
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEfficient detection of bilirubin in human serum through a displacement approach†
Nancy
Singla
 ,
Manzoor
Ahmad
,
Vishal
Mahajan
,
Prabhpreet
Singh
and
Subodh
Kumar
*
,
Manzoor
Ahmad
,
Vishal
Mahajan
,
Prabhpreet
Singh
and
Subodh
Kumar
*
Department of Chemistry, Centre for Advanced Studies, Guru Nanak Dev University, Amritsar, Punjab, India. E-mail: subodh_gndu@yahoo.co.in; subodh.chem@gndu.ac.in
First published on 2nd October 2023
Abstract
The present study provides, for the first time, a method for the fluorescence “Turn ON” quantification of bilirubin (BR) in human serum through a displacement approach. The fluorescent probe THYQ exhibited a 55-fold increase in fluorescence intensity at 555 nm with HSA. The green fluorescent THYQ–HSA complex was highly selective towards bilirubin (BR) (subdomain IB) and exhibited >90% fluorescence quenching (log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) β = 4.83 ± 0.12) through the displacement of THYQ, whereas the subdomain IIA and subdomain IIIA selective drugs warfarin and ibuprofen did not displace THYQ. The non-fluorescent BR–HSA complex displayed a linear increase in fluorescence with THYQ (logβ = 4.81 ± 0.13) to form THYQ–HSA complex. The slope of titration of BR–HSA with THYQ was linearly dependent on [bilirubin] in HSA, thus enabling the fluorescence-based detection of bilirubin below normal (0.7 mg dL−1, 12 μM) to hyperbilirubinemia conditions (12.6 mg dL−1, 216 μM), with the results in good agreement with the clinical findings. The LOD for the detection of BR was 0.004 mg dL−1 (68 nM).
β = 4.83 ± 0.12) through the displacement of THYQ, whereas the subdomain IIA and subdomain IIIA selective drugs warfarin and ibuprofen did not displace THYQ. The non-fluorescent BR–HSA complex displayed a linear increase in fluorescence with THYQ (logβ = 4.81 ± 0.13) to form THYQ–HSA complex. The slope of titration of BR–HSA with THYQ was linearly dependent on [bilirubin] in HSA, thus enabling the fluorescence-based detection of bilirubin below normal (0.7 mg dL−1, 12 μM) to hyperbilirubinemia conditions (12.6 mg dL−1, 216 μM), with the results in good agreement with the clinical findings. The LOD for the detection of BR was 0.004 mg dL−1 (68 nM).
1. Introduction
The levels of bilirubin (BR) need to be maintained for the wellbeing of humans and thus the monitoring of BR is critical, and has attracted increasing attention as the number of deaths due to hyperbilirubinemia has doubled over the past two decades.1 Its increased levels, such as in jaundice, are associated with acute and chronic health burden, while low levels have been linked to iron deficiency (anaemia) and coronary artery diseases.2,3 The normal concentration of bilirubin in adult serum lies between 0.2 and 1.2 mg dL−1. In the condition of hyperbilirubinemia, it can go up to 9.9 mg dL−1 (170 μM) in an adult and even to >24.8 mg dL−1 (425 μM) under critical neonatal and other hyperbilirubinemia conditions.At present, the clinical quantification of bilirubin depends on the diazotization reaction between bilirubin and sulfanilic acid, which was devised more than 100 years ago.4 However, the complexity of the chemical reactions and inhomogeneity of biological samples make this procedure unreliable.5 A fluorescence-based assay depending on clean chemical events could be expected to be more advantageous.
Several fluorescence methods, using carbon dots,6–8 metal nanoclusters,9 MOFs,10,11 organic molecules,12–17 have been developed for the detection of bilirubin. However, these are limited due to the “Turn OFF” signal and poor sensitivity and dependence on a spiking method for clinical detection. Recently reported coumarin-based N-oxide–Fe3+ mixtures have revealed “Turn ON” fluorescence in the presence of bilirubin owing to the deoxygenation process by the in situ generated Fe2+ (ref. 16 and 17) and have been tested and even used for the detection of bilirubin from human serum. In another approach, in non-fluorescent resorcinol-sucrose CDs and Cu2+ complex, the competitive binding of Cu2+ with bilirubin released the fluorescence of the CDs.8 Most of these methods for the detection of bilirubin from human serum have a myriad of limitations and in general have low sensitivity and depend on a spiking approach.
The human serum albumin (HSA) concentration in human serum is between 500 and 700 μM and bilirubin remains bound to it with high affinity, and this formed BR–HSA complex acts as a buffer to prevent the transfer of bilirubin from blood to the tissues and thus plays a critical role in impairing the development of bilirubin encephalopathy.18,19 In the literature, numerous probes for the fluorescence-based detection of HSA have been reported, but most of these fluorescence probes bind at Site I and Site II, i.e. the respective warfarin and ibuprofen sites of HSA. We envisaged that a fluorescent probe (i) that could selectively bind at Site IB – the bilirubin site of HSA – and (ii) that has a binding constant similar to that of bilirubin with HSA so that it could displace BR from the BR–HSA complex, with a resultant change in fluorescence intensity (FI), could be useful for the detection of bilirubin present in human serum without altering the physiological conditions.
The displacement approach is based on the competition between a probe and the analyte of interest for the binding site. Upon the addition of the analyte, the probe is displaced from the binding pocket, causing a signal change, such as in UV or fluorescence. This approach can enable the easy and real-time determination of an analyte without harming any of the substrate,20 which is often not achieved by traditional chemosensors. Recently reported Site IB selective fluorescent probes, however, lack the reversible displacement of BR and the probe with HSA under physiological conditions.21
Now, we report a fluorescent probe THYQ that can enable the highly selective displacement of bilirubin from THYQ–HSA complex under physiological concentrations of HSA and that could be applied for the detection of bilirubin from human serum. The THYQ probe displayed a 55-fold increase in fluorescence intensity at 555 nm with HSA. The fluorescent THYQ–HSA complex was highly selective towards bilirubin and revealed >90% fluorescence quenching with logβ = 4.83 ± 0.12, while the non-fluorescent BR∩HSA displayed a linear increase in fluorescence with THYQ with logβ = 4.81 ± 0.13. All the experiments for the detection of bilirubin (BR) were performed in HEPES buffer with ∼500 μM HSA. The slope of titration of the BR–HSA complex with THYQ was found to be linearly dependent on [BR], enabling the fluorescence-based detection of BR below normal (0.7 mg dL−1, 12 μM) to hyperbilirubinemia conditions (12.6 mg dL−1, 216 μM). The LOD for the detection of BR was 0.004 mg dL−1 (68 nM).
2. Experimental
2.1. Materials and methods
All chemicals and solvents were purchased from Aldrich and Spectrochem India, and were used without any purification. The organic solvents, including DMSO, DMF, and THF, were of HPLC grade. The 1H and 13C NMR spectra were recorded on a JEOL 400 MHz FT NMR machine using CDCl3 as solvent and tetramethylsilane (TMS) as the internal standard. The chemical shift values of the peaks were recorded in ppm relative to TMS as the internal standard, with the coupling constant J in Hz, and multiplicity also noted (s = singlet, d = doublet, t = triplet, m = multiplet). High-resolution mass spectra were recorded on an Agilent 6520 Q-TOF mass spectrometer with the Agilent 1200 HPLC system. Deionized water was used throughout the experiments for preparing all solutions and obtained using an ultra UV/UF Rions lab water system ultra 370 series. UV–visible studies were performed on a Shimadzu UV-2450 instrument using a slit width of 1.0 nm and matched quartz cell with a 1 cm path length, thermo stated at 25.0 ± 0.1 °C. The fluorescence spectra were recorded on a Horiba Fluorolog-3 spectrophotometer with a quartz cuvette of 1 cm path length. DLS experiments were performed at 25.0 ± 0.1 °C by using a zeta sizer instrument (Nano ZS, Malvern LTD, UK).2.2. Synthesis
2.3. Preparation of the stock solutions
A stock solution of the fluorescent probe THYQ (1 mM) was prepared in THF and was further diluted for preparing solutions of the desired concentrations. For the preparation of 10 μM solution, 30 μL of stock solution was added in to 3 ml of the desired solvent, like THF–HEPES mixture or HEPES buffer. Every time, only a freshly prepared stock solution of bilirubin was used and this was prepared in DMSO. All the proteins, amino acids, anions, and thiols were prepared by dissolving commercial reagents in Millipore double-distilled water.2.4. Detection limit22
The lowest detection limit was calculated from fluorescence studies. The standard deviation was calculated by measuring the fluorescence data of THYQ three times. The slope was calculated from the graph of the fluorescence intensity vs. the concentration of the analyte added. The lowest limit of detection (LOD) was determined by using the following equation:| LOD = 3σ/k |
2.5. Quantum yield calculation23
The quantum yield (Φ) of the solutions of THYQ was measured with an integrated sphere on an FL-1039A/40A machine by using the excitation wavelength at 420 nm.2.6. Binding constants24
In order to determine the formation of various stoichiometric complexes and their binding constants, the whole spectral data of the titration experiments were evaluated using the software SPECFIT-32. This software is able to perform a global analysis of the equilibrium and kinetic systems with a singular value of decomposition and using nonlinear-regression modelling by the Levenberg–Marquardt method. The program simulates the fluorescence data obtained experimentally. The stoichiometry of the species formed, distribution of species, and their association constants are determined through the fitted model.2.7. Measurement of circular dichroism25
The alterations in the secondary structure of HSA in the presence of THYQ were studied by measuring the circular dichroism (CD) on a BIO-Kine 32 MOS 500 spectrophotometer using a quartz cuvette of path length 1 cm at 0.25 nm data pitch intervals. All the CD spectra were taken in the wavelength range 200–250 nm. The concentration of HSA was 0.5 μM. The helicity content was calculated from the MRE values at 222 nm using the following equation: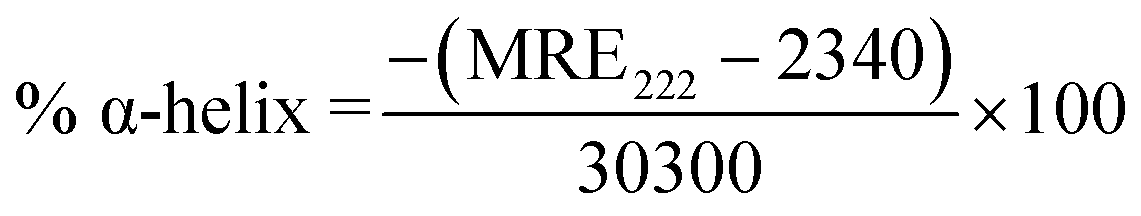
Here, the mean residue ellipticity (MRE) values were assessed from the recorded ellipticity values (θobs in mdeg at 222 nm) using the above equations, where n designates the number of amino acid residues (585 for HSA), and Cp and l are the molar concentration of HSA and the path length of the cell (here 1 cm), respectively.
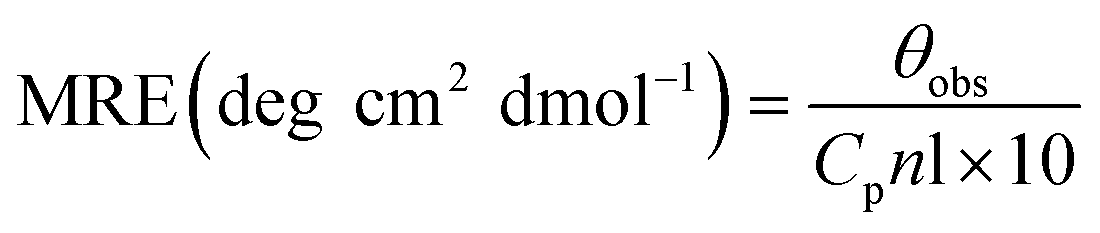
2.8. DLS sample preparation
The solvents THF and HEPES buffer were separately filtered through a 0.02 μM filter to remove any suspended particles. The DLS samples were prepared by diluting the 30 μL of stock solution into 3 ml of either THF or deionized water or their mixture and then allowed to stand for 3 h so that the solutions became homogenous. For recording the DLS spectra, 1 ml of solution was taken into a glass cuvette of 1 cm path length and allowed to stand for 3 min before recording the spectra at 25 °C. Five measurements of each sample were recorded and the mean of these are presented herein.2.9. Collection of human blood serum samples for the quantification of bilirubin
The human blood serum samples were collected from Dr Gill Path's laboratory. The values of bilirubin (calculated by the diazo method) present in human serum were provided by the laboratory and are mentioned in Table 1 (section 5). Prior to the fluorescence analysis, all the samples were centrifuged at 7000 rpm for 10 min. The supernatant liquid obtained was used and stored immediately at 4 °C. All the fluorescence measurements were carried out in a 1 ml quartz cuvette of 1 cm path length.| Sample no. | Diazo method (mg dL−1) | Present method (mg dL−1) | % recovery |
|---|---|---|---|
| 1 | 0.7 | 0.66 ± 0.02 | 95 ± 3 |
| 2 | 1.2 | 1.12 ± 0.03 | 94 ± 3 |
| 3 | 1.9 | 1.71 ± 0.05 | 90 ± 2 |
| 4 | 2.9 | 2.71 ± 0.05 | 93 ± 2 |
| 5 | 3.2 | 3.12 ± 0.10 | 97 ± 3 |
| 6 | 12.6 | 13.37 ± 0.25 | 106 ± 2 |
2.10. Determination of bilirubin in clinical samples
To the solutions of HSA (500 μM) and bilirubin (0, 10, 20, 40 μM) THYQ was added in varying amounts as 10, 20, 30, and 40 μM and the slope of the FI vs. [THYQ] graphs was determined. The plot of the slopes of these graphs vs. [bilirubin] was linear, and served as a calibration curve for the quantification of BR from the real samples. The real samples after diluting with HSA (500 μM) were titrated with THYQ (10, 20, and 40 μM) and the slopes were calculated. These slope values were then marked on the calibration curve and the corresponding BR concentration was noted.3. Results and discussion
3.1. Synthesis and characterization of THYQ
NCl-1 was synthesized by Duff's reaction of N,N-dimethylacetophenone. The reaction of NCl-1 with chloroacetaldehyde in DMF and Na2S yielded Th-Y in an 80% yield. The reaction of Th-Y with 2-methylquinoline gave THYQ (Scheme 1) in an 84% yield; HRMS (M + 1) 357.1418 (spectral data ESI† Fig. S1–S5).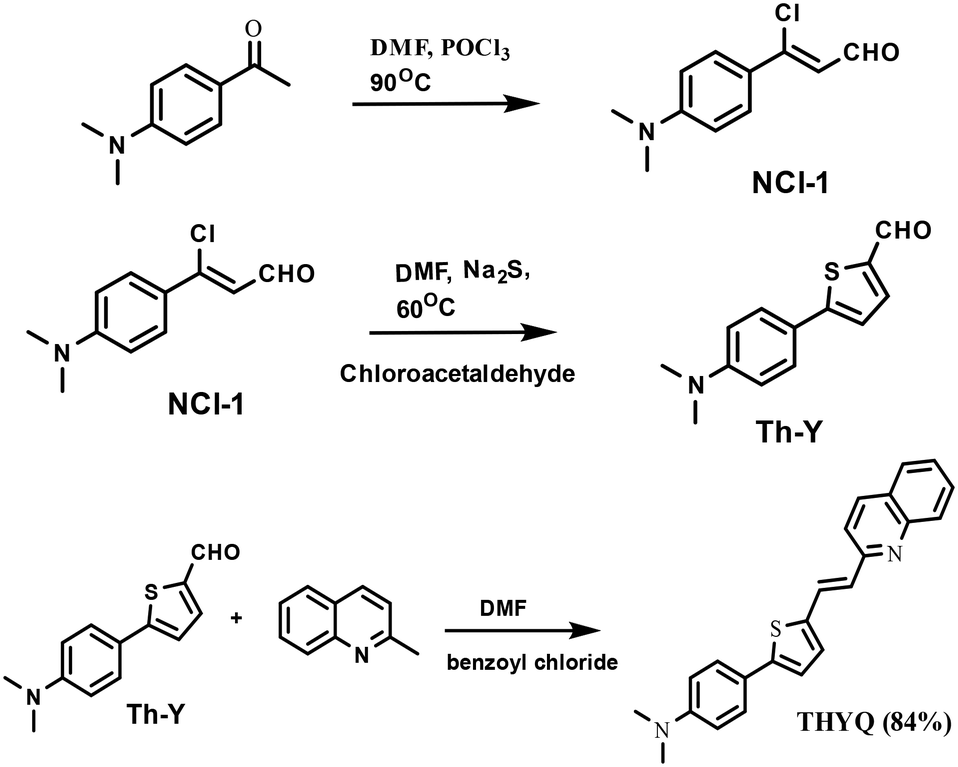 | ||
| Scheme 1 Synthetic schemes for the syntheses of NCl-1, Th-Y, and THYQ. | ||
3.2. Photophysical behaviour of THYQ in the THF-HEPES buffer binary mixtures
Fluorescent probes for the recognition of HSA, in principle, may undergo aggregation-induced quenching in water and then, upon moving the probe in to the cavity of HSA, the combined effect of de-aggregation and the restriction in rotation results in a fluorescence enhancement. Therefore, as a first step, the optical behaviour of THYQ in THF-H2O binary mixtures was investigated.The UV–vis spectrum of THYQ (10 μM) in THF exhibited an absorption maximum at ∼420 nm. The absorption of THYQ displayed only a small change in its ∈ value (∈ = 26000 ± 1200 M−1 cm−1) upon increasing fw (HEPES buffer) from 10% to 70% (Fig. 1A). The THYQ solution with fw 80% showed a dramatic decrease in absorption to 7400 M−1 cm−1 and its maximum was blue-shifted to 415 nm. Upon further increasing the fw to 90% and 99.9%, the absorption value increased to >10000 M−1 cm−1 with the maximum at 410 nm and a shoulder at ∼465 nm. The tail of the absorbance band was extended to 650 nm, pointing to Mie scattering by the aggregates of THYQ (Fig. 1A). This blue-shift in the absorbance maximum and decrease in the absorbance with increasing amounts of water suggests the formation of H aggregates.26 The appearance of the Tyndall effect by striking a red laser beam to the solution of THYQ in 99.9% fw further supports the existence of aggregates (see inset Fig. 1A).
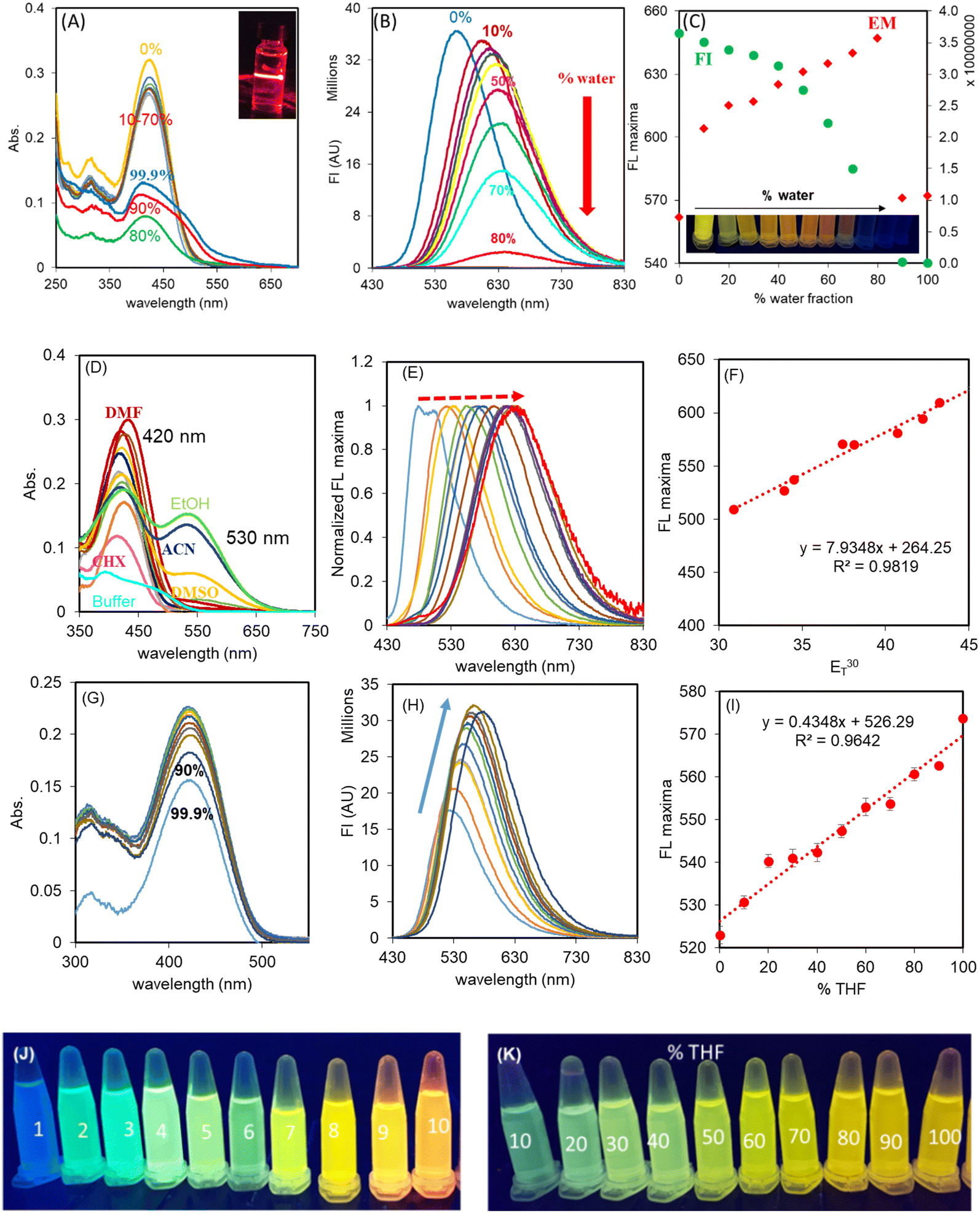 | ||
| Fig. 1 (A) Absorption spectra of THYQ (10 μM) in THF–HEPES buffer binary mixtures, inset shows the Tyndall effect in the solution of THYQ in 99.9% HEPES buffer, representing the aggregation; (B) emission spectra of THYQ solutions in THF-buffer binary mixtures; (C) plot of fwvs. the emission intensity and the maxima for the solutions of THYQ. Inset shows the change in emission colours with increasing fw in THF; (D) absorption spectra and (E) normalized emission spectra of THYQ in organic solvents with increasing polarity (ET30 value); (F) linear relationship between the emission wavelength at the maximum and ET30 values; (G) absorption spectra and (H) emission spectra of THYQ in THF–toluene binary mixtures; (I) linear relationship between the emission wavelength at maxima and the fraction of THF in toluene. [THYQ] = 10 μM, λex 420 nm; (J) fluorescence colours of THYQ solutions in various solvents under illumination at 365 nm: 1. cyclohexane, 2. toluene, 3. ether, 4. THF, 5. ethyl acetate, 6. acetone, 7. DMF, 8. DMSO, 9. CH3CN, 10. ethanol; (K) fluorescence colours of THYQ in THF–toluene binary mixtures under illumination at 365 nm. | ||
The fluorescence spectrum of THYQ (10 μM, THF) (λex 420 nm) showed the emission maximum at 562 nm. The solutions with increasing fw (HEPES buffer) exhibited a gradual red-shift of their emission maxima reaching the largest value at 647 nm in THF–HEPES buffer mixture with 80% fw, but upon further raising the fw to ≥90%, a very weak fluorescence with the maximum reversed to 572 nm was noted. The red-shift in the emission maximum from 562 nm to 647 nm (∼Δ85 nm) suggests a steady stabilization of the excited state of THYQ with increasing the amount of the water fraction and a decrease in the energy gap between the ground and excited states27 (Fig. 1B and C). The inset in Fig. 1C shows the change in colour of the solutions of THYQ in THF-water binary mixtures under illumination of 365 nm light. The decrease in fluorescence intensity of THYQ with fw ≥ 90% suggests aggregation-induced emission quenching (AIEQ) occurred.
3.3. Effect of the microenvironment, such as the polarity, viscosity, and pH, on the spectroscopic properties of THYQ
The microenvironment in the cavity of HSA and in viscous aqueous medium (Lysosome, Mitochondria of cells) have a contrasting difference between low polarity in the HSA cavity and high polarity in a viscous aqueous medium. To validate the effect of polarity of the medium on the fluorescence colour of THYQ, solutions of THYQ (10 μM) were made in solvents of varied polarity (Table S1†) with ET30 values changing from 30.9 to 55.4, and their UV–vis and fluorescence spectra were recorded. THYQ in non-polar solvents with an ET30 value ranging from 30.9 to 42.2 displayed an absorbance band at 420 nm. In DMSO, CH3CN, and ethanol, as solvents with a higher ET30 value, THYQ displayed an additional low-energy absorbance band at 530 ± 2 nm, probably due to a strong stabilization of the ground state (Fig. 1D). The fluorescence spectra of these solutions (λex 420 nm) exhibited strong positive solvatochromism with a red-shift in the emission maximum with the increase in polarity (ET30 value) (Fig. 1E) and a change in the fluorescence colour from blue in cyclohexane to yellow-orange in ethanol (Fig. 1J). Significantly, the plot of the ET30 values of these solvents against their maximum emission wavelengths showed a linear relationship, with a slope of 7.89 nm/ET30 unit (Fig. 1F). Such a steep change in the wavelength of the emission maximum with an increase in ET30 value is less known (Table S2†).The UV–vis and fluorescence spectra of THYQ in binary mixtures of toluene and THF further validated the sensitivity of the probe to the polarity of the medium. In most of the fractions of toluene–THF (10–90%), there was only a small change in absorbance, but upon increasing the fraction of THF from 90% to 99.9%, the absorbance decreased from 0.211 to 0.177 (Fig. 1G). The emission spectrum of THYQ in THF displayed a maximum at 572 nm, the emission maximum of the HSA–THYQ complex. Upon steadily increasing the amount of toluene, the fluorescence intensity gradually decreased and the emission maximum gradually shifted from 572 to 527 nm (Fig. 1H and I) and the fluorescence colour of the solutions changed from green to yellow (Fig. 1K). Therefore, this resulted in a small range of ET30 values, for THF (ET30 37.4) to toluene (ET30 33.9), and showed the viability of the probe even in a narrow range.28
The fluorescence intensity (FI) of THYQ (at 572 nm) in water–glycerol binary mixtures at up to 50% of glycerol increased only by 1.6 times without any shift in emission maximum, but further increase with the amount of glycerol, especially in 80% and 90% glycerol solutions, where the fluorescence intensity increased sharply. In the 90% glycerol–water mixture, the FI increased by 18 times in comparison to that in water and the fluorescence maximum was shifted from 572 nm to 640 nm (Fig. S6†). The restriction in rotation (RIR) at higher viscosity led to the increase in fluorescence.29 Further, between pH 5–11, both the UV–vis and fluorescence spectra of THYQ remained stable, indicating its potential use under physiological conditions (Fig. S7†).
3.4. Selectivity and sensitivity of THYQ towards HSA
The probe THYQ upon moving into the cavity of a protein, a hydrophobic environment, could be expected to cause an enhancement in its fluorescence intensity and display a blue-shift in its emission maximum compared to that observed in water. In order to evaluate the selectivity and sensitivity of THYQ towards HSA in comparison to different biomolecules, 5 equivalents each of these were added to THYQ (10 μM, HEPES buffer, pH 7.4) and the fluorescence spectra of these solutions were recorded. It was noticed that the presence of HSA caused a 55-fold increase in the fluorescence intensity at 555 nm, whereas the addition of other biomolecules, like proteins, amino acids, anions, thiols, etc., caused practically no change in fluorescence (Fig. 2A and S8†). Thus, the emission of THYQ in the presence of HSA (555 nm, green) and glycerol (640 nm, red) differentiated the two situations (Fig. 2B). In addition to this, the presence of HSA in solution could be distinctly differentiated without interference from any cations, anions, thiols, amines, or amino acids (Fig. S9†).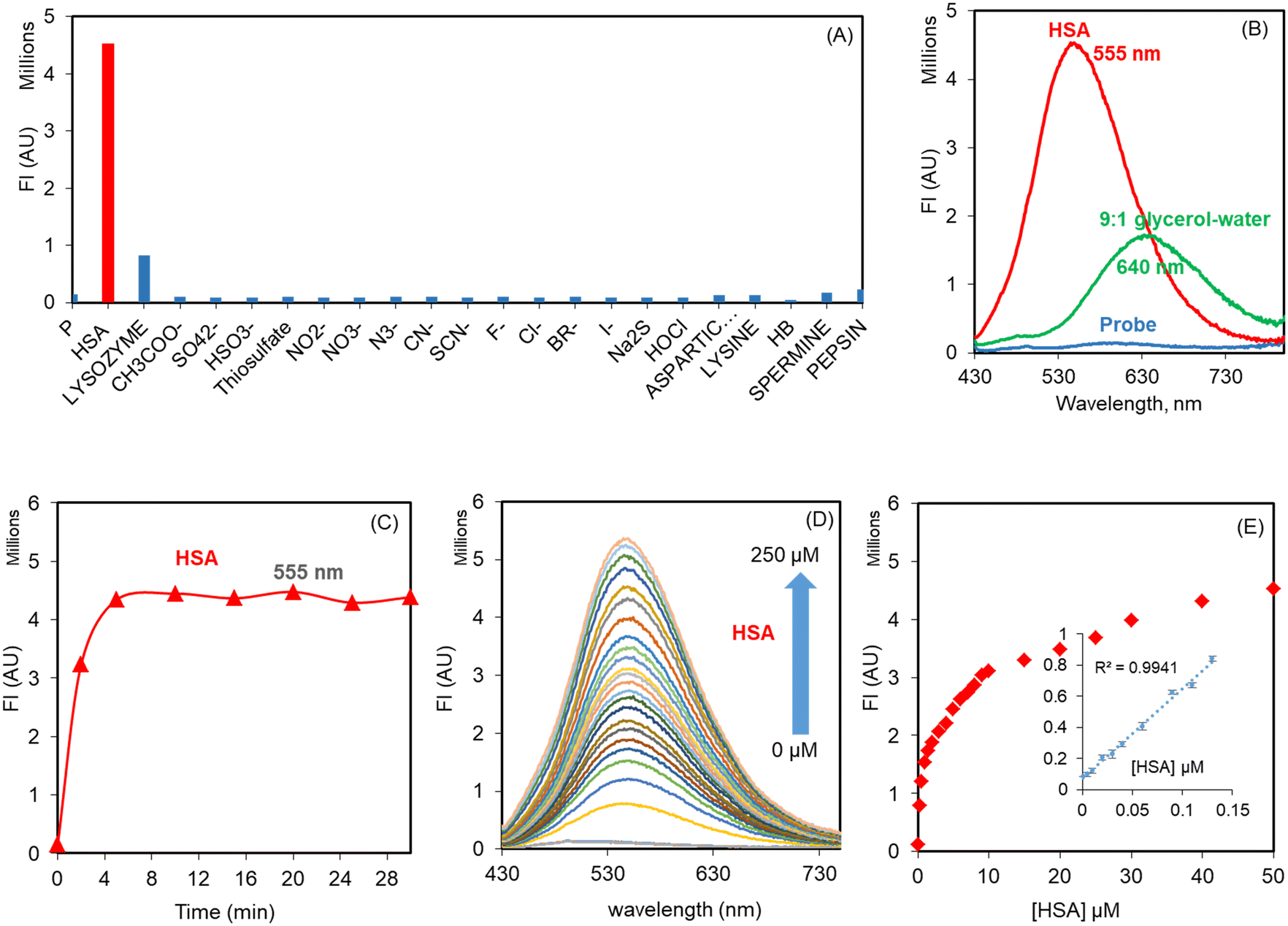 | ||
| Fig. 2 (A) Change in fluorescence intensity of THYQ (10 μM, HEPES buffer, pH 7.4) with 5 equivalents each of HSA, anions, thiols, amines, and amino acids; (B) fluorescence spectra of THYQ upon the addition of HSA and glycerol; (C) time-dependent change in fluorescence upon the addition of 30 μM HSA to THYQ (10 μM, HEPES buffer, 0.1% THF); (D) fluorescence spectra of THYQ (10 μM) with the incremental addition of HSA; (E) plot of the fluorescence intensity vs. [HSA] at 555 nm; inset shows the linear range and LOD for HSA, where I0 and I are the fluorescence intensities of THYQ in the absence and presence of different amounts of HSA. | ||
THYQ (10 μM, HEPES buffer, 0.1% THF) upon excitation at 420 nm displayed weak fluorescence with a maximum at 570 nm. The fluorescence intensity of THYQ upon the addition of 50 μM HSA achieved a plateau within 5 min, indicating its rapid response towards HSA (Fig. 2C). The gradual addition of aliquots of HSA to THYQ led to a steady increase in emission intensity with the maximum at 555 nm. After the addition of 50 μM HSA, only a residual change in fluorescence intensity was observed. However, the UV–vis spectrum of THYQ showed only a small change upon the addition of HSA (Fig. S13†). The plot of [HSA] vs. the fluorescence intensity showed a linear change between 0 to 0.13 μM HSA. The LOD for HSA was 2.8 nM as per IUPAC norms (Fig. 2D and E). Since, in human serum, the [HSA] is 500–700 μM, we were interested in knowing the binding stoichiometry of THYQ (10 μM) with HSA under these conditions. Here, multivariate global regression analysis of the spectra for the titration of THYQ with HSA showed that at [HSA] ≥ 200 μM, one molecule of THYQ binds with two molecules of HSA to form the THYQ∩(HSA)2 complex with logβ 9.52 and no formation of any other species at this concentration of HSA was observed. Significantly, at [HSA] = <20 μM, the (THYQ)2HSA. complex was the major species (Fig. S10A†).
Therefore, THYQ in buffer existed as aggregates, which may be in equilibrium with its monomers. Upon interaction with HSA, the monomers bind in the cavity of HSA to give a high fluorescence and the equilibrium is shifted towards the monomers. The high fluorescence could be attributed to the decreased polarity in the cavity of HSA and the restriction in rotation.30 The participation of a FRET mechanism was negligible (Fig. S14†) as the UV–vis spectrum of HSA had a poor overlap with the fluorescence of THYQ.
3.5. Mechanism of interaction of THYQ with HSA
:200) was titrated with warfarin, ibuprofen, bilirubin, and hemin. Quite interestingly, with 100 μM bilirubin/hemin (both subdomain IB specific), the fluorescence intensity of the THYQ–HSA complex was completely quenched. However, warfarin (subdomain IIA specific) and ibuprofen (subdomain IIIA specific) did not cause any substantial change in the fluorescence intensity of the THYQ–HSA complex (Fig. 3A). Therefore, in the THYQ–HSA complex, THYQ binds at the bilirubin site only and was displaced upon the addition of bilirubin and hemin only. The logβ value for this displacement process is logβ = 4.83 ± 0.12.
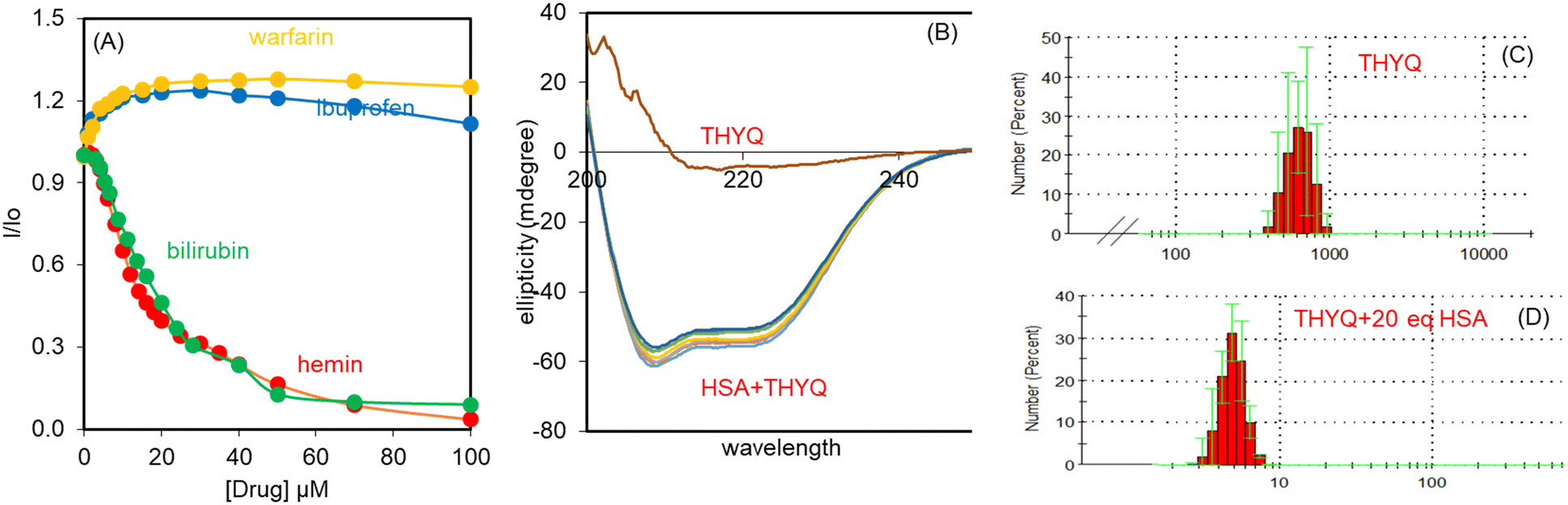 | ||
| Fig. 3 (A) Effect of the increasing concentrations of site markers on the fluorescence of THYQ–HSA (10:200) complex; (B) CD spectra of HSA with an increasing amount of THYQ; (C and D) DLS of (C) THYQ and (D) THYQ + 200 μM HSA. | ||
Thus, all these studies revealed that THYQ in the presence of excess (>200 μM) HSA preferably binds at subdomain 1B of HSA – the binding domain of bilirubin. THYQ exhibited differential changes in fluorescence with HSA, and with the viscosity and polarity of the medium, and has potential for the development of a strategy for the detection of bilirubin.
4. Protocol for the determination of bilirubin in HSA
The preceding results showed that in the THYQ–HSA (1:20) complex the addition of bilirubin resulted in an efficient quenching of fluorescence, i.e. bilirubin was able to displace the THYQ molecule from the THYQ–(HSA)2 complex (Fig. 4A and B). For the displacement of THYQ, the logβ value was 4.83 ± 0.12. We envisaged that if the fluorescence probe THYQ would be able to displace the bilirubin from the cavity of HSA, the equilibrium state between the complexes of HSA with THYQ and bilirubin would be validated (eqn 1, Scheme 2). The addition of THYQ to the weakly fluorescent bilirubin–HSA (1:20) displaced bilirubin with THYQ to form a fluorescent complex (Fig. 4C and D). The logβ value was 4.81 ± 0.13. It was noteworthy that the addition of bilirubin to THYQ or HSA alone did not lead to any significant change in fluorescence intensity under these conditions. This provides a method for the detection of BR from the HSA–BR mixture. Further, proteins, like pepsin, lysozyme, and bromoaniline, did not interfere in detection of BR (Fig. S12†).
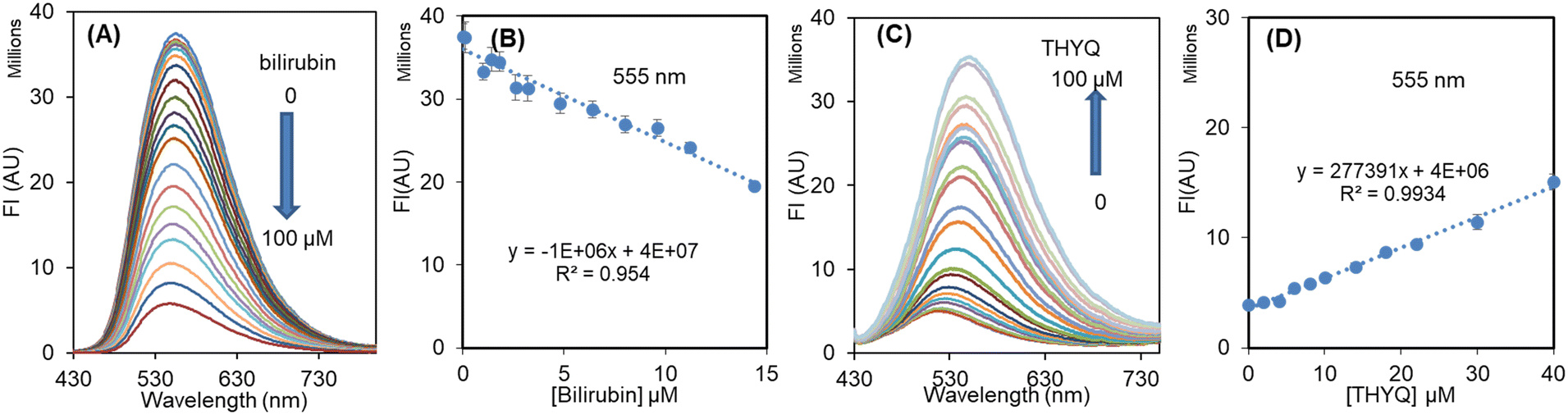 | ||
| Fig. 4 (A) Change in fluorescence spectrum of THYQ–HSA (10:200) with increasing the concentration of bilirubin; (B) linear plot showing the decrease in fluorescence intensity at 555 nm with [bilirubin]; (C) change in fluorescence spectrum of BR–HSA (20:200) complex with [THYQ]; (D) linear plot showing the increase in fluorescence intensity at 555 nm with [THYQ]. | ||
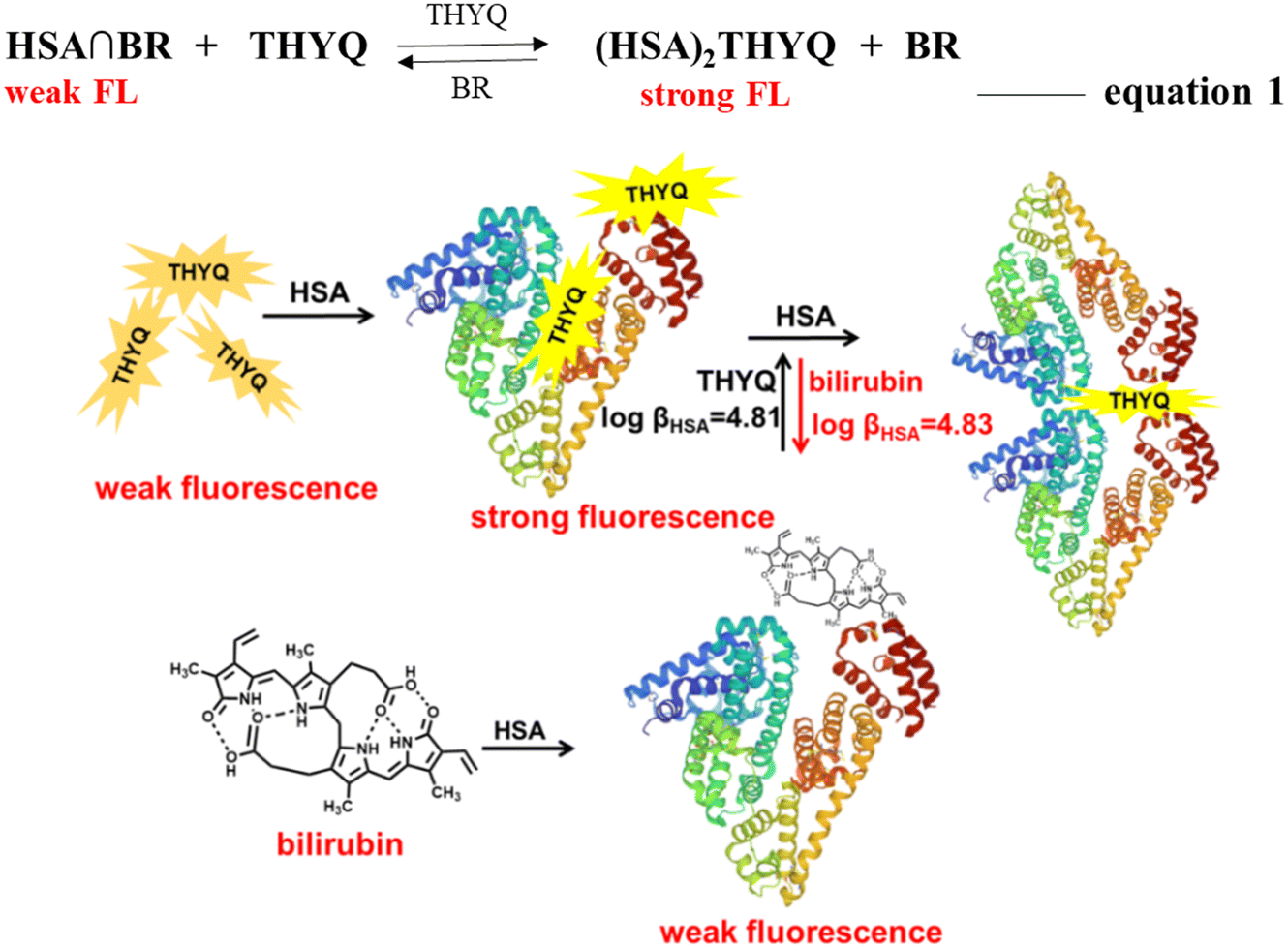 | ||
| Scheme 2 Schematic presentation of the reversible displacement of THYQ by bilirubin and vice versa from its respective complex with HSAs. | ||
The normal level of HSA in human serum is in the range of 35–50 g L−1 or approx. 500–700 μM. Therefore, keeping the [HSA] = 500 μM, different concentrations of bilirubin, i.e. 0, 20, 40, 60, 80, 200, and 300 μM, were added and these solutions were titrated with THYQ. As expected, it was found that as the concentration of BR increased in HSA, it required a greater and greater amount of THYQ to displace it. This resulted in a decrease in the slope of the linear plot of the fluorescence intensity vs. [THYQ]. Consequently, the slope for the titration of the BR–HSA complex was linearly dependent on the [BR] in Fig. 5A. The plot of the slope of these linear plots vs. [bilirubin] (Fig. 5B) revealed that the value of the slope changed linearly between 0–80 μM [bilirubin], and thus may have application potential in determining bilirubin from human serum.
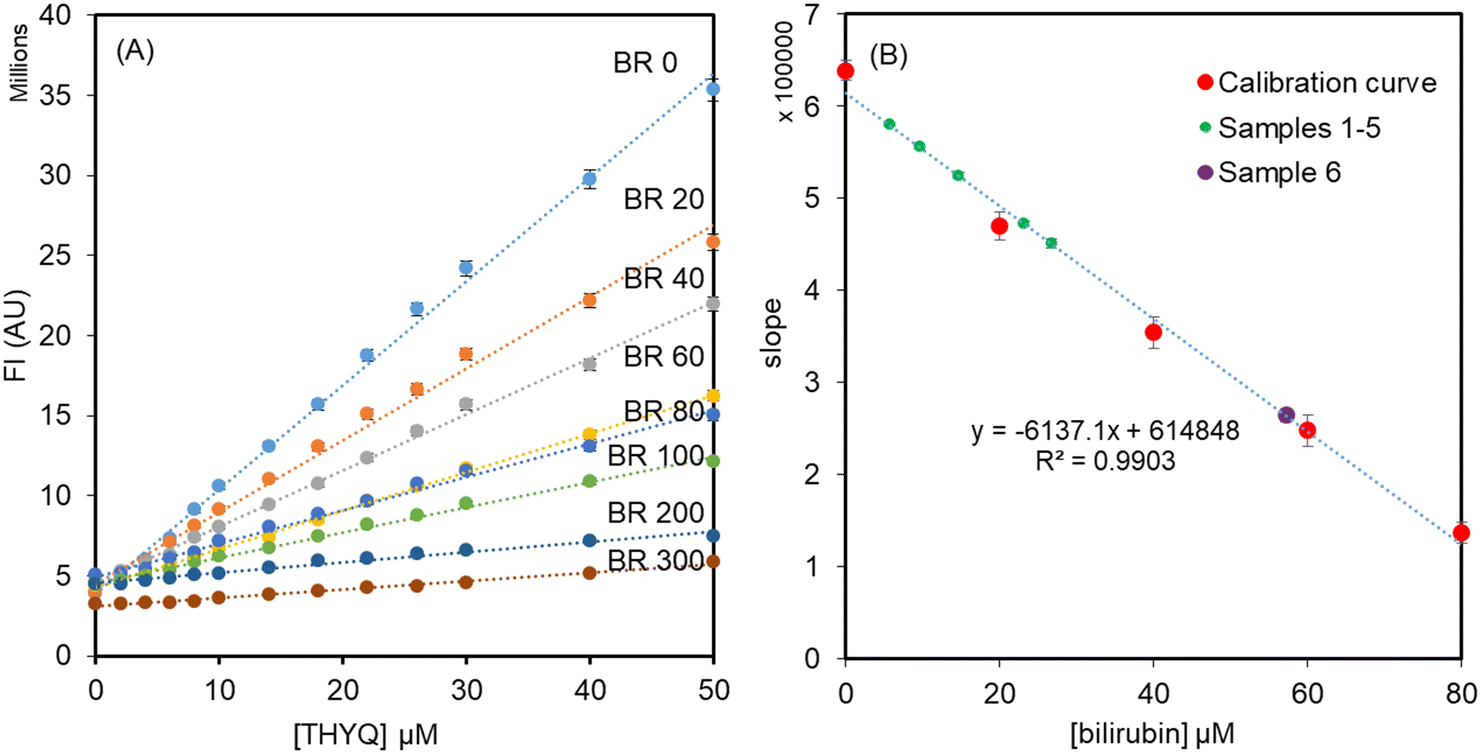 | ||
| Fig. 5 (A) Titration of solutions of HSA (500 μM) + bilirubin (0–300 μM) with the probe THYQ; (B) linear relationship between the slopes and [bilirubin]. | ||
5. Analysis of bilirubin in clinical samples
Further to validate the practical application of the fluorescent probe THYQ, the detection of bilirubin levels was performed in human blood serum samples. For samples 1–5, 400 μl human serum was diluted with 400 μl of HSA (500 μM, 0.01 M HEPES buffer) The fluorescence spectrum of the solution was recorded and then aliquots of THYQ (8 μl, 10 μM) were added twice and each time the fluorescence spectrum of the solution was recorded. The plot of the fluorescence intensity vs. [THYQ] was linear, for which the slope was calculated. The [bilirubin] was calculated from the calibration curve as in Fig. 5B and then multiplied by 2 to compensate for the dilution factor, before finally being converted to mg dL−1 (Table 1). For serum sample 6, with a higher amount of bilirubin (as apparent from the dark yellow colour), 200 μl serum sample was diluted with HSA (600 μl, 500 μM, 0.01 M HEPES buffer) to four times its volume and the slope for titration was determined by adding two aliquots of THYQ. The [bilirubin] was calculated from the calibration curve and then multiplied by 4 to compensate for the dilution factor. The amount of bilirubin determined by this fluorescence method was within ±10% of that determined by a clinical method.Therefore, the present fluorescence-based quantification of bilirubin is unique in that does not involve any chemical reaction and primarily depends on the displacement of bilirubin in HSA–BR complex by THYQ to cause fluorescence “Turn ON” under physiological conditions and straightforwardly determines BR from serum. This approach enables the real-time determination of bilirubin without harming any of the substrate,20 which is not achieved by the chemosensors discussed below.
The fluorescent probes reported earlier for the detection of bilirubin (BR) were based on organic molecules and nanomaterials and most of them show “Turn OFF” processes (Table S3,† ref. 1, 4–7 and 10–13). Even, the fluorescent systems showing “Turn ON” processes have a number of limitations. The nanomaterials with “Turn ON” mechanism towards BR are active only under strong acidic (pH 2.5, 5.0) (Table S3,† ref. 8 and 14) and basic (pH 11.4) (Table S3,† ref. 9) conditions and depend on the release of fluorescence upon the coordination of BR with Fe3+. Moreover, BR is detected through spiking methods and does not represent a clinical detection. In the case of “Turn ON” fluorescent organic molecular systems16,17 (Table S3,† ref. 2 and 3), the redox reaction of Fe3+ (added in excess) with free bilirubin produces Fe2+, which causes deoxgenation of the probe N-oxide to release the fluorescence. The two mechanisms are contradictory to each other in terms of the respective coordination and oxidation of BR with Fe3+ in nanomaterials and organic systems.
6. Conclusions
THYQ was found to bind selectively at subdomain IB of HSA and exhibited a 55-fold increase in fluorescence intensity at 555 nm and an LOD for HSA of 2.8 nM. The reversible displacement of THYQ in THYQ–HSA complex with bilirubin (logβHSA = 4.83 ± 0.12) and that of BR in the non-fluorescent BR∩HSA complex with THYQ (logβHSA = 4.81 ± 0.13) provides an opportunity for the detection of bilirubin from human serum through a displacement approach, with an LOD for BR of 0.004 mg dL−1 (68 nM). The slope of titration of BR–HSA with THYQ was found to be linearly dependent on [bilirubin] in HSA and enables a fluorescence-based direct method for the detection of bilirubin below normal (0.7 mg dL−1, 12 μM) to hyperbilirubinemia conditions (12.6 mg dL−1, 216 μM). The results for the detection of BR by this displacement approach in real samples were in good agreement with clinical findings.
Human sample statement
The permission for the experiments with blood samples was provided by the institutional ethical committee, Guru Nanak Dev University, and in accordance with the Declaration of Helsinki. Informed consents were obtained from all participants in this study.Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
SK thanks UGC, New Delhi for UGC-BSR faculty fellowship F.4-5(11)/2019 (BSR). We also thank UGC for PURSE, UPE and CAS programs and DST for FIST program.References
- T. Slusher, T. Zamora, D. Appiah, J. Stanke, M. Strand, B. Lee, S. Richardson, E. Keating, A. Siddappa and B. Olusanya, BMJ Paediatr. Open, 2017, 1, 000105 CrossRef PubMed.
- R. P. Wennberg, Clin. Chem., 2012, 58, 811–813 CrossRef CAS PubMed.
- S. Ullah, K. Rahman and M. Hedayati, Iran. J. Public Health, 2016, 45, 558–568 Search PubMed.
- A. A. H. Van den Bergh and P. Mueller, Bilirubin, Biochem. Z., 1916, 77, 90–103 Search PubMed.
- A. Westwood, Ann. Clin. Biochem., 1991, 28, 119–130 CrossRef CAS PubMed.
- R. R. Anjana, J. S. Anjali Devi, M. Jayasree, R. S. Aparna, B. Aswathy, G. L. Praveen, G. M. Lekha and G. Sony, Microchim. Acta, 2018, 185, 1–11 CrossRef CAS PubMed.
- K. Abha, J. Nebu, J. S. A. Devi, R. S. Aparna, R. R. Anjana, A. O. Aswathy and S. George, Sens. Actuators, B, 2019, 282, 300–308 CrossRef CAS.
- B. Barik and S. Mohapatra, Anal. Biochem., 2022, 654, 114813 CrossRef CAS PubMed.
- W. Xiao, J. Liu, Y. Xiong, Y. Li and H. Li, Anal. Bioanal. Chem., 2021, 413, 7009–7019 CrossRef CAS PubMed.
- K. Yi, H. Li, X. Zhang and L. Zhang, Inorg. Chem., 2021, 60, 3172–3180 CrossRef CAS PubMed.
- C. Xia, Y. Xu, M.-M. Cao, Y.-P. Liu, J.-F. Xia, D.-Y. Jiang, G.-H. Zhou, R.-J. Xie, D.-F. Zhang and H.-L. Li, Talanta, 2020, 212, 120795 CrossRef CAS PubMed.
- S. Ellairaja, K. Shenbagavalli, S. Ponmariappan and V. S. Vasantha, Biosens. Bioelectron., 2017, 91, 82–88 CrossRef CAS PubMed.
- V. Srinivasan, M. A. Jhonsi, N. Dhenadhayalan, K.-C. Lin, D. A. Ananth, T. Sivasudha, R. Narayanaswamy and A. Kathiravan, Spectrochim. Acta, 2019, 221, 117150 CrossRef CAS PubMed.
- Y. Hisamatsu, K. Otani, H. Takase, N. Umezawa and T. Higuchi, Chem. – Eur. J., 2021, 27, 6489–6499 CrossRef CAS PubMed.
- S. Qi, X. He, S. Zhang, P. Xu, M. Su, B. Dong and B. Song, Anal. Chim. Acta, 2023, 1238, 340657 CrossRef CAS PubMed.
- E. Ahmmed, A. Mondal, A. Sarkar, S. Chakraborty, S. Lohar, N. C. Saha, K. Dhara and P. Chattopadhyay, ACS Appl. Bio Mater., 2020, 3, 4074–4080 CrossRef CAS PubMed.
- E. Ahmmed, A. Mondal, N. C. Saha, K. Dhara and P. Chattopadhyay, Anal. Methods, 2021, 13, 5651–5659 RSC.
- C. E. Petersen, C.-E. Ha, K. Harohalli, J. B. Feix and N. V. Bhagavan, J. Biol. Chem., 2000, 275, 20985–20995 CrossRef CAS PubMed.
- G. H. Beaven, A. D'Albis and W. B. Gratzer, Eur. J. Biochem., 1973, 33, 500–509 CrossRef CAS PubMed.
- (a) A. C. Sedgwick, J. T. Brewster II, T. Wu, X. Feng, S. D. Bull, X. Qian, J. L. Sessler, T. D. James, E. V. Anslyn and X. Sun, Chem. Soc. Rev., 2021, 50, 9–38 RSC; (b) B. T. Nguyen and E. V. Anslyn, Coord. Chem. Rev., 2006, 250, 3118–3127 CrossRef CAS.
- S. Dhiman, R. Kour, G. Kumar, S. Kaur, V. Luxami, P. Singh and S. Kumar, Mater. Chem. Front., 2022, 6, 2651–2660 RSC.
- J. Mocak, A. M. Bond, S. Mitchell and G. Scollary, Pure Appl. Chem., 1997, 69, 297–328 CrossRef CAS.
- G. A. Crosby and J. N. Demas, J. Phys. Chem., 1971, 75, 991–1024 CrossRef CAS.
- H. Gampp, M. Maeder, C. J. Meyer and A. D. Zuberbühler, Talanta, 1985, 32, 257–264 CrossRef CAS PubMed.
- M. Sasmal, A. S. M. Islam, R. Bhowmick, D. Maiti, A. Dutta and M. Ali, ACS Appl. Bio Mater., 2019, 2, 3551–3561 CrossRef CAS PubMed.
- (a) F. C. Spano, Acc. Chem. Res., 2010, 43, 429 CrossRef CAS PubMed; (b) E. C. Kwok, D. P. Tsang, M. Chan and V. W. Yam, Chem. – Eur. J., 2013, 19, 2757–2767 CrossRef CAS PubMed; (c) N. J. Hestand and F. C. Spano, Chem. Rev., 2018, 118, 7069–7163 CrossRef CAS PubMed.
- H. Xiao, P. Li and B. Tang, Coord. Chem. Rev., 2021, 427, 213582 CrossRef CAS; Z. Zhang, Z. Gou, B. Dong and M. Tian, Sens. Actuators, B, 2022, 355, 131349 CrossRef; C. Wu, X. Li, T. Zhu, M. Zhao, Z. Song, S. Li, G. Shan and G. Niu, Anal. Chem., 2022, 94, 3881–3887 CrossRef PubMed.
- (a) K. Kudo, A. Momotake, Y. Kanna, Y. Nishimura and T. Arai, Chem. Commun., 2011, 47, 3867–3869 RSC; (b) J. Yin, M. Peng, Y. Ma, R. Guo and W. Lin, Chem. Commun., 2018, 54, 12093–12096 RSC; (c) R. Chen, Z. Li, C. Peng, L. Wen, L. Xiao and Y. Li, Anal. Chem., 2019, 91, 1928–1935 CrossRef PubMed.
- (a) R. D. Telore, M. A. Satam and N. Sekar, Dyes Pigm., 2015, 122, 359–367 CrossRef CAS; (b) H. S. Kumbhar, S. S. Deshpande and G. S. Shankarling, ChemistrySelect, 2016, 1, 2058–2064 CrossRef CAS.
- (a) S. Dhiman, R. K. G. Kumar, S. Kaur, V. Luaxmi, P. Singh and S. Kumar, Mater. Chem. Front., 2022, 6, 2651–2660 RSC; (b) S. Samanta, S. Halder and G. Das, Anal. Chem., 2018, 90, 7561–7568 CrossRef CAS PubMed; (c) S. I. Reja, I. A. Khan, V. Bhalla and M. Kumar, Chem. Commun., 2016, 52, 1182–1185 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sd00157a |
| This journal is © The Royal Society of Chemistry 2023 |