
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Atomically accurate site-specific ligand tailoring of highly acid- and alkali-resistant Ti(IV)-based metallamacrocycle for enhanced CO2 photoreduction†
Yi-Qi
Tian‡
a,
Lin-Fang
Dai‡
a,
Wen-Lei
Mu
a,
Wei-Dong
Yu
 b,
Jun
Yan
*a and
Chao
Liu
*a
b,
Jun
Yan
*a and
Chao
Liu
*a
aHunan Provincial Key Laboratory of Chemical Power Sources, College of Chemistry and Chemical Engineering, Central South University, Changsha 410083, Hunan, P. R. China. E-mail: chaoliu@csu.edu.cn
bChina College of Science, Hunan University of Technology and Business, Changsha 410000, P. R. China
First published on 30th November 2023
Abstract
Skillfully engineering surface ligands at specific sites within robust clusters presents both a formidable challenge and a captivating opportunity. Herein we unveil an unprecedented titanium-oxo cluster: a calix[8]arene-stabilized metallamacrocycle (Ti16L4), uniquely crafted through the fusion of four “core–shell” {Ti4@(TBC[8])(L)} subunits with four oxalate moieties. Notably, this cluster showcases an exceptional level of chemical stability, retaining its crystalline integrity even when immersed in highly concentrated acid (1 M HNO3) and alkali (20 M NaOH). The macrocycle's surface unveils four specific, customizable μ2-bridging sites, primed to accommodate diverse carboxylate ligands. This adaptability is highlighted through deliberate modifications achieved by alternating crystal soaking in alkali and carboxylic acid solutions. Furthermore, Ti16L4 macrocycles autonomously self-assemble into one-dimensional nanotubes, which subsequently organize into three distinct solid phases, contingent upon the specific nature of the four μ2-bridging ligands. Notably, the Ti16L4 exhibit a remarkable capacity for photocatalytic activity in selectively reducing CO2 to CO. Exploiting the macrocycle's modifiable shell yields a significant boost in performance, achieving an exceptional maximum CO release rate of 4.047 ± 0.243 mmol g−1 h−1. This study serves as a striking testament to the latent potential of precision-guided surface ligand manipulation within robust clusters, while also underpinning a platform for producing microporous materials endowed with a myriad of surface functionalities.
Introduction
Amidst the evolving landscape of advanced materials, titania nanomaterials have garnered significant interest, particularly for their promising applications as semiconductor photocatalysts in solar energy conversion.1–3 The allure of structurally precise titanium-oxo clusters (TOCs) stems from their tunable geometry and intriguing photoelectrical properties. Emerging as a captivating model, these attributes not only unravel intricate structure–property correlations within bulk TiO2 phases at the atomic scale, but also offer valuable structural and spectroscopic insights into the surface properties of TiO2.4–10 Unlike conventional TiO2, TOCs exhibit a rich diversity of structural types, carrying with them a distinctive palette of traits such as stability, semiconductor-like attributes, light absorption capacity, and band structure. These features are expertly modulated by introducing various functionalized organic ligands to achieve tailored modifications.11–21 Yet, the pursuit of precision in ligand modifications on specific TOCs comes with its challenges, requiring the careful negotiation of unstable coordination sites while upholding the core's inorganic integrity. Regrettably, altering protective ligands often triggers partial or complete dissection, resection, or structural reconstruction of the clusters.22,23 To surmount this hurdle, the creation of exceptionally stable TOCs emerges as a pivotal prerequisite. Enter the realm of macrometallocycles (MMCs), renowned for their formidable stability rooted in robust ring structures. This inherent resilience safeguards the chemical essence of TOCs, even under rigorous conditions, paving the way for subsequent adaptations. Additionally, another approach involves enveloping the cluster core with bulky polydentate macrocyclic ligands, exemplified by calixarenes.24–26 These intricate macrocycles, composed of multiple phenol units, have a storied history in orchestrating diverse metallo-supramolecular architectures.27–33 By fusing TOCs with a macrocyclic scaffold shielded by calixarene ligands, an uncharted territory opens to explore the impact of ligand tailoring at precise sites on their physicochemical attributes. While cyclic clusters abound,34–40 the realm of TiIV-based rings remains relatively untapped due to the hydrolysis susceptibility of Ti4+ ions. Notably, calixarene-stabilized TiIV-MMCs have yet to grace the literature. The envisioned assembly of atomically precise MMCs of TiIV, secured by calixarene guardians, holds immense promise for ushering rational design and optimizing performance across future applications.Herein, an exceptionally stable p-tert-butylcalix[8]arene-protected TiIV-based metallamacrocycle (MMC), denoted as [H4Ti16O8(TBC[8])4(Oa)4(Ac)4(iPrO)8], was synthesized. This MMC showcases remarkable resilience, displaying resistance against an array of organic solvents, concentrated acid (1 M HNO3), and alkali (20 M NaOH), thus establishing itself as a paramount example of cluster stability. The gigantic cluster has a “donut” shape with an inner diameter of 12 Å, an outer diameter of 30 Å, and a height of 18 Å, making it the largest known TOC in the metal-calixarene system. A pivotal discovery lies in the identification of four modifiable coordination sites on the Ti16L4 surface, which has led to further exploration of its exchange activity and applications (Scheme 1). These sites, intriguingly, offer two distinct avenues for functionalization: (1) utilizing a one-pot synthesis method, the μ2-bridging sites can be occupied by acetate and Cl−. The ratio of these ligands can be precisely tailored from 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0 to 2:2, and further to 0:4. Noteworthy is the emergence of a 3D network in Ti16L4–3, modified with four Cl−, yielding infinite cylindrical channels with diameters of approximately 2 nm. (2) An alternative post-modification strategy involves a sequential immersion of the cluster in concentrated alkali and carboxylic acid solutions, facilitating reversible exchange of the μ2-bridging sites by OH− and diverse carboxylates. The molecular-level understanding of this exchange process is unveiled through crystal-to-crystal diffraction studies, shedding light on structural transformations and ligand dynamics. Significantly, modifying the Ti16L4 shell distinctly influences physicochemical attributes, encompassing photocurrent response, hydrophilicity and energy levels, with ultimate implications for the photocatalytic CO2 reduction potential of the clusters. The ability to organically modify the shell while preserving macrocycle integrity introduces a pioneering avenue for fine-tuning chemical properties, setting the stage for these promising TiIV-based MMCs to be harnessed across diverse applications.
:0 to 2:2, and further to 0:4. Noteworthy is the emergence of a 3D network in Ti16L4–3, modified with four Cl−, yielding infinite cylindrical channels with diameters of approximately 2 nm. (2) An alternative post-modification strategy involves a sequential immersion of the cluster in concentrated alkali and carboxylic acid solutions, facilitating reversible exchange of the μ2-bridging sites by OH− and diverse carboxylates. The molecular-level understanding of this exchange process is unveiled through crystal-to-crystal diffraction studies, shedding light on structural transformations and ligand dynamics. Significantly, modifying the Ti16L4 shell distinctly influences physicochemical attributes, encompassing photocurrent response, hydrophilicity and energy levels, with ultimate implications for the photocatalytic CO2 reduction potential of the clusters. The ability to organically modify the shell while preserving macrocycle integrity introduces a pioneering avenue for fine-tuning chemical properties, setting the stage for these promising TiIV-based MMCs to be harnessed across diverse applications.
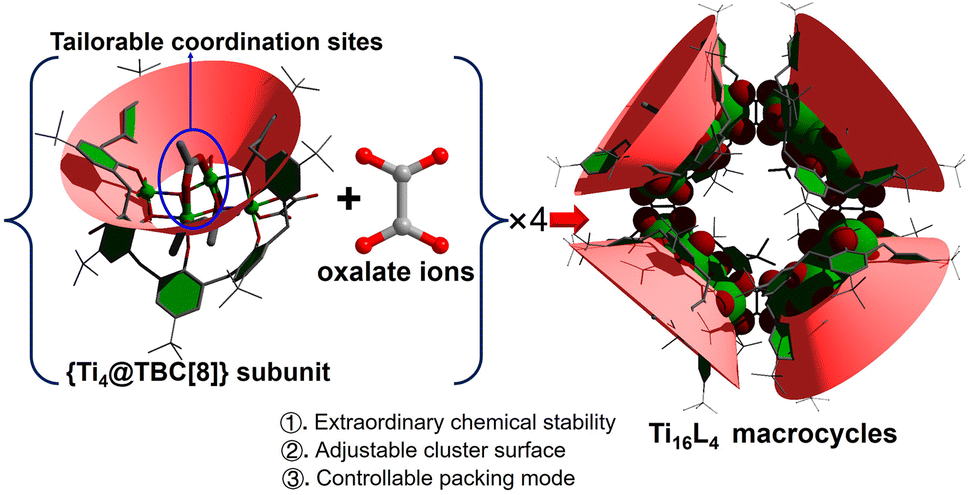 | ||
| Scheme 1 Illustration of the assembly of the Ti16L4 macrocycle with specific tailorable sites. | ||
Results and discussion
The synthesis of Ti16L4–1 entailed a one-pot solvothermal reaction involving TBC[8], oxalic acid (H2Oa), and Eu(Ac)3 in conjunction with Ti(OiPr)4, conducted within acetonitrile (CH3CN) at 120 °C for 72 hours. This procedure yielded red crystals with a notable 65% yield and consistent reproducibility. The ensuing single-crystal X-ray diffraction (SCXRD) analysis unveiled Ti16L4–1's monoclinic system crystallization in space group C2/c.41 The composition was precisely identified as [H4Ti16O8(TBC[8])4(Oa)4(Ac)4(iPrO)8]. X-ray photoelectron spectroscopy (XPS) analysis further validated the presence of Ti4+. The Ti 2p spectrum exhibited distinct regions at Ti 2p1/2 (ca. 464.9 eV) and Ti 2p3/2 (ca. 459.0 eV) with a binding energy separation of 5.9 eV, confirming the existence of Ti4+ (Fig. S32†). A comprehensive structural analysis elucidated the intricate architecture of the cluster. It emerged as a “core–shell” assembly comprising four {Ti4@TBC[8]} subunits interconnected by four oxalate ligands (Fig. 1A and B). Within each {Ti4@TBC[8]} subunit, four Ti4+ ions formed a central {Ti4O2} core connected by μ3-O2− ions, encased within the TBC[8] cavity (Fig. 1C). The TBC[8] molecule exhibits two distinct bonding modes for its eight phenoxide groups. Six of these groups are individually bonded to one Ti4+, while the remaining two groups are bridged by two Ti4+ in a μ2–κ2(O) fashion. The {Ti4O2} core showcased two distinct sets of Ti4+ sites. The Ti(2) and Ti(3) sites possess identical coordination environments, each coordinated by two phenol O, two μ3-O2− ions, and one iPrO− group. An additional acetate linked these two Ti4+ in a μ2-(O,O′) bridging fashion. Additionally, Ti(1) and Ti(4) exhibited equivalent coordination environments immobilized within the TBC[8] cavity. TBC[8]'s flexibility allowed the attachment of an Oa2− ligand to Ti(1) and Ti(4), while the oxalate ligand adopted a μ2-(O,O,O′,O′) mode, bridging two {Ti4@TBC[8]} subunits into a dimeric structure. This unique oxalate binding played a pivotal role in forming the closed-loop structure. Four oxalate ligands vertically bridged four {Ti4@TBC[8]} units, culminating in a quadrilateral macrocycle characterized by a 90° internal angle (Fig. 1D). Notably, this macrocycle exhibited an outer diameter of 30 Å and a height of 18 Å, with internal voids measuring an inner diameter of 12 Å and a window size of 7.5 Å, all determined by structural features.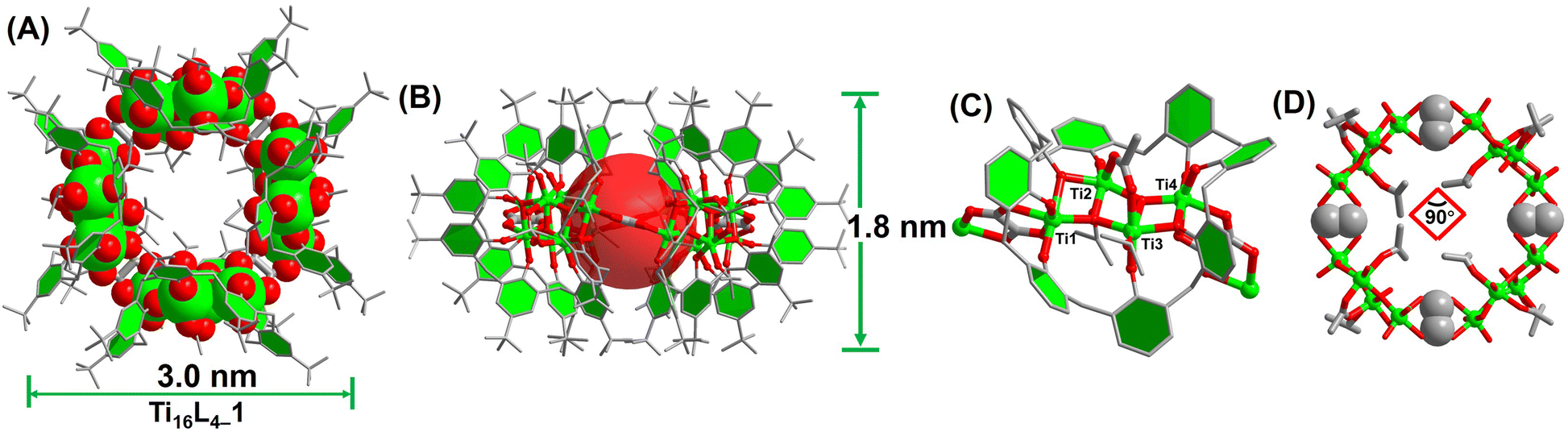 | ||
| Fig. 1 Structural depictions of Ti16L4–1. (A) and (B) Overhead and lateral perspectives; (C) configuration of the {Ti4@TBC[8]} subunit; (D) arrangement of the {Ti16} nanoring. | ||
Ligand modification
:0 ratio in Ti16L4–1, to a balanced 2:2 ratio in Ti16L4–2 (Fig. 2A), culminating in an exclusive 0:4 ratio in Ti16L4–3 (Fig. 2B).
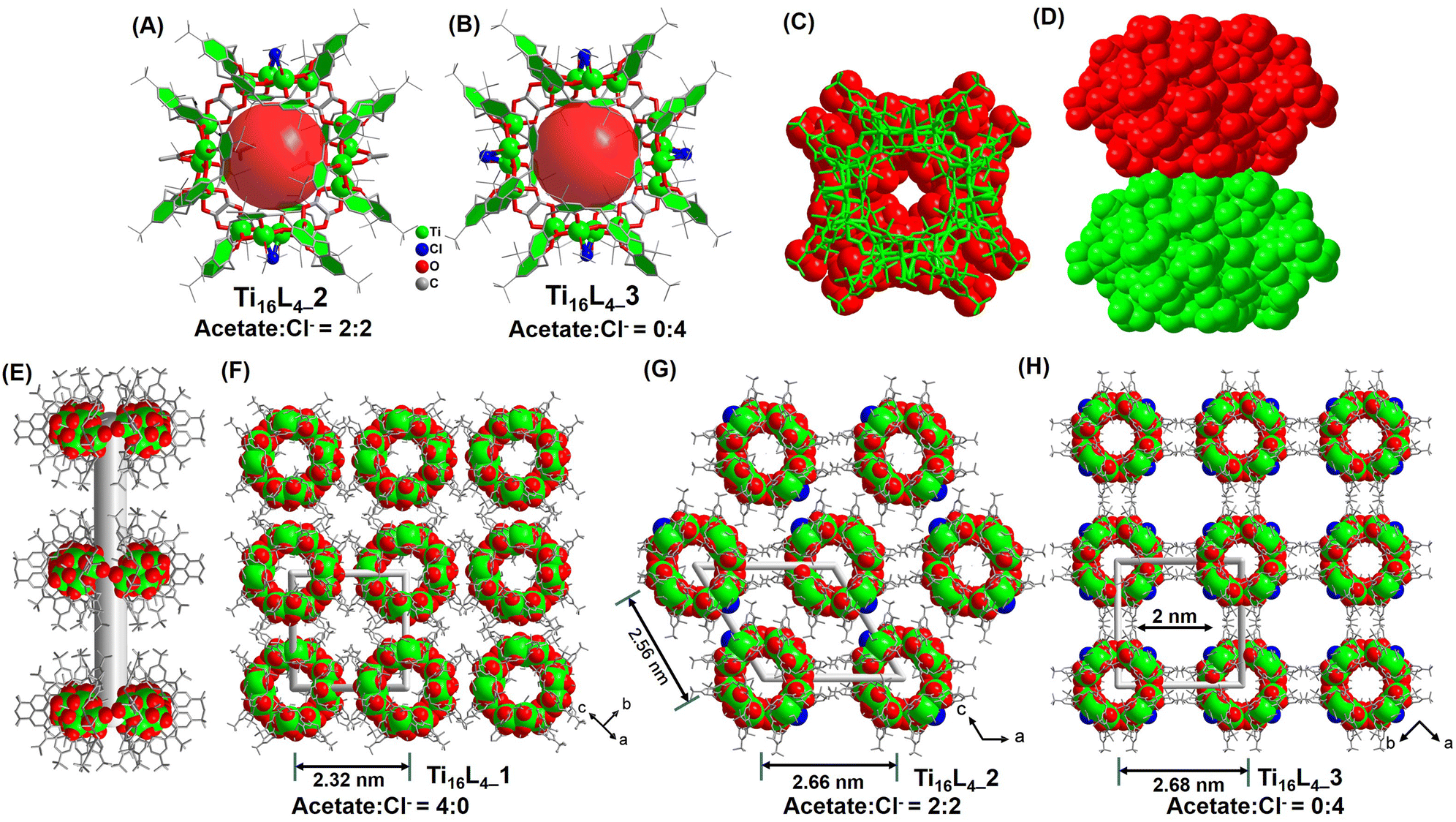 | ||
| Fig. 2 (A) and (B) Configuration of the Ti16L4–2/3 macrometallocycles (MMCs); (C) and (D) proximity arrangement of two Ti16L4 macrocycles; (E) formation of the nanotube within Ti16L4; (F)–(H) three distinct patterns of nanotube arrangement, including square and hexagonal arrays. | ||
Beyond the intricate geometries that clusters adopt, the interplay between surface ligands emerges as a pivotal determinant of their crystalline packing and consequential material attributes. Mastery over these multifaceted factors remains a formidable task. Our study, however, illuminates a path to precision control in this arena. Strikingly, while the shared macrocyclic scaffold remains constant, variations in surface ligands orchestrate disparate packing configurations within the crystal lattice of Ti16L4–1/2/3. Within all phases, the metallacycles ingeniously arrange face-to-face, thus sculpting 1D nanotubes that harmonize through van der Waals forces, culminating in their assemblage (Fig. 2C and D). The meticulous orchestration of this assembly begins by staggering neighboring nanotubes to alleviate steric hindrance stemming from tert-butyl moieties of adjacent macrocycles (Fig. 2E). This choreographed dance then evolves into a parallel stacking, fashioning an assorted array of 3D architectures. Remarkably, Ti16L4–1's nanotubes organize into a square matrix (Fig. 2F), whereas Ti16L4–2's nanotubes adopt a hexagonal pattern (Fig. 2G). This intriguing divergence traces its origins back to the precise occupancy of the four μ2-bridging sites. The substitution of acetate with Cl− profoundly impacts C–H⋯C–H interactions among the clusters. Additionally, the presence of polar Ti–Cl bonds endows the surface of Ti16L4–2, now embellished with two Cl−, with a discernible negative charge. This newfound electrostatic repulsion between the nanotubes impels their separation within the lattice. Evidently, the centroid distance between adjacent nanotubes escalates from 2.32 nm (as observed in Ti16L4–1) to 2.56 and 2.66 nm in succession. This captivating phenomenon finds further exemplification in the assembly motif of Ti16L4–3, wherein all four μ2-bridging sites embrace Cl−, ensuing in yet another square arrangement of macrocycles. Notably, a subtle 45° rotation between neighboring nanotubes in Ti16L4–1 mitigates the dominant electrostatic repulsion, paving the way for the emergence of 1D channels along the c-axis. These channels, a noteworthy 2 × 2 nm in dimension, define Ti16L4–3's unique character (Fig. 2H). The resplendent novelty of these findings lies in the precise manipulation of ligand dynamics to orchestrate such diverse and intricate packing phenomena.
The intricate interlocking of Ti16L4 macrocycles intricately assembles into meticulously ordered nanotubular frameworks across all three distinct phases. To validate the porosity of these architectures, we conducted N2 adsorption/desorption analyses that unveiled characteristic type I isotherms for each phase, unmistakably indicating their inherent porous nature. Concretely, our evaluation yielded Brunauer–Emmett–Teller (BET) surface areas of 209.24 m2 g−1 for Ti16L4–1, 389.92 m2 g−1 for Ti16L4–2, and a remarkable 813.46 m2 g−1 for Ti16L4–3 (Fig. 3A). The underlying absorbance potential was corroborated by diffuse reflectance spectra, revealing robust light absorption in the visible spectrum (Fig. S39†). Capitalizing on this light absorption attribute, we ventured into assessing the visible-light-driven photocurrent responses of Ti16L4. Through repeated irradiation cycles, steady photocurrent responses were uniformly observed across all clusters. Notably, the substitution of acetate with Cl− yielded an enhancement in photocurrent density. Among the clusters, Ti16L4–3 emerged as the standout, demonstrating the most optimal photocurrent response. Its recorded current density surged to an impressive 3.2 μA cm−2, which was approximately threefold higher than that of Ti16L4–1 (1.0 μA cm−2) (Fig. 3B). Interestingly, the hydrophobicity of Ti16L4 crystals emerged as an outcome intricately linked to their specific stacking arrangements. Evidently, water droplets exhibited distinct behaviors on the surfaces of Ti16L4–1/2 powders, maintaining rounded shapes with contact angles measuring 138.5° and 117.5°, respectively (Fig. 3C). In a striking departure, the contact angle observed for Ti16L4–3 was notably reduced to a mere 49.5°, indicative of its pronounced hydrophilic character. This intriguing contrast owes its origins to the presence of polar Ti–Cl bonds and the substantial presence of expansive cylindrical channels within the structural matrix, collectively orchestrating this distinctive behavior.
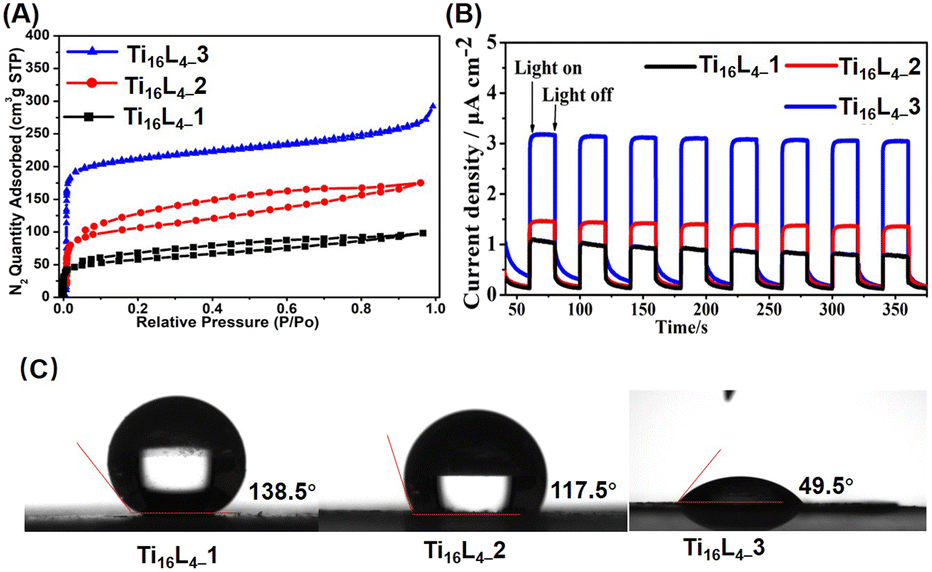 | ||
| Fig. 3 (A) N2 adsorption/desorption isotherm recorded at 77 K; (B) transient photocurrent responses of Ti16L4 captured during Xe lamp irradiation in a 0.2 M Na2SO4 electrolyte solution; (C) comparative assessment of the wettabilities across Ti16L4–1, Ti16L4–2 and Ti16L4–3. | ||
The stability of the nanocluster emerges as a pivotal facet, particularly in the context of post-synthetic modifications, warranting meticulous consideration. Significantly, the calixarene modification of the Ti16L4 confers a profound enhancement in chemical stability. Evidently, our PXRD analysis substantiated the robust stability of the Ti16L4–1 crystal across an array of solvents such as toluene, methanol, CH3CN, and DMF (Fig. S26†). To further probe its solution behavior, we harnessed matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF-MS) on Ti16L4–1 dissolved within a mixed solvent of CHCl3 and MeOH (Fig. 4A). Encouragingly, the dominant peaks in the spectrum unequivocally corresponded to species like [H4Ti16O8(TBC[8])4(Oa)4(Ac)4(iPrO)x(MeO)y]+ (x + y = 7). This finding underscores that, while retaining the overall architecture of Ti16L4, the iPrO− on the cluster exhibit high exchangeability with MeO− ions, while the chelating acetate sites remain resolutely stable. Dynamic light scattering measurements further confirmed a singular size distribution with an average diameter of 4.5 nm, a fitting match to the cluster size, concretely affirming the presence of well-defined Ti16L4 entities in solution (Fig. S37†). Another distinctive feature of Ti16L4–1 crystals comes to the fore in the realm of water stability—an attribute often fraught with challenges in many cluster crystals. In stark contrast, the exceptional pH stability of Ti16L4–1 crystals prevails over a broad pH values spanning from 1 to 14 (Fig. S27†). Subsequent testing reinforces this extraordinary stability, with the crystals remaining unaltered even in the face of concentrated acids (1 M HCl, 1 M H2SO4, and 1 M HNO3) and alkali (20 M NaOH) over a 24 hour period (Fig. 4B). This robust endurance finds its basis in the hydrophobic calixarene outer chamber enveloping Ti16L4, serving as an effective barrier against acid/alkali corrosion of the hydrophilic Ti4+. This ingeniously designed spatial protection imparts a remarkably high level of water stability to the cluster.
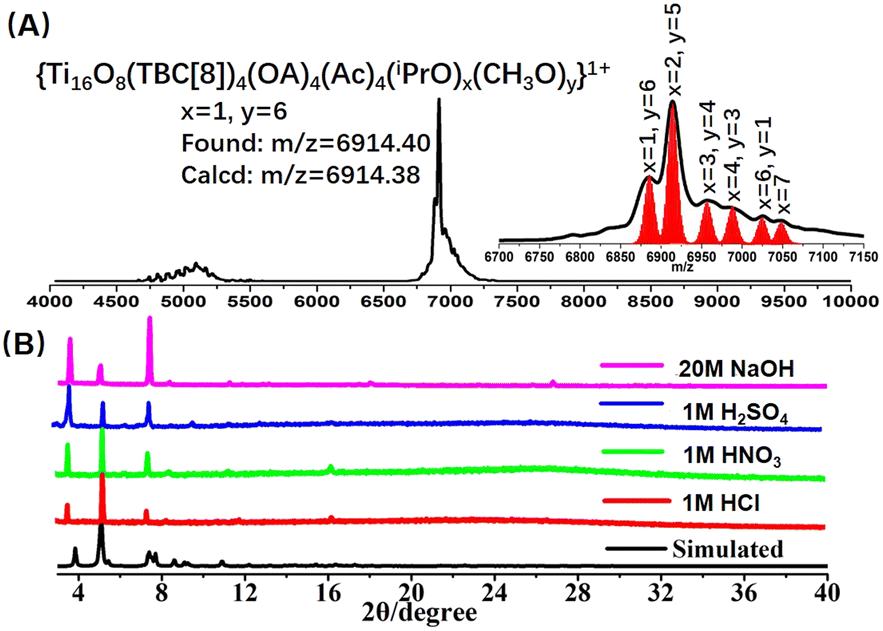 | ||
| Fig. 4 (A) Positive-mode MALDI-TOF-MS spectrum depicting Ti16L4–1 in CHCl3 solution; (B) PXRD patterns of Ti16L4–1 acquired during acid- and base-stability assessments. Remarkably, Ti16L4–1 exhibits robustness even after prolonged exposure to 1 M HCl, HNO3, H2SO4, and 20 M NaOH for 24 hours. | ||
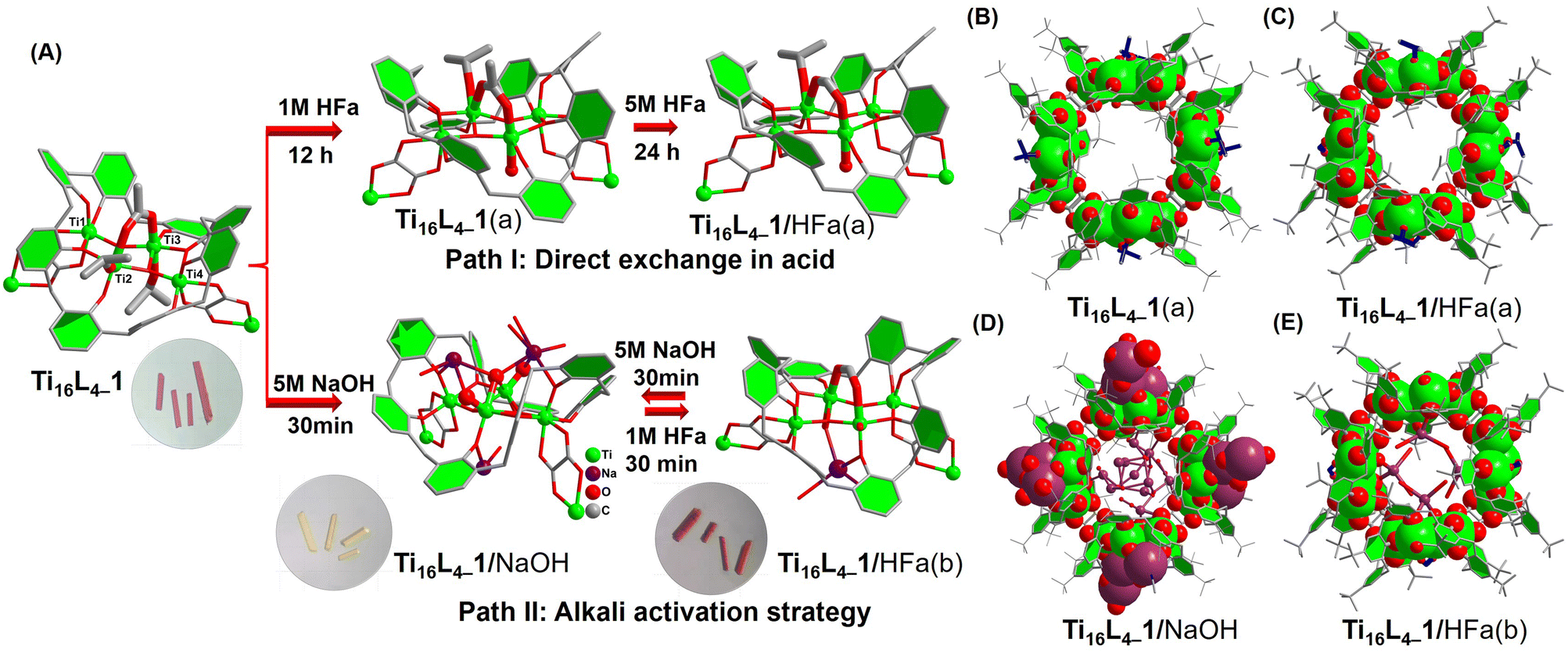 | ||
| Fig. 5 (A) Schematic representation of two different post-modification paths: (B–E) crystal structures of the derived clusters of Ti16L4–1. | ||
Drawing from the above insightful findings, the substitution of the four μ2-acetate sites within Ti16L4 emerges as a dynamic process, amenable to two distinct pathways. The first, path I, involves the direct immersion of crystals in a concentrated formic acid solution for a minimum of 24 hours. The second, path II, unfolds as a sequential immersion, commencing with a solution of NaOH, followed by a carboxylic acid. This latter path capitalizes on acid–base neutralization reactions, rendering the exchange process swift, completed within a mere hour. Evaluating the efficacy of these approaches, we embarked on a mission to extend the carboxylate modifications on the Ti16L4 surface. Encouragingly, the gamut encompassed successful modifications: chloroacetate (Cla−) and bromoacetate (Bra−) through path I, and acetate (Ac−),47 aminoacetate (Ama−), glycinate (Ga−),48 and oxalate (Oa2−)49via path II. A comprehensive SCXRD analysis discerned the occupation of all four sites by these carboxylates. Delving into specifics, the bridging modes of Ac−, Ama−, Cla−, and Bra− ligands aligned uniformly, establishing μ2-(O,O′) bridges connecting two Ti sites (as depicted in Fig. 6A–D). In marked contrast, Oa2− and Ga− ligands exhibited an intriguingly different binding pattern (as showcased in Fig. 6E andF), with the ligands forming a chelating arrangement around the Ti3 site in a bidentate μ1-(O,O) mode, concomitantly coordinating the Ti2 site with two OH− ions. Notably, the structural analysis of Ti16L4–1 modified with trifluoroacetate (Tfa−) encountered challenges due to poor crystal quality. This phenomenon can be attributed to the introduction of a significant number of F atoms to the cluster's surface, engendering repulsion among clusters within the tetragonal phase and leading to lattice displacement. However, this issue was effectively surmounted in the case of the Tfa− modified Ti16L4–2 cluster, revealing a definitive structural determination within the hexagonal lattice of Ti16L4–2 (Fig. 6G), underscoring the adaptability of the Tfa− to occupy these sites.
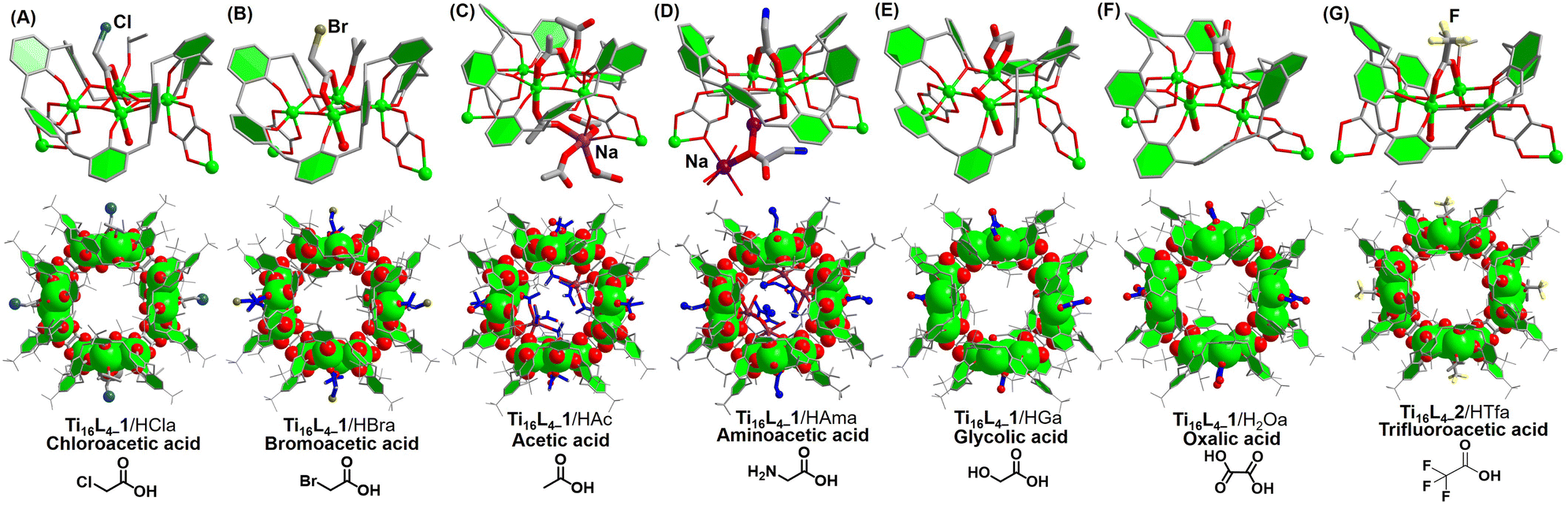 | ||
| Fig. 6 (A)–(G) Crystal structures of modified clusters derived from Ti16L4via post-modification pathways. | ||
Interestingly, our explorations extended to the realm of inorganic oxyanions. When Ti16L4–1/NaOH crystals were immersed in 1 M H3PO4 for 30 minutes, red crystals of Ti16L4–1/H3PO4 were obtained, retaining crystallinity. SCXRD analysis brought forth an intriguing revelation—PO43− did not interact with the clusters. Instead, OH− persisted as occupants within the 12 labile sites, with Na+ ions being entirely dislodged from the structure. Notably, the cumulative occupancy rate of the four μ2-OH− sites reached 2.80 (Fig. S22†). This intriguing outcome underscores our capacity to effect partial ligand removal from these four μ2-sites through a meticulous interplay of two acid–base neutralization reactions. This multifaceted exploration, together with the systematic elucidation of intricate ligand exchange pathways, reflects the groundbreaking dimension of our research endeavor.
CO2 photoreduction activities
The promising avenue of photocatalytically reducing CO2 into reusable chemicals looms large as a critical step toward carbon neutrality.50 However, the reported TOCs for driving such reactions remain limited, evoking a pressing need for innovative solutions.51–55 Harnessing the unparalleled stability, controllable ligands, and adaptable packing modes inherent to Ti16L4, we ventured into their application as heterogeneous photocatalysts for CO2 reduction. Conceiving the intricate setup, CO2 photoreduction over Ti16L4 unfolded under visible light irradiation (λ ≥ 420 nm) in the presence of [Ru(bpy)3]Cl2·6H2O as a photosensitizer, triethanolamine (TEOA) as a sacrificial agent, and a CH3CN/H2O (4:1, v/v) solvent composition. A comprehensive analysis of the product, employing gas chromatography (GC, Fig. S47†), ion chromatography (IC, Fig. S51†) and 1H NMR (Fig. S52†), lucidly affirmed the exclusive generation of CO via CO2RR, with a marginal presence of H2. It's noteworthy that no products were observed in the absence of irradiation, CO2 or clusters, or the yields were exceptionally low (Table S4†). This unequivocally underscores the orchestrated interplay of each component within the photocatalytic system. Comparative assessment of the catalytic activity of Ti16L4–1/2/3 in CO2 photoreduction revealed a remarkable hierarchy in CO yields. Ti16L4–1 produced a modest CO yield of 11.97 ± 0.67 μmol within 5 hours, with Ti16L4–2 and Ti16L4–3 exhibiting substantially enhanced CO yields of 29.97 ± 2.17 μmol and 60.70 ± 3.65 μmol, respectively (Fig. 7A). Ti16L4–3 emerged as the frontrunner in catalytic prowess, displaying an impressive CO production rate of 4046.67 ± 243.33 μmol g−1 h−1, coupled with an exceptional selectivity of 96.28%. These activity and selectivity levels surpassed those of many cluster or MOF-based materials for CO2 to CO conversion.56–61 The catalytic proficiency of this macrocyclic series aligns with the potentially catalytically active TiIV sites boasting flexible coordination spaces (specifically the Ti2 and Ti3 sites), which stand poised for CO2 adsorption. In Ti16L4–3, Cl− exhibit a greater tendency to vacate compared to acetates in Ti16L4–1. Consequently, the Ti sites in Ti16L4–3 can showcase higher catalytic activity. This assessment of catalytic activity, supported by Table S5,† revealed that each potential Ti4+ catalytic site (TONTi) in Ti16L4–3 exhibited higher activity than those in Ti16L4–1/2. Additionally, the strategic advantage of the 2 nm macrochannel within Ti16L4–3 unveiled a larger surface area, fostering heightened CO2 adsorption and exposure to additional catalytic enclaves. Experimental findings in CO2 adsorption bolstered these revelations (Fig. S36†), where Ti16L4–3 showcased superior performance over Ti16L4–1/2, with CO2 uptake values of 57.38 and 48.79 cm3 g−1, respectively, at 273 K and 298 K under 1 bar pressure. Intriguingly, functionalization of ligands on Ti16L4 also had a notable influence on catalytic activity (Fig. 7B). In the case of Ti16L4–1/NaOH, all potential catalytic sites on the cluster's surface are occupied by OH− ions. Due to the presence of strong Ti–OH bonds, CO2 is unable to access the catalytic Ti sites, resulting in a decrease in catalytic activity (2.35 ± 0.89 μmol for 5 hours). Noteworthy examples include Ti16L4–1/HBra and Ti16L4–1/HCla, unveiling CO yields of 36.25 ± 2.59 μmol and 39.48 ± 3.16 μmol, respectively, after 5 hours, approximately threefold higher than pristine Ti16L4–1 (Fig. 7C). Similarly, Ti16L4–2/HTfa exhibited double the CO production of its untouched counterpart (58.18 ± 3.10 μmol for 5 hours), Ti16L4–2. The higher photocatalytic performance of those derived clusters is mainly due to their modification with halogenated carboxylates that have the ability to quickly transfer charges. These enhancements find resonance in the modification-induced shifts within the cluster's bandgap structures, catalyzing accelerated electron transfers, and amplifying catalytic efficiency.
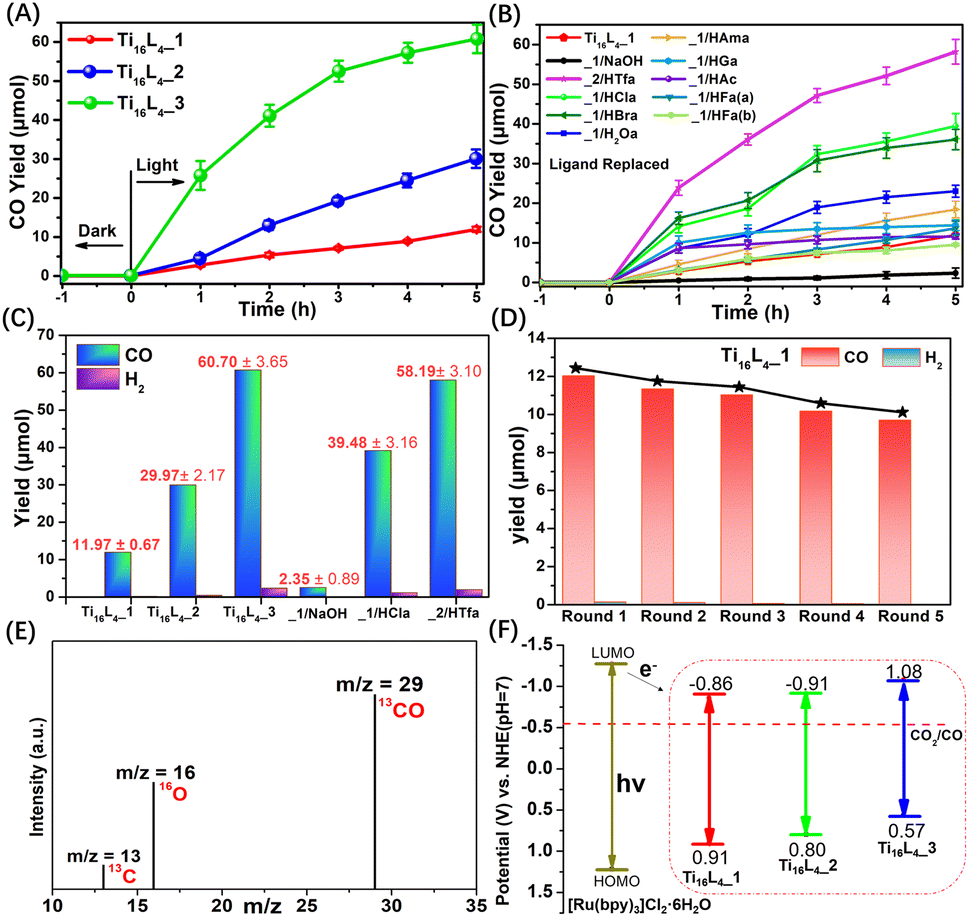 | ||
| Fig. 7 (A) Time-dependent CO generation profiles for Ti16L4–1/2/3; (B) time-dependent CO generation profiles for Ti16L4 with various ligand modifications; (C) CO production rates over Ti16L4; (D) durability assessments of Ti16L4–1. (E) 13CO recorded under 13CO2 atmosphere; (F) the energy band structure diagram for Ti16L4. Reaction conditions: 3 mg of Ti16L4, 10 mg [Ru(bpy)3]Cl2·6H2O, 5 mL triethanolamine (TEOA), acetonitrile (8 mL), H2O (2 mL), CO2 (1 atm), λ ≥ 420 nm, 25 °C, 5 h. | ||
Collectively, these revelations converge to spotlight the photocatalytic acumen inherent within the Ti16L4 macrocycles, unfurling their supremacy in CO2 reduction and accentuating their proclivity for selectively generating CO. What's more, the strategic orchestration of the Ti16L4's spatial configuration, coupled with tailored enhancements infused into its outer ligands, emerges as a potent avenue for catapulting its catalytic performance to new heights. A tangible testament to the robustness of these findings is the recyclability assessment, which resoundingly affirmed the enduring vigor of Ti16L4–1's photocatalytic activity even across five successive cycles (Fig. 7D). It's worth highlighting that Ti16L4–1 not only retained its remarkable photocatalytic stability but also withstood scrutiny through XPS, MALDI-TOF-MS and TEM morphology studies (Fig. S58–S60†). Subsequent SCXRD analysis of Ti16L4–1 post photocatalysis revealed subtle changes in the coordination environment of potential catalytic sites (Ti2 and Ti3) within the cluster, which further substantiates the catalytic activity of these flexible coordination sites (Fig. S57†). Following the culmination of five cycles, the catalyst could be efficiently reclaimed from the reaction milieu. A analysis of the resultant supernatant via Inductively-Coupled Plasma (ICP) revealed an astonishingly low Ti leakage rate from Ti16L4–1, accounting for a mere 0.01% of the total Ti content. This revelation underscores not only the catalyst's enduring stability but also its marked potential for pragmatic, real-world applications. To unravel the origin of the carbon in the generated CO, an isotopic experiment leveraging 13CO2 as the carbon source was conducted. The gas chromatography-mass spectrometry (GC-MS) identified the production of 13CO, confirming the unequivocal derivation of CO from CO2 (Fig. 7E).
The investigation into electron transfer in the catalytic process unveiled the flat band potentials of Ti16L4–1/2/3via Mott–Schottky plots, pinpointing their calculated conduction band minimum (CBM) values. These CBM values resided at −0.86, −0.91, and −1.08 V vs. the normal hydrogen electrode (NHE), respectively. Strikingly, all LUMO potentials exhibited a notably more negative profile in contrast to CO2/CO (−0.51 V vs. NHE), signifying their inherent suitability for CO2RR (Fig. 7F). Previous reports have indicated that the LUMO of [Ru(bpy)3]Cl2·6H2O is −1.27 V vs. NHE.62 Under irradiation, [Ru(bpy)3]Cl2·6H2O transits to an excited state, subsequently reductively quenched by TEOA, yielding a reducing photosensitizer. Given that the CBM values of Ti16L4–1/2/3 lie below the LUMO of [Ru(bpy)3]Cl2·6H2O, the excited electrons emanating from the reduced photosensitizer migrate to Ti16L4, setting off the activation of adsorbed CO2 on its surface. This orchestrated sequence culminates in the reduction of CO2 to CO, subsequently liberating the product. We further substantiated the photocatalytic mechanism through electron paramagnetic resonance (EPR) experiments (Fig. S53†). The experimental findings reveal that in the absence of light irradiation within an N2 atmosphere, the reaction system involving Ti16L4–1 and the sacrificial agent did not exhibit any ESR signals. Nevertheless, when subjected to visible light irradiation, distinct Ti3+ signals were observed, corresponding to g value of 1.945. This observation implies that the photoexcited electrons transfers to Ti4+, leading to their reduction to Ti3+. Concurrently, TEOA serves as the sacrificial agent to neutralize the photogenerated holes. The intensity of the Ti3+ signal gradually increased with extended irradiation time. Upon exposure of the reaction system to a CO2 atmosphere, the ESR signal of Ti3+ diminished, signifying the involvement of photogenerated Ti3+ in CO2RR. ESR results affirm that Ti4+ within Ti16L4–1 function as the active centers for photocatalytic CO2RR, providing further support for this mechanism.
To probe the CO2 radical and other reaction intermediates in the photocatalytic reaction, the Ti16L4–1 is investigated by in situ diffuse reflectance infrared Fourier transform spectroscopy (Fig. 8). Under dark conditions post CO2 pretreatment, Ti16L4–1 displayed prominent peaks around 2348 cm−1, which is associated with the asymmetric stretching of absorbed CO2.59 Exposure to a CO2 atmosphere under light for 10 minutes resulted in several new peaks: monodentate carbonate (m-CO32−) at 1351, 1451, and 1508 cm−1; bidentate carbonate (b-CO32−) at 1290 and 1543 cm−1; and bicarbonate (HCO3−) at 1406 and 1656 cm−1. These carbonates and bicarbonates likely formed from CO2 reacting with water vapor. Notably, the CO2˙− signal at 1713 cm−1 intensified with prolonged irradiation, indicating the formation of the CO2 radical, a key intermediate in CO2 photoreduction to *COOH.52,53 Furthermore, *COOH groups, crucial in CO2 reduction to CO, were identified at 1338 and 1584 cm−1, with increasing peak intensities under light exposure, suggesting light-induced formation.63 Additionally, absorption peaks for *CO and gaseous CO at 1708 and 2116 cm−1 respectively, provided further evidence of the reaction pathway.64 Therefore, according to the above analysis, a rational CO2 photoreduction mechanism was proposed (Fig. S54†): CO2 was initially adsorbed on the Ti3+. Subsequently, the adsorbed *CO2 received electrons from Ti3+ and with protons to form the *COOH during irradiation. Thereafter, the deprotonation of the *COOH intermediate further generation of CO, and ultimately desorbs to obtain CO molecules.
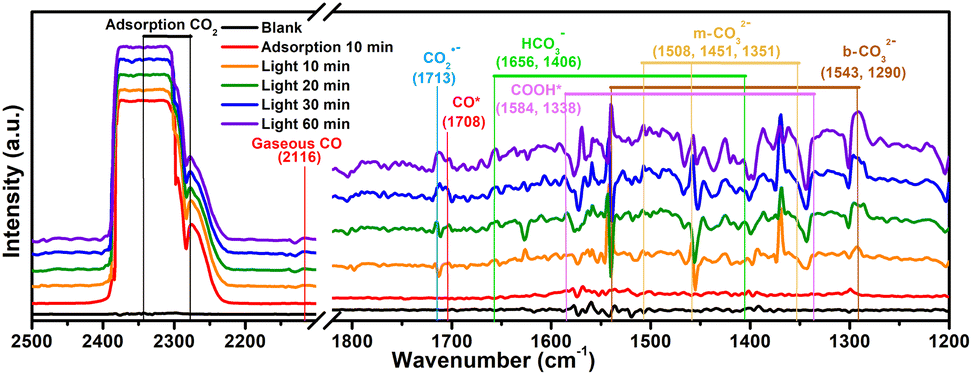 | ||
| Fig. 8 In situ FTIR spectra on Ti16L4–1. | ||
Conclusions
In summary, this research marks a pioneering exploration into the realm of TiIV-based metallamacrocycles, unveiling a host of novel insights and underlining its significance in the broader landscape of scientific inquiry. Foremost, the unparalleled stability demonstrated by the Ti16L4 stands out as a groundbreaking revelation. Its exceptional resilience against a spectrum of challenges, encompassing organic solvents, concentrated acids, and alkali, attests to a robustness that guarantees structural integrity and endurance across diverse environmental contexts. This stability not only solidifies the cluster's foundation but also paves the way for its application across a multitude of conditions. Equally groundbreaking is the abundance of coordination anchors inherent on the Ti16L4's surface, effectively serving as a fertile ground for subsequent ligand adaptations. The dual pathways for facile ligand exchange form the bedrock of a versatile platform, facilitating the creation of microporous materials endowed with a plethora of surface functionalities. These microporous materials with coordinating sites on their surfaces are promising as carriers for loading noble metal nanoparticles, constructing heterogeneous catalysts with highly efficient catalytic activity. Another revelation lies in the crystallization potential of the macrocycle, giving rise to three distinct phases contingent upon the employed surface ligands. This revelation casts a revelatory spotlight on the predominant forces orchestrating the formation of diverse cluster-packing modes within the crystal lattice. The capability to manipulate surface functionalities and packing arrangements not only broadens our fundamental understanding but also unlocks a trove of opportunities for finely honing chemical and physical attributes to align with specific applications.Data availability
The data that support the findings of this study are available in the main text and the ESI.†Author contributions
C. L. supervised the project and conceived the idea. Y. Q. T. and L. F. D carried out synthesis, characterization and photocatalytic experiment of clusters. C. L. and Y. Q. T. wrote the manuscript. All authors discussed the experimental results.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was supported by the Natural Science Foundation of Hunan Province (2023JJ30650) and Central South University Innovation-Driven Research Programme (2023CXQD061).Notes and references
- M. Dahl, Y. D. Liu and Y. D. Yin, Chem. Rev., 2014, 114, 9853–9889 CrossRef CAS PubMed.
- Y. Ma, X. L. Wang, Y. S. Jia, X. B. Chen, H. X. Han and C. Li, Chem. Rev., 2014, 114, 9987–10043 CrossRef CAS PubMed.
- X. Chen, L. Liu, P. Y. Yu and S. S. Mao, Science, 2011, 331, 746–750 CrossRef CAS PubMed.
- P. Coppens, Y. Chen and E. Trzop, Chem. Rev., 2014, 114, 9645–9661 CrossRef CAS PubMed.
- L. Rozes and C. Sanchez, Chem. Soc. Rev., 2011, 40, 1006–1030 RSC.
- P. D. Matthews, T. C. King and D. S. Wright, Chem. Commun., 2014, 50, 12815–12823 RSC.
- J. B. Benedict, R. Freindorf, E. Trzop, J. Cogswell and P. Coppens, J. Am. Chem. Soc., 2010, 132, 13669–13671 CrossRef CAS PubMed.
- W. H. Fang, L. Zhang and J. Zhang, Chem. Soc. Rev., 2018, 47, 404–421 RSC.
- Q. Y. Zhu and J. Dai, Coord. Chem. Rev., 2021, 430, 213664 CrossRef CAS.
- S. Yuan, J. S. Qin, H. Q. Xu, J. Su, D. Rossi, Y. P. Chen, L. L. Zhang, C. Lollar, Q. Wang, H. L. Jiang, D. H. Son, H. Y. Xu, Z. H. Huang, X. D. Zou and H. C. Zhou, ACS Cent. Sci., 2018, 4, 105–111 CrossRef CAS PubMed.
- J. D. Sokolow, E. Trzop, Y. Chen, J. Tang, L. J. Allen, R. H. Crabtree, J. B. Benedict and P. Coppens, J. Am. Chem. Soc., 2012, 134, 11695–11700 CrossRef CAS PubMed.
- G. Zhang, C. Liu, D.-L. Long, L. Cronin, C. H. Tung and Y. Wang, J. Am. Chem. Soc., 2016, 138, 11097–11100 CrossRef CAS PubMed.
- Y. Guo, J. L. Hou, W. Luo, Z. Q. Li, D. H. Zou, Q. Y. Zhu and J. Dai, J. Mater. Chem. A, 2017, 5, 18270–18275 RSC.
- D. H. Zou, L. N. Cui, P. Y. Liu, S. Yang, Q. Y. Zhu and J. Dai, Inorg. Chem., 2019, 58, 9246–9252 CrossRef CAS PubMed.
- Y. Lv, J. Cheng, A. Steiner, L. Gan and D. S. Wright, Angew. Chem., Int. Ed., 2014, 53, 1934–1938 CrossRef CAS PubMed.
- L. Zhang, X. Fan, X. F. Yi, X. Lin and J. Zhang, Acc. Chem. Res., 2022, 55(21), 3150–3161 CrossRef CAS PubMed.
- L. Geng, C. H. Liu, S. T. Wang, W.-H. Fang and J. Zhang, Angew. Chem., Int. Ed., 2021, 133, 21596–21603 CrossRef.
- M.-Y. Gao, F. Wang, Z. G. Gu, D. X. Zhang, L. Zhang and J. Zhang, J. Am. Chem. Soc., 2016, 138, 2556–2559 CrossRef CAS PubMed.
- X. Fan, J. Wang, K. Wu, L. Zhang and J. Zhang, Angew. Chem., Int. Ed., 2019, 58, 1320–1323 CrossRef CAS PubMed.
- W. H. Fang, L. Zhang and J. Zhang, J. Am. Chem. Soc., 2016, 138, 7480–7483 CrossRef CAS PubMed.
- G. Zhang, W. Li, C. Liu, J. Jia, C. H. Tung and Y. Wang, J. Am. Chem. Soc., 2018, 140, 66–69 CrossRef CAS PubMed.
- J. X. Liu, M. Y. Gao, W. H. Fang, L. Zhang and J. Zhang, Angew. Chem., Int. Ed., 2016, 55, 5160–5165 CrossRef CAS PubMed.
- L. Geng, C. H. Liu, S. T. Wang, W. H. Fang and J. Zhang, Angew. Chem., Int. Ed., 2020, 59, 16735–16740 CrossRef CAS PubMed.
- Y. Q. Tian, Y. S. Cui, W. D. Yu, C. Q. Xu, X. Y. Yi, J. Yan, J. Li and C. Liu, Chem. Commun., 2022, 52, 6028–6031 RSC.
- Y. Q. Tian, Y. S. Cui, J. H. Zhu, C. Q. Xu, X. Y. Yi, J. Li and C. Liu, Chem. Commun., 2022, 58, 9034–9037 RSC.
- S. Y. Zhou, C. P. Li, H. Fu, J. Cao, J. Zhang and L. Zhang, Chem.–Eur. J., 2020, 26, 6894–6898 CrossRef CAS PubMed.
- M. Liu, W. P. Liao, C. Hu, S. C. Du and H. J. Zhang, Angew. Chem., Int. Ed., 2012, 51, 1585–1588 CrossRef CAS PubMed.
- W. Y. Pei, G. Xu, J. Yang, H. Wu, B. Chen, W. Zhou and J. F. Ma, J. Am. Chem. Soc., 2017, 139, 7648–7656 CrossRef CAS PubMed.
- H. T. Han, L. Kan, P. Li, G. S. Zhang, K. Y. Li, W. P. Liao, Y. L. Liu, W. Chen and C. H. T. Hu, Sci. China: Chem., 2021, 64, 426–431 CrossRef CAS.
- X. X. Hang, B. Liu, X. F. Zhu, S. T. Wang, H. T. Han, W. P. Liao, Y. L. Liu and C. H. Hu, J. Am. Chem. Soc., 2016, 138, 2969–2972 CrossRef CAS PubMed.
- S. Wang, X. Gao, X. Hang, X. Zhu, H. Han, X. Li, W. Liao and W. Chen, J. Am. Chem. Soc., 2018, 140, 6271–6277 CrossRef CAS PubMed.
- Z. Wang, H. F. Su, Y. W. Gong, Q. P. Qu, Y. F. Bi, C. H. Tung, D. Sun and L. S. Zheng, Nat. Commun., 2020, 11, 308 CrossRef CAS PubMed.
- B. Hou, H. Y. Zheng, K. H. Zhang, Q. Wu, C. Qin, C. Y. Sun, Q. H. Pan, Z. H. Kang, X. L. Wang and Z. M. Su, Chem. Sci., 2023, 14, 8962–8969 RSC.
- A. J. Tasiopoulos, A. Vinslava, W. Wernsdorfer, K. A. Abboud and G. Christou, Angew. Chem., Int. Ed., 2004, 43, 2117–2121 CrossRef CAS PubMed.
- H. Kitagawa, H. Ohtsu and M. Kawano, Angew. Chem., Int. Ed., 2013, 52, 12395–12399 ( Angew. Chem. , 2013 , 125 , 12621–12625 ) CrossRef CAS PubMed.
- Z. M. Zhang, Y. G. Li, S. Yao, E. B. Wang, Y. H. Wang and R. Clerac, Angew. Chem., Int. Ed., 2009, 48, 1581–1584 CrossRef CAS PubMed.
- R. A. Scullion, A. J. Surman, F. Xu, J. S. Mathieson, D. L. Long, F. Haso, T. Liu and L. Cronin, Angew. Chem., Int. Ed., 2014, 53, 10032–10037 CrossRef CAS PubMed.
- C. Zhao, Y.-Z. Han, S. Dai, X. Chen, J. Yan, W. Zhang, H. Su, S. Lin, Z. Tang, B. K. Teo and N. Zheng, Angew. Chem., Int. Ed., 2017, 56, 16252–16256 CrossRef CAS PubMed.
- S. Øien-Ødegaard, C. Bazioti, E. A. Redekop, O. Prytz, K. P. Lillerud and U. Olsbye, Angew. Chem., Int. Ed., 2020, 59, 21397–21402 CrossRef PubMed.
- X. Y. Zheng, Y. H. Jiang, G. L. Zhuang, D. P. Liu, H. G. Liao, X. J. Kong, L. S. Long and L. S. Zheng, J. Am. Chem. Soc., 2017, 139, 18178–18181 CrossRef CAS PubMed.
- Y. Q. Tian, L. F. Dai, W. L. Mu, W. D. Yu, J. Yan and C. Liu, CCDC 2174429: Experimental Crystal Structure Determination, DOI:10.5517/ccdc.csd.cc2bznww.
- Y. Q. Tian, L. F. Dai, W. L. Mu, W. D. Yu, J. Yan and C. Liu, CCDC 2174430: Experimental Crystal Structure Determination, DOI:10.5517/ccdc.csd.cc2bznxx.
- Y. Q. Tian, L. F. Dai, W. L. Mu, W. D. Yu, J. Yan and C. Liu, CCDC 2174431: Experimental Crystal Structure Determination, DOI:10.5517/ccdc.csd.cc2bznyy.
- Y. Q. Tian, L. F. Dai, W. L. Mu, W. D. Yu, J. Yan and C. Liu, CCDC 2174432: Experimental Crystal Structure Determination, DOI:10.5517/ccdc.csd.cc2bznzz.
- Y. Q. Tian, L. F. Dai, W. L. Mu, W. D. Yu, J. Yan and C. Liu, CCDC 2174433: Experimental Crystal Structure Determination, DOI:10.5517/ccdc.csd.cc2bzp01.
- Y. Q. Tian, L. F. Dai, W. L. Mu, W. D. Yu, J. Yan and C. Liu, CCDC 2174435: Experimental Crystal Structure Determination, DOI:10.5517/ccdc.csd.cc2bzp23.
- Y. Q. Tian, L. F. Dai, W. L. Mu, W. D. Yu, J. Yan and C. Liu, CCDC 2174434: Experimental Crystal Structure Determination, DOI:10.5517/ccdc.csd.cc2bzp12.
- Y. Q. Tian, L. F. Dai, W. L. Mu, W. D. Yu, J. Yan and C. Liu, CCDC 2174437: Experimental Crystal Structure Determination, DOI:10.5517/ccdc.csd.cc2bzp45.
- Y. Q. Tian, L. F. Dai, W. L. Mu, W. D. Yu, J. Yan and C. Liu, CCDC 2174438: Experimental Crystal Structure Determination, DOI:10.5517/ccdc.csd.cc2bzp56.
- J. Zhou, J. Li, L. Kan, L. Zhang, Q. Huang, Y. Yan, Y. Chen, J. Liu, S. L. Li and Y. Q. Lan, Nat. Commun., 2022, 13, 4681 CrossRef CAS PubMed.
- N. Li, J.-J. Liu, J.-W. Sun, B.-X. Dong, L.-Z. Dong, S.-J. Yao, Z. F. Xin, S.-L. Li and Y.-Q. Lan, Green Chem., 2020, 22, 5325–5332 RSC.
- N. Li, J.-M. Lin, R.-H. Li, J.-W. Shi, L.-Z. Dong, J. Liu, J. He and Y.-Q. Lan, J. Am. Chem. Soc., 2023, 145(29), 16098–16108 CrossRef CAS PubMed.
- J.-J. Liu, N. Li, J.-W. Sun, J. Liu, L.-Z. Dong, S.-J. Yao, L. Zhang, Z.-F. Xin, J.-W. Shi, J.-X. Wang, S.-L. Li and Y.-Q. Lan, ACS Catal., 2021, 11, 4510–4519 CrossRef CAS.
- N. Li, J. Liu, J. J. Liu, L. Z. Dong, S. L. Li, B. X. Dong, Y. H. Kan and Y. Q. Lan, Angew. Chem., Int. Ed., 2019, 58, 17260–17264 CrossRef CAS PubMed.
- Y. Fu, D. Sun, Y. Chen, R. Huang, Z. Ding, X. Fu and Z. Li, Angew. Chem., Int. Ed., 2012, 51, 3364–3367 CrossRef CAS PubMed.
- R. Li, J. H. Hu, M. S. Deng, H. L. Wang, X. J. Wang, Y. L. Hu, H. L. Jiang, J. Jiang, Q. Zhang, Y. Xie and Y. J. Xiong, Adv. Mater., 2014, 26, 4783–4788 CrossRef CAS PubMed.
- S. B. Wang, W. S. Yao, J. L. Lin, Z. X. Ding and X. C. Wang, Angew. Chem., Int. Ed., 2014, 53, 1034–1308 CrossRef CAS PubMed.
- J. Zhao, Q. Wang, C. Y. Sun, T. T. Zheng, L. K. Yan, M. T. Li, K. Z. Shao, X. L. Wang and Z. M. Su, J. Mater. Chem. A, 2017, 5, 12498 RSC.
- H.-X. Zhang, Q.-L. Hong, J. Li, F. Wang, X. S. Huang, S. M. Chen, W. G. Tu, D. S. Yu, R. Xu, T. H. Zhou and J. Zhang, Angew. Chem., Int. Ed., 2019, 58, 11752–11756 CrossRef CAS PubMed.
- X. X. Li, L. Zhang, L. Yuan, T. Wang, L. Z. Dong, K. Huang, J. Liu and Y. Q. Lan, Chem. Eng. J., 2022, 442, 136157 CrossRef CAS.
- J. Du, Y. Ma, X. Xin, H. Na, Y. Zhao, H. Tan, Z. Han, Y. Li and Z. Kang, Chem. Eng. J., 2020, 398, 125518 CrossRef CAS.
- P. P. Bag, X. S. Wang, P. Sahoo, J. H. Xiong and R. Cao, Catal. Sci. Technol., 2017, 7, 5113–5119 RSC.
- G. Fan, R. X. Li, S. Z. Yang, X. Y. Zhang, H. Y. Jian, J. J. Urban and W.-Y. Sun, Angew. Chem., Int. Ed., 2023, 62, e202216232 CrossRef PubMed.
- J.-P. Dong, Y. Xu, X.-G. Zhang, H. Zhang, L. Yao, R. Wang and S.-Q. Zang, Angew. Chem., Int. Ed., 2023, e202313648 CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: X-ray crystallographic file in CIF format, full experimental and computational details. CCDC 2174429–2174432, 2180762, 2287667, 2287668, 2240657, 2174433–2174435, 2240528, 2174438, 2174437, 2240658 and 2287594. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc06046b |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2023 |