
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Enantioselective and collective total synthesis of pentacyclic 19-nor-clerodanes†
Zhi-Mao
Zhang
,
Junliang
Zhang
 and
Quan
Cai
*
and
Quan
Cai
*
Department of Chemistry and Research Center for Molecular Recognition and Synthesis, Fudan University, 220 Handan Rd., Shanghai 200433, China. E-mail: quan_cai@fudan.edu.cn
First published on 23rd October 2023
Abstract
We report herein the collective asymmetric total synthesis of seven pentacyclic 19-nor-clerodane diterpenoids, namely (+)-teucvin (+)-cracroson A, (+)-cracroson E, (+)-montanin A, (+)-teucvisin C, (+)-teucrin A, and (+)-2-hydroxyteuscorolide. An ytterbium-catalyzed asymmetric inverse-electron-demand Diels–Alder reaction of 4-methyl-2-pyrone with a chiral C5-substituted cyclohexa-1,3-dienol silyl ether is the key feature of the synthesis, which provides the common cis-decalin intermediate with five continuous stereocenters in excellent yield and stereoselectivity. From this diversifiable intermediate, the total synthesis of (+)-teucvin and (+)-2-hydroxyteuscorolide was realized in thirteen and eighteen steps, respectively. From (+)-teucvin, five other pentacyclic 19-nor-clerodanes were divergently and concisely generated through late-stage oxidation state adjustments.
Introduction
Clerodane diterpenoids and their 19-nor variants are a diverse group of natural products; over 1300 family members have been isolated.1 Fascinating biological and pharmacological activities are shown by this class of compounds, including insect antifeedant,2 selective κ opioid receptor agonist,3 anti-cancer,4 anti-inflammatory5 and antibiotic activities.6 Consequently, the total synthesis of clerodanoids has attracted extensive attention from the synthetic community for decades.7 Among these molecules, pentacyclic 19-nor-clerodanes are attractive synthetic targets owing to their intriguing structures, which feature a compact and densely-functionalized decalin core, a spiro γ-lactone unit, and a fused α,β-unsaturated-γ-lactone moiety (Fig. 1). This unique type of polycyclic structure provides a great platform for the development of new synthetic strategies. For instance, the Williams group explored a creative 6π-electrocyclization for the construction of ABC tricyclic ring scaffold in a concise fashion.8 Collaborating with Ley et al., the Williams group also developed a late-stage Diels–Alder approach for the synthesis of the main decalin scaffold with the furanospiro-γ-lactone moiety and most of the required functionalities.9 Despite these encouraging achievements, total syntheses of pentacyclic 19-nor-clerodanes are still quite challenging due to their structural complexities; only two research groups have succeeded in this area to date.10,11 In 2003, the Liu group reported the first total synthesis of teucvin (1) in a racemic form via the normal-electron-demand Diels–Alder reaction,10a and then realized the racemic total synthesis of teuscorolide and montanin A (3) by the same strategy.10b In 2012, the Lee group furnished the first enantioselective total synthesis of (−)-teucvidin (2) through an elegant Michael/Conia-ene cascade cyclization based on the chiral pool strategy.11 However, to the best of our knowledge, the total synthesis of more complex pentacyclic 19-nor-clerodanes with higher oxidation states (e.g.4–8), especially in an enantioselective manner, has yet to be realized.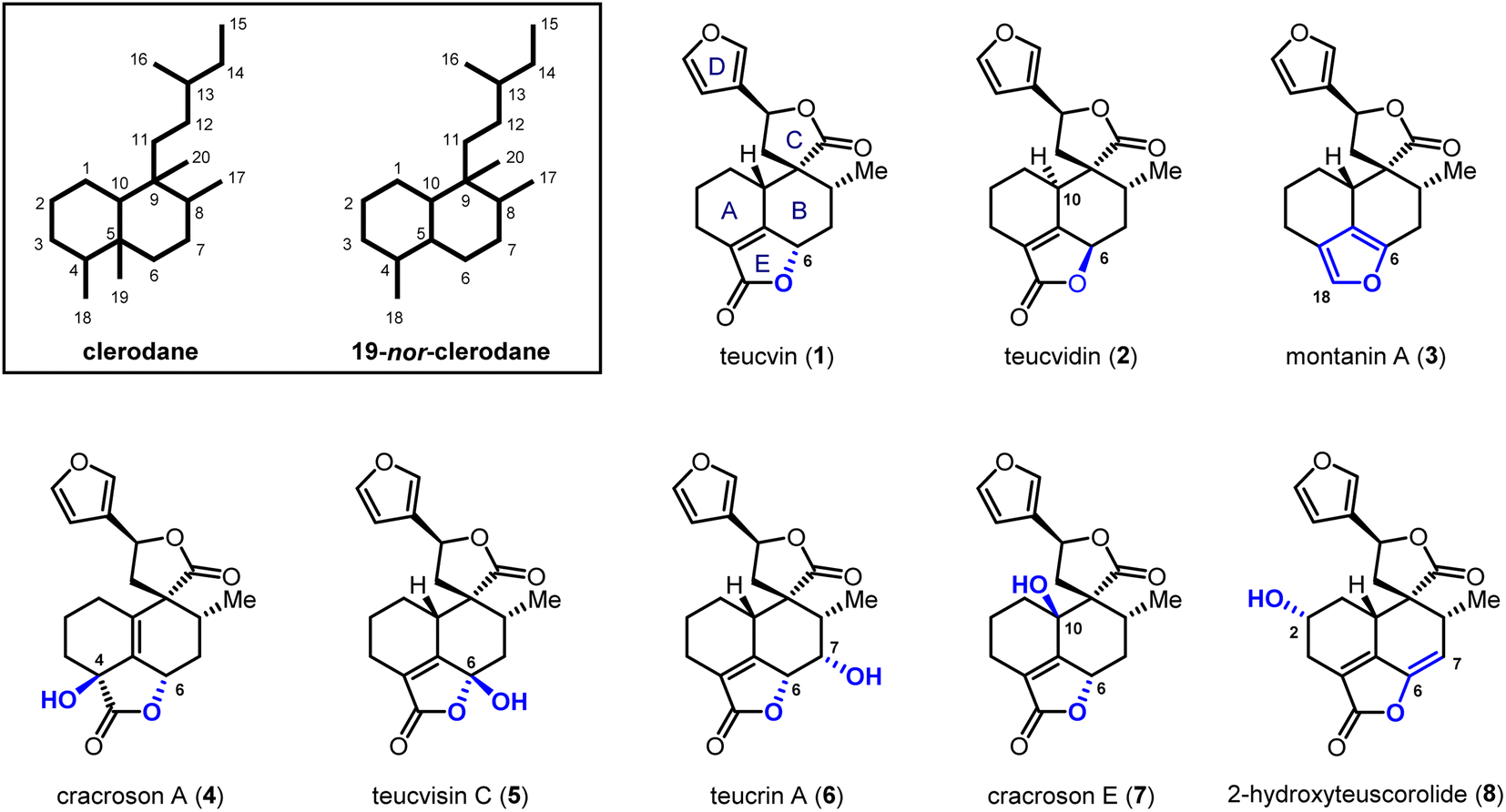 | ||
| Fig. 1 Representative pentacyclic 19-nor-clerodane diterpenoids. | ||
Compared with the traditional “single-target” strategy, the collective total synthesis of a range of structurally diverse natural products from a common intermediate has emerged as a powerful technique in organic synthesis.12 This strategy would not only trace biosynthetic relationships within targeted molecules, but also provide great opportunities for their comprehensive biological evaluation. Structurally, teucvin (1) is one of the most representative family members of pentacyclic 19-nor-clerodanes.13 Decorating hydroxyl groups at different positions (C2–C10) on the decalin moiety of teucvin (1) generates various types of family members (4–8). Based on this structural analysis, we plan to develop a unified approach for the collective total synthesis of pentacyclic 19-nor-clerodane diterpenoids. First, an efficient method for the synthesis of a decalin intermediate with multiple contiguous stereogenic centers and most of the required functionalities should be developed. From this common intermediate, the total synthesis of teucvin (1) and other family members could be accomplished concisely. Going forward, further transformations to adjust the oxidation states of teucvin (1) and introduce oxygenated functionalities regioselectively and stereoselectively are required to realize the divergent synthesis of clerodane congeners with higher oxidation states.
2-Pyrone-based Diels–Alder reactions are a prominent method for the construction of six-membered carbocycles.14 Recently, catalytic asymmetric inverse-electron-demand Diels–Alder (IEDDA) reactions of 2-pyrones have attracted increasing attention owing to their great potential in the synthesis of bioactive natural products, which was demonstrated by Markó,15 Posner,16 Gademann,17 de la Torre18 and our group.19 In 2020, our group has developed an efficient method for the construction of densely functionalized cis-decalin scaffolds by ytterbium-catalyzed asymmetric IEDDA reactions of 2-pyrones and then applied it to the total synthesis of terpenoids 4-amorphen-11-ol and cis-crotonin.19b Based on this reaction, we designed a general and unified approach for the collective synthesis of pentacyclic 19-nor-clerodane diterpenoids. As shown in Scheme 1, the key diversifiable intermediate 9 bearing five continuous stereogenic centers was generated through the catalytic asymmetric IEDDA reaction between 4-methyl-2-pyrone 10 and C5-substituted cyclohexa-1,3-dienol silyl ether 11 with a suitable absolute configuration. From tricyclic lactone 9, both teucvin (1)13 and 2-hydroxyteuscorolide (8)20 could be generated by downstream transformations involving the stereoselective 1,2-addition to attach the 3-furyl group, and the cyclization step to construct the α,β-unsaturated-γ-lactone E ring. From teucvin (1), we proposed that by sophisticated late-stage manipulations of the oxidation state on the decalin scaffold, five other 19-nor-clerodanoids, including montanin A (3),21 cracroson A (4),22 teucvisin C (5),23 teucrin A (6),24 and cracroson E (7),25 could be synthesized divergently.
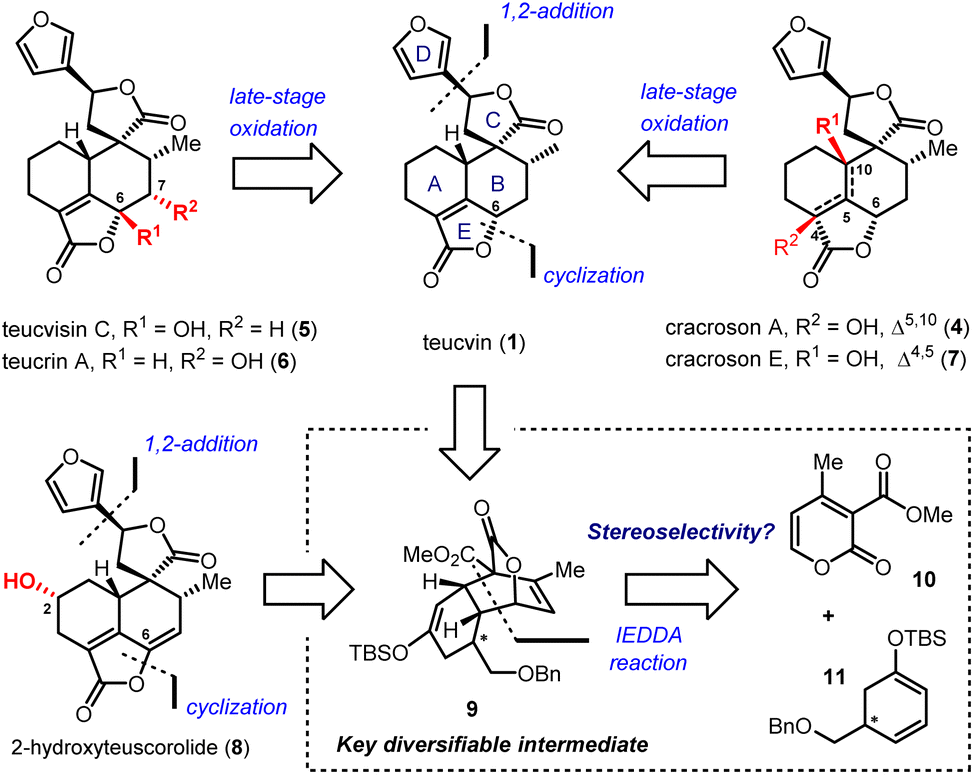 | ||
| Scheme 1 Retrosynthetic analysis based on the 2-pyrone-based IEDDA reaction and late-stage oxidative transformations. | ||
Results and discussion
Preparation of the common synthetic intermediate anti-9
Our synthesis began with the preparation of enantiopure cyclohexa-1,3-dienol silyl ether (S)-11 and (R)-11. As shown in Scheme 2, the Robinson annulation of α,β-unsaturated aldehyde 12 with tert-butyl acetoacetate 13 catalyzed by (S)-cat. Followed by the decarboxylation under acidic conditions gave cyclohexenone (R)-14 in 65% yield and 93% ee.26 (R)-14 was then treated with LiHMDS and TBSCl to provide (S)-11 in 69% yield. Enantiomeric (R)-11 was prepared in 42% yield and 92% ee over two steps using (R)-cat. as the catalyst. Furthermore, (rac)-11 was also synthesized by the same reaction sequence (see ESI for details†).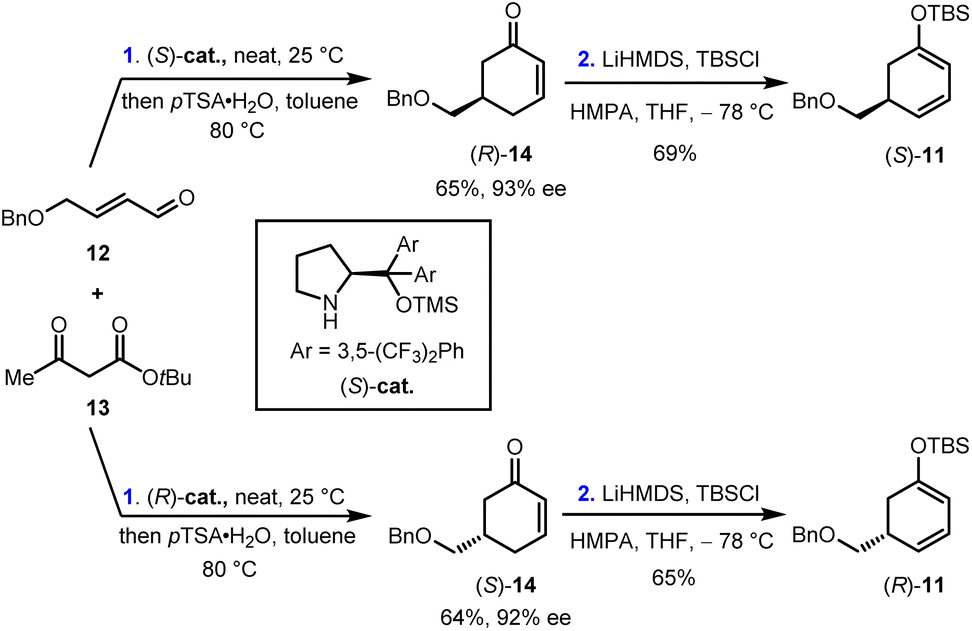 | ||
| Scheme 2 Synthesis of enantiopure cyclohexa-1,3-dienol silyl ether (S)-11 and (R)-11 by an organocatalyzed asymmetric Robinson annulation. pTSA, p-toluenesulfonic acid; LiHMDS, lithium bis(trimethylsilyl)amide; TBSCl, tert-butyl dimethylsilyl chloride; HMPA, hexamethylphosphoramide. | ||
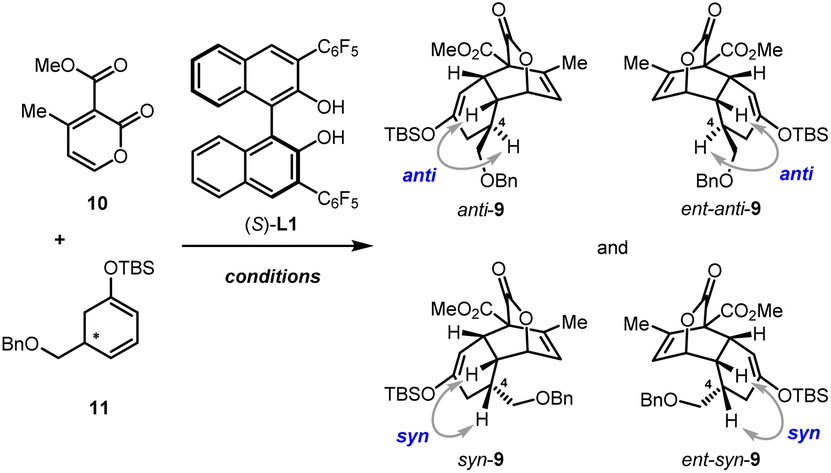
With these C5-substituted cyclohexa-1,3-dienol silyl ethers in hand, the pivotal IEDDA reaction was investigated. Initially, the IEDDA reaction of 4-methyl-2-pyrone 10 with enantiopure (S)-11 (93% ee) was attempted in the presence of catalytic amounts of Yb(OTf)3 without a chiral ligand (Table 1, entry 1). This reaction proceeded smoothly but showcased only moderate substrate-control for the stereoselectivity, giving anti-9 and ent-syn-9 as a 3.1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture in 78% yield and 94% ee. Then the effect of kinetic resolution in the IEDDA reaction was investigated (Table 1, entry 2). Upon treating 2-pyrone 10 (1.0 equiv.) and (rac)-11 (2.0 equiv.) with Yb(OTf)3 and (S)-L1, a mixture of anti-9 (78% ee) and syn-9 (90% ee) was generated in 73% yield and 1.2:1 dr. Although the diastereoselectivity was not good, the high ee of syn-9 bearing a sterically hindered substituent on the concave side of the cis-decalin scaffold indicated that very good stereocontrol might be induced by the chiral catalyst. To confirm this hypothesis, we investigated the reaction using sterically mismatched (R)-11 (92% ee) as the dienophile (Table 1, entry 3). Intriguingly, the thermodynamically disfavored product syn-9 was afforded as the main product with >99% ee and 3.8:1 dr. Encouraged by these findings, to improve the performance of the IEDDA reaction, sterically matched (S)-11 (93% ee) and Yb(OTf)3/(S)-L1 were used (Table 1, entry 4). To our great delight, through synergistic stereo-control of the substrate and the chiral catalyst, anti-9 with the right stereochemistry was afforded as the main product with a dramatically increased 98% ee, 10:1 dr, and 87% total yield in a gram scale (4.34 g).
:1 mixture in 78% yield and 94% ee. Then the effect of kinetic resolution in the IEDDA reaction was investigated (Table 1, entry 2). Upon treating 2-pyrone 10 (1.0 equiv.) and (rac)-11 (2.0 equiv.) with Yb(OTf)3 and (S)-L1, a mixture of anti-9 (78% ee) and syn-9 (90% ee) was generated in 73% yield and 1.2:1 dr. Although the diastereoselectivity was not good, the high ee of syn-9 bearing a sterically hindered substituent on the concave side of the cis-decalin scaffold indicated that very good stereocontrol might be induced by the chiral catalyst. To confirm this hypothesis, we investigated the reaction using sterically mismatched (R)-11 (92% ee) as the dienophile (Table 1, entry 3). Intriguingly, the thermodynamically disfavored product syn-9 was afforded as the main product with >99% ee and 3.8:1 dr. Encouraged by these findings, to improve the performance of the IEDDA reaction, sterically matched (S)-11 (93% ee) and Yb(OTf)3/(S)-L1 were used (Table 1, entry 4). To our great delight, through synergistic stereo-control of the substrate and the chiral catalyst, anti-9 with the right stereochemistry was afforded as the main product with a dramatically increased 98% ee, 10:1 dr, and 87% total yield in a gram scale (4.34 g).
|
|
|||
|---|---|---|---|
| Entry | Conditions | Results | Products |
| a Reaction conditions: 10 (0.10 mmol), 11 (0.15 mmol), Yb(OTf)3 (10 mol%), (S)-L1 (12 mol%), DIPEA (24 mol%), 4 Å M.S. (25 mg), DCM (0.25 mL) at 25 °C. b (S)-L1 was not added. DCM, dichloromethane; DIPEA, N,N-diisopropylethylamine; M.S., molecular sieves. | |||
| 1b | (S)-11 (1.5 equiv.) | 78%, 3.1:1 dr |
anti-9, 94% ee |
| Yb(OTf)3 | ent-syn-9, 94% ee | ||
| 2 | (rac)-11 (2.0 equiv.) | 73%, 1.2:1 dr |
anti-9, 78% ee |
| Yb(OTf)3, (S)-L1, DIPEA | syn-9, 90% ee | ||
| 3 | (R)-11 (1.5 equiv.) | 75%, 3.8:1 dr |
syn-9, >99% ee |
| Yb(OTf)3, (S)-L1, DIPEA | ent-anti-9, 72% ee | ||
| 4 | (S)-11 (1.5 equiv.) | 87%, 10:1 dr (4.34 g) |
anti-9, 98% ee |
| Yb(OTf)3, (S)-L1, DIPEA | ent-syn-9, 19% ee | ||
Total synthesis of (+)-teucvin
Having succeeded in preparing the key intermediate anti-9 with high enantiopurity (98% ee), the stage was now set for the total synthesis of teucvin (1), as summarized in Scheme 3a. Thus, anti-9 was hydrolyzed under acidic conditions to afford ketone 15 in 83% yield. The stereoselective hydrogenation (Pd/C, 1 atm H2) of the trisubstituted olefin through the convex side of the cis-decalin framework of 15, with simultaneous deprotection of the benzyl group, gave lactone 16 in 99% yield with the required configuration at C8. Then, the C2-carbonyl group of 16 was removed by treatment with tosylhydrazine followed by the reduction of catecholborane to furnish 17 in 76% yield.27 Ring opening of the lactone group of 17 (K2CO3, MeOH) led to the diol intermediate, which was immediately protected with silyl groups (TBSOTf, 2,6-lutidine) to furnish diester 18 in 77% yield. Removal of one of the two ester groups in 18 by Krapcho decarboxylation28 gave 19 with 4:1 dr at C9 in 89% overall yield. Then, the addition of the anion generated from 19 and LDA to allyl iodide followed by ozonolysis (O3, Et3N) proceeded smoothly to afford aldehyde 20 in 65% yield over two steps. To construct the C12 stereogenic center, several approaches for the 1,2-addition of 3-furanyl metal reagent to aldehyde 20 were examined (see ESI for details†). To our great delight, it was found that the stereoselective BINOL-catalyzed 1,2-addition of (3-furyl)Ti(OiPr)3 to aldehyde 20 along with spontaneous lactonization constructed the spiro γ-lactone motif very efficiently,29 generating 22 in 90% yield and 15:1 dr at C12. Global deprotection of the silyl groups by TBAF followed by sequential Swern oxidation [(COCl)2, DMSO]30 and Pinnick oxidation (NaClO2, NaH2PO4)31 led to γ-keto acid 24 in 76% yield over three steps. From 24, the α,β-unsaturated-γ-lactone E ring was constructed by treatment with NaOAc/Ac2O under reflux, thus affording (+)-teucvin (1) in 53% yield.
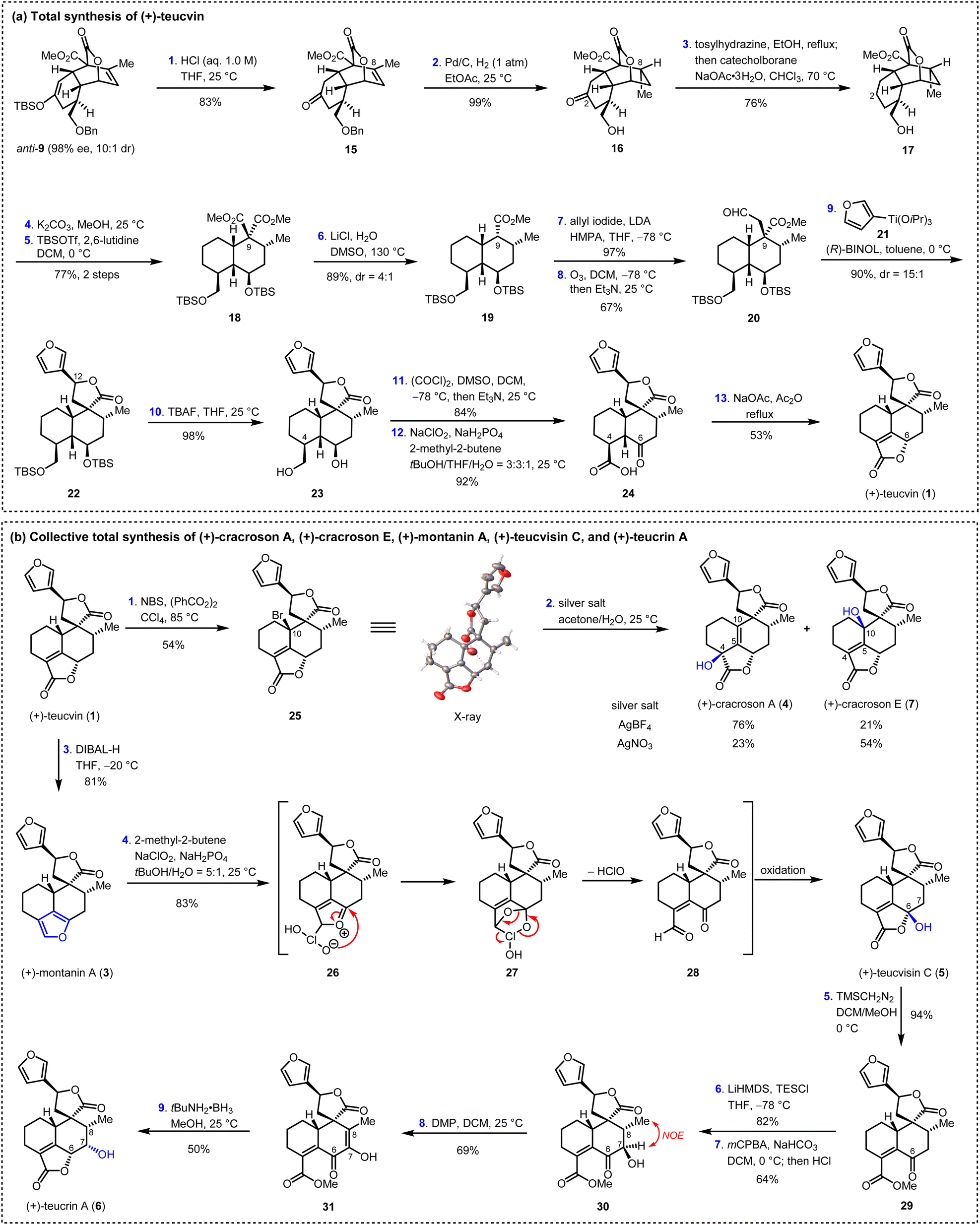 | ||
| Scheme 3 Total synthesis of six pentacyclic 19-nor-clerodane diterpenoids. THF, tetrahydrofudran; DMSO, dimethyl sulfoxide; TBSOTf, tert-butyldimethylsilyl trifluoromethanesulfonate; LDA, lithium diisopropylamide; BINOL, 1,1′-binaphthol; TBAF, tetrabutylammonium fluoride; NBS, N-bromosuccinimide; DIBAL-H, diisobutylaluminum hydride; mCPBA, meta-chloroperoxybenzoic acid; DMP, Dess–Martin periodinane. | ||
Total synthesis of (+)-cracroson A, (+)-cracroson E, (+)-montanin A, (+)-teucvisin C, and (+)-teucrin A
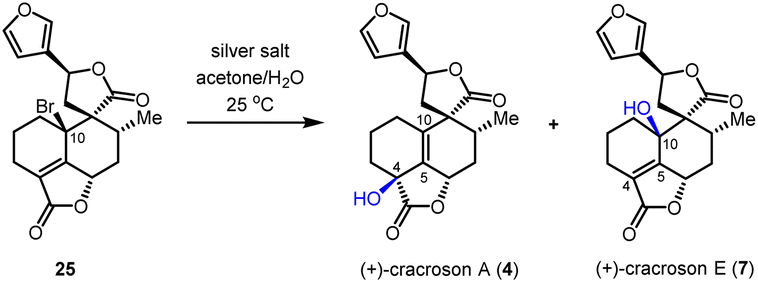
Based on the reliable synthetic route to (+)-teucvin (1), we now embarked on the collective total synthesis of more complex pentacyclic 19-nor-clerodanes, including (+)-cracroson A (4), (+)-cracroson E (7), (+)-montanin A (3), (+)-teucvisin C (5), and (+)-teucrin A (6), by oxidative transformations (Scheme 3b). Initially, we planned to obtain (+)-cracroson A (4) and (+)-cracroson E (7) by direct allylic oxidation of (+)-teucvin (1). However, this strategy was unfruitful (see ESI for details†). Intriguingly, it was discovered that bromination of 1 by NBS in the presence of benzoyl peroxide in refluxing CCl4 regioselectively and stereoselectively generated allylic bromide 25 in 54% yield.32 The structure of 25 was unambiguously confirmed by X-ray crystallographic analysis (CCDC 2274928). With 25 in hand, we decided to accomplish the synthesis of (+)-cracroson A (4) and (+)-cracroson E (7) by the Ag-promoted hydrolysis.32,33 To our delight, in the presence of AgOTf (3.0 equiv.), hydrolysis of 25 in acetone/H2O at room temperature occurred to afford (+)-cracroson A (4) in 66% yield and (+)-cracroson E (7) in 28% yield (Table 2, entry 1). Since the hydroxyl group was installed with a reserved configuration in both (+)-cracroson A (4) and (+)-cracroson E (7), we envisaged an allylic carbocation intermediate was generated. Therefore, to improve the regioselectivity of this Ag-promoted hydrolysis, we investigated various silver salts with different counterions, which might influence the distribution of products owing to the different electronic or steric effect (Table 2, entries 1–7). Of note, when AgBF4 was used as the promoter, (+)-cracroson A (4) was generated as the major product in 76% yield, along with (+)-cracroson E (7) as the minor product in 21% yield (Table 2, entry 4). Very interestingly, the utilization of AgNO3 could reverse the regioselectivity, giving (+)-cracroson E (7) as the major product in 54% yield and (+)-cracroson A (4) as the minor product in 23% yield (Table 2, entry 6). The structure of (+)-cracroson E (7) was confirmed by X-ray crystallographic analysis (CCDC 2279407).|
|
||||
|---|---|---|---|---|
| Entry | Silver salt | Cracroson Ab (%) | Cracroson Eb (%) | Ratio |
|
a Reaction conditions: 25 (0.01 mmol), silver salt (0.03 mmol), acetone/H2O (v/v = 9:1, 0.70 mL) at 25 °C.
b Isolated yield.
|
||||
| 1 | AgOTf | 66% | 28% | 2.4:1 |
| 2 | AgNTf2 | 49% | 33% | 1.5:1 |
| 3 | AgSbF6 | 47% | 15% | 3.1:1 |
| 4 | AgBF4 | 76% | 21% | 3.6:1 |
| 5 | AgClO4 | 70% | 17% | 4.1:1 |
| 6 | AgNO3 | 23% | 54% | 1:2.3 |
| 7 | Ag2CO3 | Trace | Trace | — |
Additionally, selective reduction of the α,β-unsaturated-γ-lactone group in (+)-teucvin (1) to hemiacetal, which was followed by aromatic elimination, gave (+)-montanin A (3) in 81% yield (Scheme 3b).10b,34 After judicious selection of the oxidative reaction conditions, it was found that treatment of (+)-montanin A (3) with NaClO2 and NaH2PO4 directly afforded (+)-teucvisin C (5) in 83% yield.35 We proposed this transformation involved a [4 + 2] cycloaddition between 3 and in situ generated HClO2 to give intermediate 27. The latter underwent rapid ring opening to afford the γ-keto aldehyde intermediate 28, which was then converted to (+)-teucvisin C (5) through Pinnick oxidation and cyclization.
From (+)-teucvisin C (5), the total synthesis of (+)-teucrin A (6) with five continuous stereogenic centers was realized for the first time. As shown in Scheme 3b, the reaction of tecuvisin C (5) with TMSCH2N2 efficiently gave γ-keto ester 29 in 94% yield. Treating 29 with LiHMDS and TESCl followed by the Rubottom oxidation36 by mCPBA gave 30 in 52% yield over two steps, in which the C7-hydroxyl group exhibited the opposite configuration compared with (+)-teucrin A (6) (determined by NOE spectrum). To reverse the configuration, Dess–Martin periodinane was applied to oxidize the C7-hydroxyl group. The resulting intermediate 31 was then exposed to tBuNH2 BH3,37 and total synthesis of (+)-teucrin A (6) was accomplished by stereoselective reduction of the α-keto enol group followed by spontaneous lactonization.
Total synthesis of (+)-2-hydroxyteuscorolide
We then turned our attention to the total synthesis of (+)-2-hydroxyteuscorolide (8) to demonstrate the versatility of our synthetic route. Compared with other family members, 2-hydroxyteuscorolide possesses a hydroxyl group on the C2 position of the decalin scaffold, which was located far away from the convertible α,β-unsaturated-γ-lactone structural motif. Therefore, late-stage installation of this essential hydroxyl group from (+)-teucvin (1) by oxidative transformations might require lengthy steps. Consequently, our synthesis commenced with the utilization of 16 as the key intermediate, of which the C2 carbonyl group served as a handle for the introduction of the hydroxyl functionality. As depicted in Scheme 4, protection of the carbonyl group in 16 with 2-methyl-2-ethyl-1,3-dioxolane resulted in the formation of ketal 32 in 84% yield. Ring opening of the lactone group and subsequent protection of the diol with TBS groups led to diester 33 in 70% yield. Krapcho decarboxylation of 33 gave 34 in 71% yield. Then, allyl substitution followed by ozonolysis of the resulting olefin afforded aldehyde 35 in 78% yield over two steps. The reaction of 35 with (3-furyl)Ti(OiPr)3 in the presence of a BINOL ligand provided the desired spiro γ-lactone intermediate, which was then treated with TBAF to furnish 36 in 78% yield over two steps. Subsequent Swern oxidation and Pinnick oxidation led to γ-keto acid 37 (80% yield, two steps). Sequential exposure of the latter to NaOAc in refluxing Ac2O (ring closure) and then HCl (deprotection) led to 38 in 63% overall yield. Interestingly, treatment of 38 with DIBAL-H reduced the C2 ketone and the α,β-unsaturated-γ-lactone simultaneously to install the C2 hydroxyl group with the desired configuration and form the furan moiety. The resulting intermediate was then protected with the TBS group to afford 39 in 68% yield over two steps. Finally, oxidation of the latter with NaClO2 and NaH2PO4 followed by dehydration and concomitant deprotection with pTSA accomplished the first total synthesis of (+)-2-hydroxyteuscorolide (8). | ||
| Scheme 4 The first and enantioselective total synthesis of (+)-2-hydroxyteuscorolide (8). | ||
Conclusions
In conclusion, asymmetric total synthesis of seven pentacyclic 19-nor-clerodanes was achieved through a divergent approach. Among them, the total synthesis of four members, including (+)-cracroson A, (+)-cracroson E, (+)-teucrin A and (+)-2-hydroxyteuscorolide, was realized for the first time and enantioselectively. The efficient catalytic asymmetric inverse-electron-demand Diels–Alder reaction of 3-carbometh-oxyl-2- pyrone with a chiral C5-substituted cyclohex-a-1,3-dienol silyl ether was developed to construct the central highly functionalized decalin ring. The key diversifiable intermediate with five continuous stereocenters was prepared with excellent yield and stereoselectivity through the synergistic stereo-control of the chiral catalyst and substrate. From this common intermediate, total synthesis of (+)-teucvin and (+)-2-hydroxyteuscorolide was accomplished in 13 and 18 steps, respectively. Five other pentacyclic 19-nor-clerodanes were synthesized collectively through late-stage oxidative transformations of (+)-teucvin. We are currently applying the IEDDA reaction of 2-pyrones to the synthesis of other related diterpenoids, which will be reported in due course.Data availability
The experimental procedures, copies of all spectra data and full characterization have been deposited in the ESI.†Author contributions
Q. C. conceived and directed the project and wrote the manuscript with assistance from Z.-M. Z.; Q. C. and Z.-M. Z. designed the synthetic route. Z.-M. Z. performed the experiments and analyzed the results. Q. C. and Z.-M. Z. interpreted the results and comments on the paper.Conflicts of interest
There is no conflict of interest to report.Acknowledgements
We acknowledge the National Natural Science Foundation of China (Grant No. 22071030, 22222104, 218010430), and Program of Shanghai Science and Technology Committee (Grant No. 22JC1401102) for financial support.Notes and references
- (a) A. T. Merritt and S. V. Ley, Nat. Prod. Rep., 1992, 9, 243–287 RSC; (b) S. F. P. Júnior, L. M. Conserva and J. M. B. Filho, Nat. Prod. Commun., 2006, 1, 319–344 CrossRef; (c) R. Li, S. L. Morris-Natschke and K.-H. Lee, Nat. Prod. Rep., 2016, 33, 1166–1226 RSC.
- (a) A. González-Coloma, C. Gutiérrez, J. M. M. del Corral, M. Gordaliza, M. L. de la Puente and A. S. Feliciano, J. Agric. Food Chem., 2000, 48, 3677–3681 CrossRef PubMed; (b) M. Bruno, F. Piozzi and S. Rosselli, Nat. Prod. Rep., 2002, 19, 357–378 RSC; (c) E. A. K. Gebbinck, B. J. M. Jansen and A. de Groot, Phytochemistry, 2002, 61, 737–770 CrossRef PubMed.
- (a) T. E. Prisinzano and R. B. Rothman, Chem. Rev., 2008, 108, 1732–1743 CrossRef CAS PubMed; (b) J. J. Roach and R. A. Shenvi, Bioorg. Med. Chem. Lett., 2018, 28, 1436–1445 CrossRef CAS PubMed.
- (a) S. Roengsumran, K. Musikul, A. Petsom, T. Vilaivan, P. Sangvanich, S. Pornpakakul, S. Puthong, C. Chaichantipyuth, N. Jaiboon and N. Chaichit, Planta Med., 2002, 68, 274–277 CrossRef CAS PubMed; (b) Y. Shuo, C. Zhang, X. Yang, F. Liu, Q. Zhang, A. Li, J. Ma, D. Lee, Y. Ohizumi and Y. Guo, J. Nat. Med., 2019, 73, 826–833 CrossRef PubMed; (c) R. Acquaviva, G. A. Malfa, M. R. Loizzo, J. Xiao, S. Bianchi and R. Tundis, Molecules, 2022, 27, 4791 CrossRef CAS PubMed.
- (a) J.-S. Zhang, Y.-Q. Tang, J.-L. Huang, W. Li, Y.-H. Zou, G.-H. Tang, B. Liu and S. Yin, Phytochemistry, 2017, 144, 151–158 CrossRef CAS PubMed; (b) C.-L. Wang, Y. Dai, Q. Zhu, X. Peng, Q.-F. Liu, J. Ai, B. Zhou and J.-M. Yue, J. Nat. Prod., 2023, 86, 1345–1359 CrossRef CAS PubMed.
- (a) A. Bisio, A. M. Schito, S. N. Ebrahimi, M. Hamburger, G. Mele, G. Piatti, G. Romussi, F. Dal Piaz and N. de Tommasi, Phytochemistry, 2015, 110, 120–132 CrossRef CAS PubMed; (b) F. Cappiello, M. R. Loffredo, C. Del Plato, S. Cammarone, B. Casciaro, D. Quaglio, M. L. Mangoni, B. Botta and F. Ghirga, Antibiotics, 2020, 9, 325 CrossRef CAS PubMed.
- (a) T. Tokoroyama, Synth., 2000, 611–633 CrossRef CAS; (b) H. Hagiwara, Nat. Prod. Commun., 2019, 14 DOI:10.1177/1934578X19843613; (c) J. R. Scheerer, J. F. Lawrence, G. C. Wang and D. A. Evans, J. Am. Chem. Soc., 2007, 129, 8968–8969 CrossRef CAS PubMed; (d) D. S. Müller, N. L. Untiedt, A. P. Dieskau, G. L. Lackner and L. E. Overman, J. Am. Chem. Soc., 2015, 137, 660–663 CrossRef PubMed; (e) Q. Ye, P. Qu and S. A. Snyder, J. Am. Chem. Soc., 2017, 139, 18428–18431 CrossRef CAS PubMed; (f) M. J. Zeiler, G. M. Connors, G. M. Durling, A. G. Oliver, L. Marquez, R. J. Melander, C. L. Quave and C. Melander, Angew. Chem., Int. Ed., 2022, 61, e202117458 CrossRef CAS PubMed; (g) W. Zhu, Q. Yin, Z. Lou and M. Yang, Nat. Commun., 2022, 13, 6633 CrossRef CAS PubMed.
- (a) R. H. Pouwer, H. Schill, C. M. Williams and P. V. Bernhardt, Eur. J. Org Chem., 2007, 4699–4705 CrossRef CAS; (b) P. M. Mirzayans, R. H. Pouwer, C. M. Williams and P. V. Bernhardt, Eur. J. Org Chem., 2012, 1633–1638 CrossRef CAS.
- A. T. Merritt, R. H. Pouwer, D. J. Williams, C. M. Williams and S. V. Ley, Org. Biomol. Chem., 2011, 9, 4745–4747 RSC.
- (a) H.-J. Liu, J.-L. Zhu, I.-C. Chen, R. Jankowska, Y. Han and K.-S. Shia, Angew. Chem., Int. Ed., 2003, 42, 1851–1853 CrossRef CAS PubMed; (b) I.-C. Chen, Y.-K. Wu, H.-J. Liu and J.-L. Zhu, Chem. Commun., 2008, 4720–4722 RSC.
- (a) X. Liu and C.-S. Lee, Org. Lett., 2012, 14, 2886–2889 CrossRef PubMed; (b) G. Du, G. Wang, W. Ma, Q. Yang, W. Bao, X. Liang, L. Zhu and C.-S. Lee, Synlett, 2017, 28, 1394–1406 CrossRef CAS.
- (a) L. Li, Z. Chen, X. Zhang and Y. Jia, Chem. Rev., 2018, 118, 3752–3832 CrossRef CAS PubMed; (b) X.-Y. Liu and Y. Qin, Green Synth. Catal., 2022, 3, 25–39 CrossRef; (c) K. Chen and P. S. Baran, Nature, 2009, 459, 824–828 CrossRef CAS PubMed; (d) S. B. Jones, B. Simmons, A. Mastracchio and D. W. C. MacMillan, Nature, 2011, 475, 183–188 CrossRef CAS PubMed; (e) S. A. Snyder, A. Gollner and M. I. Chiriac, Nature, 2011, 474, 461–466 CrossRef CAS PubMed; (f) W. Ren, Y. Bian, Z. Zhang, H. Shang, P. Zhang, Y. Chen, Z. Yang, T. Luo and Y. Tang, Angew. Chem., Int. Ed., 2012, 51, 6984–6988 CrossRef CAS PubMed; (g) O. Wagnières, Z. Xu, Q. Wang and J. Zhu, J. Am. Chem. Soc., 2014, 136, 15102–15108 CrossRef PubMed; (h) X.-Z. Huang, L.-H. Gao and P.-Q. Huang, Nat. Commun., 2020, 11, 5314 CrossRef CAS PubMed; (i) Y. Kanda, Y. Ishihara, N. C. Wilde and P. S. Baran, J. Org. Chem., 2020, 85, 10293–10320 CrossRef CAS PubMed; (j) J. H. Kim, H. Jeon, C. Park, S. Park and S. Kim, Angew. Chem., Int. Ed., 2021, 60, 12060–12065 CrossRef CAS PubMed; (k) W. Chen, Y. Ma, W. He, Y. Wu, Y. Huang, Y. Zhang, H. Tian, K. Wei, X. Yang and H. Zhang, Nat. Commun., 2022, 13, 908 CrossRef CAS PubMed.
- E. Fujita, I. Uchida and T. Fujita, J. Chem. Soc., Chem. Commun., 1973, 793–794 RSC.
- (a) K. Afarinkia, V. Vinader, T. D. Nelson and G. H. Posner, Tetrahedron, 1992, 48, 9111–9171 CrossRef CAS; (b) Q. Cai, Chin. J. Chem., 2019, 37, 946–976 CrossRef CAS; (c) D. Dobler, M. Leitner, N. Moor and O. Reiser, Eur. J. Org Chem., 2021, 6180–6205 CrossRef CAS; (d) G. Huang, C. Kouklovsky and A. de la Torre, Chem.–Euro. J., 2021, 27, 4760–4788 CrossRef CAS PubMed; (e) M.-M. Xu and Q. Cai, Chin. J. Org. Chem., 2022, 42, 698–713 CrossRef CAS; (f) I.-J. Shin, E.-S. Choi and C.-G. Cho, Angew. Chem., Int. Ed., 2007, 46, 2303–2305 CrossRef CAS PubMed; (g) P. Zhao and C. M. Beaudry, Angew. Chem., Int. Ed., 2014, 53, 10500–10503 CrossRef CAS PubMed; (h) P. Gan, M. W. Smith, N. R. Braffman and S. A. Snyder, Angew. Chem., Int. Ed., 2016, 55, 3625–3630 CrossRef CAS PubMed; (i) N. Wang, J. Liu, C. Wang, L. Bai and X. Jiang, Org. Lett., 2018, 20, 292–295 CrossRef CAS PubMed; (j) X. Yu, L. Xiao, Z. Wang and T. Luo, J. Am. Chem. Soc., 2019, 141, 3440–3443 CrossRef CAS PubMed; (k) M. Haider, G. Sennari, A. Eggert and R. Sarpong, J. Am. Chem. Soc., 2021, 143, 2710–2715 CrossRef CAS PubMed; (l) K.-H. Sim, T. ul Ansari, Y.-G. Park, Y. Jeong, S.-H. Oh, H.-W. Min, D.-Y. Jeon, H. Kim and C.-G. Cho, Angew. Chem., Int. Ed., 2022, 61, e202212016 CrossRef CAS PubMed.
- I. E. Markó and G. R. Evans, Tetrahedron Lett., 1994, 35, 2771–2774 CrossRef.
- G. H. Posner, F. Eydoux, J. K. Lee and D. S. Bull, Tetrahedron Lett., 1994, 35, 7541–7544 CrossRef CAS.
- P. Burch, M. Binaghi, M. Scherer, C. Wentzel, D. Bossert, L. Eberhardt, M. Neuburger, P. Scheiffele and K. Gademann, Chem.–Euro. J., 2013, 19, 2589–2591 CrossRef CAS PubMed.
- (a) G. Huang, R. Guillot, C. Kouklovsky, B. Maryasin and A. de la Torre, Angew. Chem., Int. Ed., 2022, 61, e202208185 CrossRef CAS PubMed; (b) G. Huang, C. Kouklovsky and A. de la Torre, J. Am. Chem. Soc., 2022, 144, 17803–17807 CrossRef CAS PubMed.
- (a) X.-W. Liang, Y. Zhao, X.-G. Si, M.-M. Xu, J.-H. Tan, Z.-M. Zhang, C.-G. Zheng, C. Zheng and Q. Cai, Angew. Chem., Int. Ed., 2019, 58, 14562–14567 CrossRef CAS PubMed; (b) X.-G. Si, Z.-M. Zhang, C.-G. Zheng, Z.-T. Li and Q. Cai, Angew. Chem., Int. Ed., 2020, 59, 18412–18417 CrossRef CAS; (c) Y. Lu, M.-M. Xu, Z.-M. Zhang, J. Zhang and Q. Cai, Angew. Chem., Int. Ed., 2021, 60, 26610–26615 CrossRef CAS PubMed; (d) M.-M. Xu, L. Yang, K. Tan, X. Chen, Q.-T. Lu, K. N. Houk and Q. Cai, Nat. Catal., 2021, 4, 892–900 CrossRef CAS; (e) J.-X. He, X.-G. Si, Q.-T. Lu, Q.-W. Zhang and Q. Cai, Chin. J. Chem., 2023, 41, 21–26 CrossRef CAS; (f) X.-G. Si, S.-X. Feng, Z.-Y. Wang, X. Chen, M.-M. Xu, Y.-Z. Zhang, J.-X. He, L. Yang and Q. Cai, Angew. Chem., Int. Ed., 2023, 62, e202303876 CrossRef CAS PubMed.
- J. L. Marco, B. Rodriguez, C. Pascual, G. Savona and F. Piozzi, Phytochemistry, 1983, 22, 727–731 CrossRef CAS.
- P. Y. Malakov, G. Y. Papanov and N. M. Mollov, Tetrahedron Lett., 1978, 23, 2025–2026 CrossRef.
- M. Qiu, D. Cao, Y. Gao, S. Li, J. Zhu, B. Yang, L. Zhou, Y. Zhou, J. Jin and Z. Zhao, Fitoterapia, 2016, 108, 81–86 CrossRef CAS PubMed.
- H.-W. Lv, J.-G. Luo, M.-D. Zhu, S.-M. Shan and L.-Y. Kong, Chem. Pharm. Bull., 2014, 62, 472–476 CrossRef CAS PubMed.
- (a) D. P. Popa and A. M. Reinbol'd, Chem. Nat. Compd., 1973, 9, 27–30 CrossRef; (b) D. P. Popa, A. M. Reinbol'd and A. I. Rezvukhin, Chem. Nat. Compd., 1973, 9, 165–169 CrossRef.
- M. Qiu, J. Jin, L. Zhou, W. Zhou, Y. Liu, Q. Tan, D. Cao and Z. Zhao, Phytochemistry, 2018, 145, 103–110 CrossRef CAS PubMed.
- (a) A. Carlone, M. Marigo, C. North, A. Landa and K. A. Jørgensen, Chem. Commun., 2006, 4928–4930 RSC; (b) B. Hong, D. Hu, J. Wu, J. Zhang, H. Li, Y. Pan and X. Lei, Chem.–Asian J., 2017, 12, 1557–1567 CrossRef CAS PubMed.
- (a) G. W. Kabalka, D. T. C. Yang and J. D. Baker, J. Org. Chem., 1976, 41, 574–575 CrossRef CAS; (b) E. C. Angell, F. Fringuelli, F. Pizzo, A. Taticchi and E. Wenkert, J. Org. Chem., 1988, 53, 1424–1426 CrossRef CAS.
- A. P. Krapcho, J. F. Weimaster, J. M. Eldridge, E. G. E. J. Jr, A. J. Lovey and W. P. Stephens, J. Org. Chem., 1978, 43, 138–147 CrossRef CAS.
- (a) S. Zhou, C.-R. Chen and H.-M. Gau, Org. Lett., 2010, 12, 48–51 CrossRef CAS PubMed; (b) K.-H. Wu, S. Zhou, C.-A. Chen, M.-C. Yang, R.-T. Chiang, C.-R. Chen and H.-M. Gau, Chem. Commun., 2011, 47, 11668–11670 RSC; (c) N. J. Line, A. C. Burns, S. C. Butler, J. Casbohm and C. J. Forsyth, Chem.–Euro. J., 2016, 22, 17983–17986 CrossRef CAS PubMed.
- A. J. Mancuso, S.-L. Huang and D. Swern, J. Org. Chem., 1978, 43, 2480–2482 CrossRef CAS.
- B. S. Bal, W. E. Childers Jr and H. W. Pinnick, Tetrahedron, 1981, 37, 2091–2096 CrossRef CAS.
- S. Iimura, L. E. Overman, R. Paulini and A. Zakarian, J. Am. Chem. Soc., 2006, 128, 13095–13101 CrossRef CAS.
- (a) J. G. Allen and S. J. Danishefsky, J. Am. Chem. Soc., 2001, 123, 351–352 CrossRef CAS PubMed; (b) U. Shah, S. Chackalamannil, A. K. Ganguly, M. Chelliah, S. Kolotuchin, A. Buevich and A. McPhail, J. Am. Chem. Soc., 2006, 128, 12654–12655 CrossRef CAS PubMed.
- M. A. Chatterjee, A. Banerjee and F. Bohlman, Tetrahedron, 1977, 33, 2407–2414 CrossRef.
- D. L. J. Clive, Minaruzzaman and L. Ou, J. Org. Chem., 2005, 70, 3318–3320 CrossRef CAS PubMed.
- G. M. Rubottom, M. A. Vazquez and D. R. Pelegrina, Tetrahedron Lett., 1974, 15, 4319–4322 CrossRef.
- G. C. Andrews and T. C. Crawford, Tetrahedron Lett., 1980, 21, 693–696 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2274928, 2275140, 2279407. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc04335e |
| This journal is © The Royal Society of Chemistry 2023 |