
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceGenetically engineered filamentous phage for bacterial detection using magnetic resonance imaging†
Raymond E.
Borg‡
a,
Harun F.
Ozbakir‡
b,
Binzhi
Xu‡
c,
Eugene
Li
b,
Xiwen
Fang
b,
Huan
Peng
d,
Irene A.
Chen
 *d and
Arnab
Mukherjee
*abef
*d and
Arnab
Mukherjee
*abef
aDepartment of Chemistry, University of California, Santa Barbara, CA 93106, USA. E-mail: arnabm@ucsb.edu
bDepartment of Chemical Engineering, University of California, Santa Barbara, CA 93106, USA
cBiomolecular Science and Engineering, University of California, Santa Barbara, CA 93106, USA
dDepartment of Chemical and Biomolecular Engineering, University of California, Los Angeles, CA 90095, USA. E-mail: ireneachen@ucla.edu
eBiological Engineering, University of California, Santa Barbara, CA 93106, USA
fNeuroscience Research Institute, University of California, Santa Barbara, CA 93106, USA
First published on 22nd June 2023
Abstract
Detecting bacterial cells with high specificity in deep tissues is challenging. Optical probes provide specificity, but are limited by the scattering and absorption of light in biological tissues. Conversely, magnetic resonance imaging (MRI) allows unfettered access to deep tissues, but lacks contrast agents for detecting specific bacterial strains. Here, we introduce a biomolecular platform that combines both capabilities by exploiting the modularity of M13 phage to target bacteria with tunable specificity and allow deep-tissue imaging using T1-weighted MRI. We engineered two types of phage probes: one for detecting the phage's natural host, viz., F-pilus expressing E. coli; and the other for detecting a different (F-negative) bacterial target, V. cholerae. We show that these phage sensors generate 3–9-fold stronger T1 relaxation upon recognizing target cells relative to non-target bacteria. We further establish a preliminary proof-of-concept for in vivo applications, by demonstrating that phage-labeled bacteria can be detected in mice using MRI. The framework developed in this study may have potential utility in a broad range of applications, from basic biomedical research to in situ diagnostics, which require methods to detect and track specific bacteria in the context of intact living systems.
1. Introduction
The ability to image bacteria in deep tissues has the potential to transform clinical practice by improving the diagnosis of bacterial infections and allowing spatiotemporal tracking of treatment outcomes within the body.1,2 With effective tools to noninvasively detect bacteria, diagnostic procedures could surpass current practice based on biopsy and cell culture, which are invasive, prone to sampling errors, and generally provide delayed results.3,4 In preclinical research, bacterial imaging tools are needed to characterize disease mechanisms and host–microbiota interactions and for evaluating new therapeutic approaches in animal models of infection.5 The most common techniques used to detect live bacteria in preclinical research are fluorescence and bioluminescence imaging. However, optical imaging is limited to shallow depths owing to absorption and scattering of light by biological tissue. Tissue-penetrant imaging techniques are therefore required to locate bacteria in opaque animals. To this end, nuclear imaging is widely used to visualize bacteria in vivo using radioactive tracers conjugated to ligands such as antibiotics, antimicrobial peptides, and sugars, which confer selectivity for bacteria over mammalian cells.6–12 Although nuclear imaging achieves high sensitivity, its spatial resolution is limited (mm-scale in rodents), and ionizing radiation is required to penetrate tissues.Compared with nuclear imaging, magnetic resonance imaging (MRI) can generate 3-dimensional images with a high anatomical resolution (∼100 μm in rodents) and excellent soft-tissue contrast, without posing any radiation risk. However, efforts to directly image bacteria using MRI have been hindered by the inadequate specificity of existing approaches: contrast agents are most often used to report on bacteria indirectly by detecting immune cells that home to sites of infection.13–15 The first example of an MRI probe targeting bacteria involved a Gd3+ chelate conjugated to zinc dipicolylamine, which binds to negatively charged phospholipids in the bacterial membrane. The presence of Gd3+ increases the spin–lattice relaxation rate of bacterial cells, thereby allowing their detection by T1 weighted MRI.16 Subsequently, similar Gd3+ chelates were conjugated with antibiotics to target pathogenic microbes for imaging using T1 weighted MRI.17–20 Contrast agents based on iron oxide nanoparticles have also been used to image bacteria by targeting cell-surface antigens; however, the negative contrast (i.e., labeled cells appear dark) generated by these probes is sometimes difficult to disambiguate from signal voids caused by magnetic field inhomogeneities and susceptibility differences in tissue.21–23 The principal limitation of the aforementioned MRI approaches (Table S1†) is that they are not capable of detecting specific bacterial species but instead broadly distinguish bacteria from mammalian cells. Given this limitation, there is a need to develop new MRI tools that can be modularly tailored to detect specific bacteria and ideally provide bright (i.e. T1 weighted) signals, which are robust to field inhomogeneities.
Here, we introduce a new MRI-based approach to detect and image bacterial cells with high specificity by exploiting a non-lytic filamentous bacteriophage, known as M13. The phage capsid is composed of five proteins (pIII, pV, pVII, pVIII, and pIX) that can be genetically and chemically modified to simultaneously achieve multiple functionalities such as binding to drug molecules and targeting specific cells. The modularity of M13 bacteriophage has been widely exploited to transport drugs and genetic payloads to various in vivo targets, including tumors,24–32 infections,33–37 gut microbiota,38,39 and even difficult-to-reach locations, such as past the blood–brain barrier.40,41 M13 phage also provides an excellent scaffold for bacterial detection due to its natural affinity for strains that express F-pili, and its ability to be genetically engineered to specifically target other F-negative bacterial species. To this end, phages have been applied for imaging specific bacteria by loading the phage capsid with fluorescent dyes and nuclear isotopes (Table S2†).42–46 However, the application of phage technology for MRI has been sparse (Table S3†), with no reported examples (to our knowledge) of using phages to detect and visualize bacteria using MRI. In this study, we explored whether phages could be adapted as MRI sensors by genetically engineering their capsid to bind Mn2+, a T1-weighted contrast agent; and whether the resulting Mn2+-loaded phage could be used to image bacteria that express F-pili, and further tuned to detect other F-negative bacterial strains.
2. Materials and methods
Detailed methods are provided in ESI.†2.1 Mn2+ loading of purified phage
To load DP1-phage with Mn2+, purified phage particles were incubated with MnCl2 at a 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (Mn2+ to DP1) molar stoichiometry, mixed briefly by vortexing (∼5 s), and then placed in a rotator (∼15 r.p.m.) for approximately 15 min. To convert the phage titer into molar concentration of pVIII, we estimated that 2172 pVIII molecules were present in individual phage particles. The phage–manganese mixture was then treated with PEG:NaCl precipitation buffer and chilled on ice for 30 min. Phages were subsequently purified by three rounds of precipitation and centrifugation (17000 × g, 10 min at 4 °C), resuspended in 200–300 μL of HEPES buffer (20 mM, pH 7.0), and quantified based on optical absorbance (ESI†). Phages were freshly loaded with Mn2+ before each experiment.
:1 (Mn2+ to DP1) molar stoichiometry, mixed briefly by vortexing (∼5 s), and then placed in a rotator (∼15 r.p.m.) for approximately 15 min. To convert the phage titer into molar concentration of pVIII, we estimated that 2172 pVIII molecules were present in individual phage particles. The phage–manganese mixture was then treated with PEG:NaCl precipitation buffer and chilled on ice for 30 min. Phages were subsequently purified by three rounds of precipitation and centrifugation (17000 × g, 10 min at 4 °C), resuspended in 200–300 μL of HEPES buffer (20 mM, pH 7.0), and quantified based on optical absorbance (ESI†). Phages were freshly loaded with Mn2+ before each experiment.
2.2 MRI characterization of phage
Plastic tubes containing phage suspensions in HEPES buffer were placed in water-filled agarose (1% w/v) moulds in a 3D printed MRI phantom designed to minimize magnetic susceptibility differences between the samples and the surrounding environment. All in vitro MRI experiments were performed using a Bruker 7 Tesla vertical-bore MRI scanner with a 66 mm diameter transceiver coil. To measure the spin–lattice relaxation rate (R1), phage suspensions were imaged using a rapid acquisition with relaxation enhancement (RARE) pulse sequence with the following parameters: echo time, TE: 12.6 ms, RARE factor: 4, field of view: 4.7 cm × 4.7 cm, matrix size: 128 × 128, slice thickness: 1.5–2 mm, and variable repetition times, TR: 173.5, 305.9, 458.6, 638.8, 858.7, 1141.1, 1536.1, 2198.4, and 5000 ms.The Fiji package of ImageJ was used to measure the average signal intensities in manually drawn regions of interest (ROI) encompassing phage samples. To estimate R1, the change in signal intensity as a function of repetition time (TR) was fitted to the equation 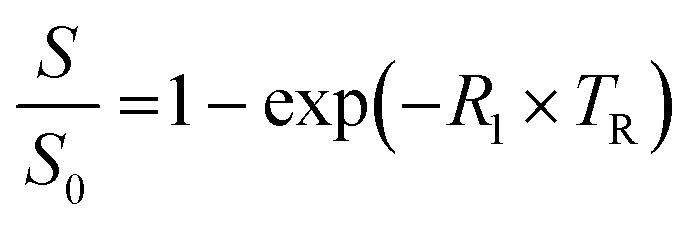 , which describes the growth of magnetization by spin–lattice relaxation following radiofrequency excitation. The molar relaxivity (r1) of phage was derived by global fitting of R1versus the concentration of pVIII and a 95% confidence interval were calculated using the nlparci() function in Matlab (R2022b). To test whether DP1-phage is selective for Mn2+, the Mn2+-loaded phage was resuspended in HEPES buffer supplemented with divalent metals at physiologically relevant (extracellular) concentrations: 1.8 mM CaCl2, 1 mM MgCl2, 20 μM FeSO4, and 10 μM ZnCl2. Phages were subsequently purified by two rounds of PEG precipitation, resuspended in 200–300 μL of HEPES buffer, quantified based on absorbance, and characterized by relaxometric titration as described above.
, which describes the growth of magnetization by spin–lattice relaxation following radiofrequency excitation. The molar relaxivity (r1) of phage was derived by global fitting of R1versus the concentration of pVIII and a 95% confidence interval were calculated using the nlparci() function in Matlab (R2022b). To test whether DP1-phage is selective for Mn2+, the Mn2+-loaded phage was resuspended in HEPES buffer supplemented with divalent metals at physiologically relevant (extracellular) concentrations: 1.8 mM CaCl2, 1 mM MgCl2, 20 μM FeSO4, and 10 μM ZnCl2. Phages were subsequently purified by two rounds of PEG precipitation, resuspended in 200–300 μL of HEPES buffer, quantified based on absorbance, and characterized by relaxometric titration as described above.
2.3 Preparation of bacterial cells for MRI
Starter cultures of bacterial cells (E. coli ER2738, E. coli Nissle, E. coli BL21, V. cholerae, and P. aeruginosa) were grown in 5 mL of lysogeny broth overnight at 37 °C. For E. coli ER2738 cells, the growth medium was supplemented with tetracycline (10 μg mL−1) to maintain episomal expression of the F-pilus. The pre-inoculum was added to fresh lysogeny broth at a 1:100 dilution (v/v) and grown for 12–16 h at 37 °C. Cell density was quantified by measuring the optical density at 600 nm. To prepare bacterial cells for phage-based MRI, the overnight culture (10 mL) was briefly incubated (∼5 min.) with Mn2+-loaded phage particles at a phage-to-cell stoichiometry of 50:1, mixed by inversion, and centrifuged at 3000 × g for 10 minutes in a cold room (4 °C). The supernatant was decanted, and the cells were resuspended in 1 mL HEPES buffer. The cells were washed again and resuspended in 150 μL HEPES in MRI-compatible plastic tubes. The resuspension was centrifuged at 3000 × g for 10 min to form pellets for MRI. In preparation for MRI, the plastic tubes were placed in water-filled agarose (1% w/v) moulds in an MRI phantom, as described above. Images were acquired in the coronal plane using a RARE sequence with the same parameters as described for phage relaxometry. R1 values were estimated by modeling the exponential growth of signal intensities in manually drawn ROIs encompassing the cell pellet.
2.4 Phage MRI in mice
V. cholerae cells were incubated with CTX–DP1 phage as described above, resuspended in 165 μL Matrigel® at 4 °C, and centrifuged at 3000 × g for 10 minutes. After decanting the supernatant, the cells were stirred into a slurry and carefully aspirated into a pre-chilled 16G syringe with the headspace pre-filled with sterile saline solution. In preparation for injection, the syringe was warmed to room temperature for ∼10 min. Female C57BL/6J mice between 5–7 weeks of age were anesthetized with 2.5% isoflurane and secured in an animal cradle with anesthetic gas (1–2% isoflurane in medical oxygen) continuously delivered through a nose cone. Approximately 100 μL cell slurry was injected subcutaneously in one flank, whereas identically prepared phage-free cells were implanted in the contralateral flank of the same animal to serve as control. Imaging was performed using a Bruker 7 Tesla vertical-bore MRI scanner with a 40 mm diameter transceiver coil. Throughout the imaging session, body temperature was maintained at ∼37 °C by circulating warm air with feedback-driven temperature control. The respiration rate (beats per minute) and body temperature were continuously monitored with a pneumatic transducer (Biopac Systems, Goleta, CA, USA) and fiber optic rectal probe (OpSens, Quebec, Canada), respectively.To measure R1, images were acquired at multiple repetition times in the axial plane using a RARE sequence with identical parameters as described for phage relaxometry. Next, T1-weighted images were acquired using a fast low-angle shot (FLASH) sequence with the following parameters: TR: 83.7 ms, TE: 3 ms, flip angle: 30 °C, averages: 10, matrix size: 128 × 128, field of view: 3.13 cm × 3.13 cm, slice thickness: 1 mm, in-plane resolution: 244 × 244 μm2. R1 values are signal intensities were estimated in manually drawn ROIs encompassing the injection site. All animal procedures were approved by the Institutional Animal Care and Use Committee of UC Santa Barbara (protocol #946).
3. Results and discussion
3.1 Display of a manganese-binding peptide on the phage coat creates MRI contrast
We hypothesized that M13 phage could be used to generate MRI contrast by engineering the pVIII coat protein to bind Mn2+. Of the five proteins in phage M13, we chose pVIII to bind Mn2+ because the latter is present in multiple copies (typically >2000 copies per phage), thus allowing a large payload of Mn2+ to be delivered by the phage to target bacterial populations. The resulting bacteria could, in turn, be detected using MRI owing to an increase in their spin–lattice relaxation rate (Fig. 1a). To load Mn2+ on the phage coat, we used a Mn2+-binding peptide, DP1, from the radiation-resistant extremophile, Deinococcus radiodurans.47,48 DP1 exhibits micromolar affinity for Mn2+ (Kd ∼ 10 μM)47,49 and a small size (10 amino acids), making it convenient to genetically fuse with pVIII, which typically tolerates 6–8 amino acid insertions.50 We truncated DP1 to eight amino acids without incurring a detectable loss of Mn2+ binding, as determined using a fluorescent dye that assays free Mn2+ (Fig. S1†). To produce the engineered phage, we used a two-plasmid system51 comprising a scaffold plasmid that harbors the M13 origin of replication and packaging sequences; and a packaging plasmid that encodes the nine proteins, pI-pIX, required for phage assembly, but is not packaged itself as it lacks the M13 replication components (Fig. S2†). We modified the pVIII sequence in the packaging plasmid to incorporate truncated DP1 at its 5′-end. We expressed DP1-functionalized phage (hereafter, DP1-phage) by transformation of E. coli with both plasmids and purified phage particles from culture supernatants by precipitation with NaCl and poly(ethylene) glycol (PEG). Phage formation was verified by Western blotting and size-exclusion chromatography (Fig. S3a and b†). To load DP1-phage with Mn2+, we incubated phage with MnCl2 in a 5:1 Mn2+-to-pVIII stoichiometry and purified the resulting phage using three additional rounds of precipitation with PEG–NaCl to remove unbound Mn2+. Using this approach, we consistently achieved a Mn2+ loading of 47.3 ± 0.9% (mean ± s.e.m, n = 3, measured relative to [pVIII]), as determined by inductively coupled plasma-mass spectrometry (Fig. 1b).
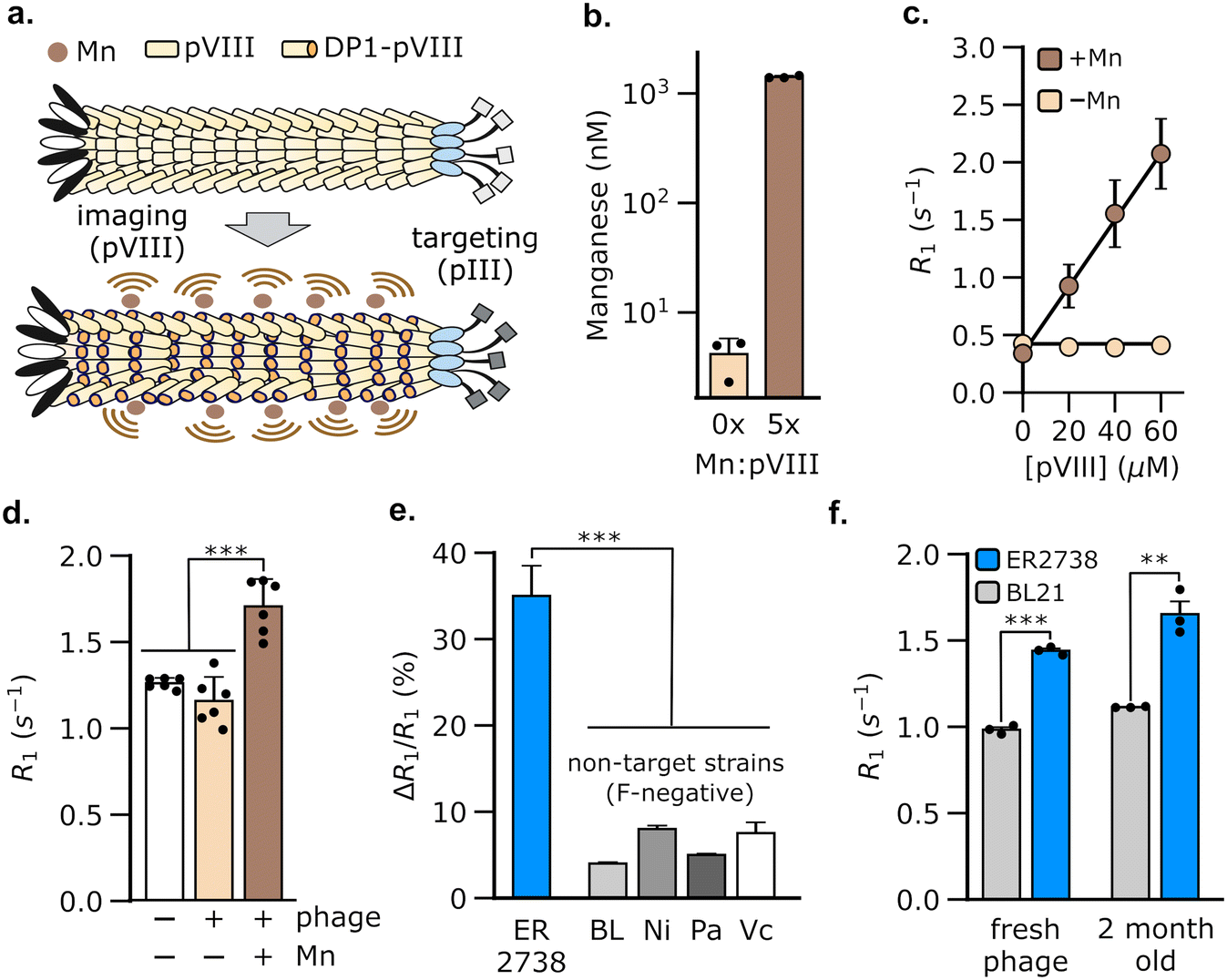 | ||
| Fig. 1 Engineering M13 phage to bind Mn2+ enables the detection of F-positive bacterial strains using MRI. (a) Schematic of our MRI approach that exploits the modularity of M13 phage for bacterial detection. A Mn2+-binding peptide (DP1) was genetically fused to a phage coat protein (pVIII), thus allowing M13 phage to be loaded with Mn2+ through multivalent binding interactions. The resulting Mn2+-loaded DP1-phage was used to detect bacterial cells that express F-pili, which are bound by the M13 pIII coat protein. Alternatively, DP1-phage may be targeted to image other (F-negative) bacterial strains by engineering pIII to bind to specific cell surface receptors in these strains. (b) Mn2+ loading in DP1-phage was confirmed using inductively coupled plasma mass spectrometry (ICP-MS). The phage titer in these experiments corresponded to ∼3 μM pVIII. (c) Mn2+-bound DP1-phage increases the spin–lattice relaxation rate (R1) of water in proportion to pVIII concentration. Based on ICP-MS studies, the solution concentration of phage-loaded Mn2+ in these experiments is estimated to be ∼47% the molarity of pVIII, viz. 0, 9, 19, and 28 μM. Phage alone, that is without Mn2+, did not change the relaxation rate of water. Error-bars that are not visible are smaller than the marker size. (d) MRI-based detection of E. coli ER2738, a prototypical F-positive strain. R1 measurements were acquired on pellets formed by centrifugation of log-phase cultures of E. coli ER2738. A significant increase in R1 relative to that of phage-free cells was only observed in cultures incubated with Mn2+-loaded DP1-phage. (e) Detection by DP1-phage is strain-specific, as incubation of F-negative bacterial strains with the phage produces 4–9× smaller changes in relaxation rate compared to E. coli ER2738. The labels refer to representative F-negative strains as follows: BL, E. coli BL21; Pa, Pseudomonas aeruginosa; Ni, E. coli Nissle; Vc, Vibrio cholerae. (f) DP1-phage was stable over at least two months of cold storage (4 °C), retaining its ability to specifically detect E. coli ER2738, similar to freshly purified phage. Relaxometry measurements were performed at room temperature and physiological pH (HEPES buffer, pH 7.4) using a 7 Tesla MRI scanner. Error bars represent standard deviation from n = 3–6 biological replicates; ** denotes P < 0.01; *** denotes P < 0.001; n.s. non-significant, P > 0.05 (2-tailed t-test, unpaired). | ||
Having formed Mn2+-loaded DP1-phage, we characterized its ability to increase the baseline spin–lattice relaxation rate of water. To this end, we performed relaxometric titration of phage suspensions in HEPES buffer using a 7 T MRI scanner. The relaxation rate (R1) of water increased proportionally with phage concentration, achieving a peak gain of 531 ± 81% at 60 μM pVIII (Fig. 1c). The contrast effect of DP1-phage can be quantified by its molar relaxivity (defined as the rate of increase in R1 with [pVIII]), which is calculated to be 29 mM−1 s−1 (95% CI: [26, 32], n = 4). On the contrary, DP1-phage that was not pre-incubated with MnCl2 did not change the relaxation rate, indicating that phage alone does not generate MRI contrast (Fig. 1c). Mn2+ ions appeared stably bound to the phage coat as evidenced by a negligible loss in the relaxation rate of phage following prolonged incubation with divalent Zn2+ ions (Fig. S3d†). Furthermore, the molar relaxivity of Mn2+-loaded phage was not significantly affected by incubation with physiologically relevant concentrations of common divalent metal ions, indicating the selectivity of DP1-phage towards Mn2+ (Fig. S3c†).
3.2 DP1-phage facilitates specific detection of E. coli (F+) strains using MRI
To evaluate whether DP1-phage can be used for bacterial detection by MRI, we exploited the ability of M13 phage to bind to the F-pilus, a cell-surface receptor that aids in the transfer of genetic material between bacteria.52,53 Notably, the F-pilus provides a site of attachment for M13 phage, which binds to the tip of the pilus using the pIII coat protein. Because only specific E. coli strains express the F-pilus, targeting this receptor provides a common testing ground for evaluating phage-based biosensors.42–44,54 To test whether DP1-phage can detect F-pilus harboring E. coli strains, we treated ER2738, a F-positive strain of E. coli, with DP1-phage (pre-loaded with Mn2+) at a phage-to-cell ratio of 50:1. After briefly (∼5 min) mixing the phage and cell suspension, we washed the cells twice to remove any unbound phage, formed pellets by centrifugation, and immediately acquired relaxation measurements at 7 T. We observed a 35 ± 1.4% (mean ± s.e.m., n = 6) increase in the spin–lattice relaxation rate of ER2738 cells following incubation with Mn2+-loaded DP1-phage (Fig. 1d). In contrast, cells treated with DP1-phage that was not loaded with Mn2+ did not show a substantial increase in their relaxation properties (ΔR1/R1 = 0.08 ± 0.004%, n = 3) (Fig. 1d), which is consistent with our relaxometric data that phage alone (i.e. without Mn2+) is incapable of generating MRI contrast (Fig. 1c).
To test whether detection was specific to the F-positive strain, we incubated Mn2+-loaded DP1-phage with representative bacterial strains that lack F-pili, including P. aeruginosa, V. cholerae, E. coli BL21, and E. coli Nissle. Compared to the F-positive ER2738 cells, we observed a 4–9× smaller change in the relaxation rates of all F-negative bacterial strains (Fig. 1e), indicating that recognition of the F-pilus is necessary for DP1-phage to generate an increase in the cell's relaxation rate. Specificity was conferred by the phage scaffold because relaxation measurements acquired after incubating E. coli ER2738 (F-positive) and BL21 (F-negative) with free Mn2+ did not reveal a significant difference between the two strains (Fig. S4†). Furthermore, DP1-phage could be stably stored at 4 °C for at least two months and retained the ability to specifically enhance the relaxation rate of the E. coli ER2738 strain to a similar extent as freshly prepared phage (Fig. 1f). Finally, mammalian cells were unaffected by incubation with DP1-phage, demonstrating neither a significant change in their relaxation rates (Fig. S5a†) nor signs of cytotoxicity, as assessed by trypan blue viability assay (Fig. S5b†).
3.3 DP1-phage can be engineered to detect V. cholerae using MRI
Having validated the ability of DP1-phage to detect its natural F-positive host, we wondered whether we could reprogram the phage probe to detect a bacterial strain that lacks F-pili. In principle, phage tropism can be tailored to any particular bacterial species by swapping out F-pilus-binding residues from pIII and replacing them with protein domains from other phages that recognize surface receptors in the targeted species.54,55 Accordingly, we constructed a chimeric DP1–CTX phage by replacing residues 18–236 of pIII with residues from phage CTXΦ, which targets the intestinal pathogen, V. cholerae, via a surface receptor.54,56,57 We purified DP1–CTX phage from E. coli and validated that the chimeric phage exhibited Mn2+ loading (56.3 ± 0.3%, n = 3) and molar relaxivity (20 mM−1 s−1, 95% CI: [17, 23], n = 4) comparable to those of DP1-phage (Fig. 2a and b). We then tested the ability of DP1–CTX phage to detect V. cholerae cells using MRI. To this end, we treated overnight cultures of V. cholerae with purified DP1–CTX phage and quantified relaxation rates as previously described. Phage-treated V. cholerae showed a 49 ± 4.9% (n = 3) increase in their spin–lattice relaxation rate relative to phage-free cells, whereas cells treated with Mn2+-free DP1–CTX phage showed no increase in their relaxation rate (Fig. 2c). Detection was specific to V. cholerae, as incubation of DP1–CTX phage with non-target strains produced markedly smaller changes in their relaxation rates relative to phage-free cells (Fig. 2d).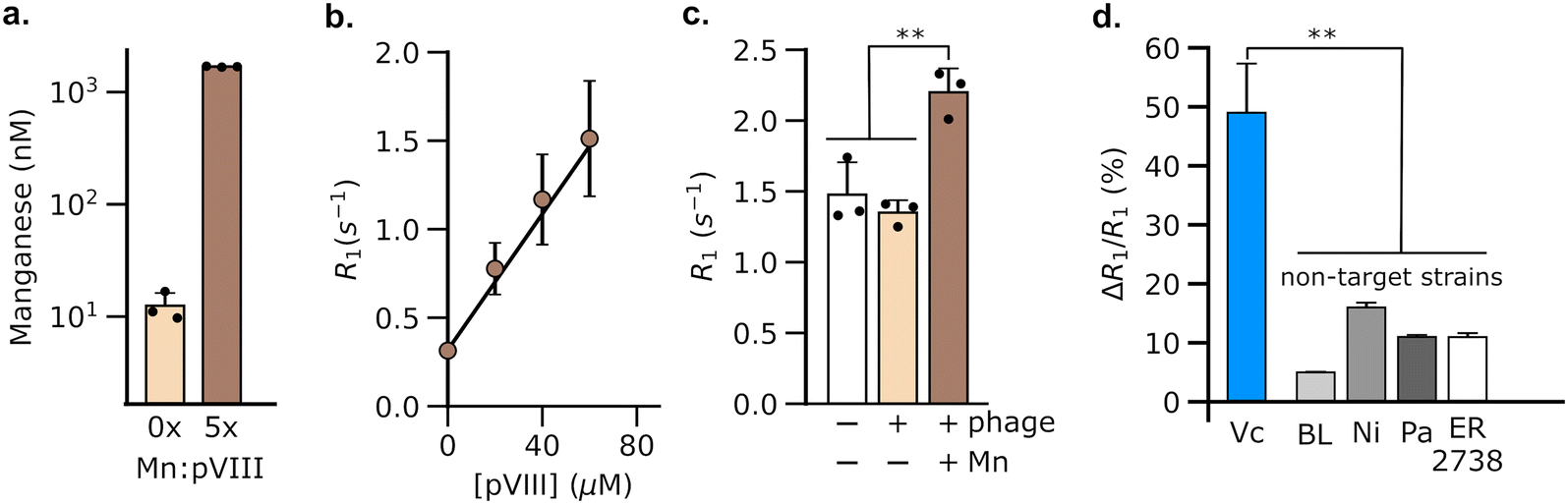 | ||
| Fig. 2 Engineering DP1-phage to target a V. cholerae cell-surface receptor enables the detection of a non-host (i.e., F-negative) strain using MRI. (a) Mn2+ loading in DP1–CTX phage, which targets V. cholerae, was confirmed using ICP-MS. The phage titer in these experiments corresponded to ∼3 μM pVIII. (b) Mn2+-bound DP1–CTX phage increases the spin–lattice relaxation rate (R1) of water in proportion to pVIII concentration. Based on ICP-MS studies, the solution phase concentration of phage-loaded Mn2+ in these experiments is estimated to be ∼56% the molarity of pVIII, viz. 0, 11, 22, and 34 μM. (c) MRI-based detection of V. cholerae. R1 measurements were acquired on pellets formed by centrifugation of log-phase cultures of V. cholerae. A significant increase in R1 relative to phage-free cells was only observed in cultures incubated with Mn2+-loaded DP1–CTX phage. (d) Detection by DP1-phage is strain-specific, as incubation of non-target bacterial strains with the phage produces smaller changes in R1 compared to V. cholerae. The labels refer to non-target strains as follows: BL, E. coli BL21; Pa, Pseudomonas aeruginosa; Ni, E. coli Nissle; and ER 2738, E. coli (F+) strain. Relaxometry measurements were performed at room temperature and physiological pH (HEPES buffer, pH 7.4) using a 7 Tesla scanner. Error bars represent the standard deviation from n = 3 biological replicates; ** denotes P < 0.01; *** denotes P < 0.001; n.s. non-significant, P > 0.05 (2-tailed t-test, unpaired). | ||
To test whether DP1–CTX phage could be imaged in vivo, we injected phage-treated V. cholerae cells into the flanks of C57/BL6 mice; the contralateral flank was injected with phage-unlabeled V. cholerae. We then measured the relaxation rates in a region of interest (ROI) placed at each injection site. As expected, the flank containing V. cholerae cells pre-labeled with phage showed a faster relaxation rate than contralateral flank in all animals analyzed (Fig. 3a). We further acquired T1-weighted images of the flanks using a fast low-flip angle pulse sequence. Consistent with the phage-induced increase in R1, the injection sites corresponding to V. cholerae labeled with DP1-phage appeared brighter compared to the control flank (Fig. 3b and c). Taken together, these results indicate that DP1-phage can be engineered to recognize a different bacterial target and provide a preliminary proof of concept that the phage probe can be used to detect and visualize bacteria in vivo using MRI.
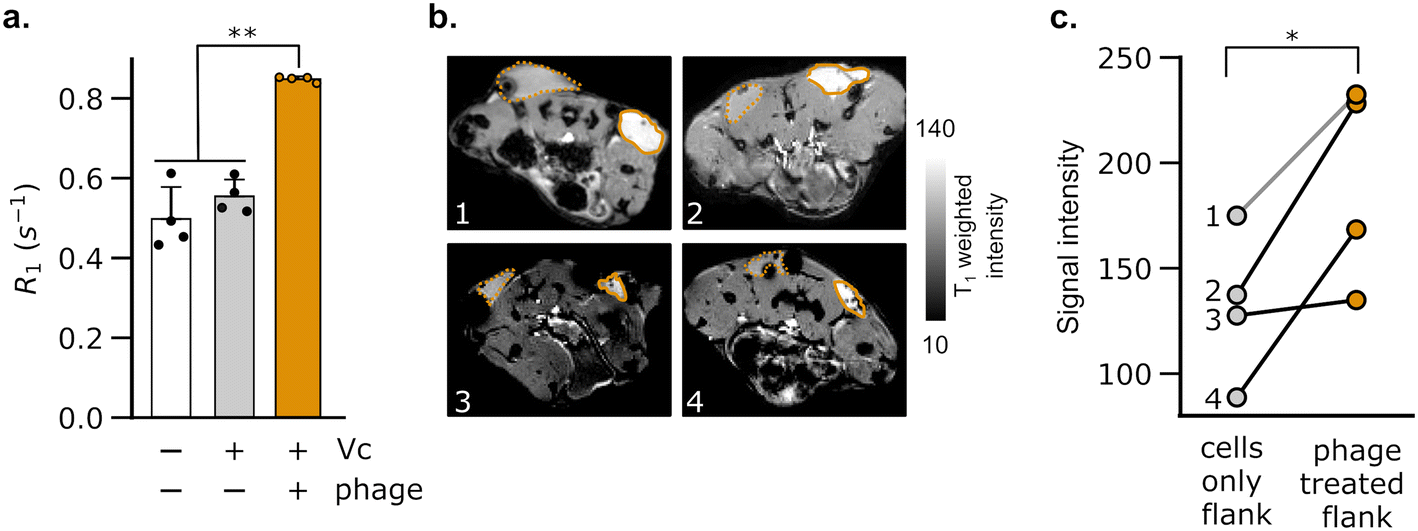 | ||
| Fig. 3 Imaging of phage-labeled bacteria in mice. (a) C57/BL6 mice were injected bilaterally in their hind flanks with V. cholerae cells pre-labeled with DP1–CTX phage and phage-unlabeled cells. The flanks harboring phage-labeled bacteria showed a significant increase in their spin–lattice relaxation rates (R1) relative to both the surrounding tissue and contralateral flank. Error bars represent the standard deviation; ** denotes P < 0.01 (2-tailed t-test, unpaired). (b) T1-Weighted images of the hind flanks of mice. Each numbered scan, 1–4, corresponds to an axial section from a distinct mouse. Regions of interest (ROIs) corresponding to flanks injected with phage-labeled V. cholerae are marked by solid orange boundaries. The phage-bearing ROIs appear hyperintense relative to the phage-free flanks (encompassed by dotted orange boundaries). (c) Lines are drawn to connect paired values of T1-weighted signal intensity measured in the same mouse. The numbers correspond to images of mice shown in (b). Phage-bearing flanks showed a statistically significant increase in signal intensity relative to phage-free flanks (P value = 0.025). * denotes P < 0.05; (2-tailed paired t-test). | ||
4. Conclusions
In summary, our findings demonstrate the potential of engineered filamentous phage nanomaterials for the targeted imaging of bacteria using MRI. This phage-based approach offers a number of advantages over existing MRI approaches to image bacteria. First, DP1-phage is composed of non-toxic and biocompatible components, which makes it suitable for imaging live bacterial cells without potential adverse effects (unlike antibiotic- and antimicrobial peptide-based probes). Second, phages are simple to produce in bulk and have high stability, allowing for multiple imaging sessions with a single batch of purified phage particles. Third, compared with existing T1 weighted MRI probes that are not specific to any particular bacterial species, DP1-phage can, in principle, be tailored to image any bacterial strain by engineering pIII to display receptor-binding domains from phages that target particular bacteria.While this study establishes a basic framework for imaging bacteria with DP1-phage, there are areas that require further optimization to develop this approach for imaging bacterial populations in real-time in vivo. First, increasing phage relaxivity would improve sensitivity, allowing smaller bacterial loads to be detected using MRI. This improvement can be achieved by screening for DP1 variants that display tighter binding to Mn2+ or by combining tandem repeats of DP1 with pVIII to increase the availability of Mn2+-binding sites on the phage coat. The size of the phage can also be reduced by genetic engineering,25,58 which would improve its ability to penetrate tighter spaces and potentially increase the amount of phage present at the target location. Second, additional studies are needed to determine and optimized the pharmacokinetics and delivery efficiency of DP1-phage for in vivo applications, as well as to investigate potential immune responses that may arise from its use.59 To this end, it is worth noting that filamentous phages have been widely used in animal models ranging from mice to non-human primates, and considerable improvements in phage delivery and biodistribution can be achieved by engineering phage surface properties.60–63 Ultimately, the approach developed in this study could have significant implications for studying bacterial infections and the microbiota in animal models by providing a modular platform for the development of targeted probes that can be used to accurately identify and localize a wide range of bacterial species using MRI.
Author contributions
Arnab Mukherjee: conceptualization, methodology, formal analysis, resources, data curation, writing – original draft, writing – review & editing, visualization, supervision, project administration, funding acquisition. Irene A. Chen: conceptualization, methodology, resources, writing – review & editing, supervision, project administration, funding acquisition. Raymond E. Borg, Harun F. Ozbakir, Binzhi Xu: investigation, validation, formal analysis, data curation. Eugene Li, Xiwen Fang: investigation, validation. Huan Peng: methodology.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Divya Iyer, Prof. Samanvaya Srivastava, and Steven Yanxi Yang (UC Los Angeles) for assistance with phage characterization. Dr. Jerry Hu (UC Santa Barbara) is acknowledged for assistance with setting up the animal MRI workflow. This research was supported by the California NanoSystems Institute (Challenge Grant to A. M. & I. A. C.); the National Institutes of Health (R35-GM133530 and R21-EB033989 to A. M.; DP2-GM123457 to I. A. C.), and the Camille & Henry Dreyfus Foundation (Teacher-Scholar Award to I. A. C.). H. F. O. gratefully acknowledges support from the Errett Fisher Foundation. B. X. was supported by the Regent's Fellowship from UC Santa Barbara. All MRI experiments were performed at the Materials Research Laboratory (MRL) at UC, Santa Barbara. The MRL Shared Experimental Facilities are supported by the MRSEC Program of the NSF under Award No. DMR 1720256; a member of the NSF-funded Materials Research Facilities Network.References
- A. A. Ordonez, M. A. Sellmyer, G. Gowrishankar, C. A. Ruiz-Bedoya, E. W. Tucker, C. J. Palestro, D. A. Hammoud and S. K. Jain, Sci. Transl. Med., 2019, 11, eaax8251 CrossRef PubMed.
- V. Hoerr and C. Faber, J. Pharm. Biomed. Anal., 2014, 93, 136–146 CrossRef CAS.
- M. Heuker, A. Gomes, J. M. van Dijl, G. M. van Dam, A. W. Friedrich, B. Sinha and M. van Oosten, Clin. Transl. Imaging, 2016, 4, 253–264 CrossRef PubMed.
- C. J. Palestro, C. Love and T. T. Miller, Cell. Microbiol., 2007, 9, 2323–2333 CrossRef CAS PubMed.
- L. A. Jelicks, M. P. Lisanti, F. S. Machado, L. M. Weiss, H. B. Tanowitz and M. S. Desruisseaux, Am. J. Pathol., 2013, 182, 296–304 CrossRef PubMed.
- M. M. Welling, A. Paulusma-Annema, H. S. Balter, E. K. Pauwels and P. H. Nibbering, Eur. J. Nucl. Med., 2000, 27, 292–301 CrossRef CAS PubMed.
- Z. Zhang, A. A. Ordonez, H. Wang, Y. Li, K. R. Gogarty, E. A. Weinstein, F. Daryaee, J. Merino, G. E. Yoon, A. S. Kalinda, R. C. Mease, J. N. Iuliano, P. M. Smith-Jones, S. K. Jain and P. J. Tonge, ACS Infect. Dis., 2018, 4, 1635–1644 CrossRef CAS PubMed.
- E. A. Weinstein, A. A. Ordonez, V. P. DeMarco, A. M. Murawski, S. Pokkali, E. M. MacDonald, M. Klunk, R. C. Mease, M. G. Pomper and S. K. Jain, Sci. Transl. Med., 2014, 6, 259ra146 Search PubMed.
- M. A. Sellmyer, I. Lee, C. Hou, C.-C. Weng, S. Li, B. P. Lieberman, C. Zeng, D. A. Mankoff and R. H. Mach, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 8372–8377 CrossRef CAS PubMed.
- G. Gowrishankar, J. Hardy, M. Wardak, M. Namavari, R. E. Reeves, E. Neofytou, A. Srinivasan, J. C. Wu, C. H. Contag and S. S. Gambhir, J. Nucl. Med., 2017, 58, 1679–1684 CrossRef CAS PubMed.
- G. Gowrishankar, M. Namavari, E. B. Jouannot, A. Hoehne, R. Reeves, J. Hardy and S. S. Gambhir, PLoS One, 2014, 9, e107951 CrossRef PubMed.
- A. Lupetti, M. M. Welling, E. K. J. Pauwels and P. H. Nibbering, Lancet Infect. Dis., 2003, 3, 223–229 CrossRef CAS PubMed.
- K.-S. Choi, S.-H. Kim, Q.-Y. Cai, S.-Y. Kim, H.-O. Kim, H.-J. Lee, E.-A. Kim, S.-E. Yoon, K.-J. Yun and K.-H. Yoon, Mol. Imaging, 2007, 6, 7290.2007.00005 CrossRef.
- T. Hertlein, V. Sturm, P. Jakob and K. Ohlsen, PLoS One, 2013, 8, e64440 CrossRef CAS PubMed.
- J. S. Lee, H. J. Kang, G. Gong, H.-D. Jung, K. H. Lim, S. T. Kim and T.-H. Lim, Radiology, 2006, 241, 142–148 CrossRef.
- L. M. Matosziuk, A. S. Harney, K. W. MacRenaris and T. J. Meade, Eur. J. Inorg. Chem., 2012, 2012, 2099–2107 CrossRef CAS PubMed.
- C. Xu, Z. Li, O. U. Akakuru, C. Pan, R. Zou, J. Zheng and A. Wu, ACS Appl. Bio Mater., 2021, 4, 3762–3772 CrossRef CAS PubMed.
- L. Zhang, Y. Liu, Q. Zhang, T. Li, M. Yang, Q. Yao, X. Xie and H.-Y. Hu, Anal. Chem., 2018, 90, 1934–1940 CrossRef CAS.
- Y. Li, H. Yu, Y. Qian, J. Hu and S. Liu, Adv. Mater., 2014, 26, 6734–6741 CrossRef CAS.
- L. Li, P. Gu, M. Hao, X. Xiang, Y. Feng, X. Zhu, Y. Song and E. Song, Small, 2021, 17, 2103627 CrossRef CAS.
- Y. Li, X. Hu, D. Ding, Y. Zou, Y. Xu, X. Wang, Y. Zhang, L. Chen, Z. Chen and W. Tan, Nat. Commun., 2017, 8, 15653 CrossRef CAS PubMed.
- C.-N. Lee, Y.-M. Wang, W.-F. Lai, T.-J. Chen, M.-C. Yu, C.-L. Fang, F.-L. Yu, Y.-H. Tsai, W. H.-S. Chang, C. S. Zuo and P. F. Renshaw, Clin. Microbiol. Infect., 2012, 18, E149–E157 CrossRef CAS PubMed.
- V. Hoerr, L. Tuchscherr, J. Hüve, N. Nippe, K. Loser, N. Glyvuk, Y. Tsytsyura, M. Holtkamp, C. Sunderkötter, U. Karst, J. Klingauf, G. Peters, B. Löffler and C. Faber, BMC Biol., 2013, 11, 63 CrossRef PubMed.
- E. Dor-On and B. Solomon, Front. Microbiol., 2015, 6, 530 Search PubMed.
- U. Tsedev, C.-W. Lin, G. T. Hess, J. N. Sarkaria, F. C. Lam and A. M. Belcher, ACS Nano, 2022, 16, 11676–11691 CrossRef CAS PubMed.
- H. Y. Wang, Y.-C. Chang, C.-W. Hu, C.-Y. Kao, Y.-A. Yu, S.-K. Lim and K. Y. Mou, ACS Synth. Biol., 2021, 10, 2087–2095 CrossRef CAS PubMed.
- S. M. DePorter and B. R. McNaughton, Bioconjugate Chem., 2014, 25, 1620–1625 CrossRef CAS PubMed.
- F. I. Staquicini, T. L. Smith, F. H. F. Tang, J. G. Gelovani, R. J. Giordano, S. K. Libutti, R. L. Sidman, W. K. Cavenee, W. Arap and R. Pasqualini, Cancer Gene Ther., 2020, 27, 301–310 CrossRef CAS PubMed.
- K. K. Palaniappan, R. M. Ramirez, V. S. Bajaj, D. E. Wemmer, A. Pines and M. B. Francis, Angew. Chem., Int. Ed., 2013, 52, 4849–4853 CrossRef CAS PubMed.
- D. Ghosh, Y. Lee, S. Thomas, A. G. Kohli, D. S. Yun, A. M. Belcher and K. A. Kelly, Nat. Nanotechnol., 2012, 7, 677–682 CrossRef CAS PubMed.
- J. Segers, C. Laumonier, C. Burtea, S. Laurent, L. Vander Elst and R. N. Muller, Bioconjugate Chem., 2007, 18, 1251–1258 CrossRef CAS PubMed.
- H.-E. Jin, R. Farr and S.-W. Lee, Biomaterials, 2014, 35, 9236–9245 CrossRef CAS.
- I. Yacoby, M. Shamis, H. Bar, D. Shabat and I. Benhar, Antimicrob. Agents Chemother., 2006, 50, 2087–2097 CrossRef CAS PubMed.
- I. Yacoby, H. Bar and I. Benhar, Antimicrob. Agents Chemother., 2007, 51, 2156–2163 CrossRef CAS PubMed.
- R. J. Krom, P. Bhargava, M. A. Lobritz and J. J. Collins, Nano Lett., 2015, 15, 4808–4813 CrossRef CAS PubMed.
- H. Peng, D. Rossetto, S. S. Mansy, M. C. Jordan, K. P. Roos and I. A. Chen, ACS Nano, 2022, 16, 4756–4774 CrossRef CAS PubMed.
- T. K. Lu and J. J. Collins, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 4629–4634 CrossRef CAS PubMed.
- X. Dong, P. Pan, D.-W. Zheng, P. Bao, X. Zeng and X.-Z. Zhang, Sci. Adv., 2020, 6, eaba1590 CrossRef CAS PubMed.
- K. N. Lam, P. Spanogiannopoulos, P. Soto-Perez, M. Alexander, M. J. Nalley, J. E. Bisanz, R. R. Nayak, A. M. Weakley, F. B. Yu and P. J. Turnbaugh, Cell Rep., 2021, 37, 109930 CrossRef CAS PubMed.
- M. R. A. Carrera, G. F. Kaufmann, J. M. Mee, M. M. Meijler, G. F. Koob and K. D. Janda, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 10416–10421 CrossRef CAS PubMed.
- A. Ksendzovsky, S. Walbridge, R. C. Saunders, A. R. Asthagiri, J. D. Heiss and R. R. Lonser, J. Neurosurg., 2012, 117, 197–203 CAS.
- N. M. Bardhan, D. Ghosh and A. M. Belcher, Nat. Commun., 2014, 5, 4918 CrossRef CAS.
- N. M. Bardhan, D. Ghosh and A. M. Belcher, J. Biophotonics, 2014, 7, 617–623 CrossRef CAS.
- S. Huang, J. Qi, D. W. deQuilettes, M. Huang, C.-W. Lin, N. M. Bardhan, X. Dang, V. Bulović and A. M. Belcher, Small, 2019, 15, 1901233 CrossRef PubMed.
- M. Rusckowski, S. Gupta, G. Liu, S. Dou and D. J. Hnatowich, J. Nucl. Med., 2004, 45, 1201–1208 CAS.
- M. Rusckowski, S. Gupta, G. Liu, S. Dou and D. J. Hnatowich, Nucl. Med. Biol., 2008, 35, 433–440 CrossRef CAS PubMed.
- M. Peana, S. Medici, H. A. Pangburn, T. J. Lamkin, M. Ostrowska, E. Gumienna-Kontecka and M. A. Zoroddu, J. Inorg. Biochem., 2016, 164, 49–58 CrossRef CAS PubMed.
- P. Gupta, M. Gayen, J. T. Smith, E. K. Gaidamakova, V. Y. Matrosova, O. Grichenko, B. Knollmann-Ritschel, M. J. Daly, J. G. Kiang and R. K. Maheshwari, PLoS One, 2016, 11, e0160575 CrossRef.
- M. Peana, E. Gumienna-Kontecka, F. Piras, M. Ostrowska, K. Piasta, K. Krzywoszynska, S. Medici and M. A. Zoroddu, Inorg. Chem., 2020, 59, 4661–4684 CrossRef CAS PubMed.
- G. Iannolo, O. Minenkova, R. Petruzzelli and G. Cesareni, J. Mol. Biol., 1995, 248, 835–844 CrossRef CAS PubMed.
- P. M. Nafisi, T. Aksel and S. M. Douglas, Synth. Biol., 2018, 3, ysy015 CrossRef CAS PubMed.
- A. Lin, J. Jimenez, J. Derr, P. Vera, M. L. Manapat, K. M. Esvelt, L. Villanueva, D. R. Liu and I. A. Chen, PLoS One, 2011, 6, e19991 CrossRef CAS PubMed.
- P. M. Silverman and M. B. Clarke, Integr. Biol., 2010, 2, 25–31 CrossRef CAS PubMed.
- H. Peng and I. A. Chen, ACS Nano, 2019, 13, 1244–1252 CAS.
- H. Peng and I. A. Chen, Curr. Opin. Biotechnol., 2021, 68, 23–29 CrossRef CAS PubMed.
- L. Houot, R. Navarro, M. Nouailler, D. Duché, F. Guerlesquin and R. Lloubes, J. Biol. Chem., 2017, 292, 13584–13598 CrossRef CAS PubMed.
- A. J. Heilpern and M. K. Waldor, J. Bacteriol., 2003, 185, 1037–1044 CrossRef CAS PubMed.
- S. Sattar, N. J. Bennett, W. X. Wen, J. M. Guthrie, L. F. Blackwell, J. F. Conway and J. Rakonjac, Front. Microbiol., 2015, 6, 316 Search PubMed.
- A. Górski, R. Międzybrodzki, J. Borysowski, K. Dąbrowska, P. Wierzbicki, M. Ohams, G. Korczak-Kowalska, N. Olszowska-Zaremba, M. Łusiak-Szelachowska, M. Kłak, E. Jończyk, E. Kaniuga, A. Gołaś, S. Purchla, B. Weber-Dąbrowska, S. Letkiewicz, W. Fortuna, K. Szufnarowski, Z. Pawełczyk, P. Rogóż and D. Kłosowska, in Advances in Virus Research, ed. M. Łobocka and W. Szybalski, Academic Press, 2012, vol. 83, pp. 41–71 Search PubMed.
- K. Dabrowska, K. Switała-Jelen, A. Opolski, B. Weber-Dabrowska and A. Gorski, J. Appl. Microbiol., 2005, 98, 7–13 CrossRef CAS PubMed.
- J. Zou, M. T. Dickerson, N. K. Owen, L. A. Landon and S. L. Deutscher, Mol. Biol. Rep., 2004, 31, 121–129 CrossRef CAS PubMed.
- K. Dąbrowska, Med. Res. Rev., 2019, 39, 2000–2025 CrossRef PubMed.
- Y. L. Yip, N. J. Hawkins, G. Smith and R. L. Ward, J. Immunol. Methods, 1999, 225, 171–178 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Borg–Ozbakir et al. SI-2022. The file contains detailed methods, supplementary Fig. S1–S5, Tables S1–S4, and associated references. See DOI: https://doi.org/10.1039/d3sd00026e |
| ‡ These authors contributed equally to the work. |
| This journal is © The Royal Society of Chemistry 2023 |