
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Laser-induced fabrication of gold nanoparticles onto paper substrates and their application on paper-based electroanalytical devices†
Iana V. S.
Arantes
 a,
Vanessa N.
Ataide
a,
Wilson A.
Ameku
a,
Juliana L. M.
Gongoni
a,
Jéssica S. G.
Selva
a,
Helton P.
Nogueira
ab,
Mauro
Bertotti
a and
Thiago R. L. C.
Paixão
*a
a,
Vanessa N.
Ataide
a,
Wilson A.
Ameku
a,
Juliana L. M.
Gongoni
a,
Jéssica S. G.
Selva
a,
Helton P.
Nogueira
ab,
Mauro
Bertotti
a and
Thiago R. L. C.
Paixão
*a
aDepartment of Fundamental Chemistry, Institute of Chemistry, University of São Paulo, São Paulo, SP 05508-000, Brazil. E-mail: trlcp@iq.usp.br; Tel: +55 11 30919150
bDepartment of Physical Chemistry, Institute of Chemistry, University of Campinas, Campinas, SP 13083-970, Brazil
First published on 25th November 2022
Abstract
A novel and quick (sub-minute) method for synthesizing gold nanoparticles (AuNPs) onto electrochemical paper-based devices (ePADs) using a CO2 laser is presented. An ePAD, fabricated by laser-scribing carbonization of kraft paper, is modified by the in situ synthesis of AuNPs onto a working electrode, driven by the CO2 laser reduction of a precursor solution (HAuCl4). Cyclic voltammograms recorded in H2SO4 and energy dispersive spectroscopy confirm the presence of Au structures embedded in the carbonized paper matrix. A gold modified electrode (LSAu-ePAD) shows an improved electrochemical response towards the ferri/ferrocyanide redox probe, with decreased peak potential separation, a 13-fold increase in peak current, and reduced charge transfer resistance (0.11 kΩ vs. 6.30 kΩ) compared to unmodified electrodes. Scanning electrochemical microscopy reveals an increase in the electrode's electron transfer rate by the Au modification, reflected in the voltammetric profile in bulk measurements. LSAu-ePAD repeatability and reproducibility are 0.4% (n = 10) and 9.7% (n = 5), with a shelf life of up to 30 days. Additionally, a novel field-deployable batch-injection analysis paper-based cell (BIA-ePAD) coupled to the LSAu-ePAD platform is designed for amperometric analyses. The BIA-ePAD is used for hypochlorite (NaClO) detection, reaching low limits of detection (6.70 μmol L−1) and quantification (22.1 μmol L−1) and linear response from 20 to 750 μmol L−1. The variability of the amperometric signals between measurements and between devices is 4.7% (n = 12) and 5.3% (n = 6), respectively, with a sampling frequency estimated as 127 h−1, which is remarkable considering the device's simple and quick fabrication. Analyses of real swimming pool and tap water samples spiked with NaClO showed good recovery values (93 ± 6%). Overall, the developed approach is promising for fast modification of ePADs with AuNPs (and possibly other metals), and coupling them with paper-based BIA cells allows for simple, inexpensive, and high-performance analyses in a fully-integrated portable platform.
1. Introduction
Electrochemical paper-based analytical devices (ePADs) have several advantages over conventional electrochemical cells, such as simple fabrication, portability, low cost of production, and miniaturization.1,2 Several methods for fabricating ePADs have been explored, including screen-printing, inkjet printing, sputtering, pencil-drawing, wire placement, drop-casting of nanomaterials, and laser-scribing, among others.1–8The laser-scribing method allows the local pyrolysis of well-defined areas in the paper substrate, forming conductive patterns, used as electrodes, in a single-step fabrication process. It is environmentally friendly (without the need for reagents), reproducible, and fast, and allows for large-scale fabrication.9,10 Electrochemical sensors fabricated via laser-scribing onto polymeric substrates, such as polyimides, have been explored.11–14 However, direct laser patterning of electrodes on cellulosic materials has attracted little attention owing to the fabrication challenges associated with inconsistent paper composition, structure, and grammage, resulting in varying electrochemical performance among fabricated devices.
Post fabrication surface treatment is a strategy to increase the electrochemical performance of electrodes and can help to increase fabrication reproducibility by pushing the device's performance towards mass transport limited conditions.10,15 Gold nanoparticles (AuNPs) are often used for modifying electrochemical sensors due to their electrocatalytic activity towards organic compounds, large surface area, and long-term stability.16,17 Hybrid materials incorporating nanoparticles in laser-scribed graphitic surfaces have demonstrated synergistic effects with improved electrochemical performance compared to individual materials.16,18–22 Most of these hybrid materials use polyimides as substrates where AuNPs are formed, and no reports were found using paper as a substrate. Here, we describe the fabrication of ePADs with laser-scribed electrodes, modified with AuNPs using paper as a substrate. Electrodes were fabricated in two steps; firstly, a CO2 laser was used for selective pyrolysis of patterns over the paper substrate, forming the electrode geometry. In the following step, AuNPs were rapidly synthesized by irradiating, with the same CO2 laser, the working electrode surface, which was covered with a small volume of HAuCl4 solution. The modified ePAD (LSAu-ePAD) exhibited lower charge transfer resistance and an electrochemical response closer to the reversible case for the ferro/ferricyanide redox probe, compared to an unmodified LS-ePAD.
We used the fabricated LSAu-ePAD to detect sodium hypochlorite (NaClO) in water samples, which is widely used in water treatment facilities. In most cases, NaClO is a precursor of hypochlorous acid (HClO), a disinfectant agent, leaving adequate chlorine residual for safe water distribution.23 For this reason, hypochlorites are added in excess, so the HClO levels are high enough through the water distribution system, ensuring potable water is delivered to the end user.24 However, HClO can be harmful at elevated concentrations,25–27 and monitoring NaClO at the end user is a key to quantifying the exposure levels.
Coupling ePADs to batch-injection analysis (BIA) in the same platform is considered a new and attractive strategy that has gained prominence due to its miniaturized design, reducing waste generation while providing low cost and portability, making it interesting for point-of-need applications.28 Briefly, BIA consists of sample injection from a micropipette tip directly onto the detector surface under controlled dispersion conditions, providing fast, automated, and accurate analyte transport to the electrode's surface.29,30 By coupling our LSAu-ePAD to the BIA method in a fully integrated kraft paper-based cell design, we developed a portable and field-deployable quantification system for NaClO with high sampling frequency, allowing multiple water samples to be tested on the location. The presence of AuNPs shifted the ClO− reduction peak to less cathodic values than unmodified ePADs, reducing the chance of interferences from other compounds in the water samples. Moreover, our method presented a limit of detection (LOD) of 6.70 μmol L−1 (∼0.5 ppm), which meets the WHO-recommended NaClO levels for swimming pool water (3–5 ppm).26,27
Therefore, we expect that the proposed paper-based BIA cell coupled with the LSAu-ePAD opens new possibilities for developing point-of-need ePADs with novel functional designs and distinct ways of electrode modification, inspiring future innovative and accessible sensing platforms.
2. Experimental section
2.1. Chemicals
All reagents used were of analytical grade, and the solutions were prepared using deionized water with a resistivity greater than 18 MΩ cm, obtained from a Millipore Direct-Q® 5 purification system. Potassium ferricyanide (K3[Fe(CN)6]), potassium ferrocyanide trihydrate (K4[Fe(CN)6]·3H2O), potassium chloride (KCl), sodium chloride (NaCl), sodium carbonate (Na2CO3), sodium bicarbonate (NaHCO3), sodium sulfate (Na2SO4), sodium nitrate (NaNO3), and sulfuric acid (H2SO4) 98% were obtained from Merck. NaClO (10–15%) was obtained from Sigma-Aldrich and tetrachloroauric(III) acid trihydrate (HAuCl4·3H2O; 99%) from Acros Organics. Britton–Robinson (BR) buffer solution (0.04 mol L−1) was prepared using phosphoric acid (H3PO4; 85%) from Synth and boric acid (H3BO3; 99.8%) and glacial acetic acid from Merck. The pH was adjusted with sodium hydroxide (NaOH; 99%), also from Merck. The HAuCl4 solution was prepared in 0.5 mol L−1 H2SO4 solution. The NaClO stock solutions and the samples were prepared daily by dilution in deionized water. Pool water samples were obtained from swimming pools in residential condominiums.2.2. LSAu-ePAD fabrication
The fabrication steps of the LSAu-ePAD and its BIA version are shown in Fig. 1. The laser-scribing procedures were performed in a cutting/engraving CO2 laser machine from Work Special Laser, with a 10.6 μm wavelength, 40 W power laser tube, and approximately 14 μs pulse duration. The devices were fabricated from two kraft paper sheets of 300 g m−2 grammage, glued together with a PVA adhesive (Cascola/Cascorez Extra). Once the glue was dry (approx. 5 minutes at room temperature), a glossy waterproof varnish (Colorgin Plastilac) was sprayed on both sides of the glued paper until the entire surfaces were coated and left to dry for 24 hours. The varnish stops the solution absorption by the paper, improving the device's durability and robustness.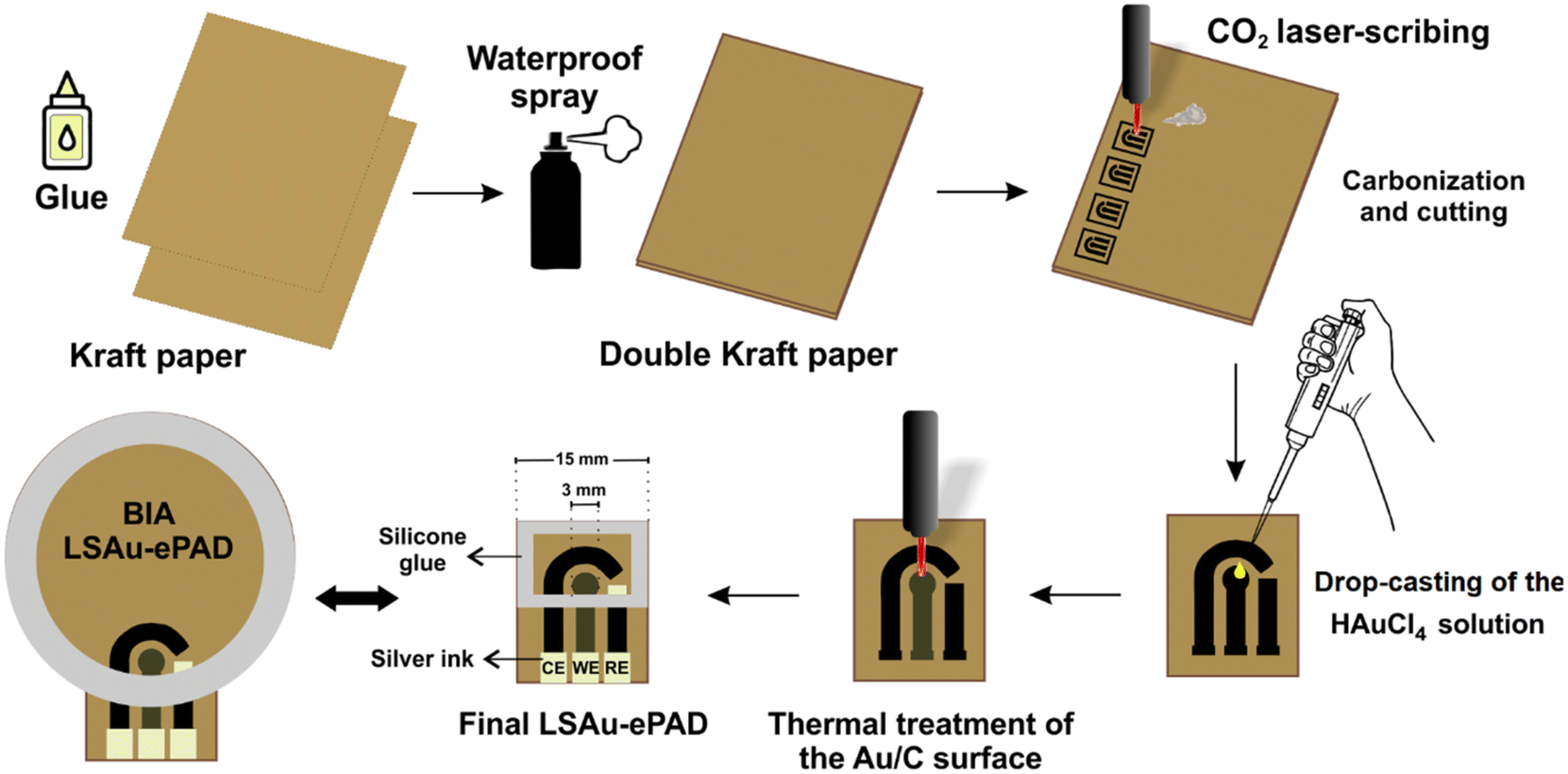 | ||
| Fig. 1 Schematic representation of the LSAu-ePAD fabrication steps. | ||
A three-electrode system consisting of 3 mm diameter disc working (WE), reference (RE), and counter (CE) electrodes was patterned over the varnished paper substrate using the CO2 laser to selectively pyrolyze the paper cellulose into the geometry of the electrodes.9 The sheet resistance of the carbonized material was measured with a digital multimeter (ICEL, model MD-6456) and used to optimize the CO2 laser parameters (vide infra). 30 μL of 10 mmol L−1 HAuCl4 solution was added to the WE surface in three 10 μL aliquots, with the electrode left to dry under an infrared lamp between each addition. The CO2 laser was scanned over the WE to reduce the Au precursor into gold structures. A conductive silver ink (Joint Metal) was used to coat the RE area, and a silicone adhesive was used to build a solution reservoir over the electrodes. The conventional LSAu-ePAD has a 15 by 20 mm rectangular layout with 400 μL solution capacity. The BIA version has a 35 mm diameter circular layout and accommodates up to 3 mL of solution. The cost of each individual BIA-ePAD was estimated at $0.11. A video (Video S1†) showing the carbonization and Au reduction processes is presented in the ESI.†
2.3. Morphological and chemical composition characterization
Scanning electron microscopy (SEM) and energy-dispersive X-ray spectroscopy (EDS) were performed on a JEOL JSM-7401F field emission gun microscope with an acceleration voltage ranging from 0.5 to 1 kV. Raman spectra were recorded on a WITEC 300R confocal microscope with a Nikon Air Lens with a magnification of 10× at an excitation wavelength of 633 nm, 18.9 mW laser power, and an integration time of 1 s.2.4. Electrochemical characterization
The electrochemical performance of the laser-scribed electrodes with (LSAu-ePAD) and without (LS-ePAD) modification was characterized by cyclic voltammetry (CV), electrochemical impedance spectroscopy (EIS), and scanning electrochemical microscopy (SECM). CVs were recorded in a 5 mmol L−1 [Fe(CN)6]3−/4– 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture in 1.0 mol L−1 KCl solution, with the potential scanned from +0.7 to −0.2 V at 20 mV s−1. EIS was carried out using the same solution, applying a sinusoidal perturbation of 10 mV over the open circuit potential (OCP) at frequencies ranging from 105 to 10−1 Hz. The electroactive area of the WE electrodes was calculated from i–t curves using Cottrell's equation.
:1 mixture in 1.0 mol L−1 KCl solution, with the potential scanned from +0.7 to −0.2 V at 20 mV s−1. EIS was carried out using the same solution, applying a sinusoidal perturbation of 10 mV over the open circuit potential (OCP) at frequencies ranging from 105 to 10−1 Hz. The electroactive area of the WE electrodes was calculated from i–t curves using Cottrell's equation.
SECM experiments were performed on a Sensolytics (Sensolytics, Bochum, Germany) SECM workstation. A two-electrode system was used with Ag/AgCl(KCl sat.) and a 10 μm radius platinum microelectrode (UME) as the RE and WE (SECM tip), respectively. Detailed information about the fabrication and characterization of the Pt UME is given in the ESI† (Fig. S1). The electrochemical activity of the LS-ePAD and LSAu-ePAD surfaces was evaluated by recording approach curves31 in 10 mmol L−1 [Fe(CN)6]3− in 0.1 mol L−1 KCl solution, with the tip polarized at −200 mV while the substrate (ePADs) was kept unbiased. Each substrate was probed in five distinct locations (Fig. S2†). CVs at the Pt UME were recorded at the same locations with a tip/substrate separation of 15 μm. The voltammograms were recorded with the substrates unbiased and biased at −200 mV.
2.5. Electroanalytical detection of hypochlorite (BIA-ePAD)
Amperometric detection of NaClO was performed with the LSAu-ePAD assembled in the BIA configuration. The device was placed over a magnetic stirrer and the cell was initially filled with 2 mL of supporting electrolyte (0.04 mol L−1 BR buffer; pH 8). Injections were performed using a Multipette stream electronic micropipette (Eppendorf), in a “wall-jet” configuration,32 with the pipette end held approximately 2 mm away from the WE surface. An image of the BIA-ePAD operation is shown in Fig. S3.† The WE potential was fixed at −0.2 V vs. Ag for reducing ClO− and injections were only performed after baseline stabilization (∼120 seconds). The injected volume was 20 μL at a rate of 135 μL s−1.3. Results and discussion
3.1. LSAu-ePAD optimization
As we have shown for other substrates,9,10 the pyrolyzed material sheet resistance can be used as a proxy for the electrode's electrochemical performance, providing a quick way to optimize the laser fabrication parameters. Table S1† shows the range of parameters evaluated. The lowest sheet resistance was obtained for 9% (∼2.7 W) laser power, 10 mm s−1 scan rate, and a laser/substrate separation (Z-distance) of 8 mm. Despite presenting the lowest resistance (170 ohms), the LS-ePAD electrochemical performance is poor, evidenced by the voltammetric profile of the redox couple ferri/ferrocyanide, displaying a large capacitive current and barely discerning faradaic processes, as exhibited in section 3.3. This is similar to what is seen for laser-scribed electrodes on cardboard surfaces,9,33 where further surface treatment/modification procedures are used to increase the electrochemical performance of the devices. Here, we focused on modifying the WE surface by reducing AuNPs using the CO2 laser.34 As the WE is exposed to the laser twice, the laser power in the first pyrolysis step was reduced to 8% (∼1.7 W), as electrodes fabricated at 9%, the optimal condition, couldn't bare a second laser exposure without sustaining physical damage.The modification step was performed by drop-casting HAuCl4 solution onto the WE, which is reduced to metallic Au by the CO2 laser, creating a hybrid Au/Carbon electrode material. For optimizing this step, the voltammetric profile of 5 mmol L−1 [Fe(CN)6]3−/4− solution was recorded for LSAu-ePADs fabricated with different laser parameters (Fig. S4†) and volumes and concentrations of the HAuCl4 solution (Fig. S5†). The anodic peak current (Ipa) and peak-to-peak separation (ΔEp) were used to assess the reversibility of the electrochemical response. Higher Ipa values were obtained for increasing laser power. Slower scan rates also favor higher Ipa values and lower ΔEp. The laser Z-distance seems to have little impact on the Ipa. However, it does affect ΔEp, with the smallest value obtained at a working distance of 11 mm. Therefore, we selected the following laser parameters, which produced electrodes with the combined largest Ipa (70 μA) and lowest ΔEp (110 mV): a laser power of 8%, scan rate of 6 mm s−1, and Z-distance of 11 mm.
At these laser parameters, the HAuCl4 solution volume and concentration were optimized also using Ipa and ΔEp values of CVs recorded in 5 mmol L−1 [Fe(CN)6]3−/4− solution. Increasing the Au content, either by increasing the volume or concentration, leads to better electrochemical performance. As HAuCl4 is an expensive reagent, we used 30 μL of a 10 mmol L−1 solution.
3.2. Morphological characterization
The ePADs were characterized at each fabrication step by Raman spectroscopy, SEM, and EDS analysis. Fig. S6-A† shows the Raman spectrum of bare kraft paper, where a large fluorescence background is seen. After the first laser fabrication step, D, G, and 2D bands appeared at 1323, 1590, and 2643 cm−1, respectively (Fig. S6-B†), indicating the formation of the carbon material.10 These bands were also observed at 1327, 1578, and 2647 cm−1, respectively, after the Au modification step (Fig. S6-C†). The relationship between the D and G bands (ID/IG ratio) was calculated to compare the effect of the second laser treatment on structural defects in the carbon matrix. The ID/IG ratio values for the LS-ePAD and LSAu-ePAD were 1.4 and 2.0, respectively, indicating that the second laser irradiation introduces more structural defects on the carbon surface.15,35The SEM image in Fig. 2-A shows the bare kraft paper surface, which was previously coated with a thin layer of sputtering gold deposition due to the insulating characteristics of this material, exhibiting a highly compact and homogeneous structure, very different from the porous surface after the first fabrication step, laser-induced pyrolysis (not coated) (Fig. 2-B). The SEM images of the electrode surface after the Au modification step, also not coated, (Fig. 2-C and D) reveal the formation of distinct, highly contrasting, nanometer-sized structures, which we assign as AuNPs. This is further supported by EDS analyses of the LSAu-ePAD (Fig. S7†), which shows a pronounced Au signal compared to the EDS spectrum of LS-ePAD. EDS elemental mapping was also performed to better show the distribution of the AuNPs synthesized over the carbon surface (Fig. S8†), offering good coverage of the entire working electrode area.
 | ||
| Fig. 2 SEM images of the (A) bare kraft paper, (B) LS-ePAD and (C and D) LSAu-ePAD in different magnifications: 250× (both A and B), 500× (C) and 10000× (D). | ||
3.3. Electrochemical characterization
The presence of Au structures in the LSAu-ePAD is confirmed by CVs recorded in 0.5 mol L−1 H2SO4 solution (Fig. 3), which show a distinct Au voltammetric profile with two oxidation peaks at +0.95 and +1.1 V, and a reduction peak at +0.7 V (red line).36,37 As expected, none of these processes are seen for the LS-ePAD (black line, inset). The current density for the Au processes obtained for the LSAu-ePAD (calculated with the geometric area) was 10-fold than for a commercial Au-SPE (blue line) (0.125 cm2), demonstrating that the ePAD has a larger amount of Au, owing to the porous structure and larger surface area (Fig. 2).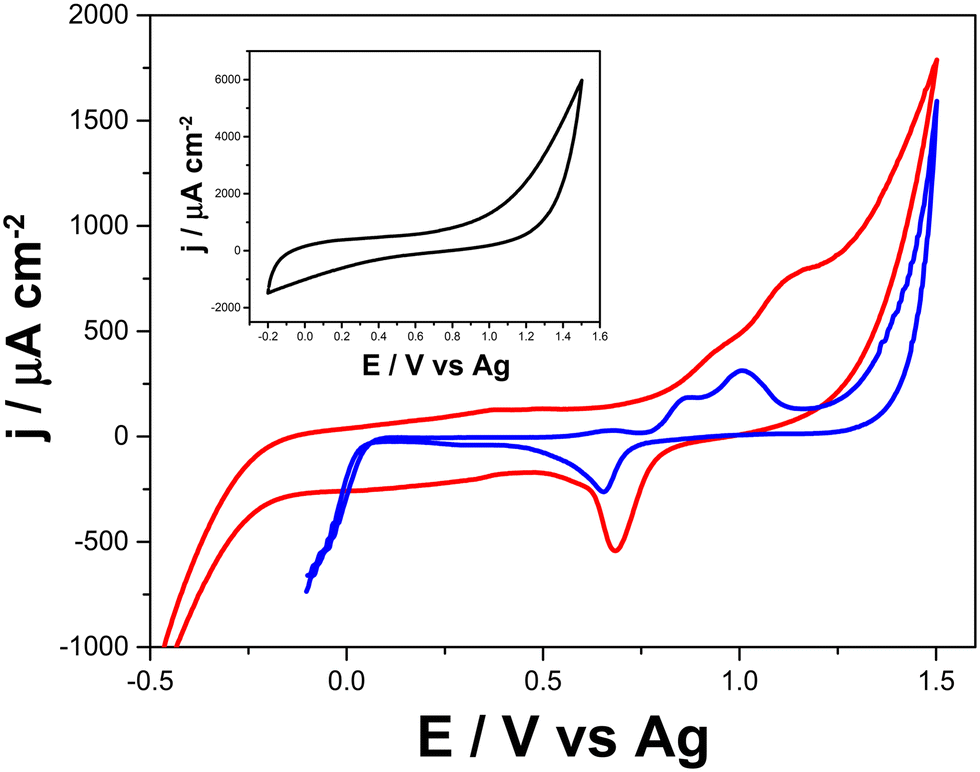 | ||
| Fig. 3 Cyclic voltammograms recorded in 0.5 mol L−1 H2SO4 with the LSAu-ePAD (red line), a commercial Au-SPE (blue line), and the LS-ePAD (black line, inset). Scan rate: 50 mV s−1. | ||
Comparing the voltammetric profile of [Fe(CN)6]3−/4− at the LSAu-ePAD (red line) with that at the LS-ePAD (black line), presented in Fig. 4-A, a 13-fold increase in cathodic and anodic peak currents and a decrease in ΔEp, from 188 to 127 mV, are seen for the LSAu-ePAD. The electroactive area for the Au modified electrode (0.052 ± 0.003 cm2), approximated by Cottrell's equation from i–t curves, is closer to the geometric area (0.070 cm2), as expected for this system,38 reflecting the quasi-reversible voltammetric behavior. The charge transfer resistance (RCT) for both the LSAu-ePAD and LS-ePAD was calculated by EIS. From the Nyquist plots in Fig. 4-B, the RCT values of 6.30 ± 3.70 kΩ for the LS-ePAD (black data) and 0.11 ± 0.04 kΩ for the LSAu-ePAD (red data) are derived, highlighting the effect of the Au structures on the electrode's electrochemical performance. This is further supported by the CVs recorded at double carbonized LS-ePADs, which were exposed to the second laser treatment step without HAuCl4 solution (Fig. S9†). Although a large peak current increase is seen, the voltammetric profile is very capacitive and poorly defined. We attribute this to an increase in the surface porosity, similar to the effect seen in Fig. 2-B, as the HAuCl4 does not absorb the laser energy to form AuNPs, further carbonizing the paper substrate.
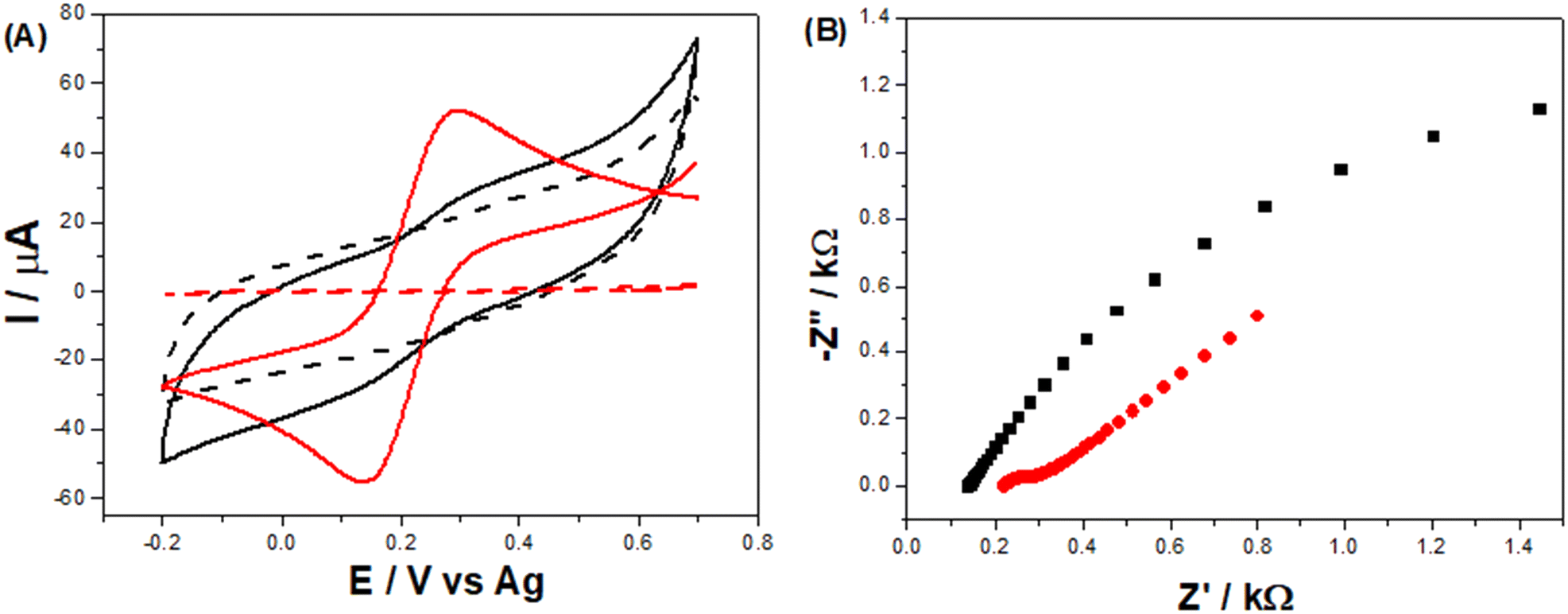 | ||
| Fig. 4 (A) Cyclic voltammograms recorded at a 20 mV s−1 scan rate and (B) Nyquist plots obtained from EIS experiments, performed in a mixture containing 5 mmol L−1 [Fe(CN)6]3−/4− in 1 mol L−1 KCl for the unmodified LS-ePAD (black data) and the LSAu-ePAD (red data). | ||
Repeatability and reproducibility studies were carried out by recording [Fe(CN)6]3−/4− CVs with the same LSAu-ePAD (Fig. S10-A†) and with five different LSAu-ePADs (Fig. S10-B†). Repeatability was found to be 0.4% (n = 10), and reproducibility was 9.7% (n = 5) RSD. The stability of the LSAu-ePADs was evaluated over 30 days by recording the CVs with six devices, fabricated on the same day over multiple days. Although marginal variations in the voltammetric profile are seen (Fig. S10-C†), there is no clear trend showing electrode degradation over time.
Normalized approach curves, where the UME current is normalized by the current recorded away from the substrate (i/iBulk) and the UME/substrate distance is normalized by the UME radius (d/a), recorded over the LS-ePAD (Fig. 5-A) show a decrease in the UME current with a decrease in the separation; however, these are much larger currents than expected for a completely insulating substrate (red dot curve).41 These profiles are characteristic of substrates with slow electron transfer kinetics, indicating that the LS-ePAD is oxidizing ferrocyanide back to ferricyanide at a low rate.40 On the other hand, the normalized approach curves over the LSAu-ePAD (Fig. 5-B) show an increase in the current with a decreased separation, indicating that the oxidation of ferrocyanide at the substrate occurs at a higher rate than what is seen for the LS-ePAD, i.e., the electron transfer kinetics is higher at the Au modified ePAD. Although the same current trend is observed, the approach curve profiles are different at each spot for both substrates, pointing to a heterogeneous electrode surface.40
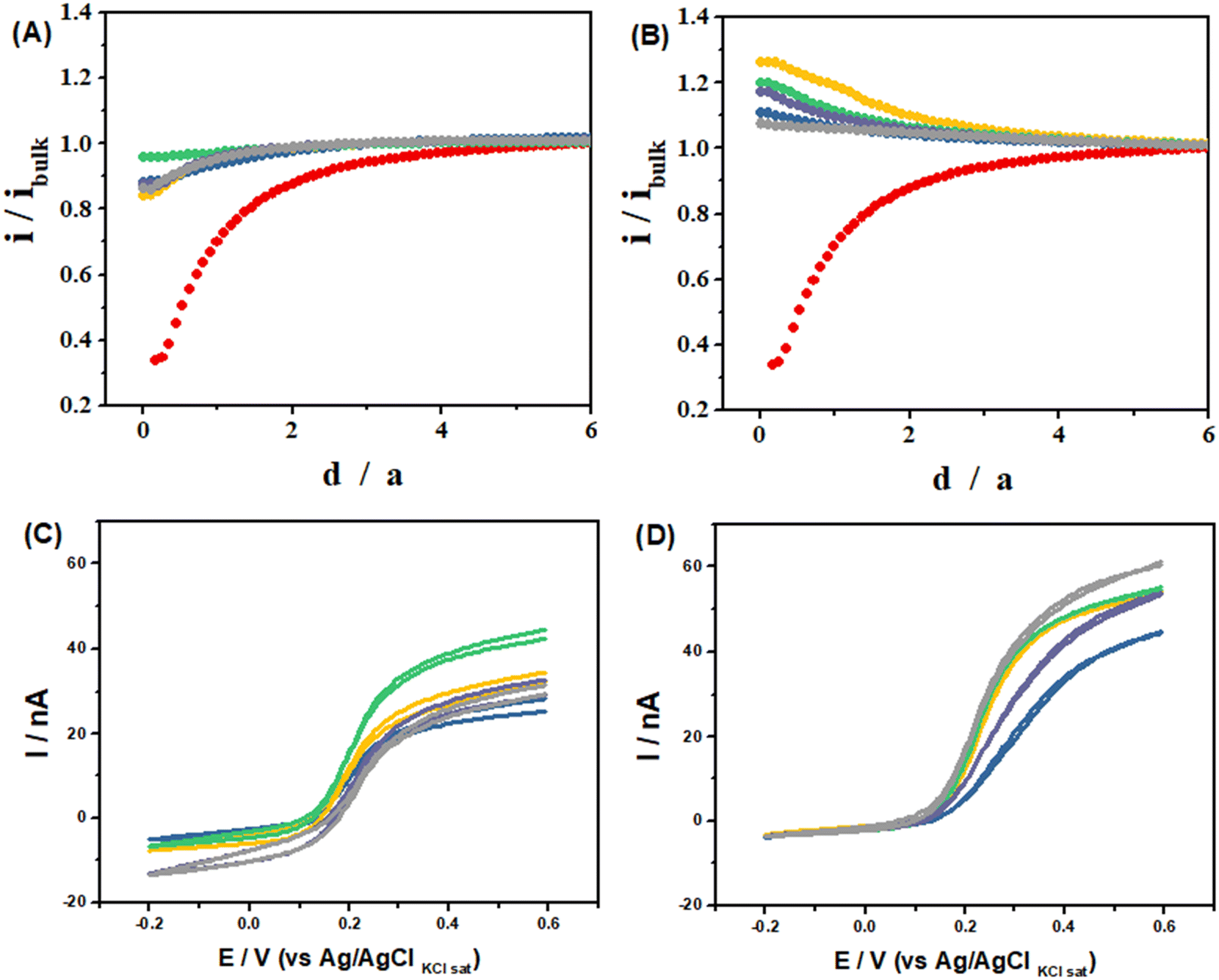 | ||
Fig. 5 (A and B): Approach curves recorded in 10 mmol L−1 [Fe(CN)6]3− in 0.1 mol L−1 KCl solution using the Pt UME as a tip (E = −200 mV) using (A) the LS-ePAD and (B) LSAu-ePAD as unbiased substrates. (C and D): CVs recorded in 10 mmol L−1 [Fe(CN)6]3− in 0.1 mol L−1 KCl solution with the Pt UME at 15 μm from the substrate after the substrate polarization (E = −200 mV) using (C) the LS-ePAD and (D) LSAu-ePAD. Each curve was recorded at a specific location on the substrate (Fig. S2†): 1( ); 2( ); 2( ); 3( ); 3( ); 4( ); 4( ); 5( ); 5( ). Curve ( ). Curve ( ) was recorded on an insulator substrate (Fig. S1-B†) using the same tip and conditions. ) was recorded on an insulator substrate (Fig. S1-B†) using the same tip and conditions. | ||
The electron transfer kinetics difference between the LSAu-ePAD and LS-ePAD is also evident when SECM is used to probe the diffusion layer composition of these substrates biased at −200 mV (ferricyanide reduction). The UME was positioned at a distance of 15 μm from the substrates, at the same 5 locations discussed above, and the CVs were recorded (Fig. 5-C and D). At this configuration, when the UME is at a cathodic potential, both the substrate and the UME will be competing for the same redox species, ferricyanide.42 The UME has a larger cathodic current over the LS-ePAD (Fig. 5-C) than when it is over the LSAu-ePAD, which is almost zero (Fig. 5-D). The UME current is proportional to the local ferricyanide concentration, and after normalizing the CV currents at each location by the negative feedback current at the same UME/substrate separation (15 μm, equivalent to a d/a of 1.5 in the red curves in Fig. 5-A and B), we compute how much ferricyanide has been consumed at this height (summarized in Table S2†), showing that the LSAu-ePAD consumed much more ferricyanide than the LS-ePAD, reflecting its faster kinetics.
3.4. Hypochlorite detection
Monitoring NaClO in water samples can be greatly beneficial to avoid overexposure to it, which can be harmful.25 Thus, the developed LSAu-ePAD was employed to quantify hypochlorite. As its content needs to be monitored in treated water supplies, quick and affordable alternatives for its quantification is necessary, as provided by our device. ClO− reduction is pH dependent,43 as seen in the voltammetric profiles of 5 mmol L−1 NaClO in 0.04 mol L−1 BR buffer in Fig. 6-A. The largest cathodic current is at pH 8, which was selected for all further measurements. In an alkaline medium, the ClO− reduces to Cl− and generates OH− intermediated by consuming H2O and two electrons for each mol of ClO− consumed.44,45 Additionally, in an alkaline solution, the gold surface is unlikely to dissolute in the presence of ClO−. Thus, it still stays preserved. On the other hand, in an acidic medium, gold goes into the solution, compromising the electrode surface.44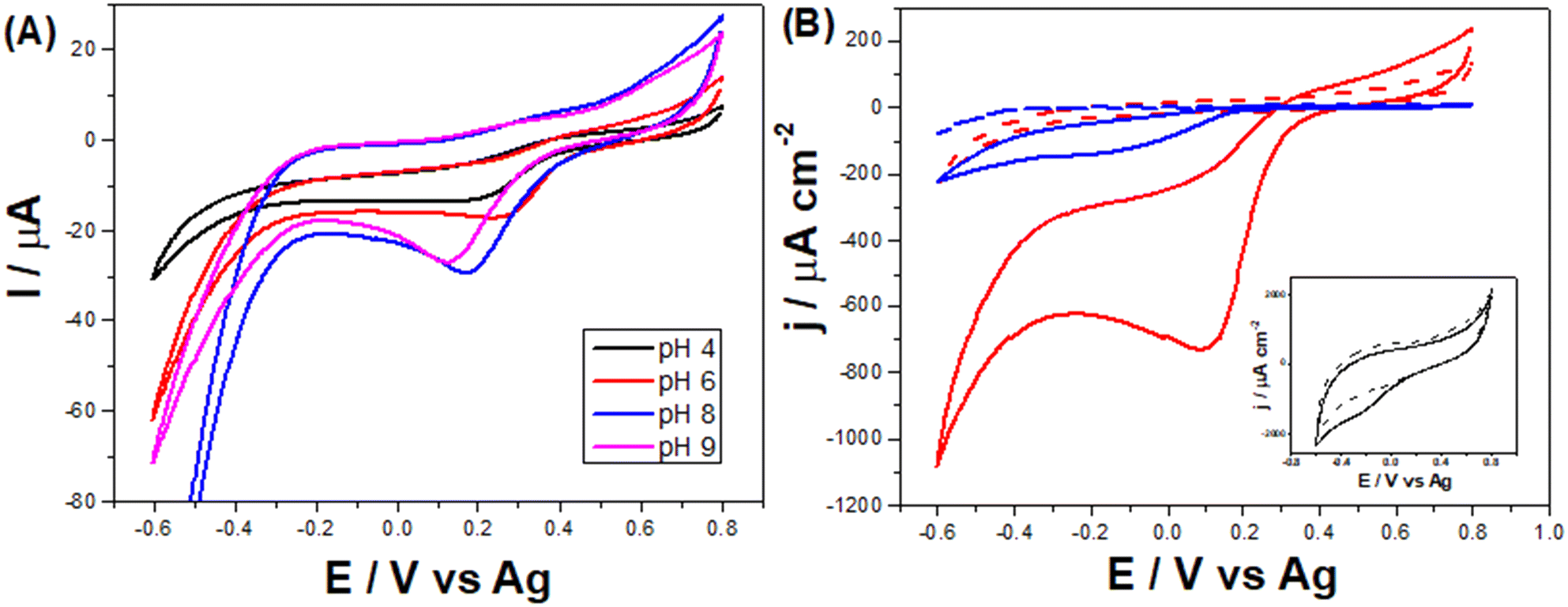 | ||
| Fig. 6 (A) Cyclic voltammograms recorded with the LSAu-ePAD in 5 mmol L−1 NaClO in 0.04 mol L−1 BR buffer at different pH values. (B) Cyclic voltammograms recorded in 5 mmol L−1 NaClO in 0.04 mol L−1 BR buffer (pH 8) with the LSAu-ePAD (red line), a commercial Au-SPE (blue line), and the LS-ePAD (black line, inset). Scan rate: 50 mV s−1. | ||
ClO− reduction seems to be catalyzed by gold nanostructures, facilitating charge transfer,43,46,47 as can be seen by comparing the voltammetric profiles of both LS-ePAD and LSAu-ePAD in NaClO solution (Fig. 6-B). In a possible mechanism, ClO− adsorbs first on the gold structure, which is eased when the structure is poorly or not negatively charged.44,47 Not only does the profile with the LS-ePAD (black line, inset) show a high capacitance, but the ClO− reduction process appears (poorly defined) approx. 300 mV more cathodic than what is seen for the LSAu-ePAD (red line). The larger amount of Au in the LSAu-ePAD, compared to a commercial Au-SPE, affords an analytical advantage to the ePAD, as the reduction wave of ClO− in the Au-SPE also happens at more cathodic potentials (blue line). The CV obtained for the twice carbonized ePAD (without Au) is very similar to that for the LS-ePAD (Fig. S11†), further supporting that Au catalyzes the ClO− reduction process.
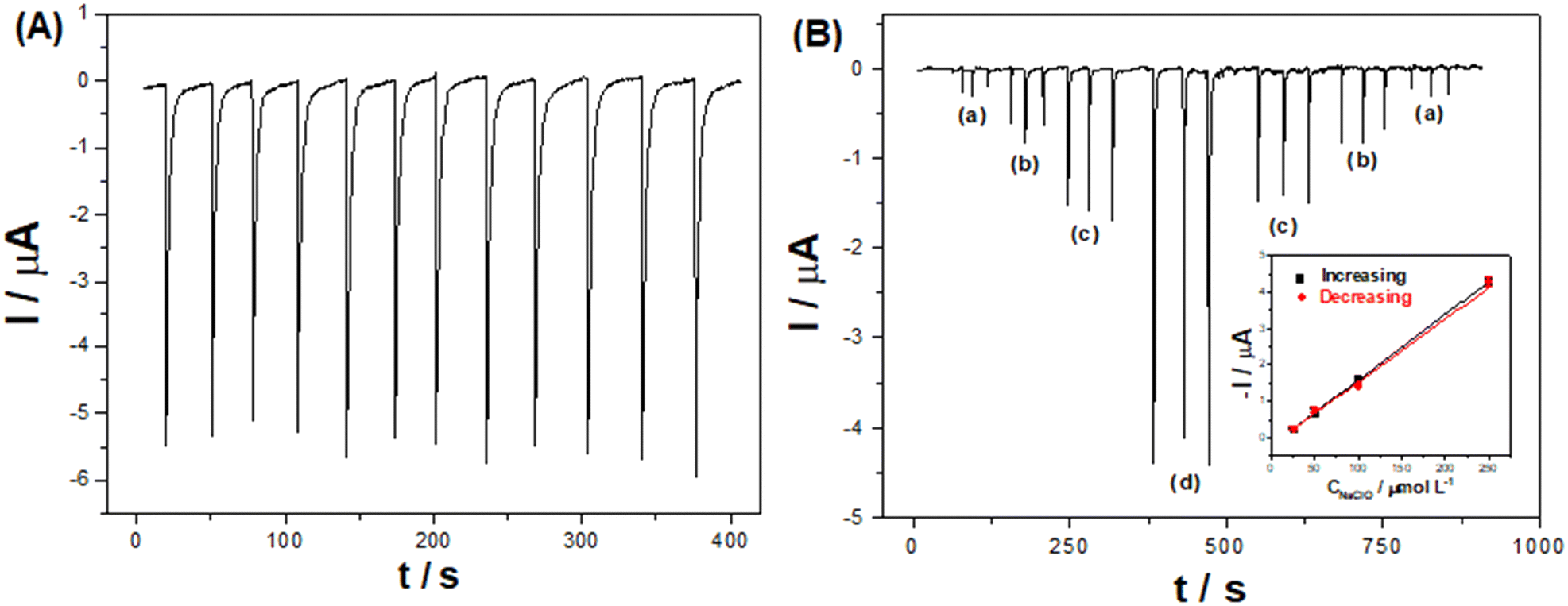 | ||
| Fig. 7 (A) Repeatability measurements of successive injections of 0.5 mmol L−1 NaClO (n = 12) in the BIA-ePAD. (B) Amperometric responses obtained from triplicate injections of NaClO in the BIA-ePAD for a memory effect study with increasing and decreasing concentrations of 25, 50, 100, and 250 μmol L−1 (a–d); and the respective calibration curves of the peak currents vs. NaClO concentration (inset). Applied potential: −0.2 V vs. Ag; injected volume: 20 μL; dispensing rate: 135 μL s−1; supporting electrolyte: 0.04 mol L−1 BR buffer (pH 8). | ||
Injections of NaClO at various concentrations (10 to 1000 μmol L−1) resulted in a linear current response (R = 0.997) between 20 and 750 μmol L−1 (Fig. S14†). The LOD and LOQ were estimated as 6.70 (∼0.5 ppm) and 22.1 μmol L−1 (1.7 ppm), calculated as 3 and 10 times the standard deviation of blank measurements, respectively. Injecting increasing and then decreasing NaClO concentrations (Fig. 7-B) indicates that there is no memory effect in the electrode, demonstrated by similar slopes of the calibration curves (inset) for increasing (0.0179 μA μmol−1 L) and decreasing (0.0173 μA μmol−1 L) concentrations. Although other electrochemical methods have reported better LODs (see Table S3†), our method can analyze real samples, for example, swimming pool water, which is recommended to have 3–5 ppm of free chlorine.26,27
As a proof-of-concept application, three swimming pool and one tap water samples were spiked with 100 μmol L−1 NaClO and analyzed with the BIA-ePAD. Table 1 shows the recovery percentages obtained for each sample, ranging from 85 to 100%. Our method is also not affected by the presence of NO3−, SO42−, CO32−, HCO3−, and Cl−, commonly found in swimming pool water,23 as revealed by the interference study (Fig. S15†) performed at a 5:1 ratio with ClO−. The results showed no appreciable differences in the ClO− current before (0.84 ± 0.02 μA) and after (0.79 ± 0.04 μA) the injection of the interferents.
| Sample | C NaClO added/μmol L−1 | Recovery/% |
|---|---|---|
| Swimming pool water 1 | 100 | 85 ± 9 |
| Swimming pool water 2 | 93 ± 1 | |
| Swimming pool water 3 | 100 ± 5 | |
| Tap water | 92 ± 2 |
4. Conclusions
Herein, we demonstrated the fabrication of Au-modified paper-based electrodes by a simple and quick two-step fabrication process. Kraft paper was used as a cellulosic substrate, where a three-electrode system was patterned using a CO2 laser to pyrolyze the carbon substrate selectively. AuNPs were rapidly synthesized onto the WE surface through the thermal reduction of HAuCl4. The presence of AuNPs in the porous carbon structure, confirmed by CVs recorded in H2SO4 solution and EDS analyses, significantly improved the electrode's electrochemical performance, leading to higher electron transfer kinetics and reduced charge transfer resistance as demonstrated by SECM and EIS. The repeatability, reproducibility, and stability studies for the electrode showed RSD values of 0.4% (n = 10), 9.7% (n = 5), and 9.7% (30 days), respectively. The incorporation of AuNPs on the electrode resulted in a catalytic effect on ClO− reduction, which we explored for high throughput analyses by integrating the electrodes into a BIA system. At the cost of $ 0.11 per device, we were able to analyze ClO− in swimming pool and tap water samples at a throughput of 127 h−1, with LOD and LOQ values of 6.70 and 22.1 μmol L−1, respectively, and recovery percentages varying from 85 to 100% for water samples.Author contributions
Iana V. S. Arantes: methodology, formal analysis, investigation, and writing – original draft. Vanessa N. Ataide: conceptualization, methodology, formal analysis, investigation, and writing – original draft. Wilson A. Ameku: conceptualization, investigation, and writing – review and editing. Juliana L. M. Gongoni: methodology, formal analysis, and writing – review and editing. Jéssica S. G. Selva: methodology, formal analysis, and writing – review and editing. Helton P. Nogueira: methodology, formal analysis, and writing – review and editing. Mauro Bertotti: supervision and writing – review and editing. Thiago R. L. C. Paixão: supervision, funding acquisition, and writing – review and editing.Conflicts of interest
The authors declare no competing interest.Acknowledgements
This research was supported by the São Paulo Research Foundation (FAPESP) (Grant numbers: 2017/10522-5, 2018/08782-1, 2018/14462-0, 2019/16491-0, and 2019/15065-7), the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES) (Grant numbers: PROEX 88887.347030/2019-00 and Pró-Forenses 3359/2014 Edital 25/2014), and the Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq) (Grant Numbers: 405620/2021-7, 141866/2016-0, 141854/2019-7, 302839/2020-8, and 465389/2014-7 – INCTBio). The authors are grateful to Central Analítica (IQ/USP) for the SEM infrastructure. We would also like to thank Dr. Gabriel N. Meloni for the graphical abstract illustration and for helping with the final article correction, scientific inputs and comments.References
- J. Mettakoonpitak, K. Boehle, S. Nantaphol, P. Teengam, J. A. Adkins, M. Srisa-Art and C. S. Henry, Electrochemistry on Paper-based Analytical Devices: A Review, Electroanalysis, 2016, 28, 1420–1436, DOI:10.1002/elan.201501143.
- V. N. Ataide, L. F. Mendes, L. I. L. M. Gama, W. R. De Araujo and T. R. L. C. Paixão, Electrochemical paper-based analytical devices: Ten years of development, Anal. Methods, 2020, 12, 1030–1054, 10.1039/c9ay02350j.
- E. Noviana, T. Ozer, C. S. Carrell, J. S. Link, C. McMahon, I. Jang and C. S. Henry, Microfluidic Paper-Based Analytical Devices: From Design to Applications, Chem. Rev., 2021, 121, 11835–11885, DOI:10.1021/acs.chemrev.0c01335.
- W. A. Ameku, W. R. de Araujo, C. J. Rangel, R. A. Ando and T. R. L. C. Paixão, Gold Nanoparticle Paper-Based Dual-Detection Device for Forensics Applications, ACS Appl. Nano Mater., 2019, 17, 24, DOI:10.1021/acsanm.9b01057.
- W. A. Ameku, J. M. Gonçalves, V. N. Ataide, M. S. F. Santos, I. G. R. Gutz, K. Araki and T. R. L. C. Paixão, Combined Colorimetric and Electrochemical Measurement Paper-Based Device for Chemometric Proof-of-Concept Analysis of Cocaine Samples, ACS Omega, 2021, 6, 594–605, DOI:10.1021/acsomega.0c05077.
- I. V. S. Arantes, J. L. M. Gongoni, L. F. Mendes, V. N. de Ataide, W. A. Ameku, P. T. Garcia, W. R. de Araujo and T. R. L. C. Paixão, Electrochemical paper-based analytical devices, in Pap. Anal. Devices Chem. Anal. Diagnostics, 2022, pp. 81–116, DOI:10.1016/b978-0-12-820534-1.00011-6.
- X. Liu, X. Gao, L. Yang, Y. Zhao and F. Li, Metal-Organic Framework-Functionalized Paper-Based Electrochemical Biosensor for Ultrasensitive Exosome Assay, Anal. Chem., 2021, 93, 11792–11799, DOI:10.1021/acs.analchem.1c02286.
- X. Liu, X. Li, X. Gao, L. Ge, X. Sun and F. Li, A Universal Paper-Based Electrochemical Sensor for Zero-Background Assay of Diverse Biomarkers, ACS Appl. Mater. Interfaces, 2019, 11, 15381–15388, DOI:10.1021/acsami.9b03860.
- W. R. De Araujo, C. M. R. R. Frasson, W. A. Ameku, J. R. J. R. Silva, L. Angnes and T. R. L. C. Paixão, Single-Step Reagentless Laser Scribing Fabrication of Electrochemical Paper-Based Analytical Devices, Angew. Chem., Int. Ed., 2017, 56, 15113–15117, DOI:10.1002/anie.201708527.
- L. F. Mendes, A. de Siervo, W. R. De Araujo and T. R. L. C. Paixão, Reagentless fabrication of a porous graphene-like electrochemical device from phenolic paper using laser-scribing, Carbon, 2020, 159, 110–118, DOI:10.1016/j.carbon.2019.12.016.
- P. Nayak, N. Kurra, C. Xia and H. N. Alshareef, Highly Efficient Laser Scribed Graphene Electrodes for On-Chip Electrochemical Sensing Applications, Adv. Electron. Mater., 2016, 2, 1600185, DOI:10.1002/AELM.201600185.
- R. R. A. Soares, R. G. Hjort, C. C. Pola, K. Parate, E. L. Reis, N. F. F. Soares, E. S. Mclamore, J. C. Claussen and C. L. Gomes, Laser-Induced Graphene Electrochemical Immunosensors for Rapid and Label-Free Monitoring of Salmonella enterica in Chicken Broth, ACS Sens., 2020, 5, 1900–1911, DOI:10.1021/acssensors.9b02345.
- W. R. P. Costa, R. G. Rocha, L. V. De Faria, T. A. Matias, D. L. O. Ramos, A. G. C. Dias, G. L. Fernandes, E. M. Richter and R. A. A. Muñoz, Affordable equipment to fabricate laser-induced graphene electrodes for portable electrochemical sensing, Microchim. Acta, 2022, 189, 1–9, DOI:10.1007/s00604-022-05294-6.
- Y. Zhao, J. Xiang, H. Cheng, X. Liu and F. Li, Flexible photoelectrochemical biosensor for ultrasensitive microRNA detection based on concatenated multiplex signal amplification, Biosens. Bioelectron., 2021, 194, 113581, DOI:10.1016/j.bios.2021.113581.
- V. N. Ataide, W. A. Ameku, R. P. Bacil, L. Angnes, W. R. De Araujo and T. R. L. C. Paixão, Enhanced performance of pencil-drawn paper-based electrodes by laser-scribing treatment†, RSC Adv., 2021, 11, 1644–1653, 10.1039/d0ra08874a.
- K. Samoson, A. Soleh, K. Saisahas, K. Promsuwan, J. Saichanapan, P. Kanatharana, P. Thavarungkul, K. H. Chang, A. F. Lim Abdullah, K. Tayayuth and W. Limbut, Facile fabrication of a flexible laser induced gold nanoparticle/chitosan/ porous graphene electrode for uric acid detection, Talanta, 2022, 243, 123319, DOI:10.1016/j.talanta.2022.123319.
- M. Mazloum-Ardakani, A. Dehghani-Firouzabadi, M. A. Sheikh-Mohseni, A. Benvidi, B. B. F. Mirjalili and R. Zare, A self-assembled monolayer on gold nanoparticles modified electrode for simultaneous determination of isoproterenol and uric acid, Meas.: J. Int. Meas. Confed., 2015, 62, 88–96, DOI:10.1016/J.MEASUREMENT.2014.11.020.
- J. Zhang, M. Ren, Y. Li and J. M. Tour, In Situ Synthesis of Efficient Water Oxidation Catalysts in Laser-Induced Graphene, ACS Energy Lett., 2018, 3, 677–683, DOI:10.1021/acsenergylett.8b00042.
- Z. You, Q. Qiu, H. Chen, Y. Feng, X. Wang, Y. Wang and Y. Ying, Laser-induced noble metal nanoparticle-graphene composites enabled flexible biosensor for pathogen detection, Biosens. Bioelectron., 2020, 150, 111896, DOI:10.1016/J.BIOS.2019.111896.
- H. Chen, W. Yang, P. Huang, C. Li, Y. Yang, B. Zheng, C. Zhang, R. Liu, Y. Li, Y. Xu, J. Wang and Z. Li, A multiple laser-induced hybrid electrode for flexible triboelectric nanogenerators, Sustain, Energy Fuels, 2021, 5, 3737–3743, 10.1039/d1se00819f.
- S. Rauf, A. A. Lahcen, A. Aljedaibi, T. Beduk, J. I. O. de Filho and K. N. Salama, Gold nanostructured laser-scribed graphene: A new electrochemical biosensing platform for potential point-of-care testing of disease biomarkers, Biosens. Bioelectron., 2021, 180, 956–5663, DOI:10.1016/j.bios.2021.113116.
- L. Ge, Q. Hong, H. Li, C. Liu and F. Li, Direct-Laser-Writing of Metal Sulfide-Graphene Nanocomposite Photoelectrode toward Sensitive Photoelectrochemical Sensing, Adv. Funct. Mater., 2019, 29, 1904000, DOI:10.1002/adfm.201904000.
- M. R. Tomei, F. Arduini, D. Neagu and D. Moscone, Carbon black-based disposable sensor for an on-site detection of free chlorine in swimming pool water, Talanta, 2018, 189, 262–267, DOI:10.1016/j.talanta.2018.07.005.
- S. T. Meyer, O uso de cloro na desinfecção de águas, a formação de trihalometanos e os riscos potenciais à saúde pública, Cad Saude Publica, 1994, 10, 99–110, DOI:10.1590/s0102-311x1994000100011.
- M. Jović, F. Cortés-Salazar, A. Lesch, V. Amstutz, H. Bi and H. H. Girault, Electrochemical detection of free chlorine at inkjet printed silver electrodes, J. Electroanal. Chem., 2015, 756, 171–178, DOI:10.1016/j.jelechem.2015.08.024.
- World Health Organization, Guidelines for safe recreational water environments: Volume 2. Swimming pools and similar environments, World Heal. Organ., 2006, vol. 2, p. 146 Search PubMed.
- World Health Organization, Guidelines for drinking-water quality, World Heal. Organ., 4th edn, 2011, DOI:10.1007/978-1-4020-4410-6_184.
- I. V. S. Arantes and T. R. L. C. Paixão, Couple batch-injection analysis and microfluidic paper-based analytical device: A simple and disposable alternative to conventional BIA apparatus, Talanta, 2021, 240, 123201, DOI:10.1016/j.talanta.2021.123201.
- M. S. M. Quintino and L. Angnes, Batch injection analysis: An almost unexplored powerful tool, Electroanalysis, 2004, 16, 513–523, DOI:10.1002/elan.200302878.
- D. P. Rocha, R. M. Cardoso, T. F. Tormin, W. R. De Araujo, R. A. A. Muñoz, E. M. Richter and L. Angnes, Batch-injection Analysis Better than ever: New Materials for Improved Electrochemical Detection and On-site Applications, Electroanalysis, 2018, 30, 1386–1399, DOI:10.1002/elan.201800042.
- J. Kwak and A. J. Bard, Scanning electrochemical microscopy. Theory of the feedback mode, Anal. Chem., 1989, 61, 1221–1227, DOI:10.1021/ac00186a009.
- J. Wang and Z. Taha, Batch Injection Analysis, Anal. Chem., 1991, 63, 1053–1056, DOI:10.1021/ac00010a025.
- A. Bezerra Martins, A. Lobato, N. Tasić, F. J. Perez-Sanz, P. Vidinha, T. R. L. C. Paixão and L. M. Gonçalves, Laser-pyrolyzed electrochemical paper-based analytical sensor for sulphite analysis, Electrochem. Commun., 2019, 107, 106541, DOI:10.1016/j.elecom.2019.106541.
- H. Zeng, X. W. Du, S. C. Singh, S. A. Kulinich, S. Yang, J. He and W. Cai, Nanomaterials via laser ablation/irradiation in liquid: A review, Adv. Funct. Mater., 2012, 22, 1333–1353, DOI:10.1002/ADFM.201102295.
- E. Vaghri and D. Dorranian, Effects of green laser fluence on the characteristics of graphene nanosheets synthesized by laser ablation method in liquid nitrogen medium, Opt. Quantum Electron., 2018, 50, 1–11, DOI:10.1007/s11082-018-1374-7.
- A. Kumar, J. S. G. Selva, J. M. Gonçalves, K. Araki and M. Bertotti, Nanoporous gold-based dopamine sensor with sensitivity boosted by interferant ascorbic acid, Electrochim. Acta, 2019, 322, 1–10, DOI:10.1016/j.electacta.2019.134772.
- L. D. Burke and P. F. Nugent, The electrochemistry of gold: I. The redox behaviour of the metal in aqueous media, Gold Bull., 1997, 30, 43–53, DOI:10.1007/BF03214756.
- T. R. L. C. Paixão, Measuring Electrochemical Surface Area of Nanomaterials versus the Randles−Ševčík Equation, ChemElectroChem, 2020, 7, 3414–3415, DOI:10.1002/celc.202000633.
- D. O. Wipf and A. J. Bard, Scanning Electrochemical Microscopy: VII. Effect of Heterogeneous Electron-Transfer Rate at the Substrate on the Tip Feedback Current, J. Electrochem. Soc., 1991, 138, 469–474, DOI:10.1149/1.2085612/XML.
- A. J. Bard, M. V. Mirkin, P. R. Unwin and D. O. Wipf, Scanning electrochemical microscopy. 12. Theory and experiment of the feedback mode with finite heterogeneous electron-transfer kinetics and arbitrary substrate size, J. Phys. Chem., 1992, 96, 1861–1868, DOI:10.1021/j100183a064.
- A. J. Bard, F. R. F. Fan, J. Kwak and O. Lev, Scanning Electrochemical Microscopy. Introduction and Principles, Anal. Chem., 1989, 61, 132–138, DOI:10.1021/ac00177a011.
- K. Eckhard, X. Chen, F. Turcu and W. Schuhmann, Redox competition mode of scanning electrochemical microscopy (RC-SECM) for visualisation of local catalytic activity, Phys. Chem. Chem. Phys., 2006, 8, 5359–5365, 10.1039/b609511a.
- I. Seymour, B. O'Sullivan, P. Lovera, J. F. Rohan and A. O'Riordan, Electrochemical Detection of Free-Chlorine in Water Samples Facilitated by in-situ pH Control using Interdigitated Microelectrodes, ChemRxiv, 2020, preprint, 325, DOI:10.26434/chemrxiv.11898126.v1.
- L. F. Kozin, V. A. Prokopenko and A. K. Bogdanova, Kinetics and mechanism of the gold corrosion dissolution in hypochlorite solutions, Prot. Met., 2005, 41, 22–29, DOI:10.1007/s11124-005-0003-6.
- J. M. Mohan, K. Amreen, A. Javed, S. K. Dubey and S. Goel, Miniaturized 3D printed electrochemical platform with optimized Fibrous carbon electrode for non-interfering hypochlorite sensing, Chemosphere, 2022, 302, 134915, DOI:10.1016/j.chemosphere.2022.134915.
- A. U. Alam, D. Clyne, W. Lush and M. J. Deen, A reusable, reagent-less free chlorine sensor using gold thin film electrode, Analyst, 2021, 146, 2626–2631, 10.1039/d1an00038a.
- Y. Wang and M. Seidel, Integration of 3D hydrodynamic focused microreactor with microfluidic chemiluminescence sensing for online synthesis and catalytical characterization of gold nanoparticles, Sensors, 2021, 21(7), 2290, DOI:10.3390/s21072290.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2sd00176d |
| This journal is © The Royal Society of Chemistry 2023 |