
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licencepara-Selective dearomatization of phenols by I(I)/I(III) catalysis-based fluorination†
Timo
Stünkel
,
Kathrin
Siebold
 ,
Daichi
Okumatsu
,
Kazuki
Murata
,
Louise
Ruyet
,
Constantin G.
Daniliuc
and
Ryan
Gilmour
*
,
Daichi
Okumatsu
,
Kazuki
Murata
,
Louise
Ruyet
,
Constantin G.
Daniliuc
and
Ryan
Gilmour
*
Organisch-Chemisches Institut, Universität Münster, Corrensstraße 36, 48149 Münster, Germany. E-mail: ryan.gilmour@uni-muenster.de
First published on 17th November 2023
Abstract
The regio- and enantio-selective dearomatization of phenols has been achieved by I(I)/I(III) catalysis enabled fluorination. The process is highly para-selective, guiding the fluoride nucleophile to the distal C4 position of the substrate to generate fluorinated cyclohexadienones in an operationally simple manner. Extensive optimization has revealed key parameters that orchestrate enantioselectivity in this historically challenging transformation. A range of diversely substituted substrates are disclosed (20 examples, up to 92![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :8 e.r.) and the reaction displays efficiency that is competitive with the current state of the art in hydroxylation chemistry: this provides a preparative platform to enable OH to F bioisosterism to be explored. Finally, the utility of the products in accessing densely functionalized cyclic scaffolds with five contiguous stereocenters is disclosed together with crystallographic analyses to unveil fluorine-carbonyl non-covalent interactions.
:8 e.r.) and the reaction displays efficiency that is competitive with the current state of the art in hydroxylation chemistry: this provides a preparative platform to enable OH to F bioisosterism to be explored. Finally, the utility of the products in accessing densely functionalized cyclic scaffolds with five contiguous stereocenters is disclosed together with crystallographic analyses to unveil fluorine-carbonyl non-covalent interactions.
Introduction
The prevalence of cyclohexadienones in the bioactive small molecule repertoire has created a powerful impetus to develop effective dearomatization strategies to facilitate direct access from abundant aromatic precursors.1 Striking examples that illustrate the functional diversity of this natural product class include the anti-thrombic agent (+)-rishirilide B (1),2 the anti-inflammatory rengyolone (2),3 and the mammalian DNA polymerase inhibitor dehydroaltenusin (3).4 Function-driven synthesis has also fuelled the development of synthetic (fluorinated) steroids such as the key anti-inflammatory drugs dexamethasone, halometasone and diflupredante (4–6):5 the former (4) is listed in the World Health Organisation's list of essential medicines.6 The remarkable success of fluorinated cyclohexadienones in pharmaceutical development stems from Fried and co-workers studies on cortisol and prednisolone fluorination in the late 1950s.7 Consequently, efficient strategies to generate these important APIs continue to be intensively pursued.5 The societal importance of fluorinated cyclohexadienones led us to explore the feasibility of a C4-regioselective dearomatization of phenols to directly access this class of materials (Scheme 1A). Cognizant of the importance of fluorine in medicinal chemistry and the potency of F to OH bioisosterism,8 it was envisaged that this transformation would be highly enabling and address a deficiency in the synthesis arsenal. To the best of our knowledge, enantioselective I(I)/I(III)-catalysis paradigms to achieve dearomatization at C4 selectively are conspicuously underdeveloped.9 Indeed, the process is limited to stoichiometric racemic protocols using either PIDA/PIFA with Olah's reagent,10 or with Selectfluor® or Accufluor™.11 Whilst this C4 selective variant has proven recalcitrant, I(I)/I(III) catalysis manifolds have a venerable history in the asymmetric dearomatization of phenols. This has enabled a broad range of diverse transformations to be executed with high levels of selectivity, whilst mitigating the need for metal catalysts. However, an important caveat that must be considered in enantioselective reaction development is the formation of phenoxenium cations through the I(III)-mediated oxidation of the phenol, which triggers a competing racemic side reaction.14 | ||
| Scheme 1 (A) Selected examples of cyclohexadienones in bioactive small molecules. (B) Dearomative hydroxylation of phenols under I(I)/I(III) catalysis conditions by Maruoka and co-workers.12 (C) Dearomative fluorination of phenols under phase-transfer conditions by Toste and co-workers.13 (D) Overview of the C4-selective dearomatization of phenols under an I(I)/I(III) catalysis manifold. | ||
Inspired by the hydroxylation-based dearomatization by Maruoka and co-workers (Scheme 1B),12 and the C4 selective fluorination of bicyclic phenols by Toste and co-workers using Selectfluor® under phase-transfer catalysis conditions (Scheme 1C)13,15 we initiated a study to explore the causative factors that orchestrate enantioinduction (Scheme 1D).
Results and discussion
To explore if the I(I)/I(III) protocol was regioselective for the C4 fluorinated isomer over the C2 species, compound 8a was selected as the model substrate for optimization. Reaction conditions that utilized pTolI as an inexpensive organocatalyst were employed,16 thereby enabling pTolIF2 to be generated in situ upon oxidation.17 A systematic study of the reaction conditions identified m-CPBA, CHCl3 and an amine:HF ratio of 1:4.0 to deliver the desired C4 isomer most effectively (please see the ESI† for full details). Variation in the amine:HF ratio proved to be detrimental, with lower ratios leading to sluggish reaction rates, whereas higher ratios led to decomposition and compromised yields. With a racemic, catalysis-based protocol having been established, attention was then focused on rendering the process enantioselective (Scheme 2. Please see the plot of ΔΔG versus yield).
 | ||
| Scheme 2 Catalyst and reaction optimization for the title dearomatization. Standard reaction conditions: phenol (0.2 mmol), catalyst ArI (20 mol%), amine:HF 1:4.0 (0.5 mL), CHCl3 (0.5 mL) and mCPBA (0.3 mmol), 24 h, rt. Yield was determined by 19F NMR using ethylfluoroacetate as the internal standard. Enantiomeric ratio (e.r.) was determined by chiral HPLC.aPerformed with 4.0 eq. mCPBA. bPerformed with 0.35 mL of solvent and 0.65 mL amine:HF 1:4.0. cPerformed with 0.75 mL of solvent and 0.25 mL of amine:HF 1:4.0. | ||
Using a lactate-based resorcinol core, a process of catalyst editing was conducted beginning with modification of the para position. This revealed a trend in efficiency that followed order p-H (11) < p-Me (10) < p-CO2Me (12), with the latter catalyst delivering product 9a with an enantiomeric ratio of 64:36 and a yield of 60%. The influence of modifications to the pendant lactate chain (13–15) were then investigated through a gradual increase in steric bulk. Unfortunately, these alterations led to a slight decrease in enantioselectivity relative to the parent structure 12. Moreover, the introduction of an aromatic Bn (16) moiety also proved detrimental.
The terminal position of the lactate chain was then adjusted through the introduction of various esters and amides (17–27): this was expedited through the development of a novel route to the free diacid of the catalyst via the tBu-ester (please see the ESI† for full details). Gratifyingly, the inclusion of an aromatic benzyl moiety (17) was accompanied by a marked increase in enantioselectivity (68:32 e.r.), but curtailing rotational freedom of the group (18) or expanding the π-system (19) did not confer any further advantage (67:33 and 63:37 e.r., respectively). A switch to simple esters revealed that comparable enantioselectivity could be reached using the iPr derivative (20; 67:33 e.r.), but further enlargement to the adamantyl ester (21) proved detrimental. When studying the influence of substituents with a second stereocenter, it was noted that incorporation of the D-menthol-derived (22) ester led to a boost in enantioselectivity (72:28 e.r.). In contrast, catalyst 23 containing the L-menthol motif gave marginally reduced selectivity (68:32 e.r.). To complete the catalyst editing study, amide-based catalysts were investigated (24 and 25), enabling enantioselectivities of up to 67:33 e.r. to be attained at ambient temperature. Notably, the L-proline-based catalyst (26) provided appreciable enantiocontrol (73:27) whereas the D-proline-based catalyst (27) proved less effective.
Having identified catalyst 26 as being a suitable candidate to facilitate the title transformation, the material was subjected to single crystal X-ray diffraction analysis.18 The perspective shown in Fig. 1 allows the C2-symmetry of the catalyst to be fully appreciated. In further optimizing the catalysis conditions, we were cognizant that the competing racemic pathway involving phenoxenium cation formation had to be suppressed.14 Zhdankin and co-workers have elegantly demonstrated that the stability of I(III) species is greatly influenced by the trans-influence of the ligands.19 Therefore, in an attempt to stabilize the iodonium-substrate complex, methanol was introduced into the reaction mixture to facilitate ligand exchange.19 This led to an increase in enantioselectivity (entry 1, 80:20 e.r.), which is also in line with observations from Ishihara and co-workers,20 but the yield was severely impacted due to methanol acting as a competing nucleophile. Unsurprisingly, similar observations were made with ethanol (entry 2), whereas fluorinated alcohols proved detrimental to both the yield and enantioselectivity of the process (entry 3). Further evaluation of reaction media led us to explore ethers, which have been shown to improve the enantioselectivity of certain I(I)/I(III)-catalyzed fluorination processes.16b,d Gratifyingly, transitioning to Et2O as the solvent (entry 4) resulted in an increased enantioselectivity of 80:20 e.r.: this could be further improved using MTBE (entry 5), leading to an enantiomeric ratio of 82:18. It is interesting to note that the beneficial effect of MTBE on the enantioselectivity was only observed for amide-based catalysts. In contrast, there was almost no change in enantioselectivity when using ester-based catalysts. Moreover, the introduction of ether solvents such as 1,4-dioxane (entry 6) had a negative impact on selectivity.
 | ||
| Fig. 1 Crystal structure of catalyst 26.18 | ||
To further improve the enantioselectivity, the reaction mixture was cooled to 0 °C and stirred for 48 h (entry 7): this enhanced the enantioselectivity to 88:12 e.r. but had a severe impact on conversion. Increasing the amount of mCPBA to 4 eq. (entry 8) enhanced catalysis efficiency, and this could be further improved by modifying the ratio of solvent to amine:HF (0.35 mL MTBE, 0.65 mL amine:HF 1:4.0, entry 9). To further increase enantioselectivity, the reaction mixture was cooled to −5 °C (entry 10) which allowed the product to be obtained in 61% yield with an enantiomeric ratio of 87:13. Moreover, the dependence of enantioselectivity on the ratio of co-solvent to amine:HF could be further leveraged to reach 90:10 e.r. (entry 11). However, due to the reduction in yield observed at lower temperatures, the conditions listed in entry 10 were selected for the remainder of the study. Further cooling of the reaction to −10 °C (entry 12) led to incomplete conversion with only modest improvements in enantioselectivity.
Having identified optimized reaction conditions for the title process, a 2.0 mmol scale transformation was performed which enabled 9a to be formed with the same yield (61%, 83:17 e.r.) which highlights the amenability of this transformation to scale-up (full details in the ESI†). To complement the bromo system 8a, the corresponding halogen series was continued (Scheme 3): this was deemed prudent given the importance of chlorinated cyclohexadienes such as the pharmaceutical halometasone (see Scheme 1A). Exposing substrates 8b and 8c to the catalysis conditions delivered the expected products 9b and 9c with enantiomeric ratios of 87:13 and 85:15, respectively. The positive impact of the electron-withdrawing C2-substituent was fully leveraged by installing a triflate handle, thereby facilitating access 9d with high levels of selectivity (92:8 e.r.). The importance of this substituent is evident from a direct comparison with the ONs derivative 9e (85:15 e.r.). The acetate derivative behaved similarly (9f, 84:16 e.r.) as did the 2,4,5-trisubstiuted phenols 8g and 8h, which furnished 9g and 9h with good levels of selectivity (85:15 e.r.) and in yields of up to 68%. Intriguingly, a switch to 2,3,4-trisubstitution patterns led to higher enantiomeric ratios as exemplified by cyclohexadienes 9i and 9j (up to 92:8 e.r. and up to 69%). Replacing the 4-methyl group with an ethyl group (8a and 8dversus8k and 8l) had no significant impact on enantioselectivity, furnishing product 9l with good levels of selectivity (up to 90:10 e.r.). Gratifyingly, it was possible to obtain crystals of the sulfone derivative 9m, allowing the absolute configuration of the product to be determined as (S) by means of X-ray diffraction.21 It is interesting to note that the C–F bond length of 142 pm is comparable to that of other tertiary C–F bonds.22 To investigate the possibility of phenoxenium cation involvement in a competing racemic side reaction, the Hammett parameters (σp) of selected ortho-substituents from the 4-methyl-subsituted substrates was plotted against ΔΔG.23 This supports the experimental observations that more electron deficient substituents suppress phenoxenium ion formation and this manifests itself in higher levels of enantioselectivity.
 | ||
| Scheme 3 Exploring the scope of the asymmetric dearomatization of phenols. General conditions: phenol (0.2 mmol), 26 (20 mol%), amine:HF 1:4.0 (0.65 mL), MTBE (0.35 mL) and mCPBA (0.8 mmol), 48 h, −5 °C. Yield was determined by 19F NMR using ethylfluoroacetate as the internal standard. Isolated yield given in parenthesis. Enantiomeric ratio (e.r.) was determined by chiral HPLC. Plot of substrate enantioselectivity versus σp. aReaction at 2 mmol scale furnished 9a in 61% yield (by 19F NMR) and 83:17 e.r. bReaction run for 72 h. | ||
To further investigate the functional group tolerance of the reaction conditions, motifs such as Weinreb amides (9n) and TIPS protected acetylenes (9o) were incorporated, enabling the desired 1,4-cyclohexadienones to be generated with 82:18 e.r. and 85:15 e.r., respectively. Given that the asymmetric dearomatization of 3,4-disubstituted phenols is generally challenging,12,24 it was gratifying to observe that products 9p and 9q could be obtained using this method. However, replacing the C4 (Me) substituent by OCF3 (9r) or a pendant alkyl chain (9s) led to a reduction in enantioselectivity. This also holds for C2 aryl substituted systems as is evident from entry 9t. Highly electron deficient substrates such as 8u proved recalcitrant to oxidation. To place these experimental observations on a structural footing, the carbon shift of the substrate para-positions [δ13C(4)] was plotted against the ΔΔG (kcal mol−1); this was performed due to the lack of Hammett parameters for many of the substrate phenols. From this plot, a loose trend can be observed that links higher enantioselectivities with an increased shift of the carbon signal. These data, together with the plot of substrate enantioselectivity versus σp shown in Scheme 3, support the involvement of a dissociative (racemic) pathway competing with the enantioselective process.14 This may explain why the inclusion of this challenging reaction in the ever-expanding I(I)/I(III) dearomatization inventory has not been achieved until now (Scheme 4, bottom), and stimulate further interest in this transformation.
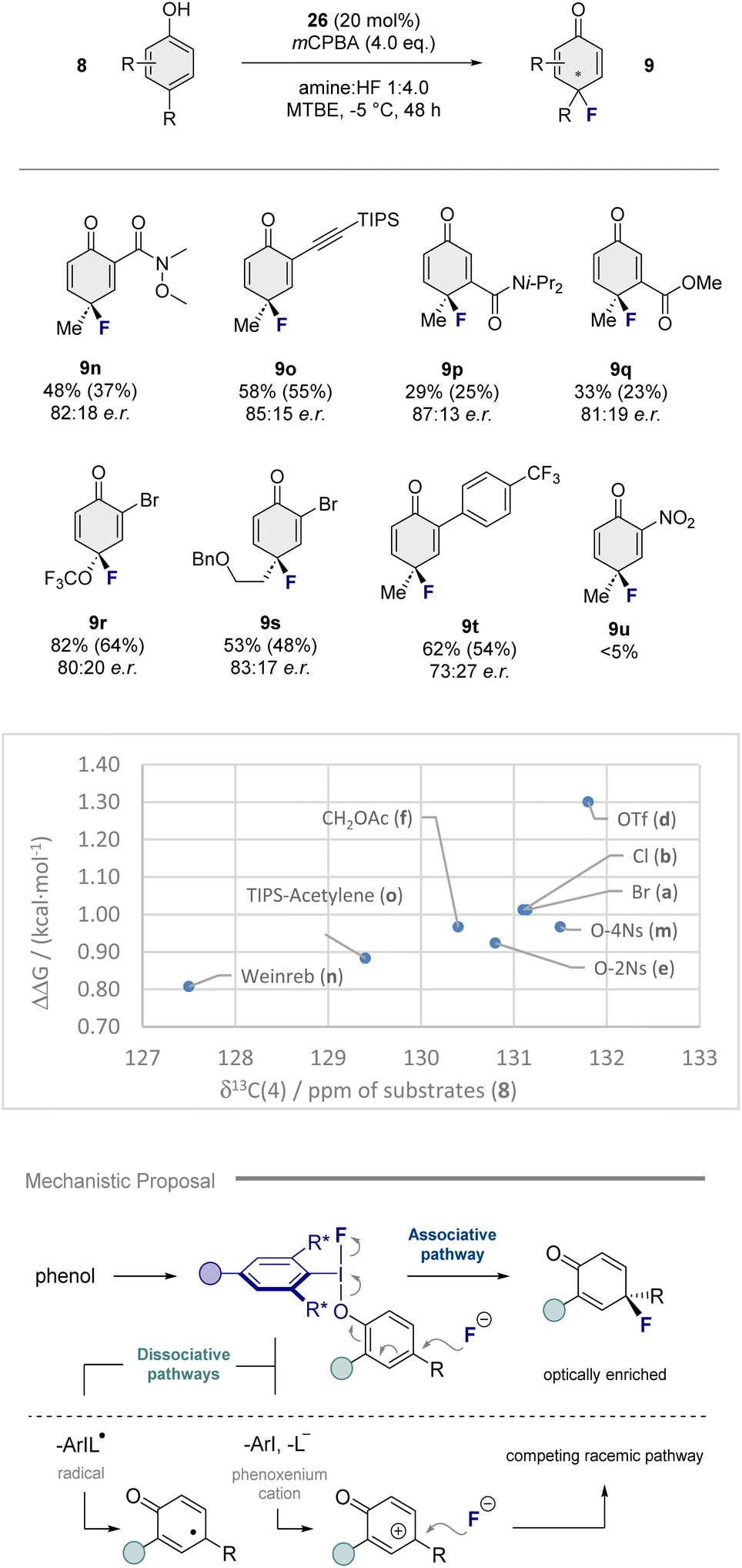 | ||
| Scheme 4 Top: Exploring the functional group compatibility of the transformation. General conditions: phenol (0.2 mmol), 26 (20 mol%), amine:HF 1:4.0 (0.65 mL), MTBE (0.35 mL) and mCPBA (0.8 mmol), 48 h, −5 °C. Yield was determined by 19F NMR using ethylfluoroacetate as the internal standard. Isolated yield given in parenthesis. Enantiomeric ratio (e.r.) was determined by chiral HPLC. Middle: A plot of ΔΔG (kcal mol−1) of the reaction against the carbon shift of the substrate para-positions [δ13C(4)]. Bottom: A postulated mechanistic scenario involving a competing dissociative pathway. | ||
To demonstrate the synthetic utility of the fluorinated cyclohexadienones generated in the study, compound 9a was processed to a set of stereochemically rich products (Scheme 5). Exposure to basic H2O2 proceeded in a highly stereoselective fashion with the methyl group ensuring facial selectivity to generate the di-epoxide 28 as the sole product of the reaction. The relative stereochemistry of the product was unambiguously established by X-ray analysis.25 Inspired by the elegant work by Pirrung and Nunn on the photochemical rearrangements of quinone monoketals,269a was irradiated at 365 nm for 24 h with the intention of inducing a di-π-methane rearrangement.27 Under similar conditions using acetic acid or methanol as the solvent, the sole products of these reactions were the densely substituted cyclopentanones 29 and 30, which were isolated as single diastereoisomers containing five contiguous stereogenic centers. The relative configuration of the product 30 was unequivocally established by single crystal diffraction.28 It is interesting to note that the C(sp3)–F of the cyclopropane occupies the concave face of the molecule and engages the carbonyl group in a manner that is highly reminiscent of n → πC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O* interactions in 1,4-dicarbonyls.29 The distance from the F atom to the C(sp2) atom is 2.556 Å (and 2.956 Å to the center of the bond). This structural analysis contributes to the growing interest in understanding fluorine interactions with proximal carbonyl groups30 and carbocations.31
O* interactions in 1,4-dicarbonyls.29 The distance from the F atom to the C(sp2) atom is 2.556 Å (and 2.956 Å to the center of the bond). This structural analysis contributes to the growing interest in understanding fluorine interactions with proximal carbonyl groups30 and carbocations.31
 | ||
| Scheme 5 Top: Oxidation and photochemical rearrangement of 9a to generate densely substituted ketones 28, 29 and 30 with five contiguous stereocenters. Bottom: Observation of a C(sp3)–F⋯CO interaction (2.556 Å). | ||
Conclusion
Of the plenum of I(I)/I(III) catalysis-based dearomatization methods, the regio- and enantio-selective fluorination of phenols at C4 has proven to be a persistent challenge. Herein, we report a strategy that enables an array of diversely substituted substrates to be processed exclusively to the corresponding (C4) fluorinated cyclohexadienones (20 examples) with good levels of enantioselectivity (up to 92:08 e.r.). The structural trends observed in the course of this work are consistent with competitive (dissociate) pathways involving phenoxenium ions which compromise selectivity. Whilst this can be mitigated by carefully adjusting the reaction conditions, the prominence of this background reaction is likely the major contributory factor that has led to a lack of methods to enable the title transformation. The method complements the current state of the art in hydroxylation-enabled desymmetrization, thereby providing a platform to study the effects of OH to F bioisosterm. We envisage that this will be highly enabling given the venerable history of fluorinated cyclohexadienones in drug discovery, and the challenges associated with generating tertiary fluorides in a more general sense.32 Finally, the synthetic utility of the products is demonstrated through a facile photochemical rearrangement to access fused cyclopentanones with five contiguous stereogenic centers. Structural analysis of this motif reveals an intriguing C(sp3)–F⋯carbonyl interactions in which the distance of the F and C atoms is lower than the sum of the van der Waals radii.
Author contributions
TS and RG designed the project and TS, KS, DO, KM, LR and CGD conducted the experimental work. X-ray crystallographic analyses were performed by CGD. TS, KS and RG wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge generous financial support from the University of Münster, the European Research Council (ERC Consolidator Grant RECON 818949) and the Deutsch Forschungsgemeischaft (DFG, German Research Foundation – GRK2678-437785492). Mr Daichi Okumatsu was a visiting scholar from Osaka University (Japan). Mr Kazuki Murata was a visiting scholar from the Tokyo Institute of Technology (Japan).Notes and references
- (a) S. P. Roche and J. A. Porco Jr, Angew. Chem., Int. Ed., 2011, 50, 4068 CrossRef CAS PubMed; (b) C.-X. Zhuo, W. Zhang and S.-L. You, Angew. Chem., Int. Ed., 2012, 51, 12662 CrossRef PubMed; (c) C. Zheng and S.-L. You, ACS Cent. Sci., 2021, 7, 432 CrossRef CAS PubMed; (d) W.-T. Wu, L. Zhang and S.-L. You, Chem. Soc. Rev., 2016, 45, 1570 RSC.
- J. G. Allen and S. J. Danishefsky, J. Am. Chem. Soc., 2001, 123, 351 CrossRef CAS PubMed.
- J. H. Kim, D. H. Kim, S. H. Baek, H. J. Lee, M. R. Kim, H. J. Kwon and C.-H. Lee, Biochem. Pharmacol., 2006, 71, 1198 CrossRef CAS PubMed.
- (a) J. Cao, Q. Deng, L. Hu, X. Zhang and Y. Xiong, Org. Biomol. Chem., 2022, 20, 8104 RSC; (b) K. Kuramochi, K. Fukudome, I. Kuriyama, T. Takeuchi, Y. Sato, S. Kamisuki, K. Tsubaki, F. Sugawara, H. Yoshida and Y. Mizushina, Bioorg. Med. Chem., 2009, 17, 7227 CrossRef CAS PubMed; (c) N. Maeda, Y. Kokai, S. Ohtani, H. Sahara, I. Kuriyama, S. Kamisuki, S. Takahashi, K. Sakaguchi, F. Sugawara, H. Yoshida, N. Sato and Y. Mizushina, Biochem. Biophys. Res. Commun., 2007, 352, 390 CrossRef CAS PubMed.
- Y. A. Jasem, T. Thiemann, L. Gano and M. C. Oliveira, J. Fluorine Chem., 2016, 185, 48 CrossRef CAS.
- World Health Organization, World Health Organization model list of essential medicines: 22nd list (2021), World Health Organization, 2021, https://iris.who.int/handle/10665/345533, License: CC BY-NC-SA 3.0 IGO Search PubMed.
- J. Fried, A. Borman, W. B. Kessler, P. Grabowich and E. F. Sabo, J. Am. Chem. Soc., 1958, 80, 2338 CrossRef CAS.
- (a) S. Meyer, J. Häfliger and R. Gilmour, Chem. Sci., 2021, 12, 10686 RSC; (b) P. Richardson, Expert Opin. Drug Discovery, 2021, 16, 1261 CrossRef CAS PubMed; (c) I. G. Molnár, C. Thiehoff, M. C. Holland and R. Gilmour, ACS Catal., 2016, 6, 7167 CrossRef; (d) E. P. Gillis, K. J. Eastman, M. D. Hill, D. J. Donnelly and N. A. Meanwell, J. Med. Chem., 2015, 58, 8315 CrossRef CAS PubMed; (e) L. E. Zimmer, C. Sparr and R. Gilmour, Angew. Chem., Int. Ed., 2011, 50, 11860 CrossRef CAS PubMed; (f) S. Purser, P. R. Moore, S. Swallow and V. Gouverneur, Chem. Soc. Rev., 2008, 37, 320 RSC; (g) D. O'Hagan, Chem. Soc. Rev., 2008, 37, 308 RSC; (h) K. Müller, C. Faeh and F. Diederich, Science, 2007, 317, 1881 CrossRef PubMed.
- (a) F. V. Singh, S. E. Shetgaonkar, M. Krishnan and T. Wirth, Chem. Soc. Rev., 2022, 51, 8102 RSC; (b) A. Parra, Chem. Rev., 2019, 119, 12033 CrossRef CAS PubMed; (c) A. Yoshimura and V. V. Zhdankin, Chem. Rev., 2016, 116, 3328 CrossRef CAS PubMed.
- (a) O. Karam, J.-C. Jacquesy and M.-P. Jouannetaud, Tetrahedron Lett., 1994, 35, 2541 CrossRef CAS; (b) O. Karam, A. Martin, M.-P. Jouannetaud and J.-C. Jacquesy, Tetrahedron Lett., 1999, 40, 4183 CrossRef CAS; (c) O. Karam, A. Martin-Mingot, M.-P. Jouannetaud, J.-C. Jacquesy and A. Cousson, Tetrahedron, 2004, 60, 6629 CrossRef CAS.
- (a) S. Stavber and M. Zupan, Synlett, 1996, 693 CrossRef CAS; (b) S. Stavber, M. Jereb and M. Zupan, Synlett, 1999, 1375 CrossRef CAS; (c) S. Stavber, M. Jereb and M. Zupan, J. Phys. Org. Chem., 2002, 15, 56 CrossRef CAS; (d) I. Pravst, M. P. Iskra, M. Jereb, M. Zupan and S. Stavber, Tetrahedron, 2006, 62, 4474 CrossRef CAS.
- T. Hashimoto, Y. Shimazaki, Y. Omatsu and K. Maruoka, Angew. Chem., Int. Ed., 2018, 57, 7200 CrossRef CAS PubMed.
- R. J. Phipps and F. D. Toste, J. Am. Chem. Soc., 2013, 135, 1268 CrossRef CAS PubMed.
- For selected examples, see: (a) A. Juneau, I. Lepage, S. G. Sabbah, A. H. Winter and M. Frenette, J. Org. Chem., 2022, 87, 14274 CrossRef CAS PubMed; (b) K. Kraszewski, I. Tomczyk, A. Drabinska, K. Bienkowski, R. Solarska and M. Kalek, Chem. – Eur. J., 2020, 26, 11584 CrossRef CAS PubMed; (c) T. Tang and A. M. Harned, Org. Biomol. Chem., 2018, 16, 8249 RSC; (d) T. Dohi, A. Maruyama, N. Takenaga, K. Senami, Y. Minamitsuji, H. Fujioka, S. B. Caemmerer and Y. Kita, Angew. Chem., Int. Ed., 2008, 47, 3787 CrossRef CAS PubMed; (e) A. Pelter, A. Hussain, G. Smith and R. S. Ward, Tetrahedron, 1997, 53, 3879 CrossRef CAS.
- For examples of C2-selective fluorinative dearomatization of phenols and related structures, see: (a) P. Wang, J. Wang, L. Wang, D. Li, K. Wang, Y. Liu, H. Zhu, X. Liu, D. Yang and R. Wang, Adv. Synth. Catal., 2018, 360, 401 CrossRef CAS; (b) H. Egami, T. Rouno, T. Niwa, K. Masuda, K. Yamashita and Y. Hamashima, Angew. Chem., Int. Ed., 2020, 59, 14101 CrossRef CAS PubMed; (c) M. Otsubo, K. Sakimoto, H. Egami and Y. Hamashima, Tetrahedron, 2021, 96, 132355 CrossRef CAS; (d) M. Thiele, T. Rose, M. Lõkov, S. Stadtfeld, S. Tshepelevitsh, E. Parman, K. Opara, C. Wölper, I. Leito, S. Grimme and J. Niemeyer, Chem. – Eur. J., 2023, 29, e202202953 CrossRef CAS PubMed.
- For selected examples, see: (a) Z.-X. Wang, K. Livingstone, C. Hümpel, C. G. Daniliuc and R. Gilmour, Nat. Chem., 2023, 15, 1515–1522 CrossRef CAS PubMed; (b) J. Häfliger, L. Ruyet, N. Stübke, C. G. Daniliuc and R. Gilmour, Nat. Commun., 2023, 14, 3207 CrossRef PubMed; (c) Y.-J. Yu, J. Häfliger, Z.-X. Wang, C. G. Daniliuc and R. Gilmour, Angew. Chem., Int. Ed., 2023, 62, e202309789 CrossRef CAS PubMed; (d) Y.-J. Yu, M. Schäfer, C. G. Daniliuc and R. Gilmour, Angew. Chem., Int. Ed., 2023, 62, e202214906 CrossRef CAS PubMed; (e) J. Häfliger, O. O. Sokolova, M. Lenz, C. G. Daniliuc and R. Gilmour, Angew. Chem., Int. Ed., 2022, 61, e202205277 CrossRef PubMed; (f) M. Schäfer, T. Stünkel, C. G. Daniliuc and R. Gilmour, Angew. Chem., Int. Ed., 2022, 61, e202205508 CrossRef PubMed; (g) S. Meyer, J. Häfliger, M. Schäfer, J. J. Molloy, C. G. Daniliuc and R. Gilmour, Angew. Chem., Int. Ed., 2021, 60, 6430 CrossRef CAS PubMed; (h) F. Scheidt, M. Schäfer, J. C. Sarie, C. G. Daniliuc, J. J. Molloy and R. Gilmour, Angew. Chem., Int. Ed., 2018, 57, 16431 CrossRef CAS PubMed; (i) I. G. Molnár and R. Gilmour, J. Am. Chem. Soc., 2016, 138, 5004 CrossRef PubMed.
- J. C. Sarie, C. Thiehoff, R. J. Mudd, C. G. Daniliuc, G. Kehr and R. Gilmour, J. Org. Chem., 2017, 82, 11792 CrossRef CAS PubMed.
- X-ray data for 26 CCDC 2288991.†.
- (a) V. V. Zhdankin, R. M. Arbit, B. J. Lynch, P. Kiprof and V. G. Young, J. Org. Chem., 1998, 63, 6590 CrossRef CAS; (b) V. V. Zhdankin, R. M. Arbit, M. McSherry, B. Mismash and V. G. Young, J. Am. Chem. Soc., 1997, 119, 7408 CrossRef CAS; (c) M. Ochiai, T. Sueda, K. Miyamoto, P. Kiprof and V. V. Zhdankin, Angew. Chem., Int. Ed., 2006, 45, 8203 CrossRef CAS PubMed.
- M. Uyanik, T. Yasui and K. Ishihara, Angew. Chem., Int. Ed., 2013, 52, 9215 CrossRef CAS PubMed.
- X-ray data for 9m CCDC 2288990.†.
- F. H. Allen, O. Kennard, D. G. Watson, L. Brammer, A. G. Orpen and R. Taylor, J. Chem. Soc., 1987, S1–S19 CAS.
- C. Hansch, A. Leo and R. W. Taft, Chem. Rev., 1991, 91, 165 CrossRef CAS.
- K. Muñiz and L. Fra, Synthesis, 2017, 49, 2901 CrossRef.
- X-ray data for 28 CCDC 2288992.†.
- M. C. Pirrung and D. S. Nunn, Tetrahedron, 1996, 52, 5707 CrossRef CAS.
- (a) H. E. Zimmerman and D. Armesto, Chem. Rev., 1996, 96, 3065 CrossRef CAS PubMed; (b) H. E. Zimmerman and J. M. Cassel, J. Org. Chem., 1989, 54, 3800 CrossRef CAS; (c) S. S. Hixson, P. S. Mariano and H. E. Zimmerman, Chem. Rev., 1973, 73, 531 CrossRef CAS.
- X-ray data for 30 CCDC 2288993.†.
- (a) K. Livingstone, K. Siebold, S. Meyer, V. Martín-Heras, C. G. Daniliuc and R. Gilmour, ACS Catal., 2022, 12, 14507 CrossRef CAS PubMed; (b) T. Neveselý, J. J. Molloy, C. McLaughlin, L. Brüss, C. G. Daniliuc and R. Gilmour, Angew. Chem., Int. Ed., 2022, 61, e202113600 CrossRef PubMed.
- S. A. Harry, M. Kazim, P. M. Nguyen, A. Zhu, M. R. Xiang, J. Catazaro, M. Siegler and T. Lectka, Angew. Chem., Int. Ed., 2022, 61, e202207966 CrossRef CAS PubMed.
- (a) K. F. Hoffmann, A. Wiesner, C. Müller, S. Steinhauer, H. Beckers, M. Kazim, C. R. Pitts, T. Lectka and S. Riedel, Nat. Commun., 2021, 12, 5275 CrossRef CAS PubMed; (b) M. D. Struble, M. G. Holl, M. T. Scerba, M. A. Siegler and T. Lectka, J. Am. Chem. Soc., 2015, 137, 11476 CrossRef CAS PubMed; (c) M. D. Struble, M. T. Scerba, M. A. Siegler and T. Lectka, Science, 2013, 340, 57 CrossRef CAS PubMed.
- (a) J.-A. Ma and D. Cahard, Chem. Rev., 2004, 104, 6119 CrossRef CAS PubMed; (b) V. A. Brunet and D. O'Hagan, Angew. Chem., Int. Ed., 2008, 47, 1179 CrossRef CAS PubMed; (c) Y. Zhu, J. Han, J. Wang, N. Shibata, M. Sodeoka, V. A. Soloshonok, J. A. S. Coelho and F. D. Toste, Chem. Rev., 2018, 118, 3887 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2288990–2288993. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc05952a |
| This journal is © The Royal Society of Chemistry 2023 |