
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceChiral cobalt(II) complex-promoted asymmetric para-Claisen rearrangement of allyl α-naphthol ethers†
Hongkun
Zeng
,
Lifeng
Wang
,
Zhishan
Su
 ,
Meijia
Ying
,
Lili
Lin
* and
Xiaoming
Feng
*
,
Meijia
Ying
,
Lili
Lin
* and
Xiaoming
Feng
*
Key Laboratory of Green Chemistry & Technology, Ministry of Education, College of Chemistry, Sichuan University, Chengdu 610064, China. E-mail: lililin@scu.edu.cn; xmfeng@scu.edu.cn
First published on 27th November 2023
Abstract
Due to experiencing a challenging dearomatization process, the aromatic sigmatropic rearrangement of allyl naphthyl ethers is a difficult yet efficient method to build useful naphthalenone skeletons. Here, we report a para-Claisen rearrangement-based asymmetric dearomatization of allyl α-naphthol ethers enabled by a N,N′-dioxide/CoII complex. A variety of naphthalenones were obtained in moderate to good yields with good to excellent ee values. Interestingly, by exchanging the allyl group on the ether and that at the para-position of the benzene ring, enantiodivergent synthesis can be achieved. Experimental studies and DFT calculations revealed that aryl allyl ethers tend to transform via a stepwise allyl π-complex migration pathway, while, alkyl allyl ethers transformed through a concerted ortho-Claisen rearrangement/Cope rearrangement sequence.
Introduction
The chiral naphthalenone skeleton is widely found in natural products and fungal species (Scheme 1a).1 For example, pereniporins A, isolated from a fungus Perenniporia sp. inhabiting the larva of Euops chinesis, shows significant antifungal activity against multiple plant pathogens.2 (+)-Rishirilides B, isolated from the culture broth of Streptomyces strain, Streptomyces rishiriensis OFR-1056, can effectively inhibit plasma α2-macroglobulin.3 (−)-5-Hydroxy-4-methoxy-1-tetralone was isolated from the roots of Engelhardia roxburghiana, exhibiting antitubercular activity.4 These compounds all have a naphthalenone skeleton and possess a chiral centre at the C4-position.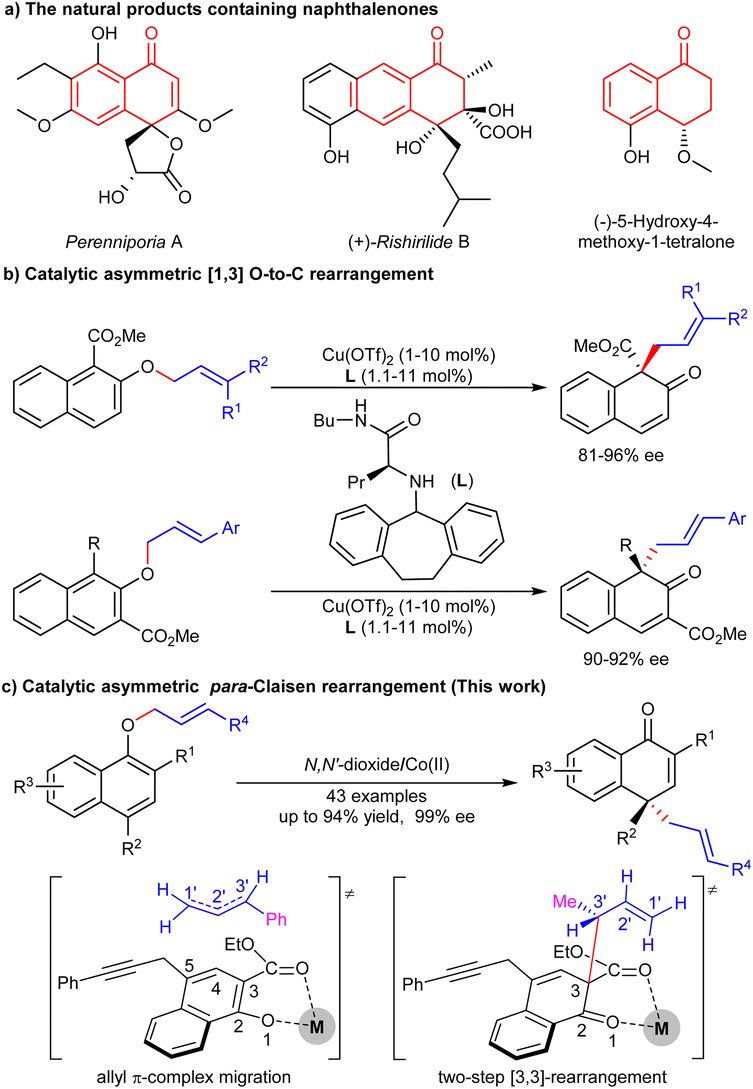 | ||
| Scheme 1 Catalytic asymmetric rearrangements of allyl naphthyl ethers. | ||
Asymmetric dearomatization of naphthol derivatives is an efficient method to build chiral naphthalenone skeletons.5 However, compared to well-established oxidative6 or transition metal mediated7 dearomatizations, rearrangement-based8 dearomatizations are relatively less explored. The aromatic [n,m]-sigmatropic rearrangement of allyl or propargyl naphthyl ethers can rapidly construct allyl or propargyl functionalized naphthalenones.9 Bandini developed a gold-catalyzed dearomatization of 2-naphthols with alkynes, and an enantioselective investigation afforded chiral dihydrofurylnaphthalen-2(1H)-one in 81% yield with 65% ee.10 Recently, our group reported a thermal [3,3]-sigmatropic rearrangement of naphthyl 1-propargyl ethers, and the enantioselective version was achieved by a chiral cobalt catalysis at 70 °C.11 For the rearrangement of allyl naphthol ethers, Ishihara and Yao used a chiral α-amino amide/Cu(II) catalyst to achieve the asymmetric [1,3] O-to-C dearomative rearrangement of alkyl 2-allyloxy-1/3-naphthoates (Scheme 1b).12 If allyl α-naphthol ethers were applied, ortho-Claisen/Cope rearrangement or an allyl π-complex migration process will occur,13 achieving para-Claisen rearrangement.14 The difficulty lies in the remote enantioselective control.
Considering that the catalytic asymmetric rearrangement-based dearomatizations are limited and further research is necessary, herein, we report a chiral cobalt(II)/N,N′-dioxide complex-promoted asymmetric para-Claisen rearrangement of allyl α-naphthol ethers. A variety of chiral naphthalenones bearing a chiral all-carbon quaternary centre were obtained in good yields with excellent ee values under mild conditions (Scheme 1c).
Results and discussion
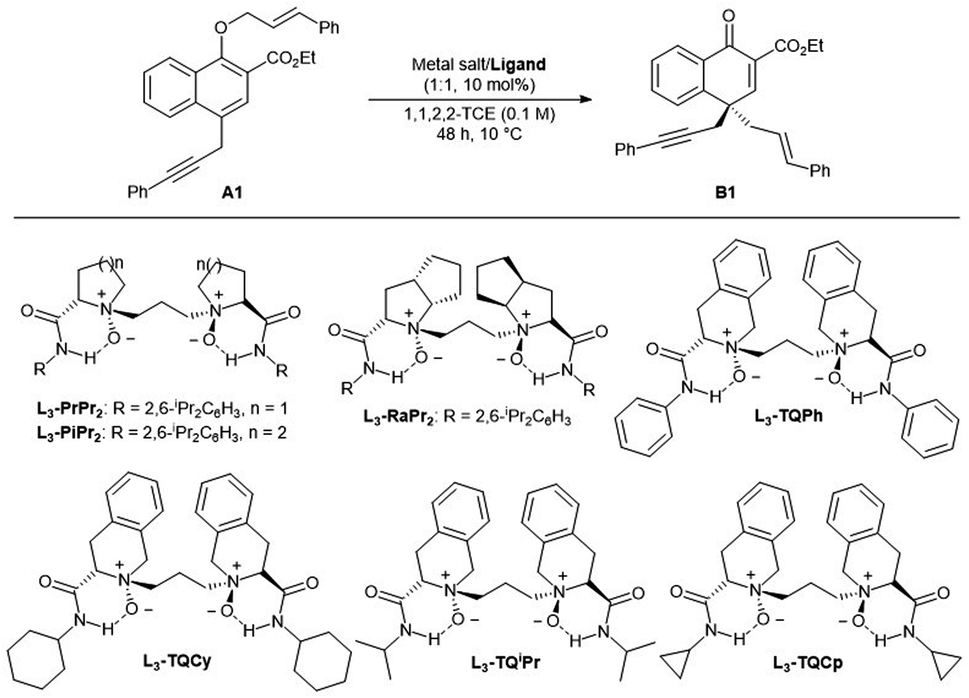
At the outset, ethyl 1-(cinnamyloxy)-4-propargyl-2-naphthoate A1 was selected as the model substrate to optimize the reaction conditions (Table 1). First, various chiral N,N′-dioxide ligands were evaluated by complexing with Co(BF4)2·6H2O in 1,1,2,2-TCE at 10 °C. It was found that the chiral skeleton affected greatly the reactivity. L-Proline derived L3-PrPr2, L-pipecolic acid derived L3-PiPr2 and L-ramipril-derived L3-RaPr2 gave very low yields albeit with moderate enantioselectivity, while (S)-tetrahydroisoquinoline-3-carboxylic acid-derived L3-TQPh could give 73% yield with 74% ee (entries 1–4). The amide moiety in the ligand, generally shielding one of the faces of prochiral substrates, also played a very important role in enantioselectivity. Switching the substituent of the amide moiety from rigid phenyl to the flexible cyclohexyl led to better yield and enantioselectivity (entry 5, 78% yield, 82% ee). When the substituent of amide was the isopropyl or cyclopropyl group, the reactivity and selectivity were further improved (entries 6–7). When the temperature was raised to 35 °C, the substrate decomposed and enantioselectivity decreased (entry 8). The solvent affected the reaction greatly. Altering 1,1,2,2-TCE to CH2Cl2 resulted in a decreased enantioselectivity (69% ee, entry 9). When THF was used as the solvent, no reaction occurred (entry 10), which might be attributed to the competitive coordination with the catalyst between the solvent and substrate. Examination of different metal salts by complexing with L3-TQCp showed that Co(OTf)2, Zn(OTf)2 and Mg(OTf)2 could also promote the reaction, and the enantioselectivities decreased a little (entries 11–13).|
|
||||
|---|---|---|---|---|
| Entry | Ligand | Metal salt | Yieldb [%] | eec [%] |
a Unless otherwise noted, all reactions were carried out with A1 (0.05 mmol) and ligand/metal salt (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, 10 mol%) in 1,1,2,2-TCE (0.1 M) at 10 °C for 48 h without the protection of inert gas.
b Yield of the isolated product.
c Determined by HPLC analysis on a chiral stationary phase.
d At 35 °C.
e CH2Cl2 (0.5 ml) as solvent.
f THF (0.5 ml) as solvent. 1,1,2,2-TCE = 1,1,2,2-tetrachloroethane. NR = no reaction. :1, 10 mol%) in 1,1,2,2-TCE (0.1 M) at 10 °C for 48 h without the protection of inert gas.
b Yield of the isolated product.
c Determined by HPLC analysis on a chiral stationary phase.
d At 35 °C.
e CH2Cl2 (0.5 ml) as solvent.
f THF (0.5 ml) as solvent. 1,1,2,2-TCE = 1,1,2,2-tetrachloroethane. NR = no reaction.
|
||||
| 1 | L3-PrPr2 | Co(BF4)2·6H2O | <5 | 63 |
| 2 | L3-PiPr2 | Co(BF4)2·6H2O | 13 | 57 |
| 3 | L3-RaPr2 | Co(BF4)2·6H2O | <5 | 55 |
| 4 | L3-TQPh | Co(BF4)2·6H2O | 73 | 74 |
| 5 | L3-TQCy | Co(BF4)2·6H2O | 78 | 82 |
| 6 | L3-TQiPr | Co(BF4)2·6H2O | 87 | 85 |
| 7 | L3-TQCp | Co(BF4)2·6H2O | 86 | 92 |
| 8d | L3-TQCp | Co(BF4)2·6H2O | <5 | 36 |
| 9e | L3-TQCp | Co(BF4)2·6H2O | 83 | 69 |
| 10f | L3-TQCp | Co(BF4)2·6H2O | NR | — |
| 11 | L3-TQCp | Co(OTf)2 | 83 | 86 |
| 12 | L3-TQCp | Zn(OTf)2 | 89 | 85 |
| 13 | L3-TQCp | Mg(OTf)2 | 90 | 89 |
With the optimized reaction conditions in hand (Table 1, entry 7), the substrate scope was explored. As illustrated in Scheme 2, the bulkier ester group was favorable for the enantioselectivity (B1–B4). Methyl ester reduced the ee value to 81%, while isopropyl ester and phenyl ester increased the enantioselectivity to 97% ee. A gram-scale synthesis of B1 by applying 3 mmol A1 furnished smoothly 1.25 g of the desired product B1 in 93% yield with 91% ee. In the absence of an ester group or when the ester group (R1) was altered to others (A41–A45), such as amide and CN, no reaction occurred. When the ester group was altered to the aldehyde or ketone group, the reaction mixture was complex, or the enantioselectivity was very low. The results demonstrated the importance of the ester group (R1), whose roles might be that the coordination ability of the ester group helped the substrates to coordinate with the catalyst in a tight bidentate manner, facilitating the C–O bond cleavage to undergo the rearrangement process, and also fixing the substrate to benefit the enantioselective control. In addition, the electron-withdrawing properties of the ester group might also be helpful for the stabilization of the transition state. Propargyl (A5), methyl ethyl ether (A6), benzyl (A7), halide (Cl, A8) and propyl (A9) groups were well tolerated on the para-position of allyl α-naphthol ether, affording the desired products B5–B9 in moderate to high yields with excellent ee values. When the cyclopropyl group (A10) or methyl ester (A11) was on the para-position, the reactivity and enantioselectivity decreased (B10 and B11). A12 with the ethoxy group transformed in only 30% yield with 5% ee. The substitution at the naphthyl ring was also investigated. Electron-donating groups, such as Me, OMe, Ph, t-Bu, even halogen Cl at the 6-position had no obvious effect on the yield and enantioselectivity (B13–B17). The electron-withdrawing group (CN) decreased the enantioselectivity (80% ee, B18). Furthermore, introducing a methyl group at the 8-position resulted in a reduced ee value (60% ee, B19), possibly due to the larger steric hindrance.
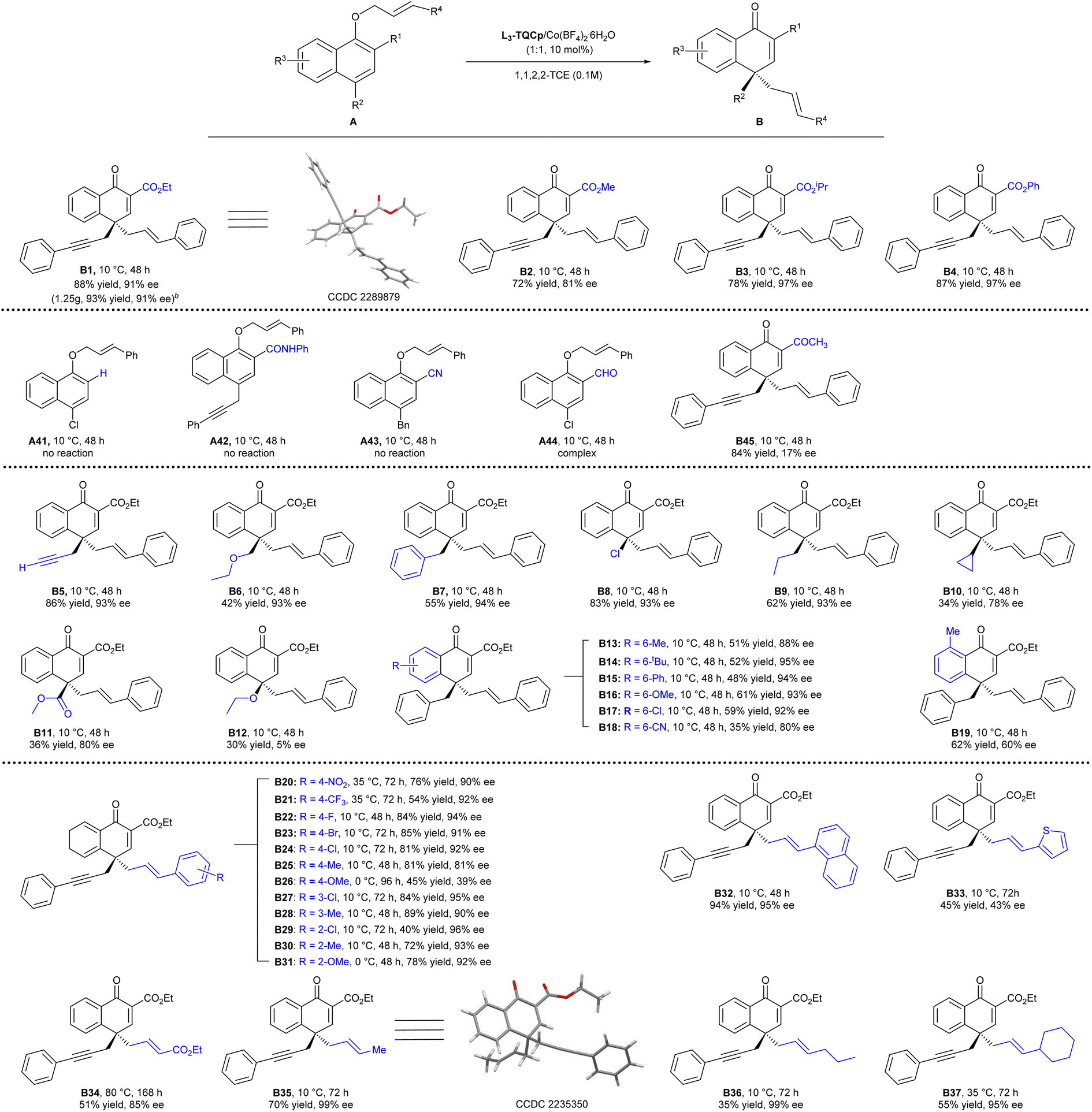 | ||
| Scheme 2 Substrate scopea. [a] Unless otherwise noted, all reactions were carried out with A (0.1 mmol) and L3-TQCp/Co(BF4)2·6H2O (1:1, 10 mol%) in 1,1,2,2-TCE (0.1 M). Yields of the isolated product. ee was determined by HPLC or UPC2 analysis on a chiral stationary phase. [b] 3.0 mmol A1 was used. | ||
A range of aryl allyl bearing either electron-donating or electron-withdrawing groups at the ortho, meta, or para position of the arene moiety were also examined. Substrates with electron-withdrawing groups at the para-position transformed in good yields with excellent enantioselectivities (B20–B24, 54–85% yield, 90–94% ee). The enantioselectivities decreased when electron-donating groups were at the para-position (B25, B26, 39–81% ee), which might be caused by a stronger background reaction due to the higher reactivity of substrates. The low yield of B26 was caused by the decomposition of A26 under the reaction conditions. Substitutes at the ortho or meta position had a little impact on the enantioselectivity, and all the tested substrates yielded the desired products with excellent ee values (B27–B31).
The 1-naphthyl allyl substrate transformed into the product B32 in 94% yield with 95% ee. Product B33 with the allyl heteroarene group was obtained in moderate yield with a moderate ee value. Moreover, unactivated allyl groups were also suitable in the asymmetric para-Claisen rearrangement. Ethyl but-2-enoate-substituted allylic ether A34 bearing an ester moiety transformed into the targeted product B34 in 51% yield with 85% ee. A35, A36 and A37 with aliphatic functional groups, furnishing the desired products B35–B37 in excellent ee values (95–99% ee) albeit with moderate yields. The absolute configurations of the products B1 and B35 were determined to be (R) by X-ray crystallography analysis.
para-Allyl substrates were compatible in the present system, giving the desired products B38, B39, and B40 in excellent ee values. Interestingly, their enantiomers (ent)-B38, (ent)-B39 and (ent)-B40 could be obtained with the same ee values by exchanging the allyl group on the ethers and that at the C4-position, which gave an interesting entry to enantiodivergent synthesis (Scheme 3).
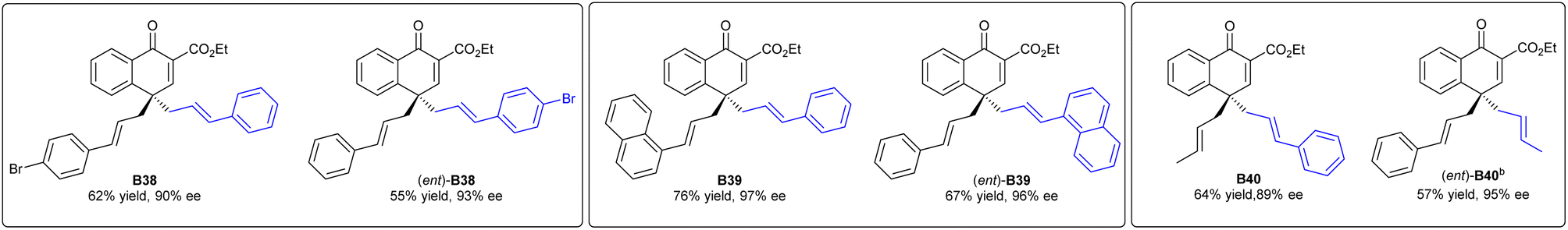 | ||
| Scheme 3 Substrate-controlled enantiodivergent synthesisa. [a] Unless otherwise noted, all reactions were carried out with A (0.1 mmol) and L3-TQCp/Co(BF4)2·6H2O (1:1, 10 mol%) in 1,1,2,2-TCE (0.1 M), at 10 °C for 48 h. Yields of the isolated product. ee was determined by HPLC analysis on a chiral stationary phase. [b] At 20 °C. | ||
To gain insight into the mechanism, the relationship between the enantiomeric excess of the product and catalyst was studied. As shown in Fig. 1a, a clear linearity was observed, indicating that the species formed by the coordination of CoII and the chiral N,N′-dioxide ligand in a 1:1 ratio might be the catalytically active species. In addition, the kinetic studies showed a first-order kinetic dependence on the chiral L3-TQCp/Co(BF4)2·6H2O catalyst and A1 (Fig. 1b). A crossover experiment was conducted using A8 and A32 as substrates (Fig. 1c). Products B8 and B32 were obtained under Lewis acid accelerated or thermal mediated conditions, and crossover products were not observed. Both kinetic studies and crossover experiment indicated that the rearrangement might involve an intramolecular reaction.
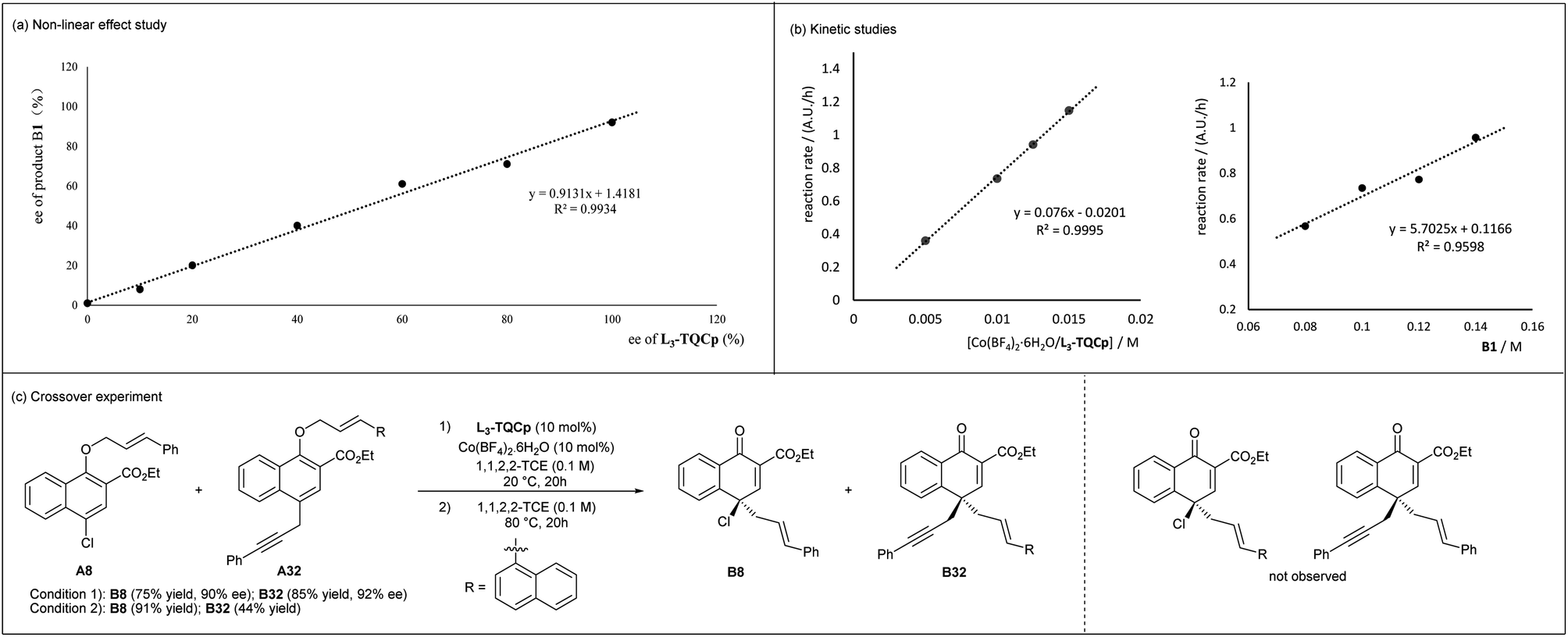 | ||
| Fig. 1 Exploration on the mechanism. | ||
In addition, to illustrate the rearrangement pathway, a density functional theory (DFT) study was performed at the B3LYP-D3/6-31G(d,p) (SMD, CCl4) level of theory with Zn(OTf)2 as the catalyst because Zn(II) gave similar results to Co(II), meanwhile reducing the computational difficulty (Fig. 2). It was revealed that the conversion from IM1-Ph to IM4-Ph involved an allyl π-complex migration mechanism (Fig. 2a). A tight ion-pair intermediate (IM2-Ph) was formed via the transition state TS1-Ph, accompanied by the cleavage of the C1′–O bond. The ΔG≠ in this step was 8.6 kcal mol−1. Then, IM2-Ph underwent a conformation change and transformed into a more stable intermediate IM3-Ph. In the following step, the C1′–C5 bond in IM4-Ph was constructed through ion-pair coupling viaTS2-Ph, with a ΔG≠ of only 1.3 kcal mol−1. For comparison, we also studied the C3–C3′ coupling viaTS2-Ph-a. The ΔG≠ of TS2-Ph-a was 1.3 kcal mol−1 less stable than that of TS2-Ph, indicating that the cation fragment preferred to interact with the C5 atom of the Zn(II)-coordinated anion for IM4-Ph.
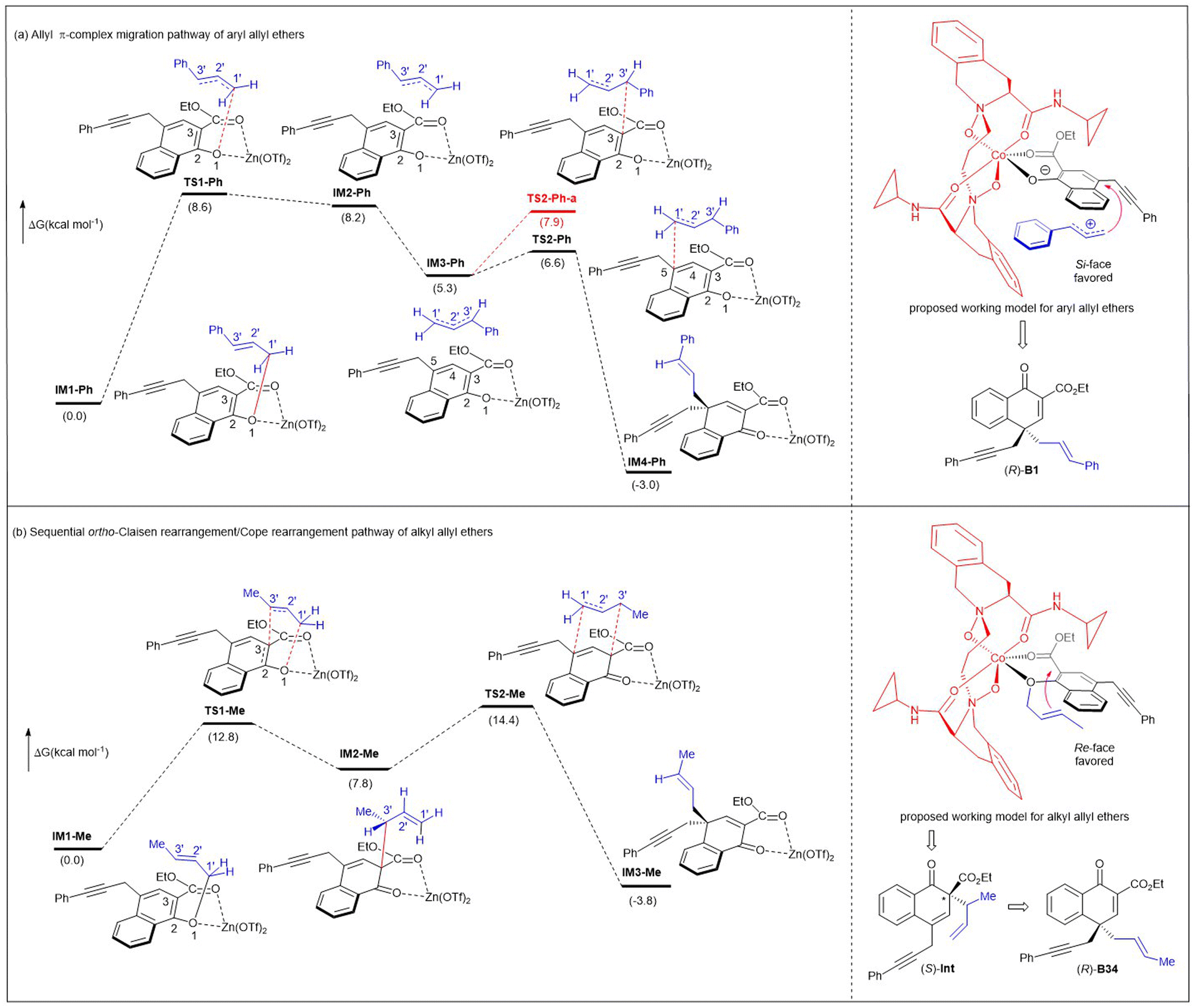 | ||
| Fig. 2 DFT calculations on rearrangement pathways and proposed asymmetric induction models for both aryl and alkyl allyl ethers. | ||
Comparatively, when the alkyl Me group replaces the aryl Ph group that can stabilize allyl cations, the conversion from IM1-Me to IM3-Me involved sequential ortho-Claisen rearrangement/Cope rearrangement (Fig. 2b). During these processes, two six-membered ring transition states (TS1-Me and TS2-Me) were generated, with the activation barriers of 12.8 kcal mol−1 and 6.6 kcal mol−1, respectively. The reaction was exergonic by 3.8 kcal mol−1. The energy barrier associated with the formation of ion-pair coupling was evaluated by a relaxed scan of the C1′–O bond in IM1-Me′ (from 1.502 to 3.022 Å). The corresponding ΔE≠ was 20.7 kcal mol−1 (see Fig. S5 in ESI† for more details).
In addition, combining our previous studies on N,N′-dioxide/metal catalysis,15,16 we proposed two catalytic modes to explain the stereochemistry of the reaction. Chiral N,N′-dioxide and naphthyl 1-allyl ether coordinated to Co(II) in tetradentate and bidentate fashions, respectively, to form a slightly distorted hexahedral complex. Aryl allyl ether A1 converts to a tight ion-pair intermediate with the assistance of the N,N′-dioxide/Co(II) complex. Since the Re-face of the enolate anion is shielded by the amide moiety of the ligand, the allyl cation attacks from its Si-face to give (R)-B1. For alkyl allyl ether A35, the allyl unit faces to the right-bottom with less steric hindrance, so it tends to approach the enolate ether from the Re-face, undergoing an ortho-Claisen rearrangement to generate the chiral (S)-Int, and then undergoing rapid Cope rearrangement to provide more stable (R)-B35.
Conclusions
In summary, we have successfully developed an efficient catalytic asymmetric para-Claisen rearrangement of naphthyl 1-allyl ethers by using a chiral N,N′-dioxide/Co(II) complex as the catalyst, providing a facile and efficient route to synthesize valuable naphthalenones with a chiral center at the C4-position under mild reaction conditions. Notably, exchanging the allyl groups at the para-position and on the ethers, substrate-controlled enantiodivergent synthesis can be achieved. DFT calculations revealed that aryl allyl ethers that can stabilize allyl cations tend to experience an allyl π-complex migration process, while alkyl allyl ethers involve concerted ortho-Claisen rearrangement/Cope rearrangement sequence processes.Data availability
Further details of the experimental procedure, 1H, 13C{1H} and 19F{1H} NMR, HPLC spectra and X-ray crystallographic data are available in the ESI.†Author contributions
H. K. Z. preformed the experiments. M. J. Y. repeated the data. Z. S. S. conducted the DFT calculation. X. M. F., L. L. L. and L. F. W. supervised the project. H. K. Z. and L. L. L. co-wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We appreciate the National Natural Science Foundation of China (no. 22171189 and 22188101), Sichuan Science and Technology Program (No. 2021YJ0561), and Fundamental Research Funds for the Central Universities for financial support. We are grateful to Dr Yuqiao Zhou from the College of Chemistry, Sichuan University for the X-ray single crystal diffraction analysis.Notes and references
- S. R. M. Ibrahim, S. A. Fadil, H. A. Fadil, B. A. Eshmawi, S. G. A. Mohamed and G. A. Mohamed, Toxins, 2022, 14, 154 CrossRef CAS PubMed.
- Y. Feng, L. Wang, S. B. Niu, L. Li, Y. k. Si, X. Z. Liu and Y. S. Che, J. Nat. Prod., 2012, 75, 1339 CrossRef CAS PubMed.
- H. Iwaki, Y. Nakayama, M. Takahashi, S. Uetsuki, M. Kido and Y. Fukuyama, J. Antibiot., 1984, 37, 1091 CrossRef CAS PubMed.
- (a) W. Y. Liu, C. F. Peng, L. L. Tsai, J. J. Chen, M. J. Cheng and I. S. Chen, Planta Med., 2005, 71, 171 CrossRef PubMed; (b) D. Chanda, D. Saikia, J. K. Kumar, J. P. Thakur, J. Agarwal, C. S. Chanotiya, K. Shanker and A. S. Negi, Bioorg. Med. Chem. Lett., 2011, 21, 3966 CrossRef CAS PubMed.
- For reviews, see: (a) C. X. Zhuo, C. Zheng and S. L. You, Acc. Chem. Res., 2014, 47, 2558 CrossRef CAS PubMed; (b) W. T. Wu, L. Zhang and S. L. You, Chem. Soc. Rev., 2016, 45, 1570 RSC; (c) J. Liu, J. T. Chen, T. T. Liu, J. Liu and Y. F. Zeng, Chin. J. Org. Chem., 2023, 43, 379 CrossRef; (d) For selected examples, see: X. H. Liu, J. Y. Zhang, L. T. Bai, L. Q. Wang, D. X. Yang and R. Wang, Chem. Sci., 2020, 11, 671 RSC; (e) H. Egami, T. Rouno, T. Niwa, K. Masuda, K. Yamashita and Y. Hamashima, Angew. Chem., Int. Ed., 2020, 59, 14101 CrossRef CAS PubMed; (f) B. G. Das, S. Shah, A. Das and V. K. Singh, Org. Lett., 2021, 23, 6262 CrossRef CAS PubMed; (g) U. Maji, B. D. Mondal and J. Guin, Org. Lett., 2023, 25, 2323 CrossRef CAS PubMed; (h) C. Parida, S. K. Dave, K. Das and S. C. Pan, Adv. Synth. Catal., 2023, 365, 1185 CrossRef CAS.
- For reviews, see: (a) R. Kumar, F. V. Singh, N. Takenaga and T. Dohi, Chem.–Asian J., 2022, 17, e202101115 CrossRef CAS PubMed; (b) M. Okumura and D. Sarlah, Eur. J. Org Chem., 2020, 10, 1259 CrossRef PubMed; (c) X. Xiao and S. E. Wengryniuk, Synlett, 2021, 32, 752 CrossRef CAS PubMed; (d) For selected examples, see: Y. Wang, C. Y. Zhao, Y. P. Wang and W. H. Zheng, Synthesis, 2019, 51, 3675 CrossRef CAS; (e) H. L. Zheng, L. Cai, M. Pan, M. Uyanik, K. Ishihara and X. S. Xue, J. Am. Chem. Soc., 2023, 145, 7301 CrossRef CAS PubMed; (f) A. H. Abazid and B. J. Nachtsheim, Angew. Chem., Int. Ed., 2020, 59, 1479 CrossRef CAS PubMed; (g) M. Uyanik, S. Ishizaki and K. Ishihara, Org. Synth., 2021, 98, 1 CrossRef CAS; (h) Y. Zhang, Y. T. Liao, X. H. Liu, X. Xu, L. L. Lin and X. M. Feng, Chem. Sci., 2017, 8, 6645 RSC.
- For reviews, see: (a) J. Z. An and M. Bandini, Eur. J. Org Chem., 2020, 27, 4087 CrossRef; (b) U. K. Sharma, P. Ranjan, E. V. Van der Eycken and S. L. You, Chem. Soc. Rev., 2020, 49, 8721 RSC; (c) For selected examples, see: C. X. Zhou and S. L. You, Angew. Chem., Int. Ed., 2013, 52, 10056 CrossRef PubMed; (d) Z. P. Yang, Q. F. Wu and S. L. You, Angew. Chem., Int. Ed., 2014, 53, 6986 CrossRef CAS PubMed; (e) X. J. Lian, L. L. Lin, G. J. Wang, X. H. Liu and X. M. Feng, Chem.–Eur. J., 2015, 21, 17453 CrossRef CAS PubMed; (f) S. L. Ge, T. F. Kang, L. L. Lin, X. Y. Zhang, P. Zhao, X. H. Liu and X. M. Feng, Chem. Commun., 2017, 53, 11759 RSC; (g) Q. X. Zhang, Q. Gu and S. L. You, Org. Lett., 2022, 24, 8031 CrossRef CAS PubMed; (h) R. Pedrazzani, J. An, M. Monari and M. Bandini, Eur. J. Org Chem., 2021, 11, 1732 CrossRef; (i) D. C. Wang, P. P. Cheng, T. T. Yang, P. P. Wu, G. R. Qu and H. M. Guo, Org. Lett., 2021, 23, 7865 CrossRef CAS PubMed; (j) J. J. Hu, S. L. Pan, S. Zhu, P. Y. Yu, R. G. Xu, G. F. Zhong and X. F. Zeng, J. Org. Chem., 2020, 85, 7896 CrossRef CAS PubMed; (k) B. M. Yang, X. J. Zhai, S. B. Feng, D. Y. Hu, Y. H. Deng and Z. H. Shao, Org. Lett., 2019, 21, 330 CrossRef CAS PubMed; (l) Q. X. Zhang, J. H. Xie, Q. Gu and S. L. You, Chem. Commun., 2023, 59, 3590 RSC; (m) Y. L. Yu, Z. H. Zhang, A. Voituriez, N. Rabasso, G. Frison, A. Marinetti and X. Guinchard, Chem. Commun., 2021, 57, 10779 RSC; (n) L. B. Han, H. Wang and X. J. Luan, Org. Chem. Front., 2018, 5, 2453 RSC; (o) J. Zheng, S. B. Wang, C. Zheng and S. L. You, J. Am. Chem. Soc., 2015, 137, 4880 CrossRef CAS PubMed.
- (a) M. T. Peruzzi, S. J. Lee and M. R. Gagne, Org. Lett., 2017, 19, 6256 CrossRef CAS PubMed; (b) S. L. Huang, L. Kötzner, C. K. De and B. List, J. Am. Chem. Soc., 2015, 137, 3446 CrossRef CAS PubMed; (c) J. Z. An, A. Parodi, M. Monari, M. C. Reis, C. S. Lopez and M. Bandini, Chem.–Eur. J., 2017, 23, 17473 CrossRef CAS PubMed; (d) L. Yao and K. Ishihara, Chem. Sci., 2019, 10, 2259 RSC; (e) A. S. Alshreimi, G. Q. Zhang, T. W. Reidl, R. L. Peña, N. G. Koto, S. M. Islam, D. J. Wink and L. L. Anderson, Angew. Chem., Int. Ed., 2020, 59, 15244 CrossRef CAS PubMed.
- For selected reviews of aromatic [n,m]-sigmatropic rearrangement, see: (a) K. Hiratani and M. Albrecht, Chem. Soc. Rev., 2008, 37, 2413 RSC; (b) Y. C. Liang and B. Peng, Acc. Chem. Res., 2022, 55, 2103 CrossRef CAS PubMed; (c) Y. B. Liu, X. H. Liu and X. M. Feng, Chem. Sci., 2022, 13, 12290 RSC; (d) For selected examples, see: F. C. Gozzo, S. A. Fernandes, D. C. Rodrigues, M. N. Eberlin and A. J. Marsaioli, J. Org. Chem., 2003, 68, 5493 CrossRef CAS PubMed; (e) H. Ito, A. Sato and T. Taguchi, Tetrahedron Lett., 1997, 38, 4815 CrossRef CAS; (f) T. S. Castro, G. F. Martins, S. F. de Alcântara Morais and D. A. C. Ferreira, Theor. Chem. Acc., 2023, 142, 40 Search PubMed; (g) T. R. Ramadhar, J. Kawakami, A. J. Lough and R. A. Batey, Org. Lett., 2010, 12, 4446 CrossRef CAS PubMed; (h) L. Song, H. Yao and R. Tong, Org. Lett., 2014, 16, 3740 CrossRef CAS PubMed; (i) C. K. Chan, Y. H. Chen, Y. L. Tsai and M. Y. Chang, J. Org. Chem., 2017, 82, 3317 CrossRef CAS PubMed; (j) T. Abe, Y. Kosaka, M. Asano, N. Harasawa, A. Mishina, M. Nagasue, Y. Sugimoto, K. Katakawa, S. Sueki, M. Anada and K. Yamada, Org. Lett., 2019, 21, 826 CrossRef CAS PubMed; (k) L. Yao, K. Takeda, K. Ando and K. Ishihara, Chem. Sci., 2023, 14, 2441 RSC.
- J. Z. An, A. Parodi, M. Monari, M. C. Reis, C. S. Lopez and M. Bandini, Chem.–Eur. J., 2017, 23, 17473 CrossRef CAS PubMed.
- L. F. Wang, Y. Q. Zhou, Z. S. Su, F. C. Zhang, W. D. Cao, X. H. Liu and X. M. Feng, Angew. Chem., Int. Ed., 2022, 61, e202211785 CrossRef CAS PubMed.
- L. Yao and K. Ishihara, Chem. Sci., 2019, 10, 2259 RSC.
- For examples about the π-complex mechanism, see: (a) M. J. S. Dewar, The Electronic Theory of Organic Chemistry, Oxford University Press, London, 1949, p. 229 Search PubMed; (b) D. Y. Curtin and H. W. Johnson Jr, J. Am. Chem. Soc., 1956, 78, 2611 CrossRef CAS; (c) J. Borgulya, R. Madeja, P. Fahrni, H. J. Hansen, H. Schmid and R. Barner, Helv. Chim. Acta, 1973, 56, 14 CrossRef CAS; (d) X. G. Lei, M. J. Dai, Z. H. Hua and S. J. Danishefsky, Tetrahedron Lett., 2008, 49, 6383 CrossRef CAS PubMed.
- For selected examples of para-Claisen rearrangements, see: (a) J. P. Ryan and P. R. O'Connor, J. Am. Chem. Soc., 1952, 74, 5866 CrossRef CAS; (b) K. Maruoka, J. Sato, H. Banno and H. Yamamoto, Tetrahedron Lett., 1990, 31, 377 CrossRef CAS; (c) Y. Okada and D. Imanari, Int. J. Org. Chem., 2012, 2, 38 CrossRef CAS; (d) Z. Hui, S. W. Jiang, X. Qi, X. Y. Ye and T. Xie, Tetrahedron Lett., 2020, 61, 151995 CrossRef CAS; (e) F. Salahi, C. B. Yao, J. R. Norton and S. A. Snyder, Nat. Synth., 2022, 1, 313 CrossRef; (f) J. A. Homer, I. D. Silvestro, E. J. Matheson, J. T. Stuart and A. L. Lawrence, Org. Lett., 2021, 23, 3248 CrossRef CAS PubMed.
- For selected reviews of N,N′-dioxide/metal catalysis, see: (a) H. X. Liu, L. L. Lin and X. M. Feng, Org. Chem. Front., 2014, 1, 298 RSC; (b) M. Y. Wang and W. Li, Chin. J. Chem., 2021, 39, 969 CrossRef CAS; (c) W. D. Cao, X. H. Liu and X. M. Feng, Chin. Sci. Bull., 2020, 65, 2941 CrossRef; (d) D. F. Chen and L. Z. Gong, Org. Chem. Front., 2023, 10, 3676 RSC; (e) X. H. Liu, L. L. Lin and X. M. Feng, Acc. Chem. Res., 2011, 44, 574 CrossRef CAS PubMed; (f) X. H. Liu, H. F. Zheng, Y. Xia, L. L. Lin and X. M. Feng, Acc. Chem. Res., 2017, 50, 2621 CrossRef CAS PubMed; (g) X. H. Liu, S. X. Dong, L. L. Lin and X. M. Feng, Chin. J. Chem., 2018, 36, 791 CrossRef CAS; (h) For selected examples, see: T. Y. Zhan, L. K. Yang, Q. Y. Chen, R. Weng, X. H. Liu and X. M. Feng, CCS Chem., 2023, 5, 2101 CrossRef CAS; (i) K. X. Wang, L. Q. Yang, Y. Li, Z. L. Liu, L. C. Ning, X. H. Liu and X. M. Feng, Angew. Chem., Int. Ed., 2023, 62, e202307249 CrossRef CAS PubMed; (j) Q. W. He, M. P. Pu, Z. Jang, H. Y. Wang, X. M. Feng and X. H. Liu, J. Am. Chem. Soc., 2023, 145, 15611 CrossRef CAS PubMed.
- For the X-ray crystal structure of the N,N′-dioxide/Co(II) complex: W. Yang, M. P. Pu, X. B. Lin, M. Chen, Y. J. Song, X. H. Liu, Y. D. Wu and X. M. Feng, J. Am. Chem. Soc., 2021, 143, 9648 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: 1H, 13C{1H} and 19F{1H} NMR, HPLC spectra (PDF). X-ray crystallographic data for B1 and B35. CCDC 2289879 and 2235350. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc05677e |
| This journal is © The Royal Society of Chemistry 2023 |