
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnabling Al sacrificial anodes in tetrahydrofuran electrolytes for reductive electrosynthesis†
Wendy
Zhang
 a,
Weiyang
Guan
b,
Yi
Wang
b,
Song
Lin
b and
Kimberly A.
See
*a
a,
Weiyang
Guan
b,
Yi
Wang
b,
Song
Lin
b and
Kimberly A.
See
*a
aDivision of Chemistry and Chemical Engineering, California Institute of Technology, Pasadena, California 91125, USA. E-mail: ksee@caltech.edu
bDepartment of Chemistry and Chemical Biology, Cornell University, Ithaca, New York 14853, USA
First published on 2nd November 2023
Abstract
Al0 is widely used as a sacrificial anode in organic electrosynthesis. However, there remains a notable knowledge gap in the understanding of Al anode interface chemistry under electrolysis conditions. We hypothesize that Al interfacial chemistry plays a pivotal role in the discernible bias observed in solvent selections for reductive electrosynthesis. The majority of existing methodologies that employ an Al sacrificial anode use N,N-dimethylformamide (DMF) as the preferred solvent, with only isolated examples of ethereal solvents such as tetrahydrofuran (THF). Given the crucial role of the solvent in determining the efficiency and selectivity of an organic reaction, limitations on solvent choice could significantly hinder substrate reactivity and impede the desired transformations. In this study, we aim to understand the Al metal interfaces and manipulate them to improve the performance of an Al sacrificial anode in THF-based electrolytes. We have discovered that the presence of halide ions (Cl−, Br−, I−) in the electrolyte is crucial for efficient Al stripping. By incorporating halide additive, we achieve bulk Al stripping in THF-based electrolytes and successfully improve the cell potentials of electrochemically driven reductive methodologies. This study will encourage the use of ethereal solvents in systems using Al sacrificial anodes and guide future endeavors in optimizing electrolytes for reductive electrosynthesis.
Introduction
Electrosynthesis has emerged as a green and efficient approach for the preparation of valuable organic molecules. In recent years, a wide array of cathodic reduction methodologies has been developed.1–5 The setup of these reductive electrosyntheses often involves a metal sacrificial anode in an undivided cell. During electrolysis, metal stripping takes place at the sacrificial anode, forming metal cations that dissolve into the reaction mixture.6–9 Al is commonly employed as a sacrificial anode material for reductive electrosynthesis. It is naturally abundant and is relatively easy to oxidize from a thermodynamic perspective (standard reduction potential, Eo = −1.67 V vs. SHE).10 The crustal abundance of Al compared with other commonly used sacrificial anodes is shown in Fig. 1a. Upon oxidation, Al releases three electrons per atom (Al → Al3+ + 3e−). As a result, Al has a high theoretical volumetric capacity of 8050 mA h cm−3 compared to other commonly used sacrificial anode materials (Fig. 1a).10–12 The significantly higher volumetric capacity of Al theoretically allows for a larger scale reductive reaction to be sustained while consuming the same volume of sacrificial anode, making Al an appealing option for reductive electrosynthesis.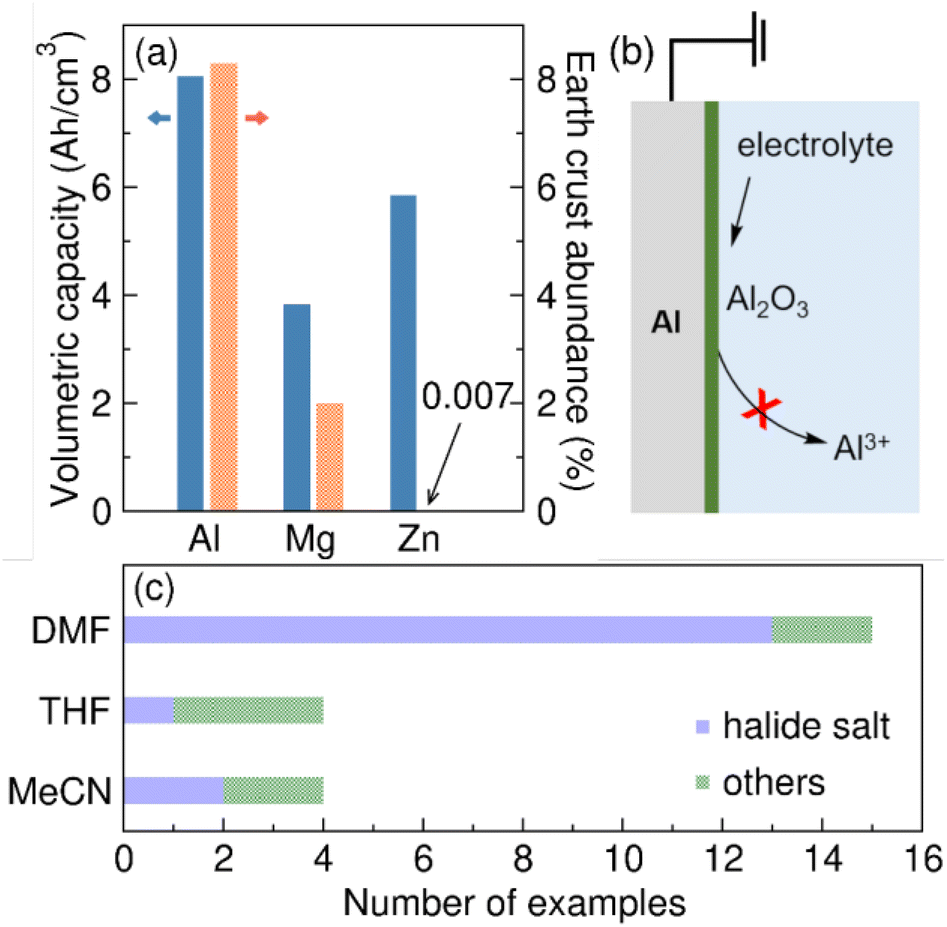 | ||
| Fig. 1 Al metal is commonly used as sacrificial anode in reductive organic electrosynthesis. (a) Compare to Mg and Zn, Al has a higher theoretical volumetric capacity and is notably more abundant in the Earth's crust. (b) However, a layer of Al2O3 may form on the surface, inhibiting electrochemical Al stripping in organic electrolytes. (c) Among the reductive electrosynthesis methodologies reported since 2000, halide salt/DMF solution is the most popular choice of electrolyte. | ||
Despite its desirable properties, Al presents a challenge as a sacrificial anode material due to its high affinity to oxygen. The standard Gibbs free energy of formation (ΔfGo) of Al2O3 at standard conditions is −378.2 kcal mol−1.13 Consequently, a layer of Al2O3 will spontaneously form on a nascent Al surface when the electrode is prepared in air or exposed to an oxidizing media.14 The layer formation is found to be essentially instantaneous.10 While the oxide layer maintains electronic conductivity, the high bond dissociation energy of Al–O (Do = 120 ± 2.5 kcal mol−1) creates a substantial barrier for Al stripping from the oxide (Fig. 1b).13 Further, we suspect that Al3+ cannot conduct through the Al2O3 surface layer, thereby shutting down any oxidative electrochemistry.
To achieve effective Al stripping counter electrode reactions in reductive electrosynthesis, it is therefore crucial to control the interface to prevent the complete passivation of Al by Al2O3 during electrolysis. High interfacial resistances can cause high cell voltages that eventually lead to premature termination of the electrolysis simply due to compliance limits on the potentiostat.15 The premature termination results in low yields, which causes the reaction conditions to be thrown out even though the organic reaction could have proceeded had the counter electrode worked.
As a result, we suggest that the effects of solvent, supporting electrolyte, and substrates on the metal sacrificial anode interfaces are critical for reaction development.9,16 Although there isn't a thorough comprehension of the chemical processes occurring at the metal interfaces, the empirical optimization approach has some success identifying conditions compatible with an Al sacrificial anode.3,6 Interestingly, there is a discernible bias observed in the solvent selection among the reported methodologies that utilize Al sacrificial anodes. As shown in Fig. 1c, most of the reactions adopt halide salt/DMF solution as the optimal electrolyte17–29 with isolated examples of TBABF4/DMF electrolyte.28,30 On the contrary, many fewer reactions were successfully optimized in THF and MeCN-based electrolytes.31–38 Since solvent can dramatically impact the efficiency and selectivity of an organic reaction, the potential limitation on solvent selection when using an Al sacrificial anode makes it challenging to expand the application of reductive electrosynthesis to various types of organic transformations.
Here, we investigate the Al interface with the aim to improve the efficacy of Al sacrificial anodes in THF-based electrolytes. THF, in comparison to DMF and MeCN, exhibits weaker coordination ability and possesses a significantly lower dielectric constant of 7.5 (vs. 38.3 for DMF and 36.6 for MeCN).13 These differences in solvent properties allow THF to stabilize distinct reaction intermediates, which may result in significant alterations in reaction rates, yields, and selectivities.39 Additionally, in organic synthesis, THF is considered as a more user-friendly solvent in comparison to DMF due to its reduced toxicity, lower environmental impact, and ease of removal from product mixtures.40,41 Therefore, it would be of great interest to enable efficient Al stripping in THF-based electrolytes for its application in reductive electrosynthesis.
Currently, the use of an Al sacrificial anode in THF-based electrolytes is commonly accompanied with extremely high overall cell potentials (>30 V) that inhibit electrolysis of the organic substrates.15,42–44 The escalation in voltage is frequently attributed to the formation of an oxide layer on the Al surface. Thus, to realize a wide application of Al sacrificial anodes in THF-based electrolytes, the ability to control the Al interface is essential. Previously, we have demonstrated that simple electrolyte tailoring strategies can change the composition of solid surface layers, often called solid electrolyte interphases (SEI), that form on Mg and improve the performance of a Mg sacrificial anode in THF-based electrolyte.9 In this work, we explore the influence of electrolyte composition on Al SEIs and stripping behavior. Linear sweep voltammetry (LSV) demonstrates that supporting electrolytes such as TBABF4, TBAClO4, and TBA bis(trifluoromethanesulfonyl)imide (TFSI) do not support efficient Al stripping in THF. X-ray photoelectron spectroscopy (XPS) reveals the formation of Al2O3 layer on the Al surface, which can inhibit Al dissolution. Inspired by Al corrosion chemistry, we use halide salts as co-supporting electrolytes to induce local disintegration of the oxide layer. With halide salts, Al stripping becomes feasible in THF, which enables bulk Al stripping under electrolysis conditions that are applicable to organic electrosynthesis. We demonstrate that addition of halides to a deoxygenative borylation reaction and a silyl cross-electrophile coupling greatly reduces the cell voltage and improves yields.
Results and discussion
Effect of the supporting electrolyte on Al stripping
To understand the Al stripping behavior in THF, we perform LSV experiments with 0.1 M TBABF4, TBAClO4, and TBATFSI supporting electrolytes. The three supporting electrolytes are chemically representative and are routinely screened for as electrolytes in reductive electrosynthesis. The LSV experiments are conducted in three-electrode cells with an Al plate working electrode (WE), graphite counter electrode (CE), and Pt|Fc/Fc+ reference electrode (RE) (Fig. 2a). We choose the Al plates from IKA, which are constructed from aluminum 5052. These particular electrodes are widely employed in electrosynthetic organic labs due to their direct compatibility with an ElectraSyn, a commonly used setup among the synthetic organic chemists. All potentials referenced in THF electrolytes are vs. the Pt|Fc/Fc+ RE unless otherwise noted. The LSV experiment starts with anodic polarization of the Al electrode from open-circuit voltage (OCV) to 0.3 V at 5 mV s−1 with 85% iR compensation. After the first LSV scan, the Al electrode has been electropolished to expose fresh Al metal. The cell is then rested at OCV for 10 min, allowing the freshly exposed Al metal to chemically react with the electrolyte. Following the rest, we repeat the LSV-OCV protocol 4 times to observe how Al stripping behavior evolves with each consecutive scan. The cell configuration and LSV protocol are adapted from our work on Mg.9 The 2nd–5th LSV scans are shown in Fig. 2b–d. The 1st LSVs of Al electropolishing are shown in the ESI.† All onset potentials and current densities of the 5th LSV scans are tabulated in Table 1.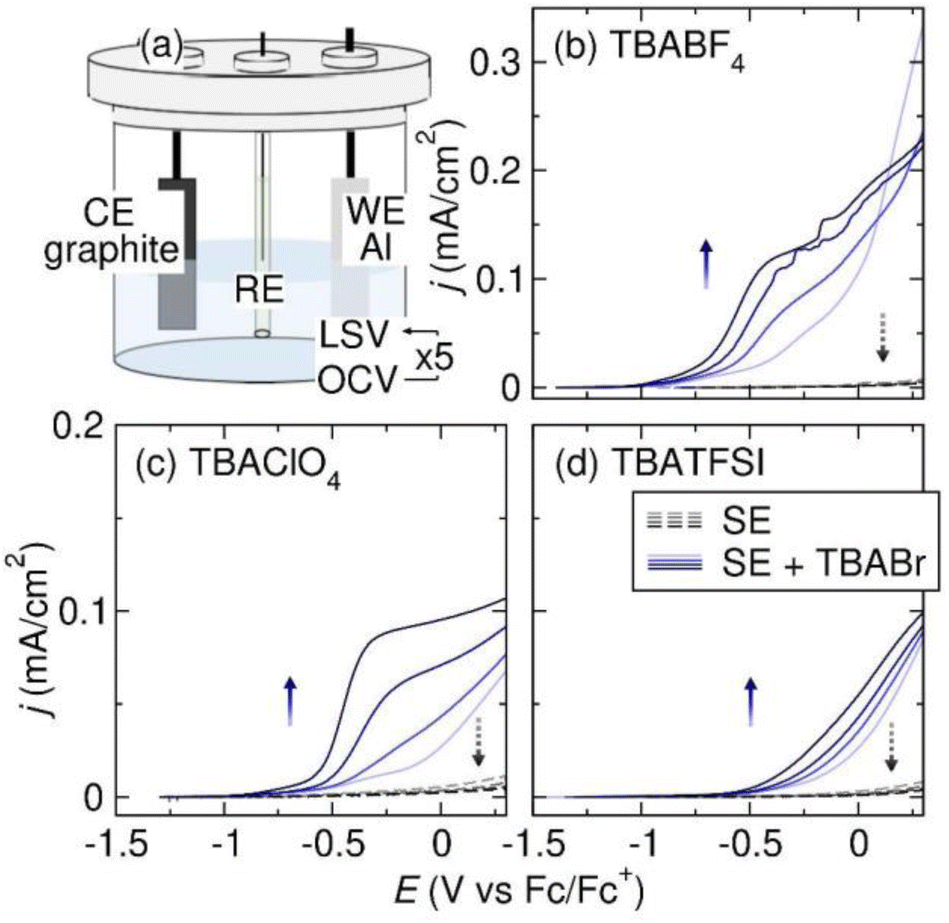 | ||
| Fig. 2 (a) A schematic of the three-electrode cell with an Al WE, graphite CE, Pt|Fc/Fc+ RE, and 0.1 M supporting electrolyte in 7 mL of THF. Linear sweep voltammograms of Al in THF with 0.1 M supporting electrolyte (SE) of interest, or 0.05 M SE + 0.05 M TBABr. The SEs are (b) TBABF4, (c) TBAClO4, and (d) TBATFSI. All voltammograms are collected at a scan rate of 5 mV s−1 with 85% iR compensation. The direction of the arrows indicates the trend of changes in current densities as the number of scans increases. | ||
| Solvent | Supporting electrolyte | E on | j (mA cm−2) | Figure ref. |
|---|---|---|---|---|
| a E on (V vs. Fc/Fc+) is defined as the potential at which dj/dE exceeds 0.1. b j at +0.5 V vs. Eon. | ||||
| THF | TBABF4 | — | <0.01 | Fig. 2b |
| TBABF4 + TBABr | −0.77 | 0.13 | Fig. 2b | |
| TBABF4 + TBACl | −0.31 | 0.30 | See ESI | |
| TBABF4 + LiI | −0.31 | 0.12 | See ESI | |
| TBAClO4 | — | <0.01 | Fig. 2c | |
| TBAClO4 + TBABr | −0.58 | 0.09 | Fig. 2c | |
| TBATFSI | — | <0.01 | Fig. 2d | |
| TBATFSI + TBABr | −0.26 | 0.09 | Fig. 2d | |
| DMF | TBABF4 | — | <0.01 | Fig. 4a |
| TBACl | −1.73 | 3.23 | Fig. 4c | |
| TBABr | −1.49 | 1.75 | Fig. 4b | |
| TBAI | −0.96 | 0.48 | Fig. 4d | |
As shown in Fig. 2b–d, the current densities for Al stripping are extremely low on the 2nd LSV scan (<0.01 mA cm−2 at 0 V) in all three electrolytes. The current densities decrease with successive LSV scans. We suspect that the poor Al stripping behavior is caused by the formation of a passivating SEI on the Al electrode surface upon contact with the electrolytes. As the Al electrode remains in the electrolyte longer, the surface layers become more passivating, leading to a progressive decline of Al stripping performance. To investigate the SEI composition, we examine the Al anodes after anodic polarization in TBABF4 and TBAClO4 electrolytes via XPS.
Fig. 3a and b shows the Al 2p region of the XPS spectra measured on the Al anode surface after the LSV experiments in THF with TBABF4 and TBAClO4 supporting electrolytes. All peak binding energies (BEs) and assignments of the Al 2p and other related regions are tabulated in the ESI.†Fig. 3a shows two Al3+-containing species after anodic polarization in the TBABF4 electrolyte. The lower BE peak at 75.1 eV is assigned to Al2O3,45 which is the major component of the Al SEI. The weak signal at 76.7 eV is assigned to AlF3,46 likely a decomposition product derived from BF4−. The sharp signal at 72.5 eV is ascribed to Al0.47 The high signal intensity indicates a thin passivating layer (<10 nm), which allows the X-ray to penetrate beneath the oxide layer to Al0. Fig. 3b shows only Al2O3 at 75.0 eV as the major component of the Al SEI formed in the TBAClO4 electrolyte. A weak signal at 199.2 eV is observed in the Cl 2p region that likely corresponds to AlClx,48 a possible decomposition product derived from ClO4− (see ESI†). However, the peak in the Al 2p region cannot be resolved from the Al2O3 due to its low intensity. A strong Al0 signal is again observed at 72.5 eV, indicating a thin SEI layer. A similar Al2O3-based SEI is observed on the surface of Al electrode polarized in TBATFSI electrolyte as well (see ESI†). Despite the SEI's formed in the three electrolytes being thin, both Al2O3 and AlF3 have been reported to inhibit Al dissolution even when present in only a thin layer.10,49 We also observe that all three electrolytes do not support Al stripping at any reasonable current density. The composition of the SEI supports the hypothesis of a passivating Al SEI inhibiting effective Al stripping in THF-based electrolytes.
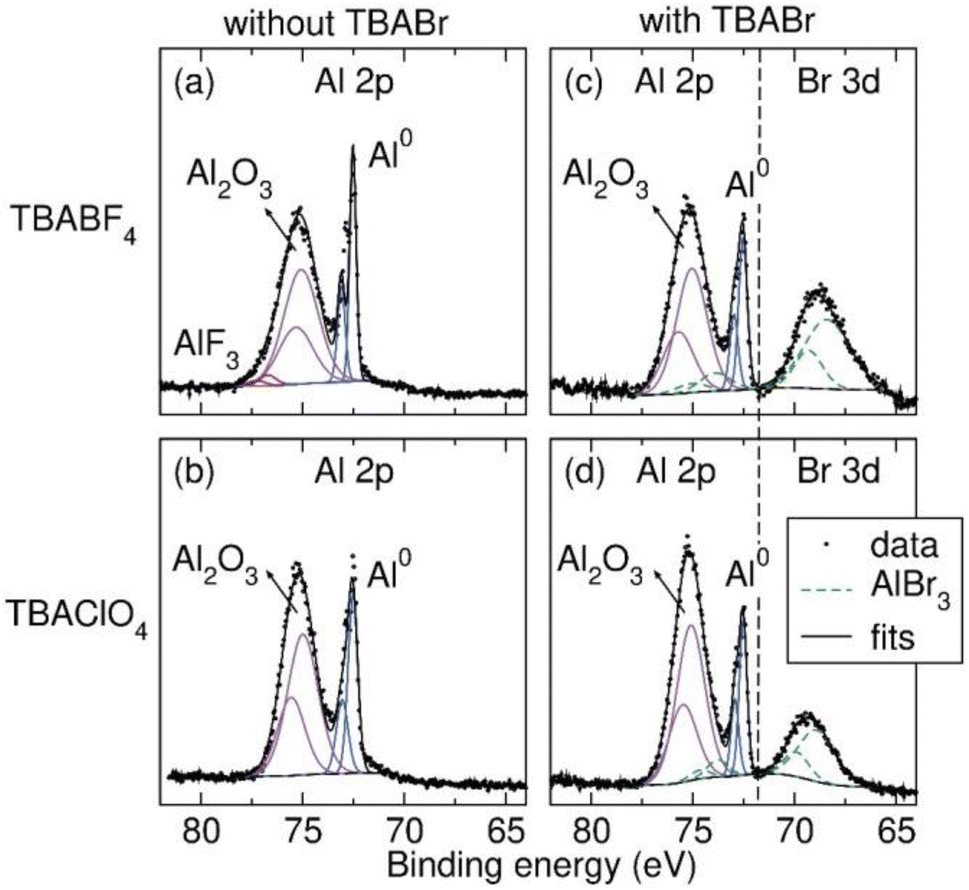 | ||
| Fig. 3 Al 2p and Br 3d regions of the XPS spectra of Al electrodes after the LSV experiments in THF with 0.1 M (a) TBABF4, (b) TBAClO4, (c) TBABF4 + TBABr, and (d) TBAClO4 + TBABr supporting electrolytes. | ||
To improve Al stripping in THF-based electrolyte, we seek to tailor the electrolyte composition to reduce the Al passivation caused by Al2O3. Research on Al corrosion has demonstrated the ability of F−, Cl−, Br−, and I− to destabilize an Al2O3 layer.50–53 Free Cl− in a solution can adsorb on the oxide layer and penetrate through the oxide film via oxygen vacancies.54 This process leads to destabilization and local degradation of the oxide layer.54 The degradation of Al2O3 by Cl− is commonly associated with Al pitting corrosion in aqueous environments. Upon the localized degradation of the Al2O3, the underlying Al becomes susceptible to dissolution in the presence of H2O, leading to pitting corrosion.53 The halide-initiated Al pitting corrosion finds utility in various applications. For instance, Al is often employed as galvanic anodes to protect metal components exposed to saltwater from corrosion.55 Additionally, researchers have used halide salts to activate Al as an efficient reducing agent for wet-chemical synthesis.53
Inspired by the Al corrosion chemistry, we attempt to improve the Al stripping behavior in THF-based electrolytes with halide additives. Previous research has also demonstrated the mobility of halides within an Al oxide film in the presence of an electric field. When subjected to an anodic potential, Cl−, Br−, and I− can migrate towards the Al2O3/Al interface, leading to localized degradation of the oxide layer.52,56–58 We hypothesize that with halide additives in the electrolyte, the halides can permeate Al2O3 upon anodic polarization, creating pathways for Al dissolution and ultimately improving the Al stripping behavior in THF-based electrolytes.
To probe the effects of halide, we add TBABr as the co-supporting electrolyte and measure the changes in Al stripping efficiency. Fig. 2b–d shows the LSVs of Al stripping in 0.05 M TBABF4, TBAClO4, and TBATFSI electrolytes with 0.05 M TBABr co-supporting electrolyte. In all cases, Al stripping improves significantly in the presence of TBABr. Additionally, the current densities increase with each successive LSV scan, indicating that the Al SEI becomes less passivating as its duration in contact with the Br− containing electrolyte is extended. TBACl as a co-supporting electrolyte improves Al stripping in THF, as well (see ESI†). It is worth noting that TBA halide salts have poor solubility in THF. TBABr and TBACl can only reach a high concentration in THF in the presence of another supporting electrolyte that is highly soluble in THF, like TBABF4 and TBAClO4. TBAI, however, is not soluble in THF even in the presence of a co-supporting electrolyte. Using LiI, we are able to demonstrate that I− can also improve Al stripping in THF-based electrolyte (see ESI†).
The improved Al stripping behavior could arise due to an impact of the halide on the composition of the Al SEI or simply due to an increase in Al3+ solubility due to the formation of Al-halide complexes. To determine the effect of the halide on the SEI, we examine the Al electrode surface after anodic polarization in TBA+ electrolytes with TBABr additive via XPS. Fig. 3c and d shows the Al 2p and Br 3d regions of the XPS spectra. Two Al3+ species are observed in the SEI after anodic polarization in TBABF4/TBABr electrolyte (Fig. 3c). In addition to the Al2O3 signal at 75.0 eV, a small amount of AlBr3 is also observed at 73.9 eV.59 The assignment is supported by the signal at 68.4 eV in the Br 3d region, which is also consistent with reported AlBr3 signal.59 The SEI still contains AlFx, as indicated by the F 1s signals (see ESI†). However, compared to the SEI formed in TBABF4 electrolyte, the SEI formed in the presence of TBABr contains comparatively less AlFx. As a result, the AlFx signal cannot be resolved from the Al2O3 signal in the Al 2p region. A similar change in the Al SEI is observed in TBAClO4 electrolyte in the presence of TBABr additive (Fig. 3d). In addition to Al2O3 (75.1 eV) being the dominant species in the SEI, the presence of AlBr3 is confirmed by the Al 2p signal at 73.8 eV and the Br 3d signal at 68.8 eV. All peak BEs and assignments of the related regions are tabulated in the ESI.† The XPS spectra reveal that addition of halide to the electrolyte can modify the composition of Al SEI. In the presence of TBABr co-supporting electrolyte, the SEIs contain Br−, which likely destabilizes the oxide layer and provides pathways for Al dissolution upon anodic polarization. Though halides are certainly changing the SEI composition, they could also play a role in solvating the Al3+ since Al halides are very soluble in THF.60
Earlier, we discussed the fact that halide salt/DMF solution stands out as the prevailing choice of electrolyte when using an Al sacrificial anode (Fig. 1c). We hypothesize that the presence of halides is crucial for Al stripping in DMF, as well. To test the effect of halide on Al stripping in DMF, we perform similar LSV of Al stripping in DMF with 0.1 M of supporting electrolyte of interest (Fig. 4). All potentials in DMF electrolytes are reported vs. Fc/Fc+ unless noted otherwise. All onset potentials and current densities of the 5th LSV scans are tabulated in Table 1.
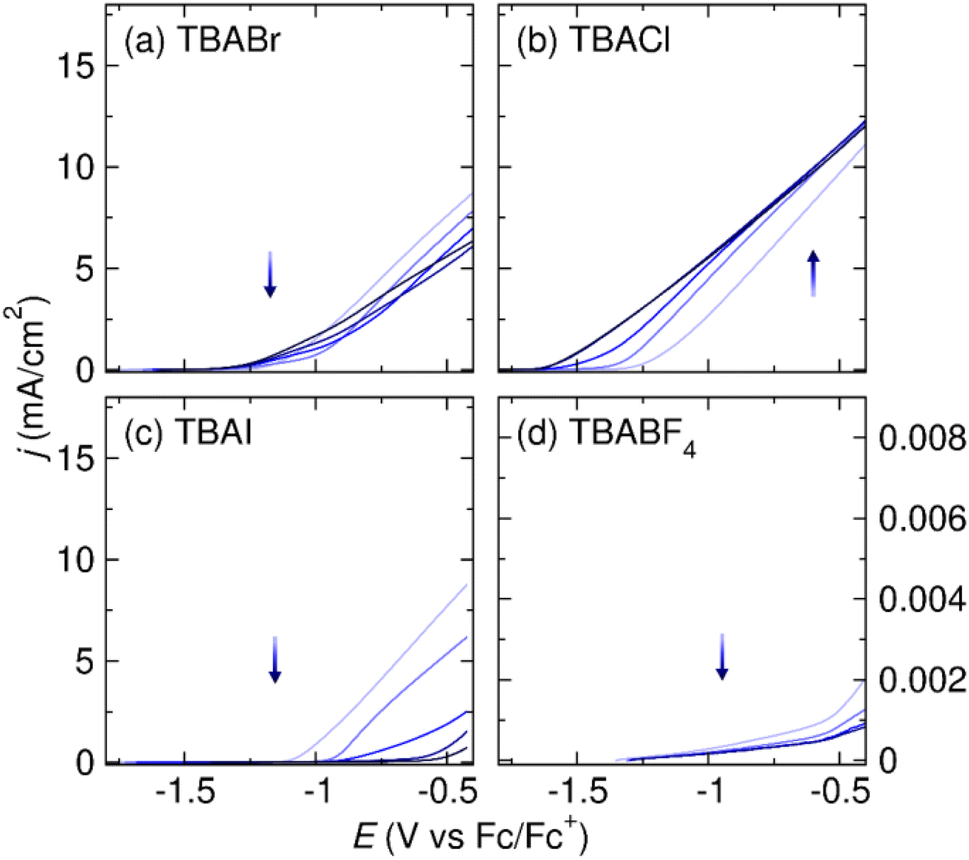 | ||
Fig. 4 Linear sweep voltammograms of Al in 7 mL DMF with 0.1 M (a) TBABr, (b) TBACl, (c) TBAI, and (d) TBABF4. All voltammograms are collected at a scan rate of 5 mV s−1. The LSVs are measured in three-electrode cells with a Al WE, graphite CE, and Ag/Ag(cryptand)+ RE (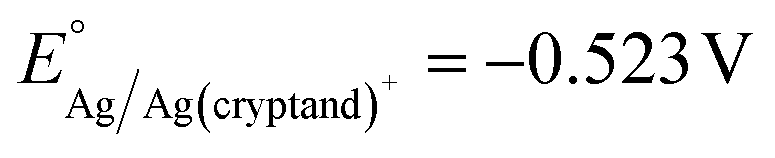 vs. Fc/Fc+). The direction of the arrows indicates the trend of changes in current densities as the number of scans increases. vs. Fc/Fc+). The direction of the arrows indicates the trend of changes in current densities as the number of scans increases. | ||
First, we test Al stripping in TBABr/DMF, the most commonly employed electrolyte for systems using an Al sacrificial anode.3 In TBABr/DMF, the current density reaches 1.75 mA cm−2 at 0.5 V vs. the Al stripping onset potential, Eon, indicating exceptional Al stripping (Fig. 4a). Fig. 4b shows that TBACl/DMF is able to support high current densities for Al stripping, as well. High current density for Al stripping can initially be achieved in TBAI/DMF. However, the current density decreases significantly with successive scans, which is likely caused by changes at the Al interface after prolonged exposure to the iodide-containing electrolyte (Fig. 4c). In the absence of halides, however, the Al stripping efficiency is extremely poor in DMF. Fig. 4d shows the Al stripping in TBABF4/DMF electrolyte. The current densities for Al stripping are extremely low within the electrolyte stability window. The LSV results in Fig. 4 demonstrate the importance of halide on Al stripping in DMF-based electrolyte as well.
Unlike THF, DMF can easily dissolve a high concentration of TBA halide salt without the aid of a co-supporting electrolyte. We believe the high solubility of TBA halide salts in DMF contributes to the observed bias in solvent selections for reductive electrosynthesis employing an Al sacrificial anode (Fig. 1c). The limited solubility of TBA halide salts in THF is likely the reason why they are not regularly considered for the optimization of reductive electrosynthesis reactions. We envision that the use of halide co-supporting electrolytes can be a simple electrolyte tailoring strategy to enable the performance of Al sacrificial anode in THF for reductive electrosynthesis.
Bulk Al stripping in halide containing THF-based electrolyte
We next probe the effect of Br− co-supporting electrolyte on bulk Al stripping in THF, a condition more relevant to reductive electrosynthesis conditions. We perform galvanostatic oxidation of Al and measure the voltage profile and impedance change at the Al electrode during electrolysis. The experiments are conducted in three-electrode cells with an Al plate WE, graphite CE, and Ag wire pseudo-RE. The electrolyte consists of 0.5 M supporting electrolyte of interest and 0.5 M tert-butyl bromide (tBuBr). tBuBr serves as the sacrificial oxidant of the galvanostatic oxidation experiment.9 The experiment begins with galvanostatic stripping of Al at j ≈ 1 mA cm−2 while the voltage profiles at the Al WE and graphite CE are recorded. After every 2 hours of galvanostatic oxidation, we rest the cell at OCV and measure the series resistance at the Al WE using electrochemical impedance spectroscopy (EIS). The galvanostatic oxidation/EIS protocol is repeated 8 times over a duration of 16 hours to observe any changes in the performance of the Al electrode during a typical time frame of reductive electrosynthesis. The cell configuration and galvanostatic oxidation/EIS protocol are adapted from our work on Mg.9Fig. 5a shows the voltage profiles at the Al WE and graphite CE in 0.5 M TBABF4/THF electrolyte. The potential at the Al electrode increases immediately after the galvanostatic oxidation starts. The cell voltage reaches the compliance limit of the potentiostat within 1 min and the experiment can no longer proceed. EIS is measured before and after, and the Nyquist plot shows no changes, likely due to the minimal charge passed during the oxidation. The series resistance, which is taken as the intercept with Z′, remains low around 74 Ω, at the end of the experiment (see ESI†). The low resistance coupled with the high cell voltage suggests that the SEI layer likely conducts electrons but not Al3+, leading to difficulties in Al stripping. The observation is consistent with our hypothesis that an Al2O3-dominant SEI is prohibiting the effective Al stripping.
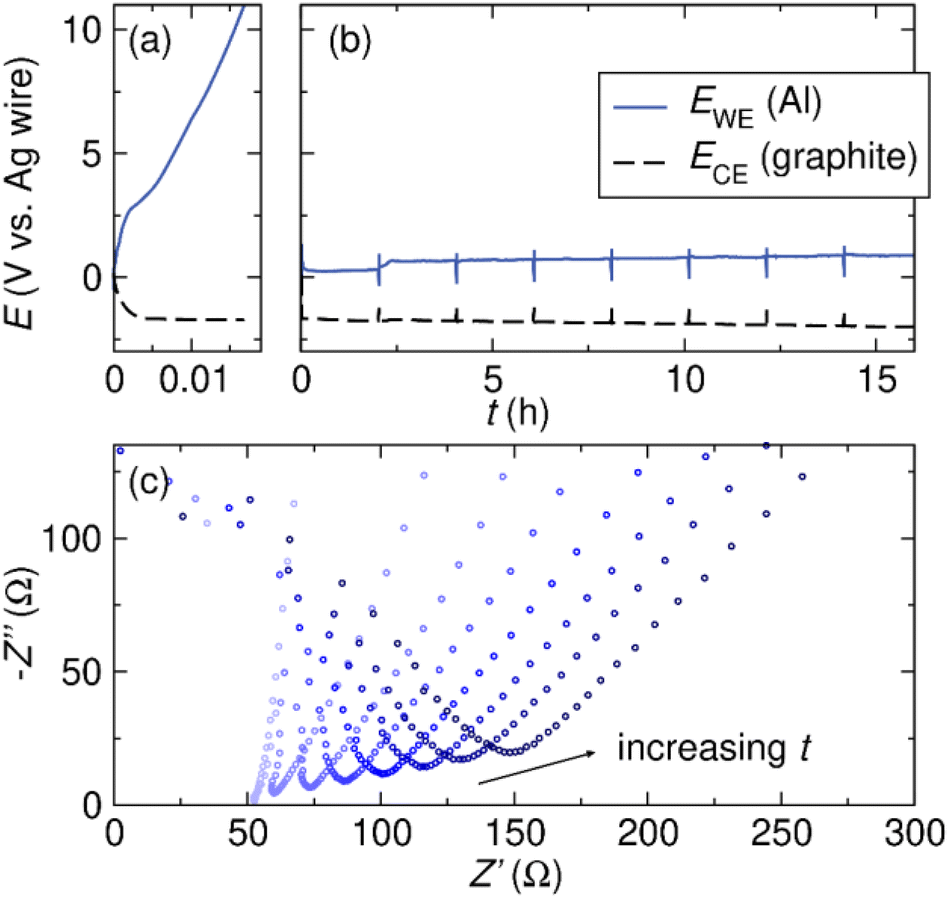 | ||
| Fig. 5 Voltage profiles of the Al WE and graphite CE during galvanostatic Al stripping (j ≈ 1 mA cm−2) experiments in THF. The experiments are performed in (a) 0.5 M TBABF4 and (b) 0.25 M TBABF4 + 0.25 M TBABr respectively in the presence of tBuBr. (c) EIS is performed every 2 hours for the galvanostatic Al stripping in 0.25 M TBABF4 + 0.25 M TBABr electrolyte. As more Al is electrochemically stripped, the series resistance increases. | ||
Earlier, we have demonstrated that Br− is beneficial for Al stripping in THF-based electrolytes (Fig. 2). To enable bulk Al stripping in THF, we perform the galvanostatic oxidation experiment in electrolyte containing 0.25 M TBABF4 and 0.25 M TBABr. With TBABr, we achieve bulk Al stripping in THF-based electrolyte. The voltage profile at the Al electrode remains stable during the 16 hours of galvanostatic Al stripping (Fig. 5b). Throughout the course of the experiment, the voltage at the Al electrode increases slightly from 0.25 V to 0.88 V vs. Ag wire. The series resistance also increases over time, which can be observed in the Nyquist plots shown in Fig. 5c. The series resistance is dominated by the electrolyte resistance, however, a change in series resistance is likely due to the introduction of another resistive element in the circuit, i.e. an SEI on the Al.
To understand the reason behind the increase in the series resistance during the galvanostatic oxidation experiment, we examine the Al electrode post electrolysis. Visually, we observe a thin layer of salt build-up on the Al electrode at the end of the experiment (Fig. 6a). The composition of the salt is investigated by energy-dispersive X-ray spectroscopy (EDS). The EDS spectrum (Fig. 6b) reveals the presence of Al, F, O, and C, indicating the formation of Al salt as the product of Al stripping. The trace amount of Mg is a result of Mg impurity present in the Al electrode. We hypothesize that during the galvanostatic Al stripping experiment, Al salt precipitates onto the surface of Al, consequently contributing to the observed increase in resistance which causes the voltage to increase at the Al electrode. Our previous study on a system using a Mg electrode has demonstrated a similar phenomenon where the accumulation of Mg salts on the surface on the Mg electrode leads to increased cell voltage.9,15 Indeed, when the electrode is removed from solution following a stripping reaction, salt is observed on the surface as shown in the photograph in Fig. 6a.
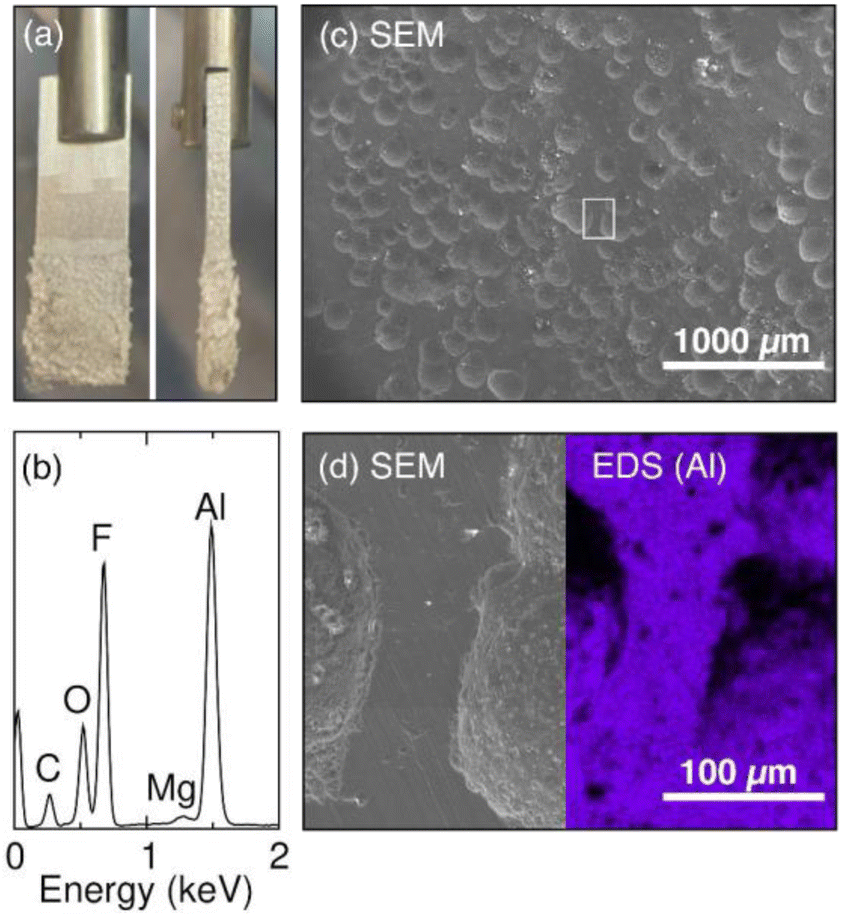 | ||
| Fig. 6 (a) Photographs of the Al electrode (front and side view) after the galvanostatic stripping experiment in the 0.25 M TBABF4 + 0.25 M TBABr electrolyte. Visually, a salt crust is formed on top of the Al surface. (b) The EDS spectrum of the salt crust. (c) The SEM image of the Al electrode upon removal of the salt crust reveals an uneven surface morphology. (d) An enlarged SEM image coupled with EDS mapping of the Al electrode. The EDS map shows the surface distribution of Al on the Al electrode after the galvanostatic Al stripping experiment. | ||
We then examine the morphology of the Al electrode surface with scanning electron microscopy (SEM) and EDS mapping. The salts are first removed with an acetone rinse to reveal the electrode surface. The SEM image in Fig. 6c reveals a pitted Al surface after the galvanostatic stripping experiment. The pitted morphology resembles the morphology that results from corrosion of Al in aqueous halide solutions.61 We hypothesize that the Br− in the electrolyte first attacks the Al SEI and incorporates itself into the Al2O3 surface layer as indicated by the XPS result in Fig. 3. The process can make the oxide layer unstable and degrade locally. As the oxide layer is not evenly degraded, Al stripping will take place at locations where the oxide layer is most fragile during electrolysis, resulting in the observed pitted morphology. The enlarged SEM image with the corresponding EDS map shows non-uniform Al distribution (Fig. 6d), which further supports an uneven Al stripping process. The pitted surface likely serve as nucleation sites for Al salts generated during electrolysis, leading to the salt build-up on the electrode surface.62 The salt build-up is responsible for the observed increase in both resistance and voltage at the Al anode.
Despite the increase in resistance caused by salt accumulation on the Al anode, the voltage profile remains low and stable during the 16 hours galvanostatic oxidation experiment (Fig. 5b). A stable voltage is crucial for reductive electrosynthesis, as it prevents the occurrence of high cell-voltage resulting from metal sacrificial anode passivation.
It is also worth noting that the Al electrode material used in this study, aluminum 5052, has a high resistance towards chloride-induced corrosion and is widely used in marine environments.63 The fact that halide co-supporting electrolyte can successfully destabilize Al2O3 formed on aluminum 5052 underscores its suitability as a robust solution for enhancing Al sacrificial anode performance. We note that several other aluminum alloys exist, most of which are more susceptible to halide-induced corrosion, and thus may show a different response to halide additives.
Using halide additive to improve the cell potentials of reductive electrosynthesis reactions employing an Al sacrificial anode
To demonstrate the efficacy of halide additive in alleviating the passivation of an Al sacrificial anode, we employ the electrolyte tailoring strategy in reported cathodic reduction methodologies. The reductive electrosynthesis are carried out in an ElectraSyn, a widely adopted setup among synthetic organic chemists. We first focus on an electrochemically driven deoxygenative borylation reaction in THF (Scheme 1), where Al was tested in a previous effort of reaction optimization but gave 0% yield of the desired product.43 As shown in entry 1, in TBABF4/THF electrolyte, the overall cell potential rapidly reaches 30 V, the upper limit of the ElectraSyn capability, and merely 0.03 F mol−1 of the charges was passed even after prolonged reaction time. The observation aligns with the high potential at the Al anode shown in Fig. 5a, where the elevated cell potential is observed at the beginning of electrolysis is likely a result of Al anode passivation. By partially substituting the supporting electrolyte with TBABr, the electrolysis progresses smoothly, maintaining a voltage range of 7–12 V throughout the reaction (entry 2). The simple electrolyte modification affords 38% yield of the desired product. When the same reaction is carried out in TBAClO4/THF electrolyte, the overall cell potential remains persistently high at 28–30 V (entry 3). Using TBABr as the co-supporting electrolyte again effectively reduces the cell potential to 10–14 V, and affords a slight improvement of yield of 36% (entry 4).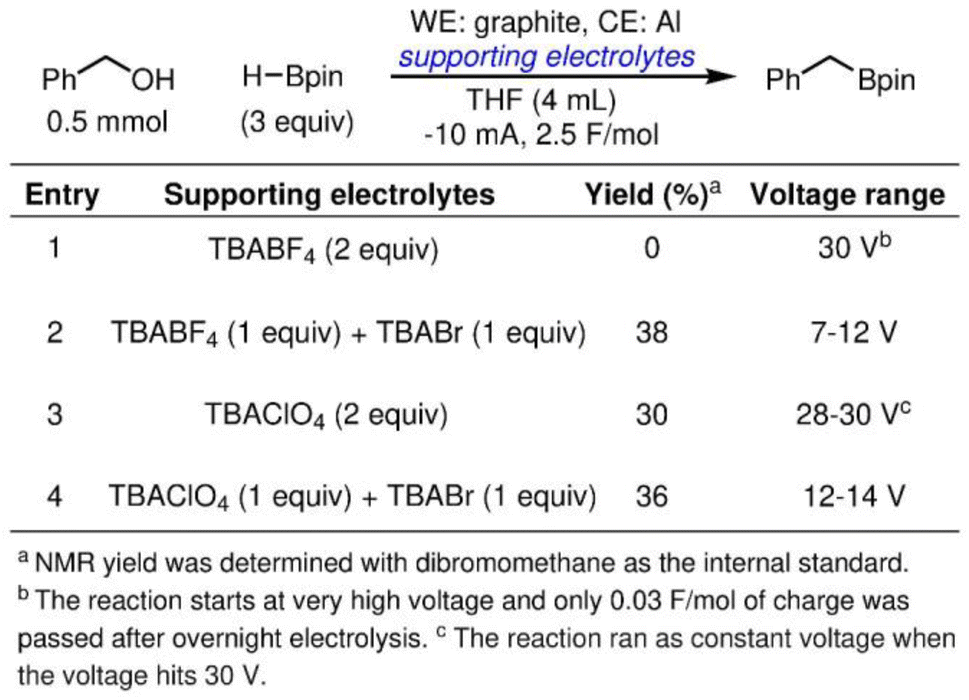 | ||
| Scheme 1 Deoxygenative borylation of benzylic alcohols. | ||
The halide additive strategy is effective at reducing the overall cell potential in other types of organic transformation, as well. Scheme 2 illustrates an electrochemically driven silyl cross-electrophile coupling reaction.44 The reaction has a notable high cell potential of 28–30 V during electrolysis when an Al sacrificial anode is used in combination with TBAClO4/THF electrolyte (entry 5). Using TBACl as a co-supporting electrolyte results in a reduction in cell potential to the range of 10–14 V with improved yield of the disilane product (entry 6). By reducing the current to −5 mA, we can further reduce the voltage range to 6–8 V and improve the yield to 41% (entry 7).
 | ||
| Scheme 2 Silyl cross-electrophile coupling. | ||
It is worth noting that both the borylation and silylation reactions have not undergone exhaustive optimizations. To further improve the yield of these two reactions, or any organic transformation of interest, various parameters (e.g. the selection of primary supporting electrolyte, the co-supporting electrolyte loading, current density, substrate concentration, etc.) must be screened thoroughly to strike a balance between good Al sacrificial anode performance and high yield of the desired product. Nevertheless, the results shown here undeniably highlight the effectiveness of halide additives in reducing the overall cell potential caused by Al sacrificial anode passivation. We hope the simple electrolyte tailoring strategy can encourage the use of Al sacrificial anodes in THF-based electrolytes in future reductive electrosynthesis endeavors.
Conclusions
The work described here provides insights into the influence of supporting electrolytes on Al stripping in THF. Supporting electrolytes that are commonly employed in organic electrosynthesis, such as TBABF4, TBFClO4, or TBATFSI, do not support Al stripping in THF. XPS indicates the presence of Al2O3 on the Al electrode surface after anodic polarization, which is passivating the Al surface. Inspired by Al corrosion chemistry, we use halide (Cl−, Br−, I−) salts to enable Al stripping. The new SEIs formed in the presence of TBABr co-supporting electrolyte contain AlBr3, which likely leads to destabilization of the Al oxide layer and allows Al stripping. Further, addition of halides will increase the solubility of Al3+ salts formed as a result of oxidation.In the presence of TBABr co-supporting electrolyte, we have successfully demonstrated the feasibility of bulk Al stripping in THF. The Al2O3 passivating layer inhibits Al stripping in TBABF4/THF electrolyte. With the introduction of Br−, Al stripping can take place at locations where the Al2O3 has been destabilized by AlBr3, resulting in a pitted morphology after prolonged metal stripping. The voltage profile at the Al anode remains stable throughout a 16 hours electrolysis, which is promising for its application in reductive electrosynthesis without causing elevated cell voltage. Using halide additives, the high overall cell potentials caused by Al anode passivation are reduced significantly in a deoxygenative borylation reaction and a silyl cross-electrophile coupling reaction. Our study demonstrates the significance of understanding the interface chemistry to enhance the performance of Al sacrificial anodes. The utilization of halide co-supporting electrolytes presents an effective electrolyte tailoring strategy and potentially opens avenues for the development of more reductive electrosynthesis reactions in THF with an Al sacrificial anode.
Methods
Materials preparation
All electrolytes for LSV and galvanostatic oxidation experiments were prepared in a N2-filled glovebox. TBATFSI (≥99.0%), TBAClO4 (≥99.0%), TBAPF6 (≥99.0%), TBABF4 (99%), TBABr (≥98.0%), TBACl (≥95.0%), and TBAI (98%) were purchased from Sigma-Aldrich. All salts were dried under vacuum at 100 °C overnight prior to use and transferred to the glovebox without exposure to air. THF (99.9%, Fischer Scientific) was dried on a solvent purification system (Pure Process Technology), transferred to the glovebox without exposure to air, and stored over dried 4 Å molecular sieves prior to use. Anhydrous DMF (99.8%, Sure/Seal™, Sigma-Aldrich) was stored in the glovebox prior to use. All electrolytes were prepared by stirring the supporting electrolyte of interest in THF or DMF until the solution turned homogeneous.Benzyl alcohol (Sigma-Aldrich, >99%), H-Bpin (Sigma-Aldrich, 97%), Ph(Me)2SiCl (TCI, >96%), H(Me)2SiCl (Sigma-Aldrich, 98%), TBABF4 (TCI, >98%), TBAClO4 (TCI, >98%), TBABr (TCI, >98%), and TBACl (TCI, >98%) were used as received for reductive electrosynthesis.
Pt|Fc/Fc+ reference electrode preparation
The Pt|Fc/Fc+ RE was prepared following literature procedure with a 0.5 mm diameter Pt wire (Sigma-Aldrich), ferrocene (Fc, 98%, Sigma-Aldrich), ferrocenium hexafluorophosphate (FcPF6, 95%, Combi-Blocks), TBAPF6, and THF.9,64–66 Fc and TBAPF6 were recrystallized prior to use. The Pt wire was first cleaned in concentrated HNO3 and heated in a H2 flame before sealed within a ceramic-fritted glass tube (inner diameter 3.5 mm, Pine Research Instrumentation). The glass tube was filled with an electrolyte of 4 mM Fc, 4 mM FcPF6, and 0.1 M TBAPF6 in THF. The RE was assembled fresh prior to each experiment.Ag/Ag(cryptand)+ reference electrode preparation
The Ag/Ag(cryptand)+ RE was prepared following literature procedure67,68 with a nonaqueous reference electrode kit purchased from BASi. The RE was filled with an electrolyte of 10 mM AgNO3 (≥99.0%, Sigma-Aldrich), 41.2 mM Cryptand 222 (Kryptofix®, 98%, Sigma-Aldrich), and 0.1 M TBAPF6 in DMF. The standard potential of the RE was determined to be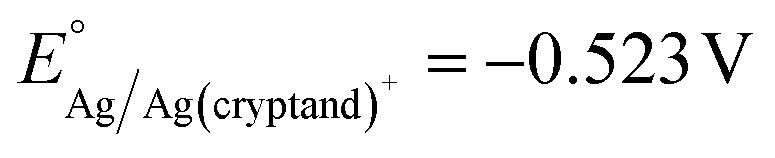 vs. Fc/Fc+, which is consistent with the reported value (see ESI†). The LSVs for Al stripping in DMF were initially measured using the Ag/Ag(cryptand)+ RE, and subqequently, the potentials were converted to be referenced against Fc/Fc+. The RE was assembled fresh prior to each experiment.
vs. Fc/Fc+, which is consistent with the reported value (see ESI†). The LSVs for Al stripping in DMF were initially measured using the Ag/Ag(cryptand)+ RE, and subqequently, the potentials were converted to be referenced against Fc/Fc+. The RE was assembled fresh prior to each experiment.
Electrochemical testing
All electrochemical experiments were performed in a N2-filled glovebox in a low volume, three-electrode cell (Pine Research Instrumentation). The Al plate electrodes (2 mm × 8 mm × 30 mm, IKA, aluminum 5052) were mechanically ablated within the glovebox prior to use. LSV experiments were performed with an Al plate as the WE, a graphite CE (Pine Research Instrumentation), the Pt|Fc/Fc+ or Ag/Ag(cryptand)+ RE, and approximately 7 mL of electrolyte. The electrolyte was prepared with 0.1 M supporting electrolyte or with 0.05 M + 0.05 M of two supporting electrolytes. The LSVs of Al stripping in THF were collected with 85% iR compensation. Galvanostatic oxidation experiments were performed with an Al WE, graphite CE, Ag wire (Pine Research Instrumentation) as a pseudo-RE, and approximately 7 mL of electrolyte. The electrolyte was prepared with 0.5 M supporting electrolyte and 0.5 M tBuBr (98%, Sigma-Aldrich) as the sacrificial oxidant. EIS experiments were performed with ±10 mV sinus amplitude from 106–1 Hz at 10 points per decade. All electrochemical experiments were conducted on a VMP3 potentiostat (Bio-Logic). The cell configuration and the electrochemical testing protocols are adapted from our work on Mg.9Sample characterization
XPS measurements were performed on Al anode surfaces after LSV experiments. After the LSV scans, the cells were disassembled inside a N2-filled glovebox and the Al electrode was removed. Each Al electrode was rinsed with 10 mL of THF and dried in ambient glovebox conditions for at least 48 h before analysis. XPS data were collected using a Kratos Axis Ultra system at a pressure <3 × 10−9 Torr. Samples were irradiated with a monochromatic Al Kα source (1486.7 eV) at 150 W. A charge neutralizer was used with a filament current of 2 A, filament bias of 1.3 V and charge balance of 3.5 V. Low-resolution survey spectra were acquired between BEs of 1–1200 eV. Higher-resolution detailed scans, with a resolution of 0.05 eV and a pass energy of 10 eV, were collected on individual XPS lines of interest. The XPS data were analyzed using CasaXPS analysis software. A Shirley background was used for all spectra. All peaks were fit using a Voigt GL(30) function with 70% Gaussian and 30% Lorentzian character. Spectra were referenced to Al0 at 72.5 eV. Al0 signal was chosen as the reference due to the complicated C 1s and O 1s signals resulting from the electrolytes.All proton nuclear magnetic resonance (NMR) spectra were recorded on VarianMercury (400 MHz) at 20 °C. Chemical shifts for proton are reported in parts per million downfield from tetramethylsilane and are reference to residual protium in the NMR solvent according to values reported in literature: δ(CDCl3) = 7.26 ppm.
Deoxygenative borylation of benzylic alcohols
Al and graphite electrodes (2 mm × 8 mm × 52.5 mm, IKA) were prepared in air by polishing with 500 grit silicon carbide sandpaper until a shiny finish was obtained. Both electrodes were rinsed with acetone and the graphite electrode was dried in oven (130 °C) for >10 min prior to use and transferred to the N2-filled glovebox. In the glovebox, the corresponding electrolytes (1 mmol, 2 equiv.) and benzyl alcohol (0.5 mmol, 1 equiv.) were added into an oven dried ElectraSyn vial (5 mL) equipped with a magnetic stir bar. Dried THF (1 mL) was then added to the mixture. H-Bpin (1.5 mmol, 3 equiv.) was slowly added to the solution. The reaction usually starts bubbling at this stage and the bubbling becomes more vigorous when stirring. The whole mixture was slowly stirred until the bubbling becomes less vigorous. Then, 3 mL dried THF was added to the mixture. The vial was sealed with the ElectraSyn vial cap equipped with anode (Al) and cathode (graphite), and then the assembly was brought out of the glovebox. A nitrogen balloon was attached to the cap, and the reaction mixture was electrolyzed at a constant current of −10 mA (j ≈ −2.38 mA cm−2) until passing 2.5 F mol−1 of charge at room temperature. After electrolysis, the reaction mixture was added to diethyl ether (10 mL) to precipitate electrolytes. The resultant mixture was then filtered through a short silica plug (8 cm thick, ca. 10 g) and flushed with diethyl ether (100 mL). The crude mixture was concentrated under vacuum and the yield was analyzed by 1H NMR using dibromoethane (0.4 equiv. added) as internal standard. The reaction procedure is adapted from ref. 43.Disilane synthesis via silyl cross-electrophile coupling
Al and graphite electrodes (2 mm × 8 mm × 52.5 mm, IKA) were prepared in air by polishing with 500 grit silicon carbide sandpaper until a shiny finish was obtained. Both electrodes were rinsed with acetone and the graphite electrode was dried in oven (130 °C) for >10 min prior to use and transferred to the N2-filled glovebox. The corresponding electrolytes (1 mmol, 2 equiv.), chlorodimethylphenyl silane (1 mmol, 1 equiv.) and chlorodimethyl silane (1.5 mmol, 1.5 equiv.) was added into an oven dried ElectraSyn vial (5 mL) equipped with a magnetic stir bar. Dried THF (4 mL) was then added to the mixture. The vial was sealed with the ElectraSyn vial cap equipped with anode (Al) and cathode (graphite), and then bring it out of the glovebox. The reaction mixture was electrolyzed at a constant current of −10 mA (j ≈ −2.38 mA cm−2) until passing 2.5 F mol−1 of charge at room temperature. After electrolysis, the reaction mixture was added to hexanes (10 mL) to precipitate electrolytes. The resultant mixture was then filtrated through a short silica plug (8 cm thick, ca. 10 g) and flushed with 5% diethyl ether/hexanes (100 mL). The crude was concentrated in vacuum and the yield was analyzed by 1H NMR using dibromoethane (0.4 equiv. added) as internal standard. The reaction procedure is adapted from ref. 44.Data availability
The data that support the findings of this study are openly available in CaltechDATA at https://doi.org/10.22002/rxsqk-8zb05.69Author contributions
W. Z. and K. A. S. designed the study. W. Z. performed the electrochemical experiments, conducted surface characterizations, and analyzed the data. W. G., Y. W. and S. L. devised the organic electrosynthetic reactions and W. G. performed the organic electrosynthesis reactions. W. Z. and K. A. S. wrote the manuscript. All coauthors participated in relevant scientific discussions and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the NSF Center for Synthetic Organic Electrochemistry, CHE-2002158. XPS data were collected at the Molecular Materials Research Center in the Beckman Institute of the California Institute of Technology. KAS acknowledges support from the David and Lucile Packard Fellowship for Science and the Alfred P. Sloan Foundation. KAS and SL acknowledge support from the Camille and Henry Dreyfus Foundation. The authors thank Jake M. Evans for the assistance with collecting XPS data.References
- C. Schotten, T. P. Nicholls, R. A. Bourne, N. Kapur, B. N. Nguyen and C. E. Willans, Making Electrochemistry Easily Accessible to the Synthetic Chemist, Green Chem., 2020, 22, 3358–3375 RSC
.
- R. D. Little and K. D. Moeller, Introduction: Electrochemistry: Technology, Synthesis, Energy, and Materials, Chem. Rev., 2018, 118, 4483–4484 CrossRef CAS PubMed
- M. Yan, Y. Kawamata and P. S. Baran, Synthetic Organic Electrochemical Methods Since 2000: On the Verge of a Renaissance, Chem. Rev., 2017, 117, 13230–13319 CrossRef CAS PubMed
- S. D. Minteer and P. Baran, Electrifying Synthesis: Recent Advances in the Methods, Materials, and Techniques for Organic Electrosynthesis, Acc. Chem. Res., 2020, 53, 545–546 CrossRef CAS PubMed
- L. F. T. Novaes, J. Liu, Y. Shen, L. Lu, J. M. Meinhardt and S. Lin, Electrocatalysis as an Enabling Technology for Organic Synthesis, Chem. Soc. Rev., 2021, 50, 7941–8002 RSC
- Y. Li, L. Wen and W. Guo, A Guide to Organic Electroreduction Using Sacrificial Anodes, Chem. Soc. Rev., 2023, 52, 1168–1188 RSC
- J. Chaussard, J.-C. Folest, J.-Y. Nedelec, J. Perichon, S. Sibille and M. Troupel, Use of Sacrificial Anodes in Electrochemical Functionalization of Organic Halides, Synthesis, 2002, 1990, 369–381 CrossRef
- D. M. Heard and A. J. J. Lennox, Electrode Materials in Modern Organic Electrochemistry, Angew. Chem., Int. Ed., 2020, 59, 18866–18884 CrossRef CAS PubMed
- W. Zhang, C. Gu, Y. Wang, S. D. Ware, L. Lu, S. Lin, Y. Qi and K. A. See, Improving the Mg Sacrificial Anode in Tetrahydrofuran for Synthetic Electrochemistry by Tailoring Electrolyte Composition, JACS Au, 2023, 8, 2280–2290 CrossRef PubMed
- T. Leisegang, F. Meutzner, M. Zschornak, W. Münchgesang, R. Schmid, T. Nestler, R. A. Eremin, A. A. Kabanov, V. A. Blatov and D. C. Meyer, The Aluminum-Ion Battery: A Sustainable and Seminal Concept?, Front. Chem., 2019, 7, 268 CrossRef CAS PubMed
-
J. Brugger, in Encyclopedia of Geochemistry, ed. W. M. White, Springer International Publishing, Cham, 2016, pp. 1–4 Search PubMed
-
W. Li and X. Wang, in Sulfide and Selenide Based Materials for Emerging Applications, ed. G. Dalapati, T. Shun Wong, S. Kundu, A. Chakraborty and S. Zhuk, Elsevier, 2022, pp. 645–678 Search PubMed
-
W. Haynes, CRC Handbook of Chemistry and Physics, CRC press, 95th edn, 2014 Search PubMed
-
C. Vargel, Corrosion of Aluminium, Oxford, Elsevier Science, 1st edn, 2004 Search PubMed
- W. Zhang, L. Lu, W. Zhang, Y. Wang, S. D. Ware, J. Mondragon, J. Rein, N. Strotman, D. Lehnherr, K. A. See and S. Lin, Electrochemically Driven Cross-Electrophile Coupling of Alkyl Halides, Nature, 2022, 604, 292–297 CrossRef CAS PubMed
- M. Dörr, M. M. Hielscher, J. Proppe and S. R. Waldvogel, Electrosynthetic Screening and Modern Optimization Strategies for Electrosynthesis of Highly Value-added Products, ChemElectroChem, 2021, 8, 2621–2629 CrossRef
- Y. Gao, D. E. Hill, W. Hao, B. J. McNicholas, J. C. Vantourout, R. G. Hadt, S. E. Reisman, D. G. Blackmond and P. S. Baran, Electrochemical Nozaki–Hiyama–Kishi Coupling: Scope, Applications, and Mechanism, J. Am. Chem. Soc., 2021, 143, 9478–9488 CrossRef CAS PubMed
- Y. Wang, S. Tang, G. Yang, S. Wang, D. Ma and Y. Qiu, Electrocarboxylation of Aryl Epoxides with CO 2 for the Facile and Selective Synthesis of β-Hydroxy Acids, Angew. Chem., Int. Ed., 2022, 61, e202207746 CrossRef CAS PubMed
- B. Zhang, Y. Gao, Y. Hioki, M. S. Oderinde, J. X. Qiao, K. X. Rodriguez, H.-J. Zhang, Y. Kawamata and P. S. Baran, Ni-Electrocatalytic Csp3–Csp3 Doubly Decarboxylative Coupling, Nature, 2022, 606, 313–318 CrossRef CAS PubMed
- C.-H. Li, X.-Z. Song, L.-M. Tao, Q.-G. Li, J.-Q. Xie, M.-N. Peng, L. Pan, C. Jiang, Z.-Y. Peng and M.-F. Xu, Electrogenerated-Bases Promoted Electrochemical Synthesis of N-bromoamino Acids from Imines and Carbon Dioxide, Tetrahedron, 2014, 70, 1855–1860 CrossRef CAS
- G. Yuan, Z. Li and H. Jiang, Electrosyntheses of α-Hydroxycarboxylic Acids from Carbon Dioxide and Aromatic Ketones Using Nickel as the Cathode, Chin. J. Chem., 2009, 27, 1464–1470 CrossRef CAS
- G. Yuan, L. Li, H. Jiang, C. Qi and F. Xie, Electrocarboxylation of Carbon Dioxide with Polycyclic Aromatic Hydrocarbons Using Ni as the Cathode, Chin. J. Chem., 2010, 28, 1983–1988 CrossRef CAS
- C. Li, G. Yuan and H. Jiang, Electrocarboxylation of Alkynes with Carbon Dioxide in the Presence of Metal Salt Catalysts, Chin. J. Chem., 2010, 28, 1685–1689 CrossRef CAS
- G.-Q. Yuan, H.-F. Jiang and C. Lin, Efficient Electrochemical Dicarboxylations of Arylacetylenes with Carbon Dioxide Using Nickel as the Cathode, Tetrahedron, 2008, 64, 5866–5872 CrossRef CAS
- G.-Q. Yuan, H.-F. Jiang, C. Lin and S.-J. Liao, Efficient Electrochemical Synthesis of 2-Arylsuccinic Acids from CO2 and Aryl-Substituted Alkenes with Nickel as the Cathode, Electrochim. Acta, 2008, 53, 2170–2176 CrossRef CAS
- R. Matthessen, J. Fransaer, K. Binnemans and D. E. D. Vos, Electrochemical Dicarboxylation of Conjugated Fatty Acids as an Efficient Valorization of Carbon Dioxide, RSC Adv., 2013, 3, 4634 RSC
- C.-H. Li, G.-Q. Yuan, X.-C. Ji, X.-J. Wang, J.-S. Ye and H.-F. Jiang, Highly Regioselective Electrochemical Synthesis of Dioic Acids from Dienes and Carbon Dioxide, Electrochim. Acta, 2011, 56, 1529–1534 CrossRef CAS
- C. Cannes, S. Condon, M. Durandetti, J. Périchon and J.-Y. Nédélec, Nickel-Catalyzed Electrochemical Couplings of Vinyl Halides: Synthetic and Stereochemical Aspects, J. Org. Chem., 2000, 65, 4575–4583 CrossRef CAS PubMed
- C. Laza, E. Duñach, F. Serein-Spirau, J. J. E. Moreau and L. Vellutini, Novel Synthesis of Arylboronic Acids by Electroreduction of Aromatic Halides in the Presence of Trialkyl Borates, New J. Chem., 2002, 26, 373–375 RSC
- M. Orsini, M. Feroci, G. Sotgiu and A. Inesi, Stereoselective Electrochemical Carboxylation: 2-Phenylsuccinates from Chiral Cinnamic Acid Derivatives, Org. Biomol. Chem., 2005, 3, 1202 RSC
- B. K. Peters,
et al., Scalable and Safe Synthetic Organic Electroreduction Inspired by Li-ion Battery Chemistry, Science, 2019, 363, 838–845 CrossRef CAS PubMed
- J. Godeau, C. Pintaric, S. Olivero and E. Duñach, Electrochemical Preparation of Pinacol Allylboronic Esters, Electrochim. Acta, 2009, 54, 5116–5119 CrossRef CAS
- M. Feroci, A. Inesi, M. Orsini and L. Palombi, Electrochemical Carboxylation of N -(2-Bromopropionyl)-4 R - Phenyloxazolidin-2-One: An Efficient Route to Unsymmetrical Methylmalonic Ester Derivatives, Org. Lett., 2002, 4, 2617–2620 CrossRef CAS PubMed
- M. Feroci, M. Orsini, L. Palombi, G. Sotgiu, M. Colapietro and A. Inesi, Diastereoselective Electrochemical Carboxylation of Chiral α – Bromocarboxylic Acid Derivatives: An Easy Access to Unsymmetrical Alkylmalonic Ester Derivatives 1, J. Org. Chem., 2004, 69, 487–494 CrossRef CAS PubMed
- K. Okamoto, S. Nagahara, Y. Imada, R. Narita, Y. Kitano and K. Chiba, Hydrosilane-Mediated Electrochemical Reduction of Amides, J. Org. Chem., 2021, 86, 15992–16000 CrossRef CAS PubMed
- S. Manabe, C. M. Wong and C. S. Sevov, Direct and Scalable Electroreduction of Triphenylphosphine Oxide to Triphenylphosphine, J. Am. Chem. Soc., 2020, 142, 3024–3031 CrossRef CAS PubMed
- Y. You, W. Kanna, H. Takano, H. Hayashi, S. Maeda and T. Mita, Electrochemical Dearomative Dicarboxylation of Heterocycles with Highly Negative Reduction Potentials, J. Am. Chem. Soc., 2022, 144, 3685–3695 CrossRef CAS PubMed
- A. A. Isse and A. Gennaro, Electrocatalytic Carboxylation of Benzyl Chlorides at Silver Cathodes in Acetonitrile, Chem. Commun., 2002, 2798–2799 RSC
- C. J. Burrows, J. B. Harper, W. Sander and D. J. Tantillo, Solvation Effects in Organic Chemistry, J. Org. Chem., 2022, 87, 1599–1601 CrossRef CAS PubMed
- K. Alfonsi, J. Colberg, P. J. Dunn, T. Fevig, S. Jennings, T. A. Johnson, H. P. Kleine, C. Knight, M. A. Nagy, D. A. Perry and M. Stefaniak, Green Chemistry Tools to Influence a Medicinal Chemistry and Research Chemistry Based Organisation, Green Chem., 2008, 10, 31–36 RSC
-
COMMISSION REFULATION (EU), 2021/2030 of 19 November 2021 Amending Annex XVII to Regulation (EC) No 1907/2006 of the European Parliament and of the Council Concerning the Restriction, Evaluation, Authorisation and Restriction of Chemicals (REACH) as Regards N,N-dimethylformamide, 2021 Search PubMed
- L. Lu, J. C. Siu, Y. Lai and S. Lin, An Electroreductive Approach to Radical Silylation via the Activation of Strong Si–Cl Bond, J. Am. Chem. Soc., 2020, 142, 21272–21278 CrossRef CAS PubMed
- W. Guan, Y. Chang and S. Lin, Electrochemically Driven Deoxygenative Borylation of Alcohols and Carbonyl Compounds, J. Am. Chem. Soc., 2023, 145, 16966–16972 CrossRef CAS PubMed
- W. Guan, L. Lu, Q. Jiang, A. F. Gittens, Y. Wang, L. F. T. Novaes, R. S. Klausen and S. Lin, An Electrochemical Strategy to Synthesize Disilanes and Oligosilanes from Chlorosilanes, Angew. Chem., Int. Ed., 2023, 62, e202303592 CrossRef CAS PubMed
- Y.-C. Kim, H.-H. Park, J. S. Chun and W.-J. Lee, Compositional and Structural Analysis of Aluminum Oxide Films Prepared by Plasma-Enhanced Chemical Vapor Deposition, Thin Solid Films, 1994, 237, 57–65 CrossRef CAS
- A. Hess, E. Kemnitz, A. Lippitz, W. Unger and D.-H. Menz, ESCA, XRD, and IR Characterization of Aluminium Oxide, Hydroxyfluoride, and Fluoride Surfaces in Correlation with Their Catalytic Activity in Heterogeneous Halogen Exchange Reactions, J. Catal., 1994, 148, 270–280 CrossRef
- M. Dürrwächter, G. Indlekofer, H.-G. Boyen, P. Oelhafen and D. Quitmann, Core Level Binding Energy Shifts in Liquid Binary Alloys: AuGa, J. Non-Cryst., 1993, 156–158, 241–245 CrossRef
- G. A. Elia, I. Hasa, G. Greco, T. Diemant, K. Marquardt, K. Hoeppner, R. J. Behm, A. Hoell, S. Passerini and R. Hahn, Insights into the Reversibility of Aluminum Graphite Batteries, J. Mater. Chem. A, 2017, 5, 9682–9690 RSC
- E. Cho, J. Mun, O. B. Chae, O. M. Kwon, H.-T. Kim, J. H. Ryu, Y. G. Kim and S. M. Oh, Corrosion/Passivation of Aluminum Current Collector in Bis(Fluorosulfonyl)Imide-Based Ionic Liquid for Lithium-Ion Batteries, Electrochem. Commun., 2012, 22, 1–3 CrossRef CAS
- N. R. Levy, M. Auinat and Y. Ein-Eli, Tetra-Butyl Ammonium Fluoride – An Advanced Activator of Aluminum Surfaces in Organic Electrolytes for Aluminum-Air Batteries, Energy Storage Mater., 2018, 15, 465–474 CrossRef
- S. S. Rehim, H. H. Hassan and M. A. Amin, Corrosion and Corrosion Inhibition of Al and Some Alloys in Sulphate Solutions Containing Halide Ions Investigated by an Impedance Technique, Appl. Surf.
Sci., 2002, 187, 279–290 CrossRef CAS
- A. Mazhar, W. Badawy and M. Abou-Romia, Impedance Studies of Corrosion Resistance of Aluminium in Chloride Media, Surf. Coat., 1986, 29, 335–345 CrossRef CAS
- W. Li, T. Cochell and A. Manthiram, Activation of Aluminum as an Effective Reducing Agent by Pitting Corrosion for Wet-chemical Synthesis, Sci. Rep., 2013, 3, 1229 CrossRef PubMed
- P. M. Natishan and W. E. O'Grady, Chloride Ion Interactions with Oxide-Covered Aluminum Leading to Pitting Corrosion: A Review, J. Electrochem. Soc., 2014, 161, C421–C432 CrossRef CAS
- A. Reese, N. Voigt, T. Zimmermann, J. Irrgeher and D. Pröfrock, Characterization of Alloying Components in Galvanic Anodes as Potential Environmental Tracers for Heavy Metal Emissions from Offshore Wind Structures, Chemosphere, 2020, 257, 127182 CrossRef CAS PubMed
- M. Skeldon, K. Shimizu, P. Skeldon, G. Thompson and G. Wood, The Migration of Cl- and I- Ions in Anodic Alumina, Corros. Sci., 1995, 37, 1473–1488 CrossRef CAS
- F. Brown and W. D. Mackintosh, The Use of Rutherford Backscattering to Study the Behavior of Ion-Implanted Atoms During Anodic Oxidation of Aluminum: Ar, Kr, Xe, K, Rb, Cs, Cl, Br, and I, J. Electrochem. Soc., 1973, 120, 1096 CrossRef CAS
- K. Shimizu, G. M. Brown, K. Kobayashi, P. Skeldon, G. E. Thompson and G. C. Wood, A Novel Approach for the Study of the Migration of Cl- Ions in Anodic Alumina, Corros. Sci., 1999, 1835–1847 CrossRef CAS
- N. A. Vinogradov, K. A. Simonov, A. A. Zakharov, J. W. Wells, A. V. Generalov, A. S. Vinogradov, N. Mårtensson and A. B. Preobrajenski, Hole Doping of Graphene Supported on Ir(111) by AlBr3, Appl. Phys. Lett., 2013, 102, 061601 CrossRef
- J. Derouault, P. Granger and M. Forel, Spectroscopic Investigation of Aluminum Trihalide-Tetrahydrofuran Complexes. 2. Solutions of Aluminum Chloride or Bromide in Tetrahydrofuran and in Tetrahydrofuran-Dichloromethanel, Inorg. Chem., 1977, 16, 3214–3218 CrossRef CAS
- K. Furumata and O. Seri, An Estimation of Chloride Ion Concentration in Pitting Cavity for Aluminum 1100 Corroded in NaCl Solution, J. Jpn. Inst. Met., 2001, 65, 880–887 CrossRef CAS
- Q. Zeng and S. Xu, Thermodynamics and Characteristics of Heterogeneous Nucleation on Fractal Surfaces, J. Phys. Chem. C, 2015, 119, 27426–27433 CrossRef CAS
- Aluminum 5052 (UNS A95052) Spec Sheet, Grimco, https://f.hubspotusercontent20.net/hubfs/5541471/Aluminum_MILL_Spec_Sheet(1)(1).pdf, accessed 2023-10-13 Search PubMed.
- C. A. Paddon and R. G. Compton, A Reference Electrode for Electrochemical and Cryoelectrochemical Use in Tetrahydrofuran Solvent, Electroanalysis, 2005, 17, 1919–1923 CrossRef CAS
- C. A. Paddon and R. G. Compton, Underpotential Deposition of Lithium on Platinum Single Crystal Electrodes in Tetrahydrofuran, J. Phys. Chem. C, 2007, 111, 9016–9018 CrossRef CAS
- T. J. Donohoe, N. M. Kershaw, R. Baron and R. G. Compton, The Effect of Ortho-Substitution on the Efficacy of Biphenyls in Mediating Electron Transfer from Lithium, Tetrahedron, 2009, 65, 5377–5384 CrossRef CAS
- A. Lewandowski, M. Osińska, A. Swiderska-Mocek and M. Galinski, A Cryptate Reference Electrode for Ionic Liquids, Electroanalysis, 2008, 20, 1903–1908 CrossRef CAS
- M. Li and M. Dincă, Reductive Electrosynthesis of Crystalline Metal–Organic Frameworks, J. Am. Chem. Soc., 2011, 133, 12926–12929 CrossRef CAS PubMed
-
W. Zhang, W. Guan, Y. Wang, S. Lin and K. See, Data for “Enabling Al Sacrificial Anodes in Tetrahydrofuran Electrolytes for Reductive Electrosynthesis”, CaltechDATA, 2023, DOI:10.22002/rxsqk-8zb05
Footnote |
| † Electronic supplementary information (ESI) available: Complete LSV scans of Al stripping, Nyquist plots during galvanostatic Al stripping, additional XPS data, and −H-NMR spectra. See DOI: https://doi.org/10.1039/d3sc04725c |
| This journal is © The Royal Society of Chemistry 2023 |