposites and Applications, Institute of Nanostructured Functional Materials, Huanghe Science and Technology College, 666 Zijing Mountain South Road, Zhengzhou, Henan 450006, China
cSchool of Basic Medical Sciences, Ningxia Medical University, Yinchuan 750004, China. E-mail: liangxm6@163.com
First published on 22nd November 2022
Abstract
Efficient water oxidation catalysts are highly desired because of their great significance for overall water splitting, which is a promising strategy to produce hydrogen on a large scale. Herein, the water oxidation catalytic ability of two five-coordinate cobalt complexes has been investigated under alkaline conditions. Stabilized by similar organic scaffolds, their catalytic behaviors are distinct in phosphate-buffered saline at pH 11.0. Complex [Co(N2Py3)](ClO4)2 (1, N2Py3 = 2,6-bis[(methyl(2-pyridylmethyl)amino)methyl]pyridine) with pentadentate pyridine-amine containing three pyridine rings homogeneously catalyzes the oxidation of water to oxygen efficiently, exhibiting satisfactory stability and high faradaic efficiency. Replacing one of the pyridine rings of the ligand with an azaalkyl structure, the corresponding complex [Co(N3Py2)(OH2)](ClO4)2 (2, N3Py2 = 2,5,8-trimethyl-1,9-bis(2-pyridyl)-2,5,8-triazanonane) shows much lower catalytic activity. Electrochemical measurements reveal that the non-innocent N2Py3 undergoes a ligand oxidation process and participates in water oxidation catalysis, further assisting the generation of Co(IV) species via intramolecular electron transfer, resulting in the higher catalytic activity of complex 1. However, consecutive oxidation of the Co(II) center to Co(V) species is required for complex 2 to achieve water oxidation. The reactivity of Co(IV) intermediates of complex 2 with substrate water molecules is much lower than that of complex 1. Therefore, the tripyridine coordination environment of N2Py3 benefits the catalytic activity of complex 1via the ligand oxidation process. This work indicates that the ligand-assisted catalytic cycle is a favorable method for the accumulation of intermediate species that account for electrochemical water oxidation.
Introduction
Excessive usage of fossil fuels has caused issues such as the greenhouse effect and environmental pollution, which have attracted much attention in recent decades. Hydrogen energy is considered a promising alternative to fossil fuels as it is environmentally friendly.1–3 Water splitting driven by electricity from wind, solar and other energy sources is a feasible pathway to produce hydrogen energy in the future.4–6 Water splitting involves both water oxidation and proton reduction, and the former is a bottleneck for water splitting owing to its high activation energy.7,8 Therefore, efficient water oxidation catalysts (WOCs) are desired for hydrogen production by water splitting.Inspired by the natural photosynthetic oxygen evolution center—the Mn4CaO5 cluster—a series of transition metal complexes with multidentate organic scaffolds have been developed as catalysts for chemical-driven, photo-driven, and electrochemical water oxidation.9–15 Among these complexes, cobalt complexes with polypyridine ligands are of interest as they usually exhibit outstanding activity for photocatalytic and electrocatalytic water oxidation.16–20 The intermediates for water oxidation catalyzed by cobalt complexes are commonly Co(IV) species, which are nucleophilically attacked by water molecules and participate in O–O bond formation.21–23 To improve the water oxidation catalytic activity of cobalt complexes, various efforts have been made to design and synthesize ligands capable of stabilizing Co(IV) intermediate species with higher reactivity.24–27 Recent literature has revealed that some non-innocent organic ligands of cobalt complexes can participate in the water oxidation catalytic cycle via ligand oxidation. Organic ligand-assisted water oxidation catalysis commonly occurs with high catalytic activity.28–30 In this work, we investigate the water oxidation electrocatalytic properties of two mononuclear cobalt complexes, [Co(N2Py3)](ClO4)2 (1) and [Co(N3Py2)(OH2)](ClO4)2 (2) (N2Py3 = 2,6-bis[(methyl(2-pyridylmethyl)amino)methyl]pyridine, N3Py2 = 2,5,8-trimethyl-1,9-bis(2-pyridyl)-2,5,8-triazanonane, Fig. 1) in phosphate-buffered saline (PBS) at pH 11.0. Experiments confirm that the two complexes as molecular WOCs. Complex 1, stabilized by a pentadentate pyridine amine ligand with a three-pyridine ring, exhibits satisfactory stability and high faradaic efficiency. Replacing one of the pyridine rings of the ligand with an azaalkyl structure, the obtained complex 2 shows much lower catalytic activity. Further electrochemical tests indicate that although the organic scaffolds of the two complexes are similar, their catalytic behaviors are distinct. The N2Py3 ligand undergoes a ligand oxidation process and participates in the water oxidation catalytic cycle, generating [·L1(CoIII![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O)]2+ (L1 = N2Py3) species, which can be converted into [L1(CoIVO)]2+ species via the intramolecular electron transfer process. However, for complex 2, consecutive stepwise oxidations of the cobalt center to form [L2(CoIVO)]2+ (L2 = N3Py2) intermediates are necessary for oxygen evolution. Catalytic tests showed that [L1(CoIVO)]2+ species exhibit higher reactivity with substrate water molecules than [L2(CoIVO)]2+ species, leading to higher catalytic activity of complex 1. This work indicates that the ligand-assisted catalytic process can be a feasible method to achieve efficient water oxidation, and the electrochemical water oxidation catalytic activity of cobalt complexes can be improved by reasonably constructing a pyridine coordination environment.
O)]2+ (L1 = N2Py3) species, which can be converted into [L1(CoIVO)]2+ species via the intramolecular electron transfer process. However, for complex 2, consecutive stepwise oxidations of the cobalt center to form [L2(CoIVO)]2+ (L2 = N3Py2) intermediates are necessary for oxygen evolution. Catalytic tests showed that [L1(CoIVO)]2+ species exhibit higher reactivity with substrate water molecules than [L2(CoIVO)]2+ species, leading to higher catalytic activity of complex 1. This work indicates that the ligand-assisted catalytic process can be a feasible method to achieve efficient water oxidation, and the electrochemical water oxidation catalytic activity of cobalt complexes can be improved by reasonably constructing a pyridine coordination environment.
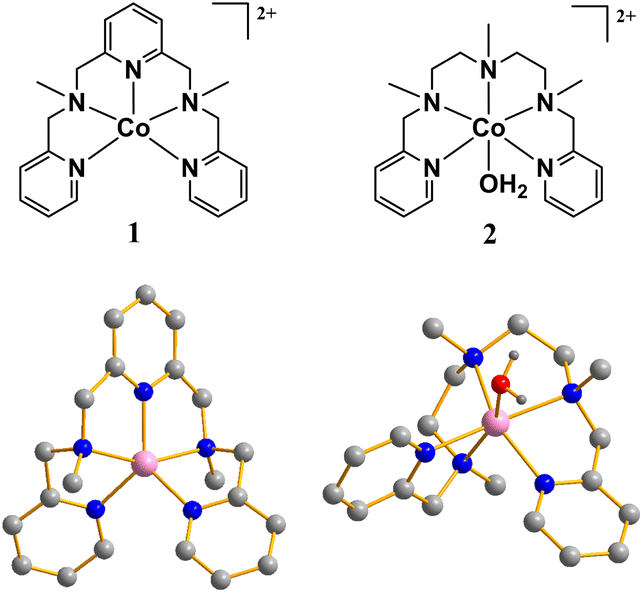 | ||
| Fig. 1 Chemical structure and ball-and-stick representation of complexes 1 and 2. ClO4− anions and H atoms of the ligand are omitted for clarity (purple, CoII; gray, C; red, O; blue, N; and dark gray, H). | ||
Experimental section
Materials and characterization
All chemicals except for the metal complexes were commercially purchased and used without further purification. Ultra-pure water (18.25 MΩ cm) used for all experiments was obtained from a water purifier.Equipment and apparatus
X-ray crystal diffraction was conducted using a Bruker D8 Venture. Infrared spectra (1 wt% of sample in KBr pellets) were obtained on a Thermo Fisher Scientific Nicolet iS5 FTIR spectrometer. UV-vis absorption spectra were obtained on a Shimadzu UV-2550 spectrometer equipped with a photomultiplier tube detector. Elemental analysis was performed on a TJA ICP-atomic emission spectrometer (IRIS Advantage ER/S). The microstructural profiles of the ITO electrode were monitored by scanning electron microscopy (Carl Zeiss Sigma 300) combined with energy dispersive X-ray spectroscopy (EDS). Dynamic light scattering measurements were conducted on a Brookhaven's NanoBrook 90Plus PALS instrument. Gas chromatography of oxygen was carried out on a gas chromatograph.Electrochemical measurements
Cyclic voltammetry (CV) was performed on a CHI660D electrochemical analyzer with a glassy carbon (0.071 cm−2), Ag/AgCl (saturated KCl) and Pt wire electrode as the working, reference, and auxiliary electrodes, respectively. All tested potentials versus Ag/AgCl were converted to potentials versus the normal hydrogen electrode (NHE) using the equation: E(Ag/AgCl) + 0.197 V. ITO electrodes were used as working electrodes for controlled potential electrolysis experiments. DPVs were obtained with the following parameters: amplitude = 50 mV, step height = 4 mV, pulse width = 0.05 s, pulse period = 0.5 s and sampling width = 0.0167 s.Synthesis of ligands and complexes
Ligands N2Py3 and N3Py2 were synthesized according to the literature.31–33 The synthetic method of the complexes is as follows.Results and discussion
General characterization
The structure of complex 1 was verified using single-crystal X-ray diffraction (Table S1†). As shown in Fig. 1, the Co center five-coordinated by the N2Py3 ligand exhibits distorted trigonal bipyramidal geometry. An equatorial plane is defined by three pyridyl nitrogen atoms, while two amino nitrogen atoms occupy the axial positions. The axial Co–N bond is evidently longer than the Co–N bonds in the equatorial plane (Table S3 and Fig. S1†). The zinc complex with the N2Py3 ligand was obtained as light yellow powder and characterized as mentioned above. The six-coordinated structure of complex 2 comprised five N atoms from N3Py2 and one O atom from coordinated water molecules, as has been confirmed in our previous work.33 Complex [Zn(N3Py2)(OH2)](ClO4)2 with a six-coordinated ZnII center is isostructural to 2 with a tetragonal biconical configuration (Tables S2, S4 and Fig. S2†). The similar infrared spectra of complex 2 and [Zn(N3Py2)(OH2)](ClO4)2 suggest their analogous structure (Fig. S3†). To investigate the coordination environment of complex 1 in PBS, the UV-vis absorption spectra of complex 1 in pure water, CH3CN and PBS at various pH were recorded. The difference in the spectra of 1 in CH3CN and water indicates its different coordination environment in the two solvents (Fig. S4†). Complex 1 is synthesized using CH3CN as a solvent, and its cobalt center is pentacoordinated. Therefore, it is believed that complex 1 is converted into [Co(N2Py3)(OH2)]2+ when dissolved in water. The absorbance of 1 in acidic conditions remained unchanged, confirming that 1 retained its structure under acidic conditions (Fig. S5†). The UV-vis spectrum of complex 1 at pH 11.0 exhibits a characteristic absorption peak at 514 nm with a shoulder peak at 474 nm, while the characteristic absorption peak of complex 2 is located at 475 nm (Fig. S6†). The intensity and position of the characteristic peak of 1 change regularly as the pH of the buffer solution changes, indicating the formation of [Co(N2Py3)(OH2)]2+via nucleophilic attack of water on the Co center of 1 and the subsequent generation of [Co(N2Py3)(OH)]+via a deprotonation process (pKa = 9.3, Fig. S7–S9†). The change of the characteristic peak of complex 2 at different pH also reveals the deprotonation process of 2 with pKa = 10.1 (Fig. S10–S12†). Therefore, the main forms of 1 and 2 dissolved in PBS at pH 11.0 should be [Co(N2Py3)(OH)]+ and [Co(N3Py2)(OH)]+, respectively. The time-independent UV-vis absorption spectra of 1 and 2 in PBS at pH 11.0 further confirm their hydrolytic stability (Fig. S13†).Electrochemical tests
Cyclic voltammetry (CV) tests were conducted to study the electrochemical properties of complexes 1 and 2. The typical CV curves of the two cobalt complexes in 0.1 M PBS at pH 11.0 are presented in Fig. 2, which exhibits an oxidation peak at Epa = 0.79 V and weak reduction peak at Epc = 0.68 V for 1 (E1/2 = 0.73 V, Fig. S14†). The oxidation peak of 2 is located at 0.74 V, accompanied by a weak reduction peak at 0.64 V; the E1/2 value of 2 is 0.69 V (Fig. S15†). The two irreversible redox couples are assigned to the coupling of CoII/CoIII species (ΔEp = 0.11 V and 0.10 V for 1 and 2, respectively). For complex 1, the catalytic current is attributable to the oxidation of water that arises at 1.28 V, exhibiting an onset overpotential of 700 mV for oxygen evolution. However, when complex 2 was tested under identical conditions, the catalytic current became relatively low and close to the background. Therefore, complex 1 shows higher water oxidation activity than complex 2.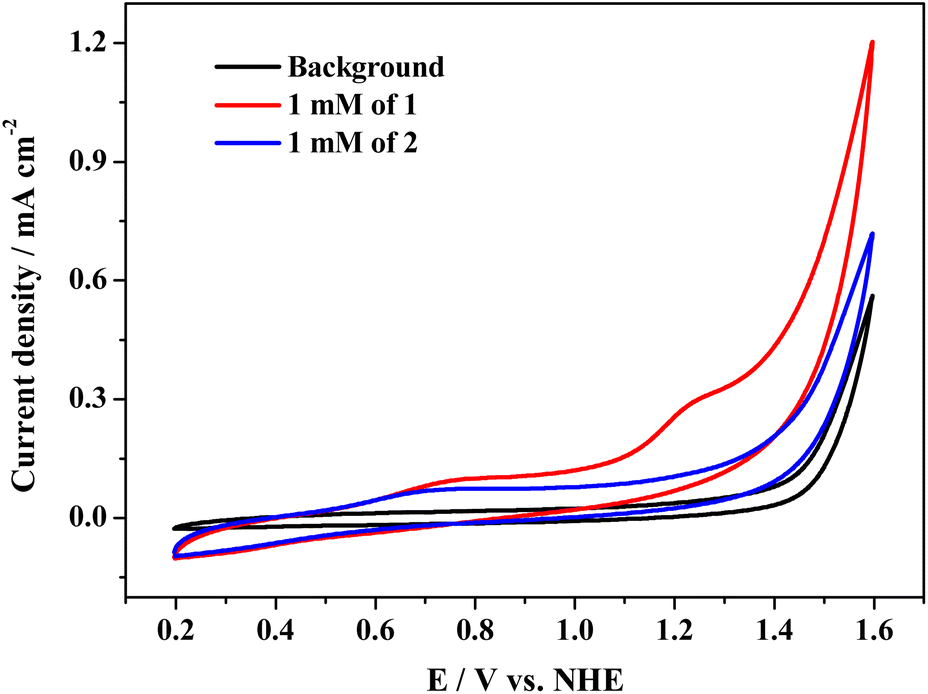 | ||
| Fig. 2 CV curves of complexes 1 and 2 at 1 mM in 0.1 M PBS at pH 11.0, scan rate = 100 mV s−1. | ||
Although there is a minor difference between the structures of 1 and 2, they perform entirely different catalytic activities toward electrochemical water oxidation. The catalytic activity and stability of the two complexes were further investigated by controlled potential electrolysis (CPE) at 1.55 V in a two-compartment cell with an ITO electrode as the working electrode. During long-term electrolysis, the catalytic current of complex 1 initially decreases and then gradually rises. It finally stabilizes at ca. 0.65 mA cm−2 (Fig. 3). The slight increasing trend and sudden fluctuation of the current curve are due to the release of accumulated oxygen bubbles on the surface of the electrode. However, the stabilized catalytic current density of complex 2 is only 0.11 mA cm−2, which is much lower than that of complex 1, indicating its lower catalytic activity (Fig. S16†). After the 4 h CPE tests of 1 and 2, the used ITO electrodes were rinsed with pure water and rested in fresh buffer solution. As shown in Fig. 3, the catalytic current of the used ITO electrodes is close to the background. Since cobalt phosphate, cobalt oxide, and cobalt hydroxide are highly active for water oxidation in PBS,34–36 the negligible catalytic current of the used ITO electrodes negates the generation of the above heterogeneous active species on the surface of the used ITO electrodes. Therefore, both complexes 1 and 2 remain intact during long-term catalytic cycling.
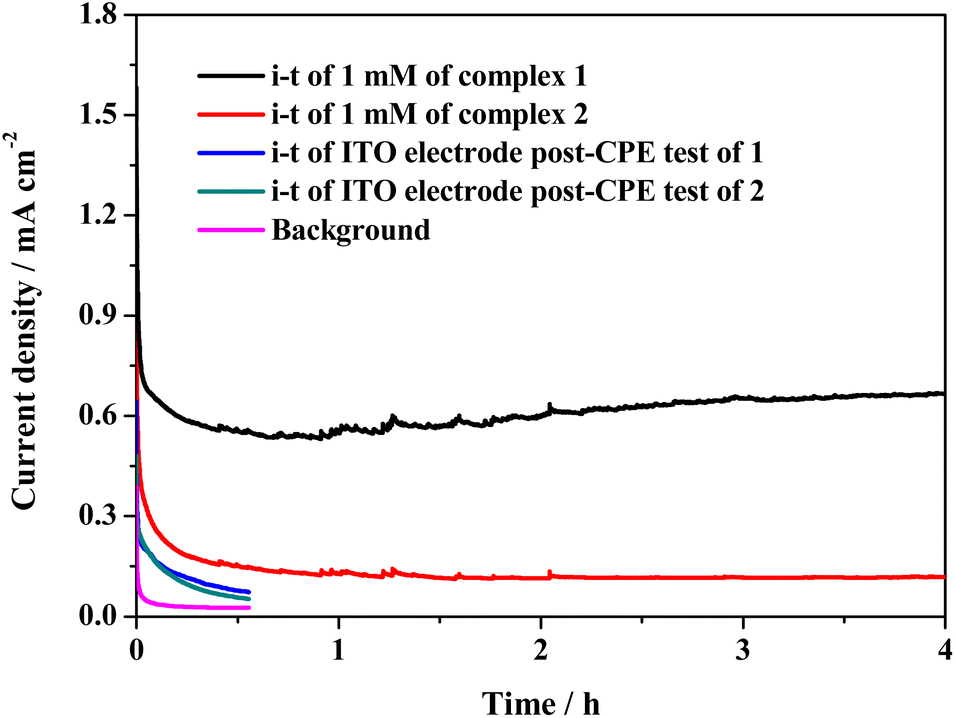 | ||
| Fig. 3 The i–t curves obtained by CPE tests of 1 mM of 1 and 2 at 1.55 V using 0.1 M PBS at pH 11.0 as the electrolyte and an ITO electrode as the working electrode (1 cm × 2 cm with 1 cm2 immersed in electrolyte). | ||
The homogeneity of the two cobalt complexes was further verified using scanning electron microscopy. According to the SEM images presented in Fig. S17,† the ITO electrodes after the CPE experiments of complexes 1 and 2 have a micro morphology similar to that of a clean ITO electrode, confirming the absence of Co-containing nanosized particles. Moreover, energy-dispersive spectroscopy (EDX) shows that there is no Co element on the surface of the two used ITO electrodes (Fig. S18†). It is hence clear that complexes 1 and 2 homogeneously catalyze water oxidation to oxygen. Dynamic light scattering (DLS) measurement of the solution after the CPE experiment was carried out to investigate the stability of 1. As shown in Fig. S19†, the DLS test indicates the absence of nanosized cobalt oxide in the electrolyte solution, confirming the stability of complex 1. Additionally, the UV-vis absorption spectrum of the post-electrolysis solution (sampled after the CPE experiment) is almost identical to that of the fresh solution, revealing the stability of complex 1 during long-term catalytic cycling (Fig. S20†). The amount of oxygen released from the reaction systems mediated by 1 and 2 was quantified using gas chromatography, and ca. 22.1 μmol of O2 for 1 and ca. 4.5 μmol of O2 for 2 were detected after the CPE experiment (Fig. S21†), affording faradaic efficiencies of 96% and 97%, respectively, which are comparable with those of previously reported homogeneous cobalt-based WOCs.37–40 The difference in the amount of oxygen production indicates that the catalytic activity of complex 2 is much lower than that of complex 1.
The catalytic kinetic process of complex 1 was revealed by CV tests with different catalyst concentrations [1] and scan rates ν. The anodic peak current density (jp) for the first irreversible redox couple of 1 exhibits a linear dependence on [1], following the diffusion-controlled electrode reaction formula of jp = 0.446nF[1](FνDcat/RT)1/2 (Fig. S22 and S23†).41 In this equation, F is the Faraday constant, R is the ideal gas constant, Dcat is the diffusion coefficient of complex 1, n is the number of transferred electrons for the CoII/CoIII redox couple, which is a diffusion-controlled non-catalytic process, and T = 298 K. The value of the diffusion coefficient Dcat is calculated to be 4.94 × 10−6 cm2 s−1 for [Co(N2Py3)(OH)]+. This calculated value is consistent with that of the diffusion-controlled process (10−6 to 10−5 cm2 s−1).42,43 Meanwhile, the water oxidation catalytic current density jcat rises linearly with increasing [1] (Fig. S24†). This result is consistent with the formula of jcat = n′F[1](Dcatkcat)1/2 (n′ = 4 is the number of electrons transferred for water oxidation to O2), suggesting that complex 1 participates in water oxidation catalytic cycles via a single-site catalytic mechanism.44 From the slope of the plot of jcat/jpversus the reciprocal of the square root of the scan rate, an apparent kcat = 0.23 s−1 was obtained (Fig. S25–S27†).
The redox processes and catalytic mechanisms of the two cobalt complexes were clarified using differential pulse voltammetry (DPV). As shown in Fig. 4, two oxidation processes are required for both complexes 1 and 2 to accomplish water oxidation. Although [Zn(N2Py3)](BF4)2 is inactive for water oxidation (Fig. S28†), its ligand N2Py3 is redox-active and can be oxidized reversibly (Fig. 4, pink curve), which is consistent with the literature.32 Therefore, for complex 1, the first redox process at 0.69 V is ascribed to the CoII/CoIII couple while the second redox process at 1.15 V is assigned to its ligand oxidation. For complex 2, the first redox process at 0.61 V has been proven to involve the CoII/CoIII couple. To identify the second oxidation process of complex 2, its analogous complex [Zn(N3Py2)(OH2)](ClO4)2, with a redox-inactive ZnII center, was synthesized. There are no oxidation peaks at a similar potential in the DPV curves of [Zn(N3Py2)(OH2)](ClO4)2 (Fig. 4). Therefore, the peak at 1.17 V for complex 2 was attributed to the oxidation of the CoII center rather than organic ligand oxidation. As complex 1 shows higher activity toward water oxidation than complex 2, the difference between the catalytic activities of both is believed to be attributed to reactivity differences of high valent intermediates produced by different mechanisms. That is, the corresponding intermediates of 1 are more active than those of 2.
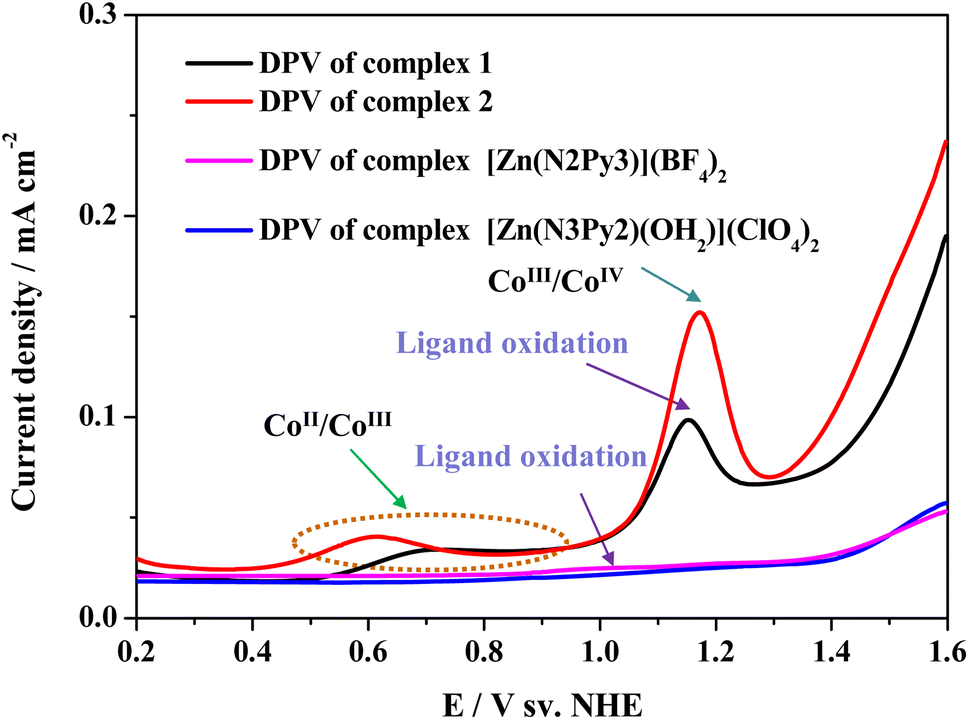 | ||
| Fig. 4 Differential pulse voltammetry (DPV) examinations of complexes 1, 2, [Zn(N2Py3)](BF4)2 and [Zn(N3Py2)(OH2)](ClO4)2 in 0.1 M PBS at pH 11.0. | ||
As shown in Fig. S29 and S30,† the redox potentials of complexes 1 and 2 vary with increasing electrolyte pH. The potentials of the first redox couple of both complexes 1 and 2 remain almost constant in the pH range of 10.0–11.5, revealing that the first oxidation process of complexes 1 and 2 is an electron transfer process without participation of a proton.37,45 The original complex will undergo one electron oxidation process to generate [LCoIII(OH)]2+ (defined as [L1CoIII(OH)]2+ and [L2CoIII(OH)]2+ for complex 1 and 2, respectively). The potential of the second redox process of both 1 and 2 decreases with increasing electrolyte pH, with a constant slope of ca. −59 mV pH−1, indicating an e−/H+ proton-coupled electron transfer (PCET) process (Fig. S31 and S32†).46,47 Therefore, for complex 1, [L1CoIII(OH)]2+ is then oxidized to [·L1(CoIIIO)]2+ species. In contrast [L2CoIII(OH)]2+ is oxidized to [L2(CoIVO)]2+ in complex 2. Since the t2g orbitals of Co(III) are fully occupied, the electron density of the CoIII center of [·L1(CoIIIO)]2+ will lead to lower reactive activity than [L2(CoIVO)]2+. However, complex 1 is more active than complex 2. Hence, it is assumed that the [·L1(CoIIIO)]2+ species will be converted into [L1(CoIVO)]2+ species via the intramolecular electron transfer process of [·L1(CoIIIO)]2+.48,49 The higher the reactivity of [L1(CoIVO)]2+ with substrate water molecules, the higher the catalytic activity of complex 1. Based on this result, the mechanism of water oxidation mediated by the two cobalt complexes has been proposed (Scheme 1). The reaction of [L1(CoIVO)]2+ or [L2(CoIVO)]2+ with a water molecule affords the peroxide species [LCoII(OOH)]2+, which participates in the oxidation process that follows to release O2 and restore the cobalt complex. It has been widely reported that for single-site catalysts, O–O formation, which is the rate-limiting step for water oxidation, is realized via nucleophilic attack of water on high valent intermediates.50–54 That is, the higher activity of [L1(CoIVO)]2+ generated via the ligand oxidation pathway makes complex 1 a more active WOC. Fig. S33† also confirms that the concussive oxidation of the CoII center and N2Py3 ligand is essential for water oxidation to O2. Therefore, the oxidation of the diamine–tripyridine ligand N2Py3 benefits the electrocatalytic water oxidation activity of complex 1. Further intensive computational studies on the catalytic mechanism will be carried out in a future work to shed light on the special catalytic pathway of complex 1.
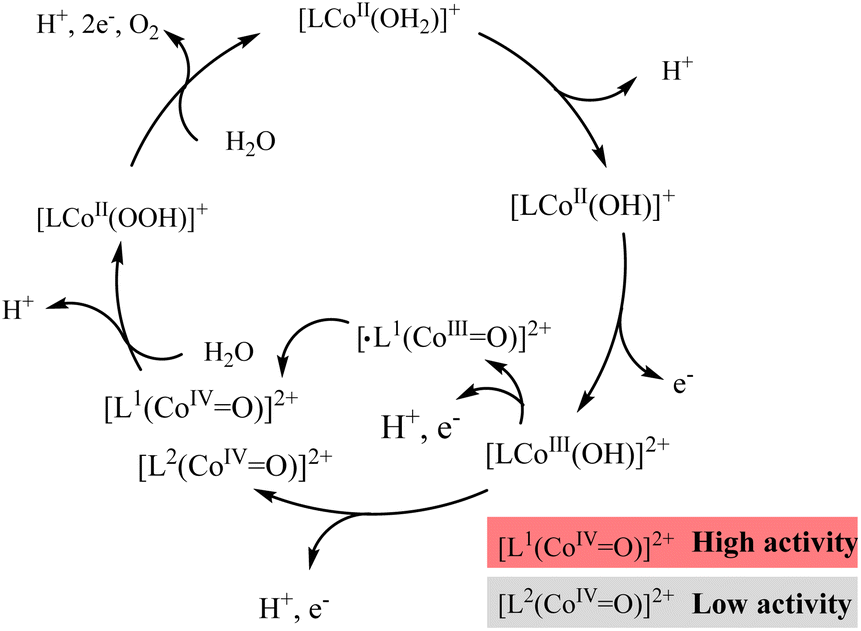 | ||
| Scheme 1 Proposed mechanism of electrochemical water oxidation catalyzed by complexes 1 and 2. | ||
Conclusions
Herein, the water oxidation catalytic mechanism and activity of two mononuclear cobalt complexes, [Co(N2Py3)](ClO4)2 and [Co(N3Py2)(OH2)](ClO4)2, were investigated at pH 11.0. The redox-innocent N2Py3 ligand with three pyridine rings enables [Co(N2Py3)](ClO4)2 to catalyze electrochemical water oxidation efficiently via a ligand-assisted catalytic mechanism. However, the similar N3Py2 ligand with two pyridine rings leads to lower reactivity in the intermediate species, affording much lower activity for water oxidation. This work reveals that the pyridine structure can affect the water oxidation activity of cobalt complexes by modulating the redox properties of the pyridine–amine ligand and tuning the reactivity of intermediate species. It also provides a new route to improve the stability and activity of molecular water oxidation catalysts.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (21501061, 21704032 and 22004044), Natural Science Foundation of Hubei Province (2017CFB223 and 2020CFB426), Research Foundation of Huanggang Normal University (2042020043), Disciplinary Team Project of Huanggang Normal University, and Hubei Key Laboratory of Low Dimensional Optoelectronic Material and Devices (HLOM222004).Notes and references
- J. Zhu, L. Hu, P. Zhao, L. Y. S. Lee and K.-Y. Wong, Chem. Rev., 2020, 120, 851–918 CrossRef CAS PubMed.
- H.-F. Wang, L. Chen, H. Pang, S. Kaskel and Q. Xu, Chem. Soc. Rev., 2020, 49, 1414–1448 RSC.
- M. Chatenet, B. G. Pollet, D. R. Dekel, F. Dionigi, J. Deseure, P. Millet, R. D. Braatz, M. Z. Bazant, M. Eikerling, I. Staffell, P. Balcombe, Y. Shao-Horn and H. Schäfer, Chem. Soc. Rev., 2022, 51, 4583–4762 RSC.
- W. Zhai, Y. Ma, D. Chen, J. C. Ho, Z. Dai and Y. Qu, InfoMat, 2022, 4, e12357 CrossRef CAS.
- D. Zhou, P. Li, X. Lin, A. McKinley, Y. Kuang, W. Liu, W.-F. Lin, X. Sun and X. Duan, Chem. Soc. Rev., 2021, 50, 8790–8817 RSC.
- X. Li, H. Lei, L. Xie, N. Wang, W. Zhang and R. Cao, Acc. Chem. Res., 2022, 55, 878–892 CrossRef CAS.
- B. You and Y. Sun, Acc. Chem. Res., 2018, 51, 1571–1580 CrossRef CAS.
- B. Das, A. Rahaman, A. Shatskiy, O. Verho, M. D. Kärkäs and B. Åkermark, Acc. Chem. Res., 2021, 54, 3326–3337 CrossRef CAS PubMed.
- M. D. Kärkäs and B. Åkermark, Dalton Trans., 2016, 45, 14421–14461 RSC.
- M. Kondo, H. Tatewaki and S. Masaoka, Chem. Soc. Rev., 2021, 50, 6790–6831 RSC.
- X.-P. Zhang, H.-Y. Wang, H. Zheng, W. Zhang and R. Cao, Chin. J. Catal., 2021, 42, 1253–1268 CrossRef CAS.
- L. Duan, F. Bozoglian, S. Mandal, B. Stewart, T. Privalov, A. Llobet and L. Sun, Nat. Chem., 2012, 4, 418–423 CrossRef CAS.
- S. M. Barnett, K. I. Goldberg and J. M. Mayer, Nat. Chem., 2012, 4, 498–502 CrossRef CAS PubMed.
- M. Okamura, M. Kondo, R. Kuga, Y. Kurashige, T. Yanai, Sh. Hayami, V. K. K. Praneeth, M. Yoshida, K. Yoneda, S. Kawata and S. Masaoka, Nature, 2016, 530, 465–468 CrossRef CAS PubMed.
- C. Liu, D. van den Bos, B. den Hartog, D. van der Meij, A. Ramakrishnan and S. Bonnet, Angew. Chem., Int. Ed., 2021, 60, 13463–13469 CrossRef CAS.
- C.-F. Leung, S.-M. Ng, C.-C. Ko, W.-L. Man, J. Wu, L. Chen and T.-C. Lau, Energy Environ. Sci., 2012, 5, 7903–7907 RSC.
- T. Ishizuka, A. Watanabe, H. Kotani, D. Hong, K. Satonaka, T. Wada, Y. Shiota, K. Yoshizawa, K. Ohara, K. Yamaguchi, S. Kato, S. Fukuzumi and T. Kojima, Inorg. Chem., 2016, 55, 1154–1164 CrossRef CAS PubMed.
- G. Ruan, L. Engelberg, P. Ghosh and G. Maayan, Chem. Commun., 2021, 57, 939–942 RSC.
- P. K. Das, S. Bhunia, P. Chakraborty, S. Chatterjee, A. Rana, K. Peramaiah, M. M. Alsabban, I. Dutta, A. Dey and K.-W. Huang, Inorg. Chem., 2021, 60, 614–622 CrossRef CAS PubMed.
- Y.-F. Su, W.-Z. Luo, W.-Q. Lin, Y.-B. Su, Z.-J. Li, Y.-J. Yuan, J.-F. Li, G.-H. Chen, Z. Li, Z.-T. Yu and Z. Zou, Angew. Chem., Int. Ed., 2022, 61, e202201430 CAS.
- L. Xu, H. Lei, Z. Zhang, Z. Yao, J. Li, Z. Yu and R. Cao, Phys. Chem. Chem. Phys., 2017, 19, 9755–9761 RSC.
- F. Xie and M.-T. Zhang, J. Energy Chem., 2021, 63, 1–7 CrossRef CAS.
- X. Zhang, Q.-F. Chen, J. Deng, X. Xu, J. Zhan, H.-Y. Du, Z. Yu, M. Li, M.-T. Zhang and Y. Shao, J. Am. Chem. Soc., 2022, 144, 17748–17752 CrossRef CAS PubMed.
- T. Nakazono and K. Sakai, Dalton Trans., 2016, 45, 12649–12652 RSC.
- S. Liu, Y.-J. Lei, Z.-J. Xin, R.-J. Xiang, S. Styring, A. Thapper and H.-Y. Wang, Int. J. Hydrogen Energy, 2017, 42, 29716–29724 CrossRef CAS.
- N. I. Neuman, U. Albold, E. Ferretti, S. Chandra, S. Steinhauer, P. Rößner, F. Meyer, F. Doctorovich, S. E. Vaillard and B. Sarkar, Inorg. Chem., 2020, 59, 16622–16634 CrossRef CAS PubMed.
- N. D. McMillion, A. W. Wilson, M. K. Goetz, M.-C. Chang, C.-C. Lin, W.-J. Feng, C. C. L. McCrory and J. S. Anderson, Inorg. Chem., 2019, 58, 1391–1397 CrossRef CAS PubMed.
- H.-Y. Du, S.-C. Chen, X.-J. Su, L. Jiao and M.-T. Zhang, J. Am. Chem. Soc., 2018, 140, 1557–1565 CrossRef CAS PubMed.
- S. Biswas, S. Bose, J. Debgupta, P. Das and A. N Biswas, Dalton Trans., 2020, 49, 7155–7165 RSC.
- W.-C. Hsu, W.-Q. Zeng, I.-C. Lu, T. Yang and Y.-H. Wang, ChemSusChem, 2022, 15, e202201317 CAS.
- M. Grau, J. England, R. Torres, M. d. Rosales, H. S. Rzepa, A. J. P. White and G. J. P. Britovsek, Inorg. Chem., 2013, 52, 11867–11874 CrossRef CAS PubMed.
- Z. Xu, Z. Zheng, Q. Chen, J. Wang, K. Yu, X. Xia, J. Shen and Q. Zhang, Dalton Trans., 2021, 50, 10888–10895 RSC.
- H. Zheng, H. Ye, T. Xu, K. Zheng, X. Xie, B. Zhu, X. Wang, J. Lin and Z. Ruan, New J. Chem., 2022, 46, 7522–7527 RSC.
- M. W. Kanan and D. G. Nocera, Science, 2008, 321, 1072–1075 CrossRef CAS PubMed.
- L. Ma, S.-F. Hung, L. Zhang, W. Cai, H. B. Yang, H. M. Chen and B. Liu, Ind. Eng. Chem. Res., 2018, 57, 1441–1445 CrossRef CAS.
- R. Qin, H. Wan, X. Liu, G. Chen, N. Zhang, R. Ma and G. Qiu, Inorg. Chem. Front., 2019, 6, 1744–1752 RSC.
- D. Das, S. Pattanayak, K. K. Singh, B. Garai and S. S. Gupta, Chem. Commun., 2016, 52, 11787–11790 RSC.
- H. M. Shahadat, H. A. Younus, N. Ahmad, S. Zhang, S. Zhuiykov and F. Verpoort, Chem. Commun., 2020, 56, 1968–1971 RSC.
- X. Yin, S. Zhang, J. Wang, J. Li, F. Chen, S. Yao, Y. Fan and M. Wang, Appl. Organomet. Chem., 2021, 35, e6371 CrossRef CAS.
- B.-H. Luo, S.-Y. Zhao, Y.-L. Pu, X.-Y. Zhou, J.-Z. Xie, L. Chen, M. Wu, M. Zhou and H.-X. Zhang, Dalton Trans., 2022, 51, 15854–15862 RSC.
- X.-J. Su, M. Gao, L. Jiao, R.-Z. Liao, P. E. M. Siegbahn, J.-P. Cheng and M.-T. Zhang, Angew. Chem., Int. Ed., 2015, 54, 4909–4914 CrossRef CAS PubMed.
- G. Ruan, P. Ghosh, N. Fridman and G. Maayan, J. Am. Chem. Soc., 2021, 143, 10614–10623 CrossRef CAS PubMed.
- J. Lin, N. Wang, X. Chen, X. Yang, L. Hong, Z. Ruan, H. Ye, Y. Chen and X. Liang, Sustainable Energy Fuels, 2022, 6, 1312–1318 RSC.
- M. K. Coggins, M.-T. Zhang, Z. Chen, N. Song and T. J. Meyer, Angew. Chem., Int. Ed., 2014, 53, 12226–12230 CrossRef CAS PubMed.
- J. Lin, P. Kang, X. Liang, B. Ma and Y. Ding, Electrochim. Acta, 2017, 258, 353–359 CrossRef CAS.
- M. Zhang, M.-T. Zhang, C. Hou, Z.-F. Ke and T.-B. Lu, Angew. Chem., Int. Ed., 2014, 53, 13042–13048 CrossRef CAS PubMed.
- J.-W. Wang, X.-Q. Zhang, H.-H. Huang and T.-B. Lu, ChemCatChem, 2016, 8, 3287–3293 CrossRef CAS.
- F. Yu, F. Li, J. Hu, L. Bai, Y. Zhu and L. Sun, Chem. Commun., 2016, 52, 10377–10380 RSC.
- D. Das, S. Pattanayak, K. K. Singh, B. Garai and S. Sen Gupta, Chem. Commun., 2016, 52, 11787–11790 RSC.
- L. Wang, L. Duan, R. B. Ambre, Q. Daniel, H. Chen, J. Sun, B. Das, A. Thapper, J. Uhlig, P. Dinér and L. Sun, J. Catal., 2016, 335, 72–78 CrossRef CAS.
- J. Shen, M. Wang, J. Gao, H. Han, H. Liu and L. Sun, ChemSusChem, 2017, 10, 4581–4588 CrossRef CAS PubMed.
- J. Shen, M. Wang, P. Zhang, J. Jiang and L. Sun, Chem. Commun., 2017, 53, 4374–4377 RSC.
- G. Ruan, N. Fridman and G. Maayan, Chem.–Eur. J., 2022, 28, e202202407 CAS.
- H. Pan, L. Duan and R. -Z. Liao, ChemCatChem, 2020, 12, 219–226 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2211200 and 2211201 for 1 and [Zn(N3Py2)(OH2)](ClO4)2. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2se01431a |
| This journal is © The Royal Society of Chemistry 2023 |