
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCurrent advance of CRISPR/Cas-based SERS technology
Huimin
Wang
ab,
Ailing
Su
a,
Jingjing
Chang
b,
Xiangguo
Liu
c,
Chongyang
Liang
 d and
Shuping
Xu
*aef
d and
Shuping
Xu
*aef
aState Key Laboratory of Supramolecular Structure and Materials, College of Chemistry, Jilin University, Changchun 130012, P. R. China. E-mail: xusp@jlu.edu.cn
bSchool of Chemistry and Environmental Engineering, Changchun University of Science and Technology, Changchun 130022, P. R. China
cInstitute of Agricultural Biotechnology, Jilin Academy of Agricultural Sciences, Changchun, 130033, P. R. China
dInstitute of Frontier Medical Science, Jilin University, Changchun, 130021, P. R. China
eCenter for Supramolecular Chemical Biology, College of Chemistry, Jilin University, Changchun 130012, P. R. China
fTheoretical Chemistry, College of Chemistry, Jilin University, Changchun 130012, P. R. China
First published on 5th May 2023
Abstract
In recent years, clustered regularly interspaced short palindromic repeats (CRISPR)-associated nuclease (Cas) has emerged as a powerful genome editing system. In addition to genome editing, CRISPR/Cas technology has attracted considerable attention in molecular diagnostics, particularly for nucleic acid detection as it has high sensitivity and specificity for target nucleic acids, which can initiate trans-cleavage actions at programmed sites or determine the target gene chain. Surface-enhanced Raman spectroscopy (SERS), which depends on the presence of plasmonic nanoparticles or nanostructures, is widely used in biosensing because of its superior sensitivity and fingerprint spectral bands. The combination of SERS with the CRISPR/Cas technique has been realized by fabricating many elaborate plasmonic nanorobots and nanodevices that respond to target genes via Cas proteins. This review summarizes the operation of CRISPR/Cas technology, the application of CRISPR/Cas technology combined with SERS technology for nucleic acid detection, elements of SERS sensors, sensitivity and sensing time, and provides an outlook on future developments.
1. Introduction
Over several decades, surface-enhanced Raman scattering (SERS) has become an ultra-sensitive tool for characterizing, detecting, and quantifying biomolecules owing to its ultra-high sensitivity and unique advantages in terms of fingerprint information.1–3 SERS depends largely on the plasmon-mediated amplification of Raman scattering from molecules located on the surface of nanostructured metal substrates to obtain their vibrational fingerprints at very low concentrations.3 The enhancement of SERS results primarily from electromagnetic and chemical enhancements.4,5 Electromagnetic enhancement is due to the excitation of surface plasmon polarization waves, leading to a strong electromagnetic field on metallic nanostructures. Chemical enhancement is mainly based on the charge transfer mechanism between chemisorbed molecules and SERS substrate surfaces.6 With advantages of easy operation, rich spectral fingerprints, good specificity, and high sensitivity,7–9 SERS has been widely employed for nucleic acid detection.10,11 The sensing strategy of SERS for nucleic acid detection is classified as direct12–15 and indirect sensing methods.16–19 Direct SERS detection is performed by obtaining intrinsic SERS vibrational fingerprints of nucleic acids. Although direct SERS detection prevents false positives, it involves pre-purification and amplification steps to isolate and obtain a certain amount of deoxyribonucleic acid (DNA) from complex biological matrices. Indirect SERS detection relies on monitoring the changes in Raman signals generated by external SERS tags. SERS reporters and oligonucleotide ligands are typically bound to nanomaterials to enable the external detection of target sequences, making indirect SERS detection highly sensitive and signal intensive. Despite the lack of extensive structural information on intrinsic nucleic acid vibrational fingerprints, indirect SERS methods are inherently more suitable for bisensing purposes in biological media, as they generally provide a more stable and selective response, while enabling multiplex detection.Clustered regularly interspaced short palindromic repeats (CRISPR) and CRISPR-associated nuclease (Cas) systems are widely found in bacteria and archaea as adaptive immune systems against foreign nucleic acid invasion. They have attracted considerable attention in recent years in biology and other related fields, such as gene editing and biosensing.20,21 CRISPR/Cas technology has also been a novel nucleic acid detection tool for molecular diagnostics owing to its unique properties of high sensitivity, specificity, rapidity, and low cost.22–27 Presently, with the ongoing global SARS-CoV-2 epidemic, nucleic acid detection is becoming a major molecular diagnostic practice. CRISPR/Cas technology, in addition to various polymerase chain reaction (PCR)-based nucleic acid detection, isothermal amplification, and nucleic acid hybridization methods, has advanced rapidly. Cas proteins tend to bind to target sequences in a site-specific manner because of the activation effect of single-stranded guide ribose nucleic acid (gRNA). Cas12a/Cas13a can initiate an unlimited shearing function for single-stranded deoxyribonucleic acids (ssDNA), enabling an amplification strategy for nucleic acid detection. The CRISPR/Cas system has sequence-specific recognition capability. In addition, gRNA can be programmed to recognize any target sequence. Thus, established CRISPR/Cas-based methods can be universally applied to any target gene and can be extended to molecular diagnostics and clinical applications. These have accelerated the progress of CRISPR/Cas-based biosensing.
Most CRISPR/Cas-based strategies adopt fluorescence signal readouts according to cleavage actions on the paired fluorescence/quencher to obtain fluorescence signal recovery.28,29 Although fluorescent signals are relatively sensitive, nucleic acid amplification is still required to improve sensitivity, and the complexity of experimental manipulation and primer design limits its application.30,31 Colorimetry (3,3′,5,5′-tetramethylben-zidine, TMB)32,33 and colorimetric changes in a paper lateral flow assay (LFA)34 have also been integrated into CRISPR/Cas biosensing owing to low-resource settings. SERS is a powerful biosensing tool because of its ultra-high sensitivity. Combining SERS with CRISPR/Cas technology further improves the sensitivity and specificity of nucleic acid detection while eliminating the need for complicated nucleic acid amplification steps and avoiding experimental operational difficulties. Here, we summarize the different strategies for nucleic acid detection using CRISPR/Cas-based SERS technology, categorizing and elaborating on the binding positions between Raman reporters and metal and DNA sequences. We also list the performances of reported CRISPR/Cas-based SERS strategies and predict the development of this technology.
2. Brief introduction of Cas proteins and their action mechanism
The CRISPR system mainly comprises Cas endonuclease and gRNA, which recognizes and directs Cas nucleic acid endonuclease to reach the target region to destroy the target sequence under the guidance of gRNA.35 Different types of CRISPR/Cas systems (Fig. 1) can target double-stranded DNA (dsDNA) (e.g., Cas9/dCas9 (ref. 36) and Cas12a37) or RNA (e.g., Cas13a38) to realize gene detections.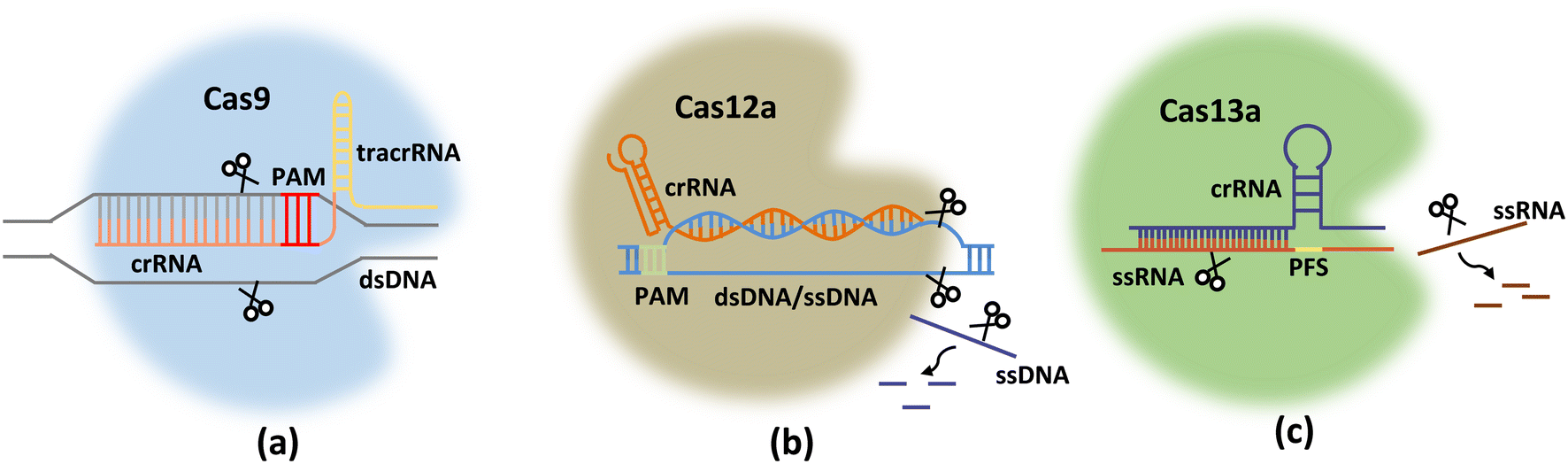 | ||
| Fig. 1 Schematic diagrams of Cas9 (a), Cas12a (b), and Cas13a (c) proteins with gRNAs for target genes. | ||
2.1 Mechanism of action
The CRISPR-Cas9 system consists of a gRNA (or sgRNA) and Cas9 nuclease, which together form a ribonucleoprotein (RNP) complex (Fig. 1a). gRNA includes both mature CRISPR/RNA (crRNA) and transduced CRISPR/RNA (tracrRNA). TracrRNA binds to a portion of the crRNA sequence via base pairing to form gRNA (crRNA: tracrRNA). With the help of base pairing of the other part of the crRNA sequence with the target DNA site, gRNA directs the Cas protein complex to bind at the target site, followed by cleavage of this site.39 The protospacer adjacent motif (PAM) sequence recognized by the Cas9 protein is 5′-NGG-3′ (Table 1), where N represents any nucleotide. If the PAM sequence matches correctly with dsDNA and the gRNA successfully binds to the target site, Cas9 performs a dsDNA cleavage approximately 3–4 nucleotides upstream of the PAM sequence. In addition to harnessing the enzymatic cleavage activity of the Cas9 protein, enzymatically deactivated Cas9 (dCas9 protein) has only sequence-specific binding capabilities, which can also be employed for building CRISPR/Cas-based sensing systems.| CRISPR protein | Guide RNA (gRNA) | PAM/PFS sequence | Target |
|---|---|---|---|
| Cas9/dCas9 | sgRNA, 100 nt (20 nt crRNA + tracrRNA) | 5′-NGG-3′ | dsDNA |
| Cas12a | crRNA, 20 nt | 5′-(T)TTN-3′ | dsDNA/ssDNA |
| Cas13a | crRNA, 24 nt | 5′-A/T/C-3′ | RNA |
The CRISPR-Cas12a system recognizes dsDNA containing PAM under the guidance of crRNA, and prompts the target dsDNA to unlink. The target chain in the target dsDNA then forms an R-loop with the crRNA. Subsequently, the active site of RuvC in Cas12a was released. RuvC further cuts the non-target chains (NTS) in the target dsDNA at the just-released active site after unwinding. This cleavage results in the unwinding of the target DNA, and the released target dsDNA is cleaved by RuvC. When Cas12a finishes cutting the target dsDNA (cis-cleavage), the dsDNA leaves. At this time, the active site of RuvC starts to cut ssDNA that enters the active site, which is trans-cleavage.
Unlike Cas9 and Cas12a, the CRISPR-Cas13a system is specific to single-stranded RNA sequences, and is more suitable for tracing RNA viruses because reverse transcription is unnecessary during sample pretreatment. After the CRISPR-Cas13a system combines with crRNA and substrate RNA to form a ternary complex, Cas13a is activated. Cas13a requires only 24 base crRNA, which interacts with the Cas13a molecule through its uracil-rich structure and facilitates the cutting of its target through a series of conformational changes in Cas13a. Similar to Cas9, Cas13a tolerates a single mismatch between the crRNA and the target sequence; however, if two mismatches exist, the cutting efficiency is greatly reduced. Its protospacer flanking sequence (PFS) (equivalent to the PAM sequence) is located at the 3′ end of the spacer region, and consists of A, U, or C bases. Once Cas13a recognizes and cuts the RNA target guided by the crRNA sequence, it switches to an enzymatic “activation” state to bind and cut other RNAs, which is important for signal amplification in sensing systems.
2.2 gRNA design
CRISPR/Cas systems target any genomic site by designing a single RNA oligonucleotide containing approximately 20–30 nt. A satisfactory gRNA is an important part of CRISPR/Cas systems, and creating gRNA sequences is considered the first step before starting a CRISPR/Cas sensing system. The search for a suitable gRNA can be divided into three main stages (Fig. 2).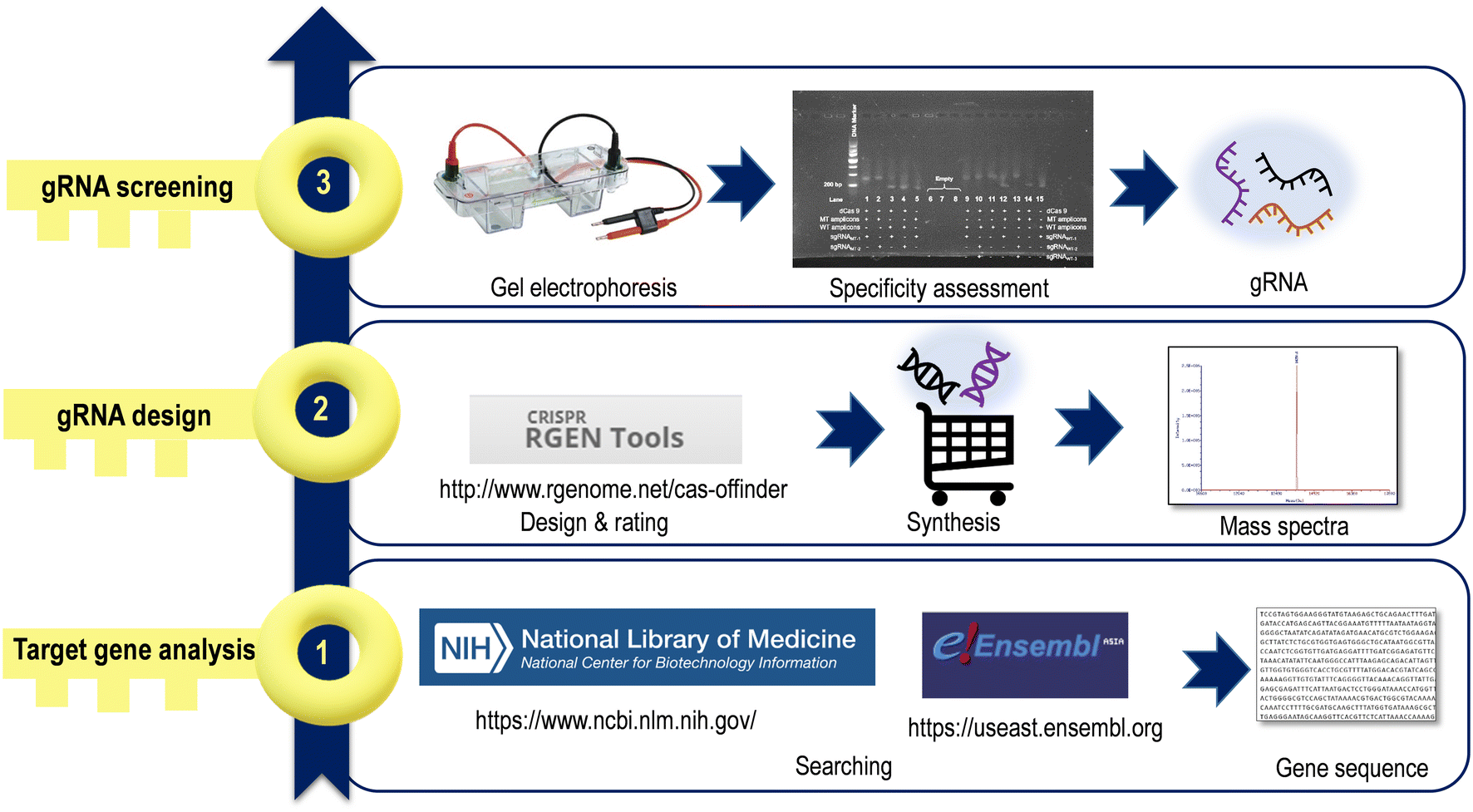 | ||
| Fig. 2 Workflow for acquiring gRNAs for CRISPR/Cas-based sensing. | ||
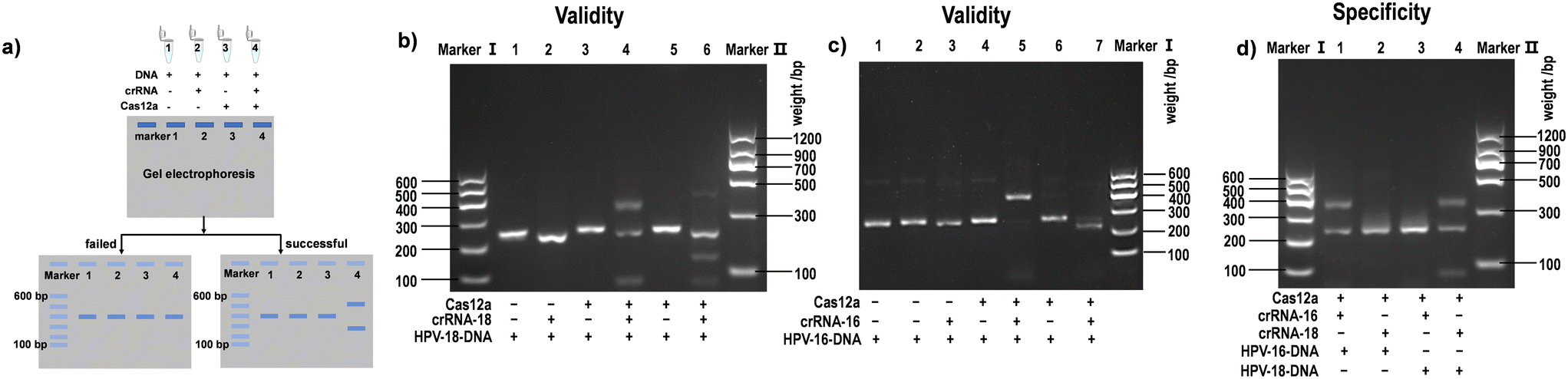 | ||
| Fig. 3 a) Schematic diagram of crRNA screening using agarose gel electrophoresis. The DNA, gRNA, and Cas protein are alternately absent (line 1–3), or all three are presented (line 4). b–d) Results of agarose gel electrophoresis screening of crRNAs for human papillomavirus (HPV)-16 and HPV-18 genes and specificity test. Reproduced from ref. 41 with permission from [ELSEVIER], copyright [2022]. | ||
3. Biosensing with CRISPR/Cas-based SERS technology
This section describes recently reported CRISPR/Cas-based SERS sensing strategies. Most reported CRISPR/Cas-based SERS methods adopt indirect SERS, that involve Raman reporter molecules with large scattering cross sections and strong SERS signals, and the bonding of Raman reporters with plasmonic substrates is considered. Thus, in order to better understand these SERS sensing methods, we first described the concept and composition of “SERS tags”. Depending on the locations of Raman reporters on different SERS substrates and their unique signal output methods, sensing strategies can be divided into three categories, as Raman reporter modifies DNA, Raman reporter modifies metal surface, and unique reporters for post-signal amplification strategies.3.1 SERS nanotags
SERS nanotags mainly include SERS substrates and Raman reporters. SERS tags are more suitable in liquid homogeneous reaction systems based on the consideration of the molecular diffusion dynamics for highly efficient mixing and reacting. In most CRISPR/Cas sensing systems, the volume of test solution used in experiments is only about dozens to hundreds of microliters due to the limited amount of nucleic acid extraction, less solution, and expensive Cas proteins. Thus, most researchers will adopt colloidal SERS tags instead of solid-supporting SERS substrates for testing.The first step in the preparation of SERS nanotags is to identify suitable SERS substrates. Noble metal colloidal nanoparticles as SERS substrates enable the tailoring of optical properties over a wide wavelength range by tuning the particle shape and size, as well as different surface functionalizations. The most widely used NPs display a simple spherical morphology, and because of their high symmetry, they exhibit moderate Raman signal enhancement as individual particles.42,43 In contrast, individual anisotropic plasmonic NPs can significantly enhance the Raman signal because of the presence of intrinsic hot spots at the edges and corners (e.g., nanorods and nanocubes) or sharp and prominent features (e.g., nanostars).44,45 Objectively, silver is a more efficient SERS substrate material, with silver NPs producing 10–100 times stronger SERS signals than gold NPs.46 However, in most cases, gold NPs are preferred as SERS substrates because of the better biocompatibility, which reduces the safety risk of in vivo detection,47 and another reason is the higher chemical activity of silver and the poor stability of SERS enhancement on silver.
The commonly used Raman reporters contain sulfhydryl, amino groups, or heterocyclic nitrogen, which have strong binding affinity owing to the direct bonding to the surface of SERS substrates via Au/Ag–S and Au/Ag–N. The binding between the Raman reporters and the SERS substrates prevents detachment of the reporters in the subsequent modification process, which is important for the signal strength and stability of the SERS nanotags.48,49 Resonance effect of some reporters with a particular absorption band overlapping the laser wavelength can bring 104 times amplification on the SERS signal, which can be involved in SERS tag fabrication for high sensitivity.50 Moreover, in the selection of Raman reporters that have multiple peaks in the fingerprint region (1000–1700 cm−1), one should try to choose Raman reporters with fewer characteristic peaks, which can effectively reduce the spectral overlap between different SERS characteristic peaks during multiplex detection and improve detection accuracy. There is a Raman-silent range (1800–2800 cm−1) in which the signals of biological species are negligible. Several molecules with special groups, such as alkynes, nitriles, azides, and deuterium, give bands in this range. Thus, SERS tags with molecules having such groups are welcome for multi-targeting and multicolor SERS bio-detection.51
Due to the adsorption and modification of reporter molecules on the surface of gold/silver colloidal nanoparticles, the stability of the colloidal particles would be broken, resulting in condensation phenomena that bother many beginners. To avoid the uncontrollable aggregation that leads to a fluctuation of SERS signals, polymer ligands can be applied to prevent aggregation by electrostatic repulsion or steric forces, for example, sodium citrate, dodecanethiol, polyethylene glycol (PEG), cetyltrimethylammonium bromide (CTAB), tannic acid, hydroxylamine hydrochloride, and polyvinylpyrrolidone (PVP), etc.
Besides the SERS substrates and reporters, their combination modes are also the core of sensor designs. The following sections will describe in categories, especially for DNA/RNA-involved sensing systems.
3.2 Raman reporter modifies DNA: SERS signal turn-on
This strategy is achieved by inserting a Raman reporter into dsDNA or modifying it at one end of ssDNA, generating a unique SERS signal when the DNA is attached to a metal for detection purposes.Negatively charged phosphates of gene chains tend to attract positively stained dyes or fluorophores owing to static interactions. For example, SYBR Green I binds nonspecifically to dsDNA. In the free state, its fluorescence was relatively weak. Once bound to dsDNA, its fluorescence increases by 1000 times. The total fluorescence signal is proportional to the amount of outgoing dsDNA, which is the basis of quantitative real-time PCR (qPCR) for the precise quantitative analysis of target sequences. In SERS sensing, some dyes that are chosen as Raman reporters own their unique SERS peaks when the plasmonic substrates quench their fluorescence completely. Kim et al.52 chose methylene blue (MB) to stain dsDNA chains and provide identifiable SERS signals. They developed a CRISPR/dCas9-mediated SERS assay for multidrug-resistant (MDR) bacterial detection for the first time (Fig. 4A). Gold-coated magnetic nanoparticles (Au MNP) bind to dCas9/gRNA, and the Au MNP-dCas9/gRNA probe can accurately identify and bind to MDR bacterial genes because dCas9/gRNA can capture complementary target DNA sites through specific sites. Next, MB was added to the system, and the Au MNP-dCas9/gRNA probes bound to MDR bacteria were separated by magnetic separation and subjected to SERS detections. A strong SERS signal can be detected when a sample contains MDR bacteriophage genes; otherwise, the SERS signal becomes relatively weak. This method restricts DNA to SERS hotspots, thereby providing high sensitivity without gene amplification. This method measures three kinds of MDR bacteria, Staphylococcus aureus, Acinetobacter baumannii, and Klebsiella pneumonia, without purification or gene amplification steps, with a detection limit down to the fM level, which can prevent serious infections by ensuring appropriate antibiotic therapy. A similar detection approach for the drug resistance gene macB was proposed by Du et al.,55 who used an Au-dCas9/gRNA as a detection probe to react with the bacterial macB gene, while MB was used as the Raman reporter. The enhanced Raman signal from the Au-dCas9/gRNA ribonucleoprotein complex was monitored. The detection probe by forming the Au-dCas9/gRNA complex was captured by macB, resulting in an enhancement on Raman signals. This method enables a simple and highly sensitive detection of the drug resistance gene macB at a fM level. This study is significant because it aims to prevent contamination with drug resistance genes in food hygiene.
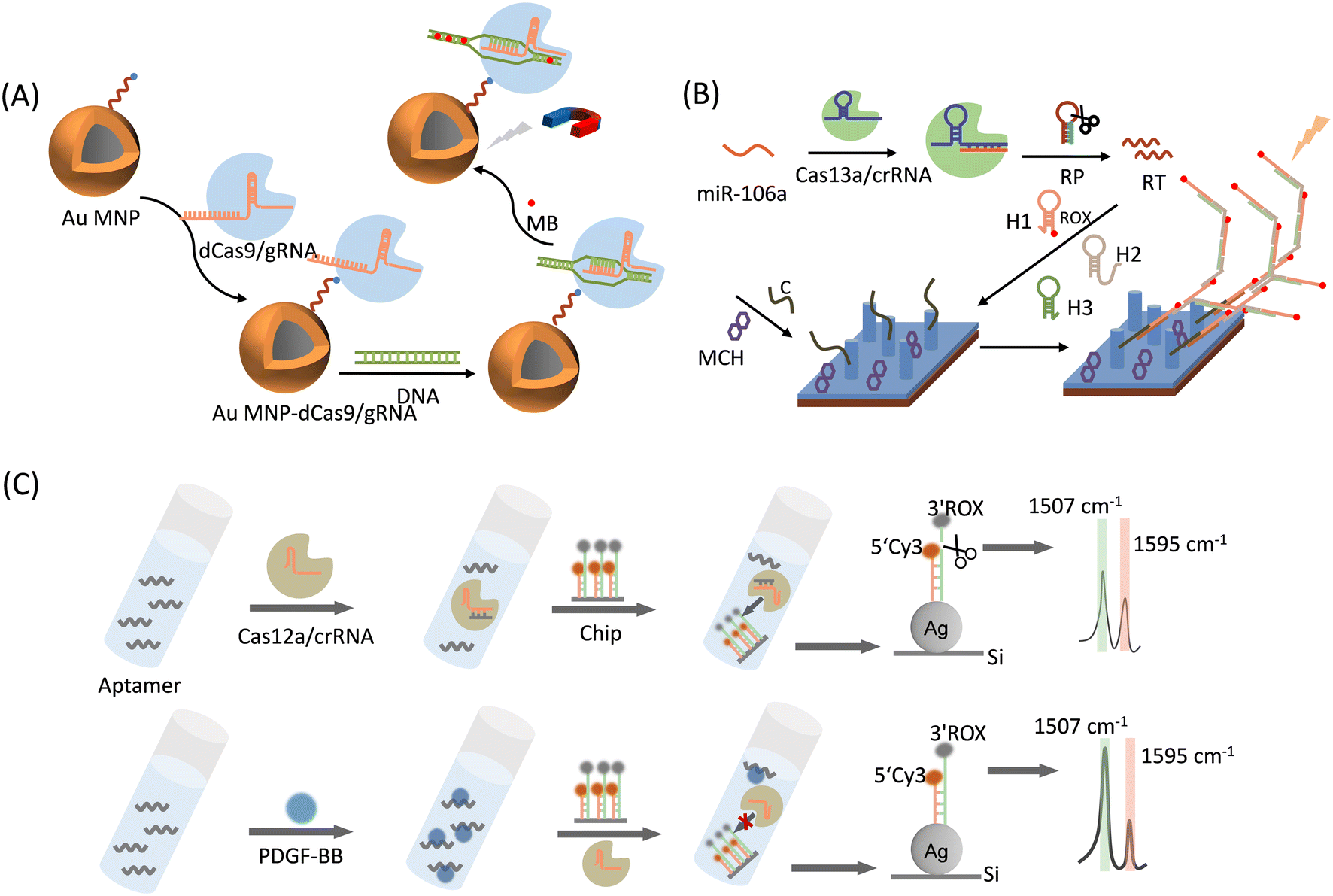 | ||
| Fig. 4 (A) Schematic representation of a superbug dsDNA detection using the CRISPR-mediated SERS assay.52 (B) Schematic illustration of the sensing mechanism of Cas13a-bHCR based cascade signal amplification strategy for the SERS sensing of miRNA.53 RP: recognition probe, RT: recombination trigger, C: capture probes, H1: hairpin probe 1, H2: hairpin probe 2, H3: hairpin probe 3, and MCH: mercaptohexanol. (C) Schematic illustration of a CRISPR/Cas12a-powered silicon SERS ratiometric sensor for the detection of platelet-derived growth factor-BB (PDGF-BB).54 | ||
In addition to the electrostatic adsorption of dyes on the DNA chains, the decoration and graft of dyes at one end of the DNA chain by the covalent bond is another option for labeling DNAs for SERS strategies. Zhang et al.53 proposed a CRISPR/Cas13a-mediated SERS assay to detect gastric-cancer-associated miRNA-106a (miR-106a) (Fig. 4B). The detection strategy can be divided into three parts: the CRISPR/Cas13a system (Cas13a), branched-strand hybridization chain reaction (bHCR), and SERS sensing chip. Cas13a/crRNA recognizes and binds to miRNA-106a, which activates the Cas13a shear recognition probe (RP) and releases many recombination trigger (RT) fragments. Next, RT can sequentially trigger three hairpin probes (H1, H2, and H3) to perform a branched-strand hybridization chain reaction, forming a bHCR product, where a Raman reporter 6′-carboxy-X-rhodamine (ROX) has been modified on H1. Finally, the bHCR product was specifically captured by the capture probe (marked as C) on the SERS sensing chip by complementary hybridization of RT with capture probe C. The branching HCR product contains a large number of ROX molecules, and the excellent surface enhancement of the Ag nanorod (AgNR) array can detect a significantly amplified ROX signal, enabling the qualitative and quantitative detection of miR-106a. The method detected miR-106a using a triple-signal amplification approach: CRISPR/Cas13-based signal amplification, bHCR-based signal amplification, and SERS sensing-based signal amplification. Therefore, the SERS detection of gastric cancer-associated miR-106a based on Cas13a-bHCR can be achieved within 1 h with a limit of detection (LOD) of 8.55 aM. In addition, nonspecific samples (i.e., miR-21, miR-155, a mixture of miR-21 and miR-155, and a mixture of miR-21, miR-155, and miR-106a) were detected using this method. The results showed that the SERS signal was only detected in the presence of miR-106a. This sensor exhibits good specificity and high reproducibility, and is a more promising detector for the detection of miR-106a in clinical samples, making it more useful in early disease clinical diagnosis.
Cao et al.54 demonstrated for the first time a CRISPR/Cas12a-driven system on a silicon chip for sensitive and quantitative SERS analysis (Fig. 4C). As a proof-of-concept application, they chose platelet-derived growth factor-BB (PDGF-BB) as the target. The SERS substrate consisted of homogeneous silver nanoparticles (AgNPs) grown in situ on a Si wafer (AgNPs@Si). Longer ssDNA labeled with ROX was attached to the AgNPs via the Ag–S bond, and its complementary DNA, which is a shorter ssDNA labeled with Cy3 (internal standard) was modified on AgNPs@Si by complementary base pairing. Cleavage of ROX-labeled non-complementary ssDNA by Cas12a was controlled by the presence or absence of PDGF-BB. The Cy3 molecule was maintained at a shorter ssDNA end with or without PDGF-BB, resulting in a scaled chip. The LOD of this system for PDGF-BB was as low as 3.2 pM, and this sensing strategy showed good reproducibility and sensitivity toward PDGF-BB.
3.3 Raman reporter modifies metal surface: SERS signal turn-off
This CRISPR/Cas-SERS sensing type is mainly based on the trans-/cis-cleavage function of the Cas12a protein, which can adjust the distance of a plasmonic ruler bridged by ssDNA (which is a Cas12a substrate), change the plasmonic coupling fields of two metal nanoparticles or output elements to the metal, and alter the enhancement ability of plasmonic robots to Raman reporters. Modifying the Raman reporter on a metal nanoparticle surface is a common strategy based on physical enhancement mechanisms and SERS tags. The SERS signal was enhanced by synthesizing metallic nanoparticles of different shapes. In some cases, a metal nanostructure and SERS tag (with a Raman reporter) are linked to ssDNA, which is the substrate for activated Cas12a to form plasmonic rulers. In other cases, one end of ssDNA is the metal nanostructure while the other end is a dye (e.g., MB) or an enzyme, which will depart from the metal surface for a “turn-off” sensing strategy in response to a positive sample.Liang et al.56 introduced a CRISPR/Cas12a-mediated SERS assay to detect SARS-CoV-2 (Fig. 5A). The Raman reporter 4-aminothiophenol (4-ATP) was first modified on the surface of the AgNPs to form the SERS tag, AgNP@4ATP, which was linked to magnetic beads via ssDNA. Target DNA activates the trans-cleavage ability of Cas12a. Once the SERS tag was removed from the magnetic beads, the SERS signal recorded above the magnetic beads will be significantly decreased. This method improves the sensitivity of nucleic acid detection without DNA amplification, and can be completed within 30–40 min with a detection limit as low as the fM level. The platform can rapidly identify SARS-CoV-2 RNA without amplification, which saves time compared with PCR-based approaches or other amplification-based methods. Based on the advantages of short waiting times and low test environment requirements, this platform is a potential nucleic acid test tool for point-of-care testing. Du et al.63 used a similar method for amplification-free detection of hepatitis B virus (HBV) DNA using AuNPs as the SERS substrate. This system could achieve rapid and highly sensitive detection of HBV DNA within 50 min with a wide detection range of 0.1 pM to 1 nM.
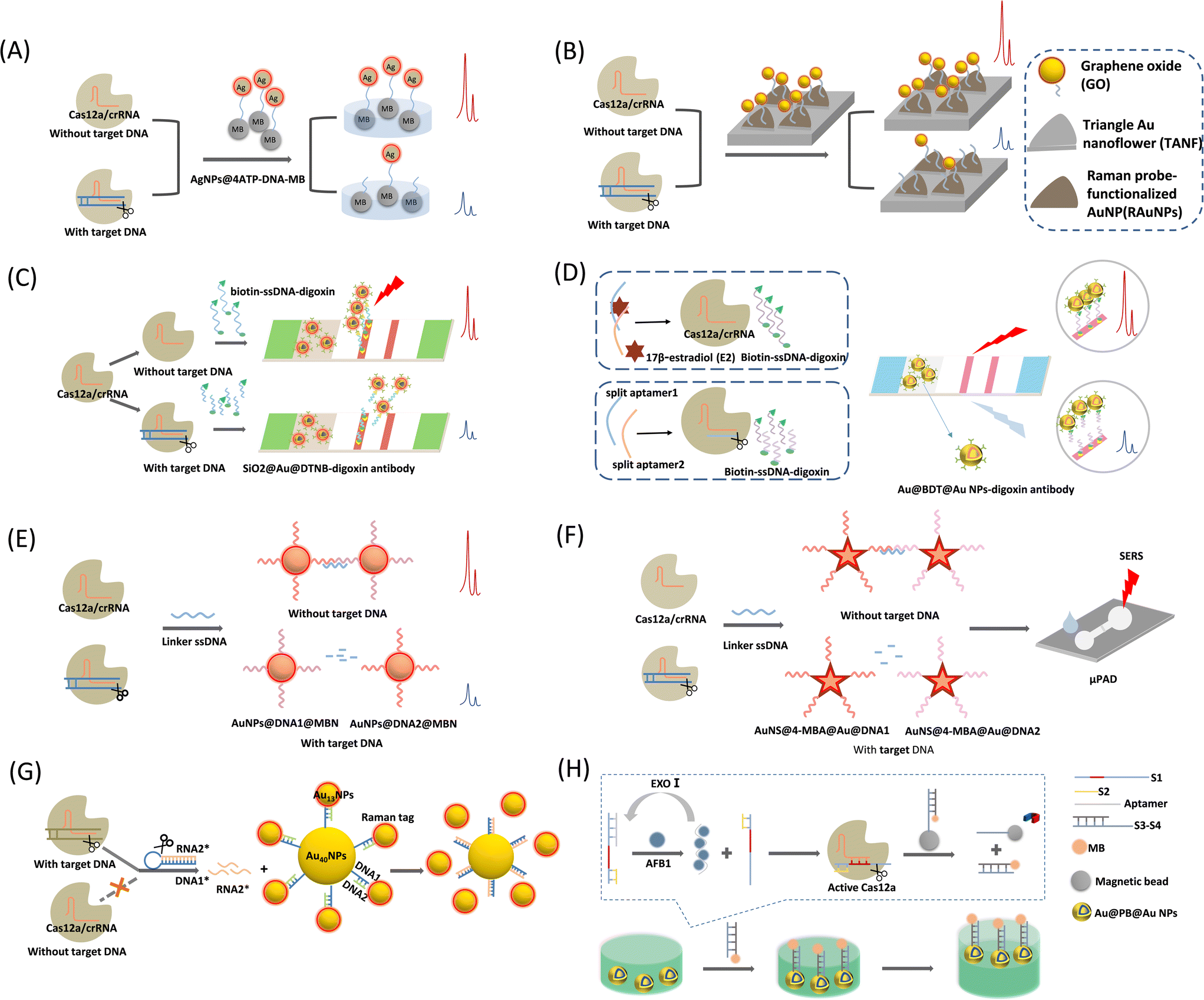 | ||
| Fig. 5 (A) Schematic illustration of CRISPR/Cas12a-mediated SERS assay for SARS-CoV-2 detection.56 (B) Schematic diagram of the CRISPR/Cas12a-assisted SERS-based analysis system for detection of viral DNA.57 (C) Schematic diagram of CRISPR/Cas12a-mediated lateral flow of SERS.58 (D) Schematic illustration of the CRISPR/Cas12a biosensing for SERS-based LFA detection of 17β-estradiol (E2).59 (E) Schematic illustration of the CRISPR/Cas12a-mediated AuNPs polymerization SERS platform for viral gene detection.41 (F) Schematic diagram of the CRISPR/Cas12a-based SERS microfluidic paper assay device (μPAD) for ultrasensitive detection of food pathogenic bacteria.60 (G) Schematic diagram of a CRISPR/Cas12a integrated SERS nanoplatform with a chimeric DNA/RNA hairpin guidance for nucleic acid detection.61 (H) Schematic diagram of CRISPR/Cas12a-based core–shell Au@PB@Au NPs SERS substrate for detection of AFB1.62 | ||
Choi et al.57 proposed a CRISPR/Cas12a-assisted SERS analysis system that is also a nucleic acid amplification-free biosensor (Fig. 5B). This system was developed using the ssDNA-modifying Raman reporter TAMRA-functionalized AuNPs on SERS-active graphene oxide (GO)/periodic triangular gold nanoflower (TANF) arrays. The TANF arrays composed of periodic triangular gold micropatterns and gold nanoflower structures significantly enhanced the SERS signal when Cas12a was activated by the target DNA. This strategy improves the detection sensitivity of multiviral DNA for HBV, HPV-16, and HPV-18, with very low detection limits and a large detection range of 1 aM to 100 pM without amplification steps.
Pang et al.58 proposed a CRISPR/Cas12a-mediated SERS lateral flow to detect dsDNA and single-base mutations (Fig. 5C). This system combines the CRISPR/Cas12a-SERS system with an LFA for simultaneous colorimetric and SERS signal detection. First, digoxin antibody-labeled SiO2@Au@5,5′-dithiobis-(2-nitrobenzoic acid) (DTNB) NPs are used as SERS tags and dispersed on the conjugation pad. In the control trial with no target DNA, Cas12a was inactivated and biotin–DNA–digoxin remained. Accordingly, the SERS tag was fixed to the test line (T-line) of the panel via a biotin–DNA–digoxin bridge. The conjugation of SERS tags to the T-line induces both strong SERS and colorimetric signals. If the target DNA is present, Cas12a activates single-strand cleavage, resulting in the shearing of biotin–DNA–digoxin. The biotin–DNA–digoxin fragment causes failure in the attachment of parts of the SERS tags to the T-line. A reduction in the number of SERS tags on the T-line decreases the SERS intensity and colorimetric fading. By combining the ultrasensitive SERS tag with the targeted signal amplification capability of CRISPR-Cas12a, the method can directly quantify HIV-1 dsDNA without any pre-amplification step with an LOD of 0.3 fM, which is nearly four orders of magnitude lower than that of the conventional colorimetric LFA method. The entire assay was completed within 1 h.
In 2022, Li et al.59 improved this CRISPR/Cas12a-based SERS LFA biosensing method for the detection of 17β-estradiol (E2) (Fig. 5D). They adopted a controlled release action of a programmed aptamer/crRNA transducer via the analyte (E2) to achieve the SERS signal from the positive sample “turn-on”. They first fabricated anti-digoxin antibody-modified Au@benzene-1,4-dithiol (BDT)@AuNPs as SERS tags, where the Raman reporter BDT was embedded between the Au nucleus and shell. Such a Raman reporter-embedded structure protects it from interference from sensing systems and environmental alterations. The method was designed separately for the split aptamers of E2, denoted as split aptamer1 and split aptamer2. Split aptamer1 is complementary to the crRNA of the Cas12a/crRNA system and acts as an activator of Cas12a. In the presence of E2, split aptamer1 and split aptamer2 bind to E2. Thus, Cas12a/crRNA is inactivated and biotin–DNA–digoxin remains intact. The SERS tag was linked to the end of the biotin–DNA–digoxin by SERS tag–DNA–biotin, and the biotin end was captured by modified streptavidin on the test line (T-line). As a result, a T-line colorimetric assay was performed owing to the assembly of the AuNPs. Simultaneously, Raman signals can be easily read out owing the high loading of the SERS tags. In contrast, in the absence of E2, splitting aptamer 1 activates the Cas12a/crRNA trans-cleavage of biotin–DNA–digoxin, resulting in an inability to anchor the SERS tags to the test line, and the colorimetric and SERS intensities become attenuated, indicating a negative result. This method can achieve dual-mode detection with a detection limit as low as 180 fM for SERS measurements.
Fig. 5E shows a CRISPR/Cas12a-mediated AuNP polymerization SERS platform for viral gene detection, which was developed by Su et al.41 They prepared two SERS tags, probe 1 (AuNPs@DNA1@MBN) and probe 2 (AuNPs@DNA2@MBN), by modifying Raman reporters (4-mercaptobenzonitrile, MBN) and ssDNAs above the AuNP surfaces. The two probes can be complementarily aggregated by a linker DNA via a hybridization reaction, achieving SERS signal amplification. The target DNA-activated Cas12a can cleave the linker DNA, causing failure in the crosslinking of the two SERS probes and decreasing the SERS signal. In contrast, if there is no target DNA, the linker DNA remains intact, causing severe crosslinking of the SERS probes by the linker DNA and a significant increase in the SERS signal intensity. This sensing method has been proven to be valid for HPV 16 and HPV 18 in pseudovirus and serum samples, respectively. The pM level of the target DNA was measured using a CRISPR/Cas12a-based SERS platform without targeting gene amplification. Target-induced trans-ssDNA cleavage triggered the aggregation of the designed SERS probe, enabling the completion of SERS gene detection within 40 min. This rapid gene detection platform exhibits potential for point-of-care tests, and may be useful for diagnosing viral infections in clinical settings.
In a recent report, Zhuang et al.60 proposed a SERS-based CRISPR/Cas12a microfluidic paper assay device for the ultrasensitive detection of food-borne pathogenic bacteria (Fig. 5F). They also chose a core–shell SERS tag with a Raman reporter inserted to prevent uncontrolled disturbances during SERS measurements, guaranteeing detection reliability. Two SERS probes were prepared by conjugating SH-ssDNAs above gold nanostar@4-mercaptobenzoic acid@gold nanoshells (AuNS@4-MBA@Au@DNA1 and AuNS@4-MBA@Au@DNA2). The trans-cleavage of CRISPR/Cas12a is activated in the presence of target DNA, leading to linker ssDNA fragmentation. This ssDNA linker cannot hybridize these two SERS probes by base complementation, and the nanoprobe remains dispersed in a colloidal solution with a weak SERS signal intensity. In the absence of the target DNA, the ssDNA linker is cleaved and the nanoprobe aggregates, enhancing the SERS signal. Different from the method proposed by Su,41 they transferred this sensing system to μPAD, and portability and simplicity enable its simple implementation in point-of-need detection or resource-limited areas.
Plasmonic nanosatellites are often found in homogeneous system detection and exhibit excellent SERS sensing performance because they support strong plasmonic coupling between the metal core and its satellite particles. Yin et al.61 recently proposed a CRISPR/Cas12a integrated SERS nanoplatform for nucleic acid detection based on a plasmonic satellite nanoconjugate. Interestingly, they adopted a chimeric DNA/RNA hairpin for gene amplification, significantly improving the detection sensitivity (Fig. 5G). First, a SERS probe consists of 40 nm AuNPs (Au40NPs-DNA1) as the core of a satellite nanocluster with a Raman reporter (4-mercaptobenzoic acid; MBA) wrapped around 13 nm AuNPs (Au13NPs-MBA/DNA2) as the surrounding satellites, both of which were linked by partially complementary DNA1/DNA2. The chimeric DNA/RNA hairpin stem was formed by fully hybridized DNA1* and RNA2*, and the loop was an A–T repetitive DNA sequence. In the presence of the target DNA, Cas12a is activated and immediately cleaves the DNA loop, releasing a large amount of RNA2. The released RNA2* is fully complementary to DNA1 and therefore replaces DNA2 and hybridizes with DNA1 on Au40NPs, resulting in the separation of Au13NP-MBA/DNA2 from Au40NPs-DNA1 and the breakdown of the hotspots, causing the sensing platform of the SERS signal to decrease sequentially. The Orf gene of the SARS-CoV-2 (Orf-cDNA) was selected as the target DNA for the sensing system. Owing to the avoidance of spatial site constraints, this approach reduces the system's SERS intensity more efficiently than the direct use of activated CRISPR-Cas12a in a turn-off sensing system; thus, a detection limit down to the aM level is obtained.
Zhang et al.62 designed a CRISPR/Cas12a-based SERS platform for the sensitive detection of AFB1. The sensor consists of EXO-I exonuclease-mediated amplification and self-assembled SERS arrays at the water–oil two-phase interface (Fig. 5H). Prussian blue (PB) was selected as the Raman reporter and inserted into the bi-metallic core–shell Au@PB@Au NPs. When the target AFB1 was present, it bound to the aptamer. Under the action of EXO-I, the target is recycled, and the released S1–S2 structure activates the trans-cleavage function of Cas12a. The dsDNA portion of S3–S4d will self-assemble at the water–oil interface by linking to the Au@PB@Au NPs via Au–S bonds. At the water–oil interface, cyclohexane was used as the oil phase solvent to form a water–oil two-phase interface, facilitating the formation of nanoparticle films at the interface, and enabling the SERS signal to be detected. The LOD of this sensing system was 3.55 pg mL−1. This study provides a new approach for the future SERS detection of non-nucleic acid targets.
3.4 Unique reporters for post-signal amplification strategies
In this category, a Raman reporter can be generated by converting specific molecules to molecules with characteristic Raman peaks under acidic or basic conditions, or by the massive release of a Raman reporter encapsulated by substances in the presence of surfactants, where the released Raman reporters are mixed with the SERS substrates, thereby generating unique SERS signal amplification. Therefore, we catalog these as post-signal amplification strategies. A typical feature of these strategies is the unique form of Raman reporters.Liu et al.64 proposed a CRISPR/Cas12a-mediated liposome amplification strategy for SERS detection of nucleic acids (Fig. 6A). First, trace amounts of target DNA were amplified using the loop-mediated isothermal amplification (LAMP). Next, biotin–ssDNA–NH2 was linked to a 96-well plate by a covalent reaction, followed by the surface block of BSA. Cas12a was activated in the presence of gRNA and target DNA to indiscriminately cleave ssDNA. In the absence of the target DNA, the trans-cleavage activity of Cas12a was inhibited, retaining the ssDNA undamaged in the wells. After the complete removal of the supernatant, only the unbroken ssDNA captured the signal molecule-loaded liposomes based on biotin–streptavidin affinity. Finally, by adding a surfactant (phosphate-buffered saline with Tween 20, PBST) to the system, the liposomes were broken up to immediately release their encapsulated signal molecules. In the SERS measurement, 4-nitrothiophenol (4-NTP) was selected as the Raman reporter, which was co-mixed with AuNPs to produce a strong SERS signal owing to the aggregation of AuNP. This method employs a unique post-signal amplification strategy with ultra-high sensitivity and specificity.
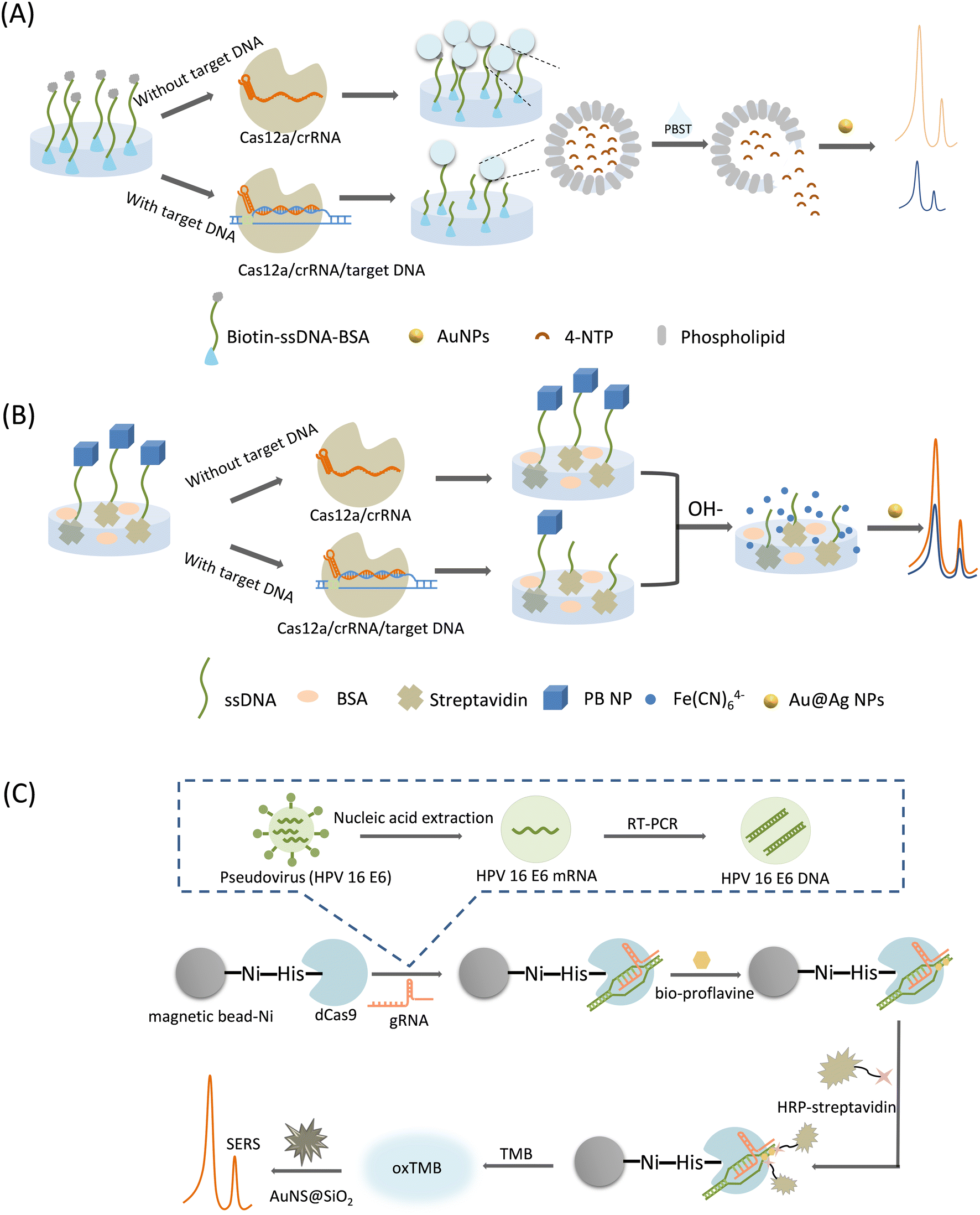 | ||
| Fig. 6 (A) Schematic illustration of CRISPR-/Cas12a-mediated liposome-amplified strategy for SERS and naked-eye detection of the target nucleic acid assay.64 (B) Scheme of CRISPR/Cas12a-driven ultra-sensitive SERS-based biosensor.65 (C) Schematic diagram of CRISPR/dcas9-based SERS sensing method for SERS detection of virus samples.66 | ||
Raman profiles of biosamples and tissues are mainly distributed in a spectral range of less than 1800 cm−1. This region is also the signal response range of many Raman probes, often causing an overlap between the biological background and probe signal peaks during biological sensing. It has been found that the range of 1800–2800 cm−1 in the Raman spectral range of biosamples has almost no background interference, and is called the Raman silence range. Although some exogenous biological groups, such as alkynes and azides produce characteristic peaks in the cell-silent region these groups usually have weak Raman signals. The cyano group of PB exhibits a single Raman peak (2156 cm−1) in the cell-silent region, significantly improving the detection signal-to-noise ratio, which was first proposed by Yin et al. in 2017.67 Using PB as a Raman reporter, Pan et al.65 developed a CRISPR/Cas12a-driven ultrasensitive SERS-based biosensor (Fig. 6B). First, PB NPs were modified on one end of ssDNA, and then the PB NP-ssDNA conjugate was immobilized on the inner surface of a 96-well plate. In the presence of the target DNA, the cleavage activity of Cas12a was activated, and the released PB NPs departed from the well surface because of the broken ssDNA linker. Then, under alkaline treatment, decomposition of the remaining PB NPs in the microtiter plate was triggered, generating a large amount of ferricyanide anions (Fe (CN)64−), which exhibit a unique Raman peak that can be detected by mixing them with the SERS substrate (Au@Ag NP). This method significantly amplified the SERS signal based on the target response function of the PB NP and the bimetallic core–shell SERS substrate. The system was used for milk adulteration detection with an LOD of down to the aM level, and can be a universal platform for applications ranging from clinical diagnostics to food safety monitoring.
Su et al.66 established a simple and ultra-sensitive SERS method based on CRISPR/dCas9 technology and an enzyme-catalyzed amplification reaction for the HPV virus detection (Fig. 6C). A reaction solution involving magnetic beads and dCas9 proteins was prepared first. The dCas9 can be activated by gRNA to specifically bind target DNAs, achieving a CRISPR/dCas9/gRNA/DNA complex. A Ni-nitrilotriacetic acid (NTA)-modified magnetic bead was utilized to settle down the His-Tag-modified dCas9 proteins, in order to realize the magnetic separation of the magnetic bead-dCas9/gRNA/DNA complex. Sample DNAs had been decorated with biotin, which can be linked with the streptavidin-conjugated horse radish peroxidase (HRP) enzyme to achieve an HRP-labelling conjugate above the magnetic bead surface. Thus, after magnetic separation, the magnetic bead with target DNA and HRP will remain, which can start a enzyme-catalyzed amplification reaction of TMB and H2O2, producing a blue-green oxidation product (oxTMB) that can give SERS signal output. Unique AuNS coated with SiO2 (AuNS@SiO2) was used as an effective SERS substrate, and the lightning rod effect at the tip significantly enhances the SERS signal of the TMB. Owing to the dual amplification effect, this method can detect the HPV virus at the ng level.
4. Summary and outlook
The CRISPR/Cas-based SERS technology has enabled the detections of drug-resistant bacteria, viruses, and single-base mutations in DNA. Because of the large variety of CRISPR/Cas proteins, including Cas9, dCas9, Cas12a, Cas12b, and Cas13a, CRISPR/Cas-based SERS technology allows the development of multiple strategies for nucleic acid detection. CRISPR/Cas-based SERS combines the advantages of both, while complementing the deficiencies present in individual systems. Table 2 lists the CRISPR/Cas-based SERS methods and their targets and performances. Almost all of these methods achieve the pM level of detection, with five applications reaching the aM level. Most of these systems do not use amplification techniques, which is a significant improvement compared to the existing fluorescent methods that need amplification techniques such as RPA, PCR, and LAMP for help.26| Classification | No. | SERS substrates | SERS reporters (Raman band position) | Laser wavelength | Time | Application | Amplification | Sensitivity | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| Cas9-based class | 1 | Au MNPs | Methylene blue (MB) (1620 cm−1) | 632 nm | <2 h | Multidrug-resistant bacteria | Free | fM | 52 |
| 2 | AuNPs | MB (1621 cm−1) | 638 nm | <2 h | Drug resistance gene macB | Free | fM | 55 | |
| 3 | AuNS@SiO2 | 3,3′,5,5′-Tetramethylben-zidine (TMB) (1605 cm−1) | 633 nm | >4 h | Human papillomavirus-16 (HPV-16) | Reverse transcription-polymerase chain reaction (RT-PCR) | ng | 66 | |
| Cas12a-based class | 4 | AgNPs@Si chip | 6′-Carboxy-X-rhodamine (ROX) (1507 cm−1) | 633 nm | 30 min | Platelet-derived growth factor-BB (PDGF-BB) | Free | pM | 54 |
| Cyanine3 (Cy3) (1595 cm−1) | |||||||||
| 5 | AgNPs | 4-Aminothiophenol (4-ATP) (1074 cm−1) | 785 nm | 30–40 min | Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) | Free | fM | 56 | |
| 6 | AuNPs | 4-ATP (1590 cm−1) | 638 nm | 50 min | Hepatitis B virus (HBV) | Free | pM | 63 | |
| 7 | AuNP-modified GO-TANF | 5-Carboxytetramethylrhodamine (5-TAMRA) (1359 cm−1) | 785 nm | 20 min | HBV, HPV-16, HPV-18 | Free | aM | 57 | |
| 8 | SiO2@Au | 5,5′-Dithiobis-(2-nitrobenzoic acid) (DTNB) (1332 cm−1) | 785 nm | 1 h | HIV-1 | Free | fM | 58 | |
| 9 | AuNPs | 4-Mercaptobenzonitrile (MBN) (1581 cm−1) | 633 nm | 40 min | HPV-16, HPV-18 | Free | pM | 41 | |
| 10 | AuNPs | 4-Mercaptobenzoic acid (MBA) (1078 cm−1) | 785 nm | ∼3 h | SARS-CoV-2 Orf-cDNA | Free | aM | 61 | |
| 11 | AuNSs@Au | 4-MBA (1075 cm−1) | 785 nm | 45 min | S. typhi | Recombinase polymerase amplification (RPA) | 3–4 CFU mL−1 | 60 | |
| 12 | Core–shell Au@Au NPs | Benzene-1,4-dithiol (BDT) (1055 cm−1) | 785 nm | 40 min | 17β-Estradiol | Free | fM | 59 | |
| 13 | AuNPs | 4-Nitrothiophenol (4-NTP) (1334 cm−1) | 785 nm | >2 h | Dehydrogenase subunit 2 (ND2) (food authenticity) | Loop-mediated isothermal amplification (LAMP) | aM | 64 | |
| 14 | Core–shell Au@Ag NPs | Prussian blue (PB) (2083 cm−1) | 785 nm | >3 h | Cytochrome b (Cyt b) (food authenticity) | LAMP | aM | 65 | |
| 15 | Core–shell Au@Au NPs | PB (2130 cm−1) | 633 nm | 9 h | Aflatoxin B1 (AFB1) | Free | 3.55 pg mL−1 | 62 | |
| Cas13a-based class | 16 | AgNRs array | ROX (1503 cm−1) | 633 nm | 1 h | miR-106a | Free | aM | 53 |
Although SERS is highly sensitive as a powerful biosensing tool, it has limitations in micro-target nucleic acid detection, and CRISPR/Cas technology can complement this deficiency well. The gRNA of the CRISPR/Cas system can precisely identify specific sites of nucleic acid sequences, amplifying the signal during nucleic acid detection and improving the detection sensitivity. The combination of SERS, with its ultrahigh surface sensitivity, and the CRISPR/Cas system, with its sequence-specific recognition capability, further improves the specificity of target gene detection.
Currently, CRISPR/Cas-based SERS technology is more based on Cas12a to design detection strategies, and Cas9/Cas13-based SERS biosensings are less studied, which encourages us to devote more attempts in new strategy designs based on different Cas proteins to enrich the CRISPR/Cas-based SERS technology. Although the CRISPR/Cas-based SERS technology has great advantages in terms of sensitivity and specificity, it can only be used to detect known DNA sequences, which is a significant limitation in practical applications. In addition, the complexity of sample preparation prior to detection, and the dependence of SERS detection on expensive and large equipment restricted us to diagnostic analysis in the laboratory at present. At this stage, CRISPR/Cas-based SERS technology is mainly used for in vitro nucleic acid detection, and it is not known whether it can maintain good sensitivity and specificity in complex biological environments. How to incorporate CRISPR/Cas-based SERS technology into the in vivo cells for analysis will be important for disease prevention, diagnosis and treatment. Although we have summarized the advantages of the CRISPR/Cas-based SERS technology, it remains to be seen whether these advantages are still available in the face of complex fields or conditions, and we look forward to the day when this technology can be transferred from the laboratory to practical applications.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by National Natural Science Foundation of China NSFC (No. 21873039 and 22173035), Changchun Science and Technology planning Project (21ZY13), the Program of Jilin Provincial Science & Technology Department (No. 20230204053YY), the Technology Development Program of Jilin Province (20220101046JC), the opening project of the State Key Laboratory of Applied Optics (SKLAO2021001A14), and the Interdisciplinary Integration Innovation Project of Jilin University (JLUXKJC2020106).References
- C. Zong, M. Xu, L.-J. Xu, T. Wei, X. Ma, X.-S. Zheng, R. Hu and B. Ren, Chem. Rev., 2018, 118, 4946–4980 CrossRef CAS PubMed.
- K. C. Bantz, A. F. Meyer, N. J. Wittenberg, H. Im, O. Kurtulus, S. H. Lee, N. C. Lindquist, S.-H. Oh and C. L. Haynes, Phys. Chem. Chem. Phys., 2011, 13, 11551–11567 RSC.
- S. Schlücker, Angew. Chem., Int. Ed., 2014, 53, 4756–4795 CrossRef PubMed.
- C. L. Haynes, J. Am. Chem. Soc., 2007, 129, 2197–2198 CrossRef CAS.
- J. Popp and T. Mayerhöfer, Anal. Bioanal. Chem., 2009, 394, 1717–1718 CrossRef CAS PubMed.
- S.-Y. Ding, J. Yi, J.-F. Li, B. Ren, D.-Y. Wu, R. Panneerselvam and Z.-Q. Tian, Nat. Rev. Mater., 2016, 1, 16021 CrossRef CAS.
- D. Cialla-May, X. S. Zheng, K. Weber and J. Popp, Chem. Soc. Rev., 2017, 46, 3945–3961 RSC.
- W. Liao, Q. Lin, Y. Xu, E. Yang and Y. Duan, Nanoscale, 2019, 11, 5346–5354 RSC.
- Y. Yang, J. Zhu, J. Zhao, G.-J. Weng, J.-J. Li and J.-W. Zhao, ACS Appl. Mater. Interfaces, 2019, 11, 3617–3626 CrossRef CAS PubMed.
- J. Chao, W. Cao, S. Su, L. Weng, S. Song, C. Fan and L. Wang, J. Mater. Chem. B, 2016, 4, 1757–1769 RSC.
- E. Garcia-Rico, R. A. Alvarez-Puebla and L. Guerrini, Chem. Soc. Rev., 2018, 47, 4909–4923 RSC.
- H. Cho, B. Lee, G. L. Liu, A. Agarwal and L. P. Lee, Lab Chip, 2009, 9, 3360–3363 RSC.
- H. Zhang, Y. Liu, J. Gao and J. Zhen, Chem. Commun., 2015, 51, 16836–16839 RSC.
- L. Guerrini and R. A. Alvarez-Puebla, Analyst, 2019, 144, 6862–6865 RSC.
- Y. Bao, X. Zhang, X. Xiang, Y. Zhang, B. Zhao and X. Guo, Phys. Chem. Chem. Phys., 2022, 24, 10311–10317 RSC.
- J. Zheng, J. Bai, Q. Zhou, J. Li, Y. Li, J. Yang and R. Yang, Chem. Commun., 2015, 51, 6552–6555 RSC.
- Z. Liang, J. Zhou, L. Petti, L. Shao, T. Jiang, Y. Qing, S. Xie, G. Wu and P. Mormile, Analyst, 2019, 144, 1741–1750 RSC.
- S. Ge, M. Ran, Y. Mao, Y. Sun, X. Zhou, L. Li and X. Cao, Analyst, 2021, 146, 5326–5336 RSC.
- M. Zhang, M. Yao, J. Gong, Z. Wang, W. Tu and Z. Dai, Chem. Commun., 2022, 58, 11665–11668 RSC.
- J. van der Oost, M. M. Jore, E. R. Westra, M. Lundgren and S. J. J. Brouns, Trends Biochem. Sci., 2009, 34, 401–407 CrossRef CAS PubMed.
- L. Cong, F. A. Ran, D. Cox, S. Lin, R. Barretto, N. Habib, P. D. Hsu, X. Wu, W. Jiang, L. A. Marraffini and F. Zhang, Science, 2013, 339, 819–823 CrossRef CAS PubMed.
- K. Abnous, N. M. Danesh, M. Ramezani, M. Alibolandi, M. A. Nameghi, T. S. Zavvar and S. M. Taghdisi, Anal. Chim. Acta, 2021, 1165, 338549 CrossRef CAS PubMed.
- S. Bu, X. Liu, Z. Wang, H. Wei, S. Yu, Z. Li, Z. Hao, W. Liu and J. Wan, Sens. Actuators, B, 2021, 347, 130630 CrossRef CAS.
- Y. Sheng, T. Zhang, S. Zhang, M. Johnston, X. Zheng, Y. Shan, T. Liu, Z. Huang, F. Qian, Z. Xie, Y. Ai, H. Zhong, T. Kuang, C. Dincer, G. A. Urban and J. Hu, Biosens. Bioelectron., 2021, 178, 113027 CrossRef CAS PubMed.
- Z. Ali, E. Sanchez, M. Tehseen, A. Mahas, T. Marsic, R. Aman, G. S. Rao, F. S. Alhamlan, M. S. Alsanea, A. A. Al-Qahtani, S. Hamdan and M. Mahfouz, ACS Synth. Biol., 2022, 11, 406–419 CrossRef CAS PubMed.
- C. Han, W. Li, Q. Li, W. Xing, H. Luo, H. Ji, X. Fang, Z. Luo and L. Zhang, Biosens. Bioelectron., 2022, 200, 113922 CrossRef CAS PubMed.
- Y. Li, S. Li, J. Wang and G. Liu, Trends Biotechnol., 2019, 37, 730–743 CrossRef CAS PubMed.
- S.-Y. Li, Q.-X. Cheng, J.-M. Wang, X.-Y. Li, Z.-L. Zhang, S. Gao, R.-B. Cao, G.-P. Zhao and J. Wang, Cell Discovery, 2018, 4, 20 CrossRef PubMed.
- P. Fozouni, S. Son, M. D. de León Derby, G. J. Knott, C. N. Gray, M. V. D'Ambrosio, C. Zhao, N. A. Switz, G. R. Kumar, S. I. Stephens, D. Boehm, C.-L. Tsou, J. Shu, A. Bhuiya, M. Armstrong, A. R. Harris, P.-Y. Chen, J. M. Osterloh, A. Meyer-Franke, B. Joehnk, K. Walcott, A. Sil, C. Langelier, K. S. Pollard, E. D. Crawford, A. S. Puschnik, M. Phelps, A. Kistler, J. L. DeRisi, J. A. Doudna, D. A. Fletcher and M. Ott, Cell, 2021, 184, 323–333.e329 CrossRef CAS PubMed.
- R. Wang, C. Qian, Y. Pang, M. Li, Y. Yang, H. Ma, M. Zhao, F. Qian, H. Yu, Z. Liu, T. Ni, Y. Zheng and Y. Wang, Biosens. Bioelectron., 2021, 172, 112766 CrossRef CAS PubMed.
- H. Wu, C. Qian, C. Wu, Z. Wang, D. Wang, Z. Ye, J. Ping, J. Wu and F. Ji, Biosens. Bioelectron., 2020, 157, 112153 CrossRef CAS PubMed.
- K. Pardee, A. A. Green, M. K. Takahashi, D. Braff, G. Lambert, J. W. Lee, T. Ferrante, D. Ma, N. Donghia, M. Fan, N. M. Daringer, I. Bosch, D. M. Dudley, D. H. O'Connor, L. Gehrke and J. J. Collins, Cell, 2016, 165, 1255–1266 CrossRef CAS PubMed.
- X.-Y. Qiu, L.-Y. Zhu, C.-S. Zhu, J.-X. Ma, T. Hou, X.-M. Wu, S.-S. Xie, L. Min, D.-A. Tan, D.-Y. Zhang and L. Zhu, ACS Synth. Biol., 2018, 7, 807–813 CrossRef CAS PubMed.
- J. H. Soh, H.-M. Chan and J. Y. Ying, Nano Today, 2020, 30, 100831 CrossRef CAS.
- H. Rahimi, M. Salehiabar, M. Barsbay, M. Ghaffarlou, T. Kavetskyy, A. Sharafi, S. Davaran, S. C. Chauhan, H. Danafar, S. Kaboli, H. Nosrati, M. M. Yallapu and J. Conde, ACS Sens., 2021, 6, 1430–1445 CrossRef CAS PubMed.
- J. Moon, H.-J. Kwon, D. Yong, I.-C. Lee, H. Kim, H. Kang, E.-K. Lim, K.-S. Lee, J. Jung, H. G. Park and T. Kang, ACS Sens., 2020, 5, 4017–4026 CrossRef CAS PubMed.
- M. Duan, B. Li, Y. Zhao, Y. Liu, Y. Liu, R. Dai, X. Li and F. Jia, Biosens. Bioelectron., 2023, 219, 114823 CrossRef CAS PubMed.
- F. Hu, Y. Liu, S. Zhao, Z. Zhang, X. Li, N. Peng and Z. Jiang, Biosens. Bioelectron., 2022, 202, 113994 CrossRef CAS PubMed.
- M. Jinek, F. Jiang, D. W. Taylor, S. H. Sternberg, E. Kaya, E. Ma, C. Anders, M. Hauer, K. Zhou, S. Lin, M. Kaplan, A. T. Iavarone, E. Charpentier, E. Nogales and J. A. Doudna, Science, 2014, 343, 1247997 CrossRef PubMed.
- S. Bae, J. Park and J.-S. Kim, Bioinformatics, 2014, 30, 1473–1475 CrossRef CAS PubMed.
- A. Su, Y. Liu, X. Cao, W. Xu, C. Liang and S. Xu, Sens. Actuators, B, 2022, 369, 132295 CrossRef CAS.
- Y. Cheng, A. C. Samia, J. D. Meyers, I. Panagopoulos, B. Fei and C. Burda, J. Am. Chem. Soc., 2008, 130, 10643–10647 CrossRef CAS PubMed.
- P. Ghosh, G. Han, M. De, C. K. Kim and V. M. Rotello, Adv. Drug Delivery Rev., 2008, 60, 1307–1315 CrossRef CAS PubMed.
- L. Jiang, J. Qian, F. Cai and S. He, Anal. Bioanal. Chem., 2011, 400, 2793–2800 CrossRef CAS PubMed.
- L. Rodríguez-Lorenzo, Z. Krpetic, S. Barbosa, R. A. Alvarez-Puebla, L. M. Liz-Marzán, I. A. Prior and M. Brust, Integr. Biol., 2011, 3, 922–926 CrossRef PubMed.
- S. Abalde-Cela, P. Aldeanueva-Potel, C. Mateo-Mateo, L. Rodríguez-Lorenzo, R. A. Alvarez-Puebla and L. M. Liz-Marzán, J. R. Soc., Interface, 2010, 7, S435–S450 CrossRef CAS PubMed.
- S. Lee, H. Chon, M. Lee, J. Choo, S. Y. Shin, Y. H. Lee, I. J. Rhyu, S. W. Son and C. H. Oh, Biosens. Bioelectron., 2009, 24, 2260–2263 CrossRef CAS PubMed.
- L. C. Martin, I. A. Larmour, K. Faulds and D. Graham, Chem. Commun., 2010, 46, 5247–5249 RSC.
- H.-L. Liu, J. Cao, S. Hanif, C. Yuan, J. Pang, R. Levicky, X.-H. Xia and K. Wang, Anal. Chem., 2017, 89, 10407–10413 CrossRef CAS PubMed.
- G. Bodelón, V. Montes-García, C. Fernández-López, I. Pastoriza-Santos, J. Pérez-Juste and L. M. Liz-Marzán, Small, 2015, 11, 4149–4157 CrossRef PubMed.
- J. Ando, M. Asanuma, K. Dodo, H. Yamakoshi, S. Kawata, K. Fujita and M. Sodeoka, J. Am. Chem. Soc., 2016, 138, 13901–13910 CrossRef CAS PubMed.
- H. Kim, S. Lee, H. W. Seo, B. Kang, J. Moon, K. G. Lee, D. Yong, H. Kang, J. Jung, E.-K. Lim, J. Jeong, H. G. Park, C.-M. Ryu and T. Kang, ACS Nano, 2020, 14, 17241–17253 CrossRef CAS PubMed.
- J. Zhang, C. Song, Y. Zhu, H. Gan, X. Fang, Q. Peng, J. Xiong, C. Dong, C. Han and L. Wang, Biosens. Bioelectron., 2022, 219, 114836 CrossRef PubMed.
- H. Cao, J. Xie, J. Cheng, Y. Xu, X. Lu, J. Tang, X. Zhang and H. Wang, Anal. Chem., 2023, 95, 2303–2311 CrossRef CAS PubMed.
- Y. Du, D. Han, Z. An, J. Wang and Z. Gao, Microchim. Acta, 2022, 189, 394 CrossRef CAS PubMed.
- J. Liang, P. Teng, W. Xiao, G. He, Q. Song, Y. Zhang, B. Peng, G. Li, L. Hu, D. Cao and Y. Tang, J. Nanobiotechnol., 2021, 19, 273 CrossRef CAS PubMed.
- J.-H. Choi, M. Shin, L. Yang, B. Conley, J. Yoon, S.-N. Lee, K.-B. Lee and J.-W. Choi, ACS Nano, 2021, 15, 13475–13485 CrossRef CAS PubMed.
- Y. Pang, Q. Li, C. Wang, S. Zhen, Z. Sun and R. Xiao, Chem. Eng. J., 2022, 429, 132109 CrossRef CAS.
- Q. Li, X. Li, P. Zhou, R. Chen, R. Xiao and Y. Pang, Biosens. Bioelectron., 2022, 215, 114548 CrossRef CAS PubMed.
- J. Zhuang, Z. Zhao, K. Lian, L. Yin, J. Wang, S. Man, G. Liu and L. Ma, Biosens. Bioelectron., 2022, 207, 114167 CrossRef CAS PubMed.
- B. Yin, Q. Zhang, X. Xia, C. Li, W. K. H. Ho, J. Yan, Y. Huang, H. Wu, P. Wang, C. Yi, J. Hao, J. Wang, H. Chen, S. H. D. Wong and M. Yang, Theranostics, 2022, 12, 5914–5930 CrossRef CAS PubMed.
- J. Zhang, L. Jiang, H. Li, R. Yuan and X. Yang, Food Chem., 2023, 415, 135768 CrossRef CAS PubMed.
- Y. Du, S. Ji, Q. Dong, J. Wang, D. Han and Z. Gao, Anal. Chim. Acta, 2023, 1245, 340864 CrossRef CAS PubMed.
- J. Liu, J. Chen, D. Wu, M. Huang, J. Chen, R. Pan, Y. Wu and G. Li, Anal. Chem., 2021, 93, 10167–10174 CrossRef CAS PubMed.
- R. Pan, J. Liu, P. Wang, D. Wu, J. Chen, Y. Wu and G. Li, J. Agric. Food Chem., 2022, 70, 4484–4491 CrossRef CAS PubMed.
- A. Su, Y. Liu, X. Cao, J. Zhao, W. Xu, C. Liang, P. Li and S. Xu, Anal. Chem., 2023, 95, 5927–5936 CrossRef CAS PubMed.
- Y. Yin, Q. Li, S. Ma, H. Liu, B. Dong, J. Yang and D. Liu, Anal. Chem., 2017, 89, 1551–1557 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2023 |