
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnhancing organic cathodes of aqueous zinc-ion batteries via utilizing steric hindrance and electron cloud equalization†
Guanzhong
Ma‡
a,
Zhengyu
Ju‡
c,
Xin
Xu
a,
Yunfei
Xu
a,
Yao
Sun
a,
Yaqun
Wang
*a,
Guoxin
Zhang
 a,
Mian
Cai
a,
Lijia
Pan
*b and
Guihua
Yu
*c
a,
Mian
Cai
a,
Lijia
Pan
*b and
Guihua
Yu
*c
aCollege of Energy Storage Technology, Shandong University of Science and Technology, Qingdao 266590, China. E-mail: yqwang@sdust.edu.cn
bSchool of Electronic Science and Engineering, Collaborative Innovation Center of Advanced Microstructures, Nanjing University, Nanjing 210093, China. E-mail: ljpan@nju.edu.cn
cMaterials Science and Engineering Program and Walker Department of Mechanical Engineering, The University of Texas at Austin, TX 78712, USA. E-mail: ghyu@austin.utexas.edu
First published on 20th October 2023
Abstract
Polyaniline (PANI), with merits of high electronic conductivity and capacity, is a promising material for zinc (Zn)-ion batteries. However, its redox window in Zn batteries is often limited, mainly due to the oxidative degradation at high potentials—in which imine groups can be attacked by water molecules. Here, we introduce phytic acid, a kind of supermolecule acid radical ion, as a dopant and electrolyte additive. Various in/ex situ analyses and theoretical calculations prove that the steric hindrance effect can prevent electroactive sites from the attack by water molecules. Meanwhile, the redox reaction can be stabilized by an even distribution of electron cloud due to the conjugated structure of phenazine groups. Accordingly, the assembled Zn–PANI battery can allow stable and long-term charge–discharge reactions to occur at a potential as high as 2.0 V with a discharged plateau of 1.5 V, and it also shows high rate performance and stable long cycle life (75% capacity retention after 1000 cycles at 10 A g−1).
Introduction
As the most widely used electrochemical energy storage systems at present, the inherent safety and environment issues of lithium-ion batteries are the primary concerns in their practical applications.1–3 Among the alternative batteries beyond lithium, aqueous zinc (Zn) batteries have shown great potential for future safe energy storage applications due to the intrinsic advantages of Zn,4–6 including high theoretical specific capacity (820 mA h g−1), low redox potential (−0.76 V vs. SHE), and good compatibility with water.7,8 However, the bottleneck that limits the practical applications of Zn-ion batteries (ZIBs) is the lack of suitable cathode materials with simultaneously high specific capacity and stability after extended cycling.9,10 The inherent merits of organic materials, such as abundant sources, tailorable structures, and environmental friendliness, make them superior to their inorganic counterparts.11–13 Among them, conductive polymers, e.g., polyaniline (PANI), that possess both a highly conjugated and porous structure for efficient electron and ion transport are of broad interest as potential cathode materials.14The unique doping mechanism makes the anion species crucial to the charge storage mechanism of PANI.15 Moreover, the stability of PANI as the ZIB cathode is highly influenced by the operating voltage. Notably, PANI typically exhibits poor stability under extended cycles at high potentials. This is mainly due to the over-oxidation-induced degradation of PANI when the potential exceeds a certain threshold.16,17 Such degradation is primarily caused by the reaction between PANI and water, which continuously breaks down the polymer's long chain and forms soluble short-chain oligomers.18,19 The shortage of protons and irreversible redox process of PANI synergistically cause a rapid decline in capacity. To this end, extensive efforts have been devoted to stabilizing PANI at various length scales, by derivatization, copolymer formation, self-doping engineering, and three-dimensional (3D) micro/nano structuring.20,21 In particular, self-doped species can function as an internal proton reservoir to maintain a highly acidic local environment for excellent electrical conductivity.22,23 In addition to molecular structure designs, other strategies have been proposed to prevent PANI degradation by establishing a stable electrode–electrolyte interface. For example, intermolecular hydrogen bonds can be formed between the gel electrolyte and PANI.24 Additionally, in order to broaden the voltage window of aqueous batteries, equally important is to inhibit water decomposition. For example, by introducing diethylenetriamine (DETA) into the electrolyte, Gong et al. formed a protective interlayer on a Zn negative electrode surface, inhibiting water molecules from entering the electrode surface to mitigate hydrogen evolution.25 It is worth noting that most reported cells in the literature are cycled within a voltage range of 0.5–1.5 V;26 with very few cases where the initial discharge plateau exceeds 1.5 V.
In addition, the benzene, quinone, and phenazine moieties on PANI polymer chains can all contribute to its redox reaction. These units, particularly the phenazine structure, are reported to be electrochemically reactive for Zn-ion storage. Shim et al. demonstrated the reversible redox of phenazine in PANI.24 Wang et al. studied ZIBs using monomeric phenazine with an output potential of 0.6 V.27,28 However, there are still no relevant studies at high voltages.
In this paper, the phytic acid radical was utilized as both a dopant and an electrolyte additive to stabilize the electrochemical redox reaction of aqueous ZIBs and expand their potential window. After doping, the introduced macromolecular phytate creates a steric hindrance effect—preventing water molecules from attacking the long chain of PANI—to inhibit peroxidative degradation. By such means, the discharge platform of ZIBs can be expanded and their energy density can be further improved. Furthermore, the incorporation of phenazine groups into PANI results in a more uniform distribution of electron clouds, enhancing the system's endurance and stabilizing redox peaks at higher potentials. This allows for the utilization of the redox activities of phenazine and safranine groups at elevated potentials. Moreover, zinc phytate is added as an additive to the commonly used zinc sulfate electrolyte. It can also bind water molecules through other phosphate groups with hydrogen bonding and inhibit the hydrogen evolution reaction of the negative electrode. In this paper, the voltage range of the PANI-based ZIB has been expanded to 0.5–2.0 V, showing an average initial discharge voltage as high as 1.7 V and a specific capacity of 140 mA h g−1 at 10 A g−1.
Results and discussion
Protic acid doping plays a crucial role in the synthesis of PANI, exerting significant influence on both its conductivity and morphology.29 The doping reaction of PANI occurs in the imine moiety of its extended chain, and the acid dissociation constants (pKa) of the imines at the terminal positions of PANI's quinone ring structure are 1.05 (pKa1) and 2.55 (pKa2), respectively. These pKa values fall within a range that enables dissociation of protons bound to the imine group, thereby meeting the doping requirements of PANI.24 Considering sulfuric acid (pKa2 = 1.99) and phytic acid (pKa1 = 1.13), they both meet the doping conditions for PANI, and thus PANI can be protonated by the acid doping effect to effectively increase its electrical conductivity (Fig. 1a). However, there is a significant difference in morphology between PANI doped with these acids, as characterized by scanning electron microscopy (SEM) (Fig. 1b and c). The phytate doped PANI (PANI-P) (Fig. 1b) shows a sponge-like structure composed of a homogeneous 3D nanowire network with hierarchical micro/nanopores. The porous structure leaves an open space between the electrolyte and active materials, facilitating ion transport kinetics and enhancing cycle stability. The morphologies of various proportions of polyaniline doped with phytic acid showed no significant differences (Fig. S1†). In contrast, the morphology of PANI by sulfuric acid doping (PANI-S) shows irregular and tightly stacked nanowires (Fig. 1c). The dense stacking of PANI-S makes it more difficult for the charge transfer process to occur at the electrode–electrolyte interface. PANI doped with other acids, such as hydrochloric acid, exhibits similar morphological characteristics to PANI-S (Fig. S2†).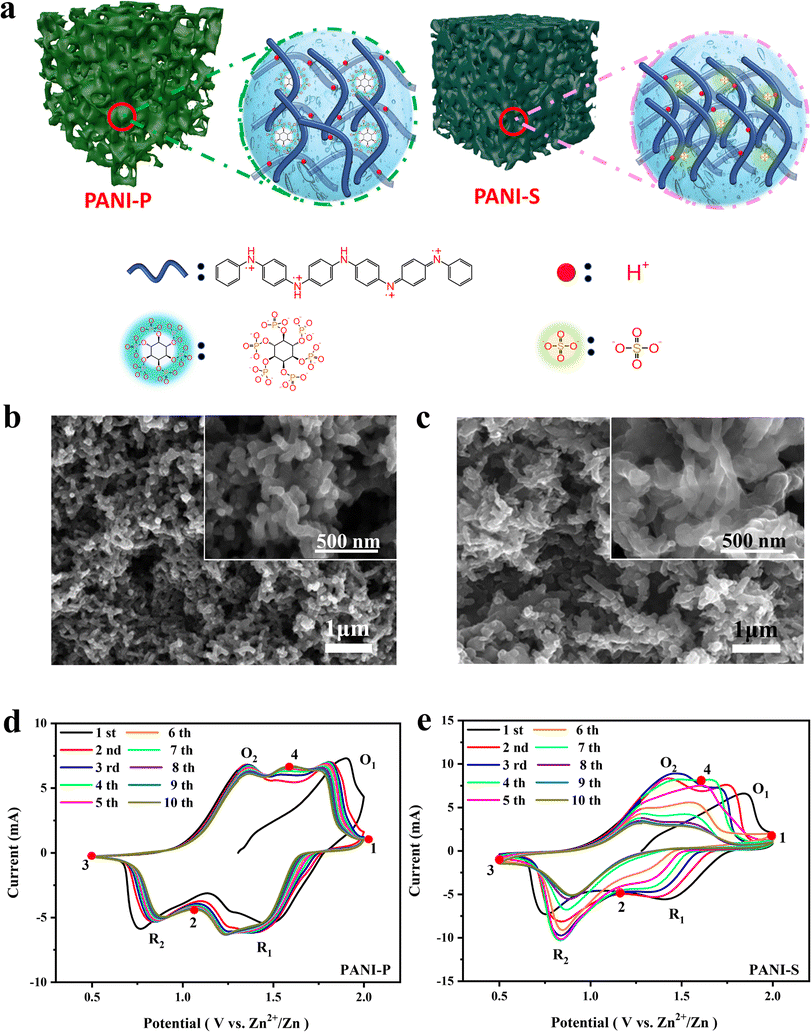 | ||
| Fig. 1 (a) Microscopic schematic diagram of different PANI doped acids; scanning electron microscopy images of different PANI doped acids: (b) doped phytic acid; (c) doped sulfuric acid; cyclic voltammetry curve (CV curve) sweep rate of 10 mV s−1 for different PANI doped acids: (d) PANI doped phytic acid electrode in 1 M zinc sulfate electrolyte with addition of zinc phytate (1%); (e) PANI doped sulfate electrode in 1 M zinc sulfate electrolyte. | ||
From the Fourier-transform infrared (FTIR) spectra of the two acid-doped PANI (Fig. S3b†), peaks located at 1602, 1459, 1305, and 1139 cm−1 represent the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C stretching vibration on the quinone ring, CC stretching vibration of the benzene ring backbone, C–N bonding vibration in the structure of the benzoquinone unit, and C–H stretching vibration on the conjugated benzene ring, respectively. Such results indicate that the phytic acid and sulfuric acid doped PANI backbones are consistent with the regularly synthesized one.30 PANI-P (red spectral line) shows higher peak intensity at the wavenumber of 1143 cm−1, which means more Q = NH+–B and B–NH+–B fragments, indicating that PANI-P has more effective proton doping. This is very helpful for cathode conductivity enhancement and optimization of electrode reactivity. Furthermore, by Raman analysis of different doped PANI (Fig. S3a†), C–H bond out-of-plane vibrations in the quinone ring and its stretching vibrations in the benzene ring at 974 and 1181 cm−1,31,32 C–N bond stretching vibrations in the benzene ring at 1257 cm−1, C–N+ stretching vibrations at 1337 cm−1, and CN bond stretching vibrations in the quinone ring at 1469 cm−1 were detected.33 These peaks also indicate that the main chain of PANI with acid doping is similar to that of the regular one.34 It is noteworthy that among them, characteristic peaks of phenazine were observed at 974, 1257, 1337, 1469, 1559 and 1634 cm−1, while there were other types of fragmentary groups produced in the acid-doped synthesis of PANI (Fig. S4†).35
C stretching vibration on the quinone ring, CC stretching vibration of the benzene ring backbone, C–N bonding vibration in the structure of the benzoquinone unit, and C–H stretching vibration on the conjugated benzene ring, respectively. Such results indicate that the phytic acid and sulfuric acid doped PANI backbones are consistent with the regularly synthesized one.30 PANI-P (red spectral line) shows higher peak intensity at the wavenumber of 1143 cm−1, which means more Q = NH+–B and B–NH+–B fragments, indicating that PANI-P has more effective proton doping. This is very helpful for cathode conductivity enhancement and optimization of electrode reactivity. Furthermore, by Raman analysis of different doped PANI (Fig. S3a†), C–H bond out-of-plane vibrations in the quinone ring and its stretching vibrations in the benzene ring at 974 and 1181 cm−1,31,32 C–N bond stretching vibrations in the benzene ring at 1257 cm−1, C–N+ stretching vibrations at 1337 cm−1, and CN bond stretching vibrations in the quinone ring at 1469 cm−1 were detected.33 These peaks also indicate that the main chain of PANI with acid doping is similar to that of the regular one.34 It is noteworthy that among them, characteristic peaks of phenazine were observed at 974, 1257, 1337, 1469, 1559 and 1634 cm−1, while there were other types of fragmentary groups produced in the acid-doped synthesis of PANI (Fig. S4†).35
To investigate the performance of PANI-based ZIBs, we assembled various Zn–PANI cells utilizing PANI-S and PANI-P as positive electrodes and Zn foil as the negative electrode. The electrolytes of PANI-P and PANI-S cells are 1 M zinc sulfate solution with zinc phytate (1%) and 1 M zinc sulfate, respectively, and the cyclic voltammetry (CV) curves were tested in the voltage range of 0.5–2.0 V (Fig. 1d and e). We found that different acid-doped PANIs in different electrolytes show two pairs of redox peaks, but their peak position shifts and intensity decay behaviours are significantly different. From the first cycle in CV curves, it is evident that both PANI-S and PANI-P display two oxidation peaks at 1.4 V and 1.9 V, as well as two reduction peaks at 1.5 V and 0.8 V, representing the intrinsic properties of PANI. However, anionic dopants show different effects on the size and retention of the reduction peaks with extended cycles. Starting from the second cycle, the reduction peak of PANI-S at 1.5 V gradually decreases and disappears at the seventh cycle. In contrast, PANI-P shows good peak retention. In fact, the electrolyte additive also does matter. By comparing the CV curves of different acid-doped PANIs in different electrolytes (Fig. S5†), it can be observed that PANI-S exhibits the poorest retention in zinc sulfate electrolyte, while its performance shows some improvement when using the electrolyte with zinc phytate. For PANI-P, good peak retention can be achieved in various electrolytes, especially the one with the zinc phytate additive. The slow decay rate suggests that protons dope into the long chain of PANI through continuous dissociation of phytate molecules to stabilize the doping and de-doping reactions at high potentials, thereby maintaining the electrical conductivity of PANI. Meanwhile, phytic acid molecules act at the surface of PANI and reinforce the interaction between its long chains, which greatly contributes to the stability of the molecular structure.
The synthesis of PANI by chemical methods produces different types of fragments, as demonstrated in Fig. S4.† Our FTIR and Raman characterizations reveal the existence of fragment structures such as phenazine and safranine in the synthesized PANI. Although these fragment structures have been reported as cathode materials in ZIBs, their discharge voltage plateau is generally lower than 1.0 V.36
To investigate the reaction mechanism of PANI, we conducted in situ Raman spectroscopy analysis on both PANI-S and PANI-P cathodes, with charge–discharge voltage profiles and data points collected at each voltage shown in Fig. 2a. Detailed Raman spectra of the PANI-S and PANI-P at different voltages are plotted in Fig. 2b and c respectively. The peak located at 1594 cm−1 (refs. 37 and 38) corresponds to the stretching vibration of the CC bond in the quinone ring. It is noteworthy that peak intensity is higher at O1, indicating an increase in quinone ring content with increasing voltage. This observation aligns with the redox reaction mechanism of PANI. The peak at 1491 cm−1 (ref. 33) represents the stretching vibration of the CN bond in the quinone ring, which is found to shift toward 1480 cm−1 at high potentials. Such a peak shift is caused by the exposed CN bond due to the breakage of the main chain when the degradation of PANI-S occurs (Fig. 2d); however, the peak shift can barely be observed in PANI-P, which is due to the inhibition of PANI degradation by the phytic acid molecules around PANI-P. The peaks at 1337 and 1368 cm−1 are related to the C–N stretching vibrations in phenazine and safranine,39,40 respectively, and the decrease of C–N at O1 can be attributed to the oxidation of C–N bonds to CN+ bonds in phenazine and safranine at high potentials. The stretching vibration of the C–N bond in the benzene ring and the characteristic peak of phenazine are at 1270 cm−1.41 The peak position at O1 is obviously shifted to a lower wavenumber, which is due to the oxidation reaction of the benzene ring connected to the C–N bond, leaving decreased electron cloud density. As a result, the bonding between the benzene ring and N atom is weakened and causes the peak shift. However, when discharge was carried out, the peak shift could return to its original position. This indicates that the redox process is still reversible. Through the in situ Raman spectroscopy analysis of the PANI positive electrodes, we found that the oxidation reaction of phenazine and safranine fragments took place at O1. It can be observed in Fig. S6a† that with the increase of the cycle number, the trend of each peak intensity in PANI-S did not change, but the rate of peak intensity alteration at each voltage did change, indicating that the degradation reaction of the PANI cathode occurred after long cycling, along with the reduced reaction activity. The change in PANI-P was not obvious (Fig. S6b†), which indicates that the doped phytate root has a certain protective effect on the PANI cathode and inhibits its degradation reaction.
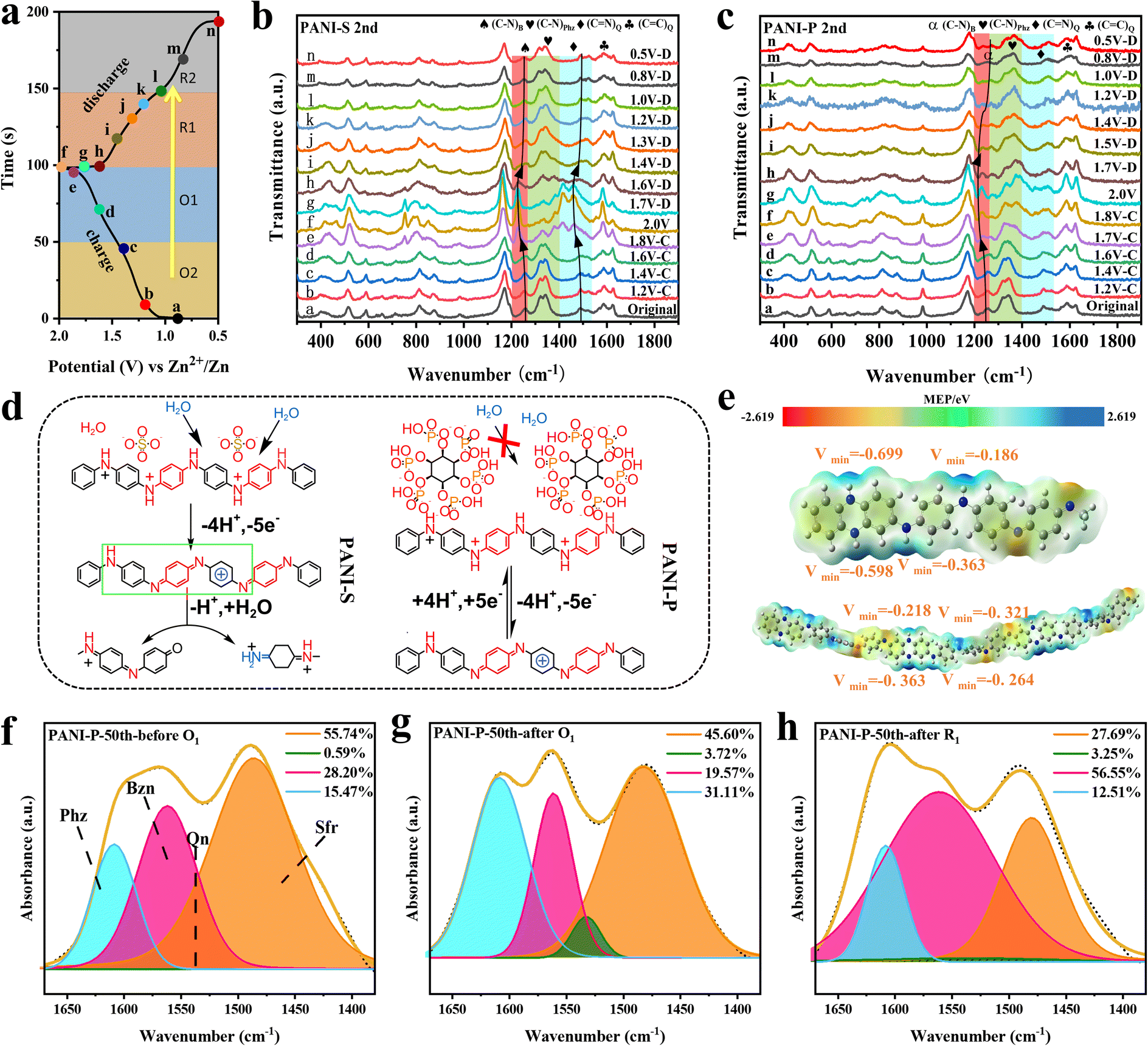 | ||
| Fig. 2 (a) Charging and discharging curves of Zn//PANI cells and in situ Raman sampling points at different voltages; in situ Raman spectra for different anodes: (b) Zn//PANI-S cell, (c) Zn//PANI-P cell. (d) Chemical equations for the degradation of polyaniline by water attack. (e) Molecular electrostatic potential (eV) on the surface of the oligomer and polymer, where the white, gray and blue balls indicate H, C and N atoms, respectively, and Vmin is the minimum value of MEP; (f–h) variation of the content of each fragment at different points of PANI-P when cycled to 50 turns. | ||
We investigated the electrophilic and nucleophilic sites of the polymer and its monomers by density functional theory (DFT) to calculate the molecular electrostatic potential (MEP) to predict the reaction sites. The optimized structures and their MEP values are shown in Fig. 2e, where the polymer consists of three monomer fragments (n = 3). The monomeric phenazine fragment exhibits a uniform distribution of electron cloud density due to its molecular symmetry. The electron cloud density around the N atom in phenazine is higher because of the attractive effect of the amino group on electrons. When an aniline fragment is present around the phenazine fragment, it alters the electron cloud distribution around both N atoms, resulting in a decrease in electron cloud density at one side and making it harder to oxidize. This feature increases the voltage plateau of the phenazine fragment in the PANI electrode, and the activity of the phenazine fragment in PANI at higher potentials can be observed in Fig. 2b and c. We performed FTIR spectroscopy analysis of the changes occurring in each fragment (Fig. S7†). The peak at 1616 cm−1 represents the CN bond stretching vibration in phenazine. It is evident that the peak intensity increases when the battery passes through O1, indicating that the oxidation of the C–N bond in phenazine to the CN bond occurs in O1, which is consistent with the in situ Raman spectroscopy results. To further demonstrate the changes of PANI during charging and discharging, we analyzed the peaks within the wavenumber range of 1300–1600 cm−1 in ex situ FTIR spectra and identified four distinct stretching modes for fragments. Among them, 1605, 1570, 1490 and 1530 cm−1 are assigned to the phenazine (Phz), quinone (Qn), benzene (Bzn), and safranine (Sfr) fragments, respectively.24,42 The peak distribution of PANI at different states (before O1, after O1, and after R1) in the 50th cycle is shown in Fig. 2f–h. A comparison of the changes in the content of each fragment as it undergoes the first oxidation peak (O1) and the first reduction peak (R1) in the PANI-P reveals that, in addition to the changes in the conventional active units such as benzene and quinone, the changes in phenazine and safranine fragments are also evident. Similarly, these fragments also show high activity in PANI-S (Fig. S8†). The analysis reveals that the phenazine and safranine fragments are active in the long chain of PANI and possess good electrochemical reversibility.
The reaction mechanism of the electrodes was studied by analyzing the X-ray photoelectron spectroscopy (XPS) in two different electrolytes. The imine group in PANI is the main reactive site,43 so we analyzed the N 1s spectra of PANI cathodes in the cell at four points of the redox process to understand the evolution of N-containing chemical bonds (Fig. 3a–d). Since the synthesized PANI is in a semi-oxidized and semi-reduced state (emerald green imine salt state) at the beginning,44 the potentials of the first cycle in Fig. 1d and e start at 1.25 V, which is also the starting point (point 4) in this XPS analysis. PANI can be found in the first stage of reaction (O1) with –N = content increased, while –NH+– and –NH– content decreases, proving that the main reactions occurring in this process are the de-doping reaction of –NH+ and the oxidation of –NH– to –N (point 4 to point 1). In the second stage (R1), it is clearly observed that the contents of –NH– and –NH+ are increased, while the content of –N decreases (point 1 to point 2) due to the reduction of –N to –NH– and the increase in the content of –NH+ is due to the partial doping reactions. In the third stage of the reaction (R2), a significant decrease of –N fragments and an increase of –NH– can be observed (point 2 to point 3), indicating that the reaction in this process is mainly the reduction of –N to –NH–. In the fourth stage of the reaction (O2) the –N content increases and the –NH– content decreases significantly (point 3 to point 4). The reaction on the positive electrode of PANI at this stage is mainly the initial oxidation stage, and the changes that occur are mainly the oxidation of –NH– to –N and the partial doping to –NH+–.
 | ||
| Fig. 3 In situ XPS analysis of N 1s in Zn//PANI cells at different voltages during the second cycle and the electron gain/loss process: (a) point 4; (b) point 1; (c) point 2; (d) point 3. | ||
The general pattern of the changes in the bonding content of the N element in PANI after 50 cycles reveals that the trend is similar to that of the 2nd loop (Fig. S9†), which indicates that the main reaction does not change after a long time of cycling, but the retention of the peak position is not optimistic in PANI, which proves that the decay of the reduction peak of PANI occurs at high potentials during cycling. Fig. S10† shows the XPS full spectrum information of PANI-S and PANI-P at different voltages, at 1022/1024 eV for the characteristic peak of Zn 2p, which indicates that Zn2+ is also involved in the reaction.45 In particular, the content change is obvious when discharging from 1.1 V to 0.5 V and charging from 0.5 V to 1.6 V, which indicates that the Zn2+ de-embedding at the cathode mainly occurs at the lower voltage platform. After cycling, we observed a decrease in pH value for the zinc sulfate electrolyte, while an increase was noted for the zinc phytate (1%) electrolyte (Table S1†). This suggests that during de-doping of the PANI-S surface, sulfate anions and H+ ions enter the electrolyte resulting in poor reversibility upon subsequent doping reactions. Moreover, oxygen evolution is more prone to occur at elevated potentials in zinc sulfate electrolytes, leading to a decreased pH level within the electrolyte. PANI-P not only exhibits reversible doping/de-doping reactions, but also benefits from continuous phytate-assisted doping by absorbing excess H+.46 Moreover, PANI-P maintains excellent redox activity in zinc phytate-doped electrolytes even after prolonged cycling.
The peak position of (Fig. 1d) O1 and R1 in PANI-S shifts rapidly to a lower potential with an increase in the number of cycles due to PANI's susceptibility to degradation during high voltage oxidation, resulting in poor cycling performance. For the PANI, the imine group next to its quinone ring is easily degraded by oxidation at high potentials to form p-benzoquinone (Fig. S11†), which eventually forms quinones with a smaller molecular weight.19 The presence of free p-benzoquinone fragments in the electrolyte renders them electrochemically inactive, thereby reducing the overall electrochemical activity of the PANI.
From Fig. 4a, it is evident that the introduction of phytic acid doping results in a significant proliferation of phytic acid clusters surrounding PANI-P. Due to their large molecular weight, these phytic acids prevent water molecules in the electrolyte from attacking imine groups due to the steric hindrance effect. At the same time, phytic acid could form hydrogen bonds with water molecules in the electrolyte to further inhibit water molecules from attacking PANI long chains. In contrast, for PANI-S, sulfuric acid, as a small molecule doped acid, did not play a significant role in inhibiting the degradation of PANI.
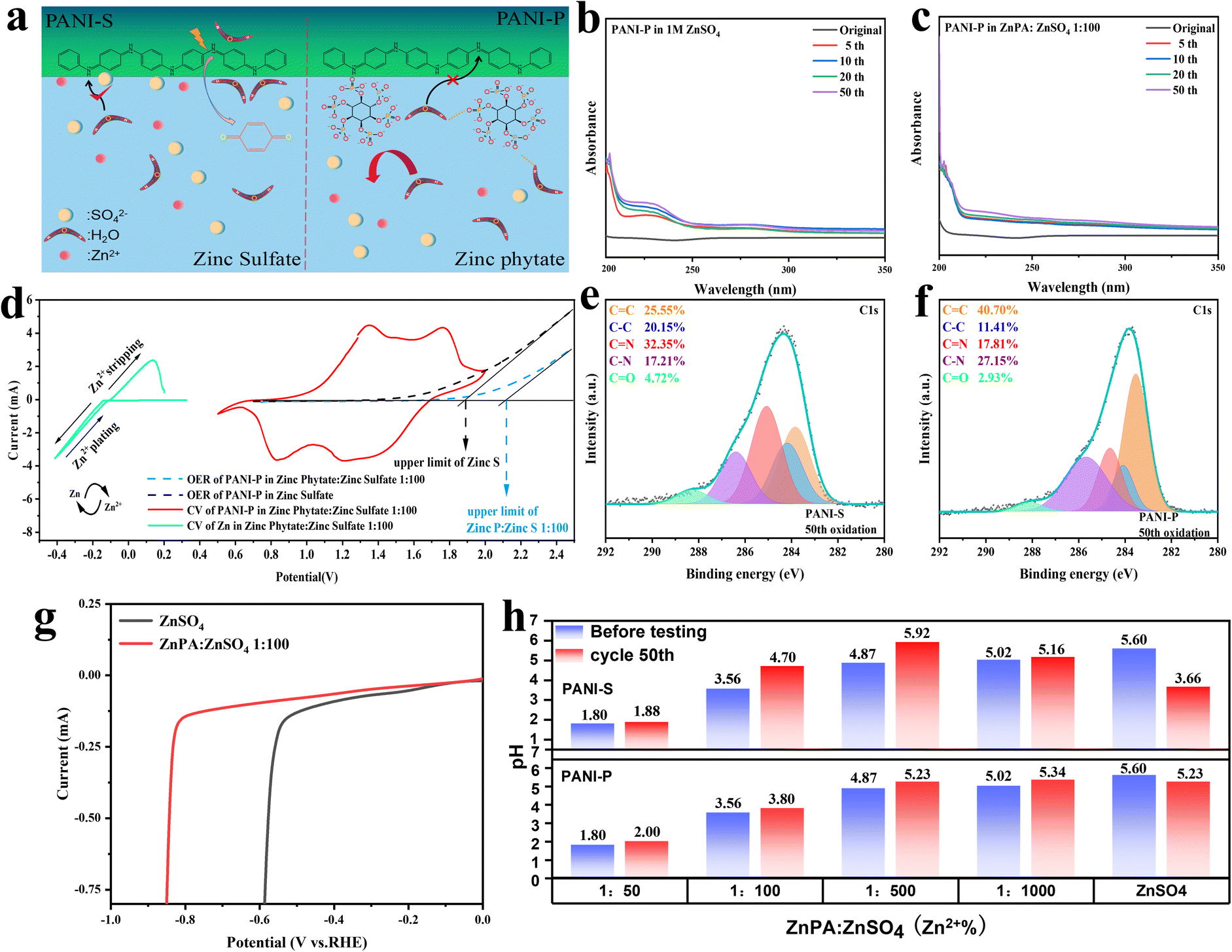 | ||
Fig. 4 (a) Schematic diagrams of PANI-S and PANI-P in different electrolytes: PANI-S in 1 M Zn sulfate, PANI-P in Zn phytate doped electrolyte; quasi-in situ UV spectral profiles of PANI-S and PANI-P in different electrolytes: (b) PANI-P in 1 M Zn sulfate; (c) PANI-P in Zn phytate doped electrolyte; (d) deposition potential of Zn and electrochemical windows of two different electrolytes; XPS fractionation results of C 1s on the surface of PANI-S and PANI-P at the 50th cell cycle in different electrolytes at point 4: (e) PANI-P in Zn-doped phytate electrolyte, (f) PANI-S in 1 M ZnSO4. (g) Comparison of hydrogen evolution overpotentials in ZnSO4 and ZnPA![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :ZnSO4 of 1:100, (h) pH values before and after 50 cycles of electrolytes with different ratios of ZnSO4 and ZnPA:ZnSO4. :ZnSO4 of 1:100, (h) pH values before and after 50 cycles of electrolytes with different ratios of ZnSO4 and ZnPA:ZnSO4. | ||
Because the degradation of PANI produces p-benzoquinone, the degree of oxidative degradation of the two positive electrodes is determined by the number of oxygen-containing functional groups in the electrolyte. In the ultraviolet (UV) spectral analysis, the absorption peaks of p-benzoquinone and other CO bond-containing groups generally exist between 200 and 300 nm. The functional groups containing CO bonds such as p-benzoquinone detected in the electrolyte were all products formed by the anodic peroxidative degradation of PANI.19 The UV spectrum analysis results of PANIP in ZnSO4 with/without zinc phytate (ZnPA) within 50 cycles are compared in Fig. 4b and c, respectively. It can be observed that the absorption peak of the CO double bond increases with the cycle of the battery in zinc sulfate electrolyte. However, in the electrolyte doped with zinc phytic acid, there was no significant change in the absorbance as the cycles increased. This indicates that the attack and hydrolysis of PANI fragments in zinc sulfate electrolyte result in the production of p-benzoquinone, which enhances the CO double bond absorbance. After doping with phytic acid, due to the steric hindrance effect and binding effect of the phytic root on water molecules, PANI can be protected from the attack by water in the zinc phytic electrolyte, and the content of CO double bond does not change significantly. Furthermore, a comparison was made between the changes in the CO content of PANI and PANI-S during cycling in electrolytes with and without additives (Fig. S12†). The study revealed that in the absence of additives, there is a varying degree of degradation of additives, which effectively suppresses the formation of CO and decelerates the degradation process. In order to verify this view, we conducted ex situ XPS analysis on polyaniline and carried out peak separation treatment on C 1s. By analyzing the change of the CO bond content of PANI in the electrolyte doped with zinc sulfate and zinc phytate at the 50th cycle, it was found that the content of CO in PANI-P in zinc phytate electrolyte increased from 2.70% to 2.93% (Fig. 4e and S13b†). The CO content of PANI-S in the zinc sulfate electrolyte increased from 4.26% to 4.72% (Fig. 4f and S13a†). We investigated the impact of different electrolytes on the hydrogen evolution reaction at the negative electrode. As shown in Fig. 4g, the hydrogen evolution overpotential in the electrolyte with additives is 0.23 V larger than that of zinc sulfate without additives, indicating that the addition of additives can inhibit the occurrence of the hydrogen evolution reaction in the electrolyte. Meanwhile, we also conducted an analysis on the pH variations of PANI-S and PANI-P before and after 50 cycles in the electrolyte with different concentrations of additives (Fig. 4h). The limited solubility of zinc phytate in systems necessitates the addition of a certain amount of phytate as a co-solvent. However, it is crucial to avoid excessive acidity in the environment for zinc electrodes, thus the additives should be used judiciously. The electrolyte with the additive exhibits a slight increase in pH after the battery cycle, whereas a sharp decrease in the pH of 1 M ZnSO4. This is due to the degradation-induced release of doped protons into the electrolyte and generation of hydrogen ions during the process, resulting in enhanced acidity.
At the same time, we observed the electrode surfaces of PANI-S and PANI-P after 200 cycles of the battery (Fig. S14†). Unsurprisingly, we found that PANI-S fell off seriously on the surface of the collector after a long cycle, while PANI-P was more evenly attached to the surface of the collector, showing a cross-linked structure and maintaining better reactivity. Various characterization methods showed that phytic acid doping in PANI has protective effects, which can effectively inhibit the degradation of PANI. Also, doping in the electrolyte can make the positive electrode of PANI maintain a long cycle life. Oxygen evolution reaction (OER) test was conducted on PANI in the two electrolytes (Fig. 4d), and it was found that it was more difficult for the electrolyte doped with zinc phytate to precipitate oxygen at the positive electrode. The addition of zinc phytate broadened the electrochemical window of the electrolyte.
The side reactions occurring in the negative electrode of a ZIB also have a great impact on its performance, including the hydrogen evolution reaction on the surface of the negative electrode of Zn, formation of insoluble zinc oxides and uneven deposition leading to dendrite formation. Many problems are closely related to the electrolyte. We observed the surface of Zn negative electrode in different electrolytes and found that the electrolyte with zinc sulfate was smooth after 200 cycles (Fig. 5b), while the Zn negative electrode surface was severely cracked after 200 cycles in the electrolyte without zinc phytate (Fig. 5a). Phytic acid macromolecules on the negative surface of zinc can effectively inhibit the proximity of water molecules due to the steric hindrance effect (Fig. 5c). Meanwhile, oxygen on the phytic root can form hydrogen bonds with hydrogen atoms in water molecules, which effectively inhibits negative side reactions by restricting the proximity of water molecules to the negative surface of Zn. We prepared Zn//Zn symmetric batteries to test the constant current polarization in different electrolytes (Fig. 5g). The addition of zinc phytate to the electrolyte resulted in a smaller and more stable polarization of the Zn negative electrode. This suggests that the doped electrolyte created an environment conducive to stable deposition and precipitation, leading to a more ordered and stable reaction at the surface of the Zn electrode. The charge and discharge curves of PANI-S and PANI-P electrodes in different electrolytes are respectively shown in Fig. 5d and e. It was obvious that the discharge platform of PANI-S at high potentials attenuated rapidly. However, a plateau can still be observed at high potentials in PANI-P after a long cycle (200 cycles), indicating that PANI-P shows better cycling performance in the electrolyte doped with zinc phytate.
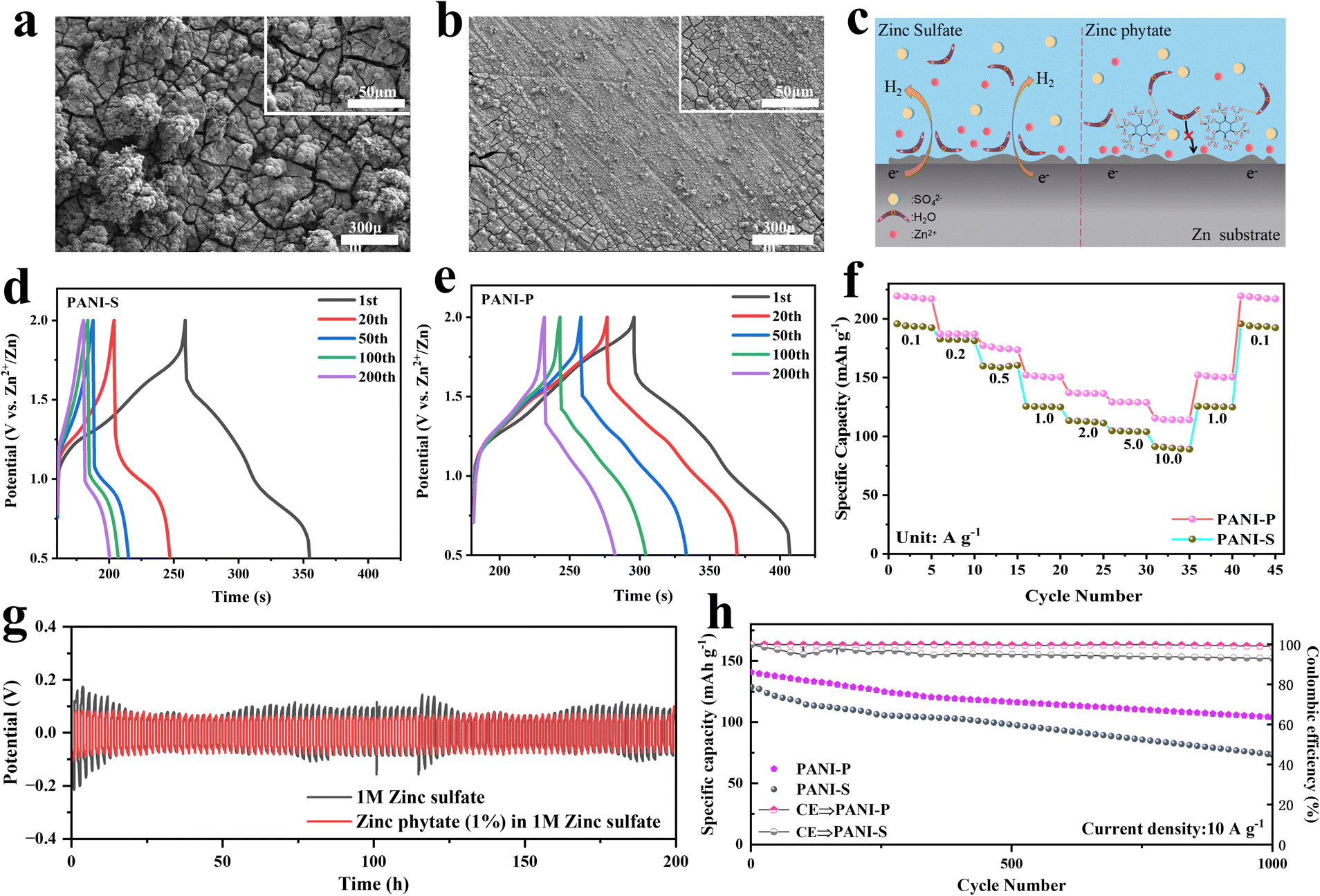 | ||
| Fig. 5 SEM images of Zn negative electrode surface after 200 cycles in different electrolytes for Zn//PANI cells: (a) Zn//PANI-S cell in zinc sulfate electrolyte; (b) Zn//PANI-P cell in zinc phytate doped electrolyte; (c) schematic diagram of Zn negative electrode in different electrolytes. Charge–discharge curves of the Zn//PANI cell in different electrolytes for 200 cycles: (d) Zn/PANI-S cell in zinc sulfate electrolyte, (e) Zn//PANI-P cell in zinc phytate doped electrolyte; (f) multiplicity curves of Zn/PANI-P and Zn/PANI-S in different electrolytes; (g) constant current polarization curves of Zn//Zn symmetric cells in different electrolytes (0.5 mA cm−2); (h) cycling curves of Zn//PANI-S and Zn//PANI-P cells at a current density of 10 A g−1 in different electrolytes. | ||
It is found that PANI-P exhibits better rate performance (Fig. 5f), and the capacity of PANI-P remains more stable when the current density is changed, while the capacity will suddenly decrease when the current density is increased suddenly in PANI-S, especially when the current density is increased from 0.5 to 1.0 A g−1. The same conclusion was also verified in the long-cycle performance of two different positive electrodes in different electrolytes: The PANI-P electrode showed better long-term cycle stability in the doped zinc phytate electrolyte. Fig. 5h shows the cycling performance plots of PANI-P in 1 M zinc sulfate added zinc phytate electrolyte and PANI-S in 1 M zinc sulfate electrolyte at a high current density (10 A g−1). Fig. S15† shows the long-cycle test of PANI-S and PANI-P in the electrolyte with and without additives at 0.2 A g−1. It can be observed that PANI-P shows significantly better cycling performance than PANI-S and achieves higher coulombic efficiency and better reversibility of charge and discharge. The decay of PANI-S is mainly due to the degradation of PANI. It can be seen that the doping of phytic acid in PANI and the addition of zinc phytic acid in the electrolyte are of great help to the overall protection of the whole battery. Both the doping of PANI by phytic acid near the positive electrode and the protection of zinc by phytic acid at the surface of the negative electrode greatly improve the battery performance.
Conclusions
We have presented a general strategy of enhancing the stability and electrochemical performance of organic PANI cathodes in Zn-ion aqueous batteries via utilizing steric hindrance and electron cloud equalization. The reversible redox reaction exists in the benzene ring of PANI at high potentials probed by in situ Raman, and we demonstrated that this reaction exists in the initial stage of PANI degradation. With phytic acid as a dopant to PANI, the steric hindrance effect of phytic acid and the formation of hydrogen bonds with water can protect PANI from the attack of water molecules and inhibit its further degradation, and the semi-self-doping property of phytic acid makes PANI maintain better electrical conductivity. In addition, we analyzed the small molecule fragment phenazine produced during the synthesis of PANI and the phenazine fragment in the long chain of PANI by DFT calculation, and found that the electron cloud density of the active unit is closer to the long chain of PANI when the small molecule phenazine fragment is present in the long chain of PANI, which improves the voltage of n-type organics. On the other hand, the electrolyte after the addition of zinc phytate formed a huge phytate network at the surface of the Zn anode, which had a protective effect on the Zn anode for favoring the uniform deposition of Zn ions. The modified PANI cathode and electrolyte have a specific capacity of 140 mA h g−1 at a high current density of 10 A g−1 and a capacity retention of 75% after 1000 cycles and show high energy density and power density (210 Wh kg−1 and 15000 W kg−1) in ZIBs. In conclusion, our work achieved the simultaneous protection of positive and negative electrodes by a simple additive modification and electrolyte method. Meanwhile, this study opens a new direction for improving the voltage platform of n-type organic compounds, and this strategy will be applicable to the design of other organic compounds for improving the voltage platform of batteries.
Data availability
All data are shown in the manuscript or the associated ESI.†Author contributions
Y. W., L. P. and G. Y. directed the project. G. M. and Y. S. were involved in material preparation, characterization and testing. Y. X. and X. X. performed the in situ XPS test and in situ Raman test. Y. W., G. M., M. C., G. Z., and Z. J. provided some constructive suggestions for the experiment. G. M. wrote the first draft, and all authors discussed the experimental results and continued to prepare the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Y. W. acknowledges the support from the National Natural Science Foundation of China (61904097) and the Program for Tsingtao Power and Energy-storage Battery Research Team in the University. G. Y. acknowledges the support from the Welch Foundation F-1861.Notes and references
- S. Rana, R. Kumar and R. S. Bharj, Chem. Eng. J., 2023, 463, 142336 CrossRef CAS.
- P. Lyu, X. Liu, J. Qu, J. Zhao, Y. Huo, Z. Qu and Z. Rao, Energy Storage Mater., 2020, 31, 195–220 CrossRef.
- T. Joshi, S. Azam, D. Juarez-Robles and J. A. Jeevarajan, ACS Energy Lett., 2023, 8, 2831–2839 CrossRef CAS PubMed.
- X. H. Chen, P. C. Ruan, X. W. Wu, S. Q. Liang and J. Zhou, Acta Phys.-Chim. Sin., 2022, 38, 2111003 Search PubMed.
- Y. Yang, S. Guo, Y. Pan, B. Lu, S. Liang and J. Zhou, Energy Environ. Sci., 2023, 16, 2358–2367 RSC.
- X. Cai, Y. Liu, J. Zha, F. Tan, B. Zhang, W. Yan, J. Zhao, B. Lu, J. Zhou and C. Tan, Adv. Funct. Mater., 2023, 33, 2303009 CrossRef CAS.
- C. Zhu, P. Li, G. Xu, H. Cheng and G. Gao, Coord. Chem. Rev., 2023, 485, 215142 CrossRef CAS.
- Y. Shang and D. Kundu, Joule, 2023, 7, 244–250 CrossRef.
- C. Li, S. Jin, L. A. Archer and L. F. Nazar, Joule, 2022, 6, 1733–1738 CrossRef.
- J. Chen, W. Zhao, J. Jiang, X. Zhao, S. Zheng, Z. Pan and X. Yang, Energy Storage Mater., 2023, 59, 102767 CrossRef.
- Y. Chen, S. Zhuo, Z. Li and C. Wang, EnergyChem, 2020, 2, 100030 CrossRef.
- H.-G. Wang, Q. Wu, L. Cheng and G. Zhu, Coord. Chem. Rev., 2022, 472, 214772 CrossRef CAS.
- T. Sun, W. Zhang, Q. Nian and Z. Tao, Chem. Eng. J., 2023, 452, 139324 CrossRef CAS.
- S. D. Kang and G. J. Snyder, Nat. Mater., 2017, 16, 252–257 CrossRef PubMed.
- M. Ballabio, T. Zhang, C. Chen, P. Zhang, Z. Liao, M. Hambsch, S. C. B. Mannsfeld, E. Zschech, H. Sirringhaus, X. Feng, M. Bonn, R. Dong and E. Canovas, Adv. Funct. Mater., 2021, 31, 2105184 CrossRef CAS.
- C. Q. Cui, X. H. Su and J. Y. Lee, Polym. Degrad. Stab., 1993, 41, 69–76 CrossRef CAS.
- A. Kabumoto, K. Shinozaki, K. Watanabe and N. Nishikawa, Synth. Met., 1988, 26, 349–355 CrossRef CAS.
- M. Gao, G. Zhang, G. Zhang, X. Wang, S. Wang and Y. Yang, Polym. Degrad. Stab., 2011, 96, 1799–1804 CrossRef CAS.
- T. Kobayashi, H. Yoneyama and H. Tamura, J. Electroanal. Chem. Interfacial Electrochem., 1984, 177, 293–297 CrossRef CAS.
- X. Fu, W. Zhang, B. Lan, J. Wen, S. Zhang, P. Luo, R. Zhang, S. Hu and Q. Liu, ACS Appl. Energy Mater., 2020, 3, 12360–12367 CrossRef CAS.
- M. I. Khan, X. Jia, Z. Wang and G. Cao, ACS Appl. Mater. Interfaces, 2023, 15, 25980–25989 CrossRef CAS PubMed.
- M. S. Rahmanifar, M. F. Mousavi and M. Shamsipur, J. Power Sources, 2002, 110, 229–232 CrossRef CAS.
- H.-Y. Shi, Y.-J. Ye, K. Liu, Y. Song and X. Sun, Angew. Chem., Int. Ed., 2018, 57, 16359–16363 CrossRef CAS PubMed.
- G. Shim, M. X. Tran, G. Liu, D. Byun and J. K. Lee, Energy Storage Mater., 2021, 35, 739–749 CrossRef.
- X. Gong, H. Yang, J. Wang, G. Wang and J. Tian, ACS Appl. Mater. Interfaces, 2023, 15, 4152–4165 CrossRef CAS PubMed.
- F. Wan, L. Zhang, X. Wang, S. Bi, Z. Niu and J. Chen, Adv. Funct. Mater., 2018, 28, 18044975 Search PubMed.
- J. Liang, M. Tang, L. Cheng, Q. Zhu, R. Ji, X. Liu, Q. Zhang, H. Wang and Z. Liu, J. Colloid Interface Sci., 2022, 607, 1262–1268 CrossRef CAS PubMed.
- Q. Wang, Y. Liu and P. Chen, J. Power Sources, 2020, 468, 228401 CrossRef CAS.
- I. Kulszewicz-Bajer, A. Proń, J. Abramowicz, C. Jeandey, J.-L. Oddou and J. W. Sobczak, Chem. Mater., 1999, 11, 552–556 CrossRef CAS.
- A. Kellenberger, E. Dmitrieva and L. Dunsch, Phys. Chem. Chem. Phys., 2011, 13, 3411–3420 RSC.
- S. Quillard, G. Louarn, J. P. Buisson, S. Lefrant, J. Masters and A. G. MacDiarmid, Synth. Met., 1992, 50, 525–530 CrossRef CAS.
- S. Quillard, G. Louarn, J. P. Buisson, S. Lefrant, J. Masters and A. G. MacDiarmid, Synth. Met., 1993, 55, 475–480 CrossRef CAS.
- J. E. P. da Silva, S. I. C. de Torresi, D. L. A. de Faria and M. L. A. Temperini, Synth. Met., 1999, 101, 834–835 CrossRef.
- M. Trchová, Z. Morávková, M. Bláha and J. Stejskal, Electrochim. Acta, 2014, 122, 28–38 CrossRef.
- G. Ćirić-Marjanović, M. Trchová and J. Stejskal, J. Raman Spectrosc., 2008, 39, 1375–1387 CrossRef.
- S. Zhang, S. Long, H. Li and Q. Xu, Chem. Eng. J., 2020, 400, 125898 CrossRef CAS.
- M. Cochet, S. Quillard, J. P. Buisson, S. Lefrant and G. Louarn, Synth. Met., 1999, 101, 793–794 CrossRef CAS.
- M. Lapkowski, K. Berrada, S. Quillard, G. Louarn, S. Lefrant and A. Pron, Macromolecules, 1995, 28, 1233–1238 CrossRef CAS.
- G. M. do Nascimento, V. R. L. Constantino, R. Landers and M. L. A. Temperini, Macromolecules, 2004, 37, 9373–9385 CrossRef CAS.
- R. Mažeikienė, V. Tomkutė, Z. Kuodis, G. Niaura and A. Malinauskas, Vib. Spectrosc., 2007, 44, 201–208 CrossRef.
- Y. Furukawa, F. Ueda, Y. Hyodo, I. Harada, T. Nakajima and T. Kawagoe, Macromolecules, 1988, 21, 1297–1305 CrossRef CAS.
- T. J. Durnick and S. C. Wait, J. Mol. Spectrosc., 1972, 42, 211–226 CrossRef CAS.
- H. Yan, X. Mu, Y. Song, Z. Qin, D. Guo, X. Sun and X.-X. Liu, Chem. Commun., 2022, 58, 1693–1696 RSC.
- L. A. P. Kane-Maguire, A. G. MacDiarmid, I. D. Norris, G. G. Wallace and W. Zheng, Synth. Met., 1999, 106, 171–176 CrossRef CAS.
- S. Bera, A. A. M. Prince, S. Velmurugan, P. S. Raghavan, R. Gopalan, G. Panneerselvam and S. V. Narasimhan, J. Mater. Sci., 2001, 36, 5379–5384 CrossRef CAS.
- A. Malinauskas, J. Power Sources, 2004, 126, 214–220 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc04766k |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |