
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInterface defects repair of core/shell quantum dots through halide ion penetration†
Changwei
Yuan
 a,
Mengda
He
a,
Xinrong
Liao
a,
Mingming
Liu
a,
Qinggang
Zhang
a,
Qun
Wan
b,
Zan
Qu
a,
Long
Kong
*a and
Liang
Li
*b
a,
Mengda
He
a,
Xinrong
Liao
a,
Mingming
Liu
a,
Qinggang
Zhang
a,
Qun
Wan
b,
Zan
Qu
a,
Long
Kong
*a and
Liang
Li
*b
aSchool of Environmental Science and Engineering, Shanghai Jiao Tong University, Shanghai 200240, P. R. China. E-mail: longmao88@sjtu.edu.cn
bMacao Institute of Materials Science and Engineering (MIMSE), Macau University of Science and Technology, Taipa, Macao 999078, P. R. China. E-mail: lli@must.edu.mo
First published on 1st November 2023
Abstract
The interface defects of core–shell colloidal quantum dots (QDs) affect their optoelectronic properties and charge transport characteristics. However, the limited available strategies pose challenges in the comprehensive control of these interface defects. Herein, we introduce a versatile strategy that effectively addresses both surface and interface defects in QDs through simple post-synthesis treatment. Through the combination of fine chemical etching methods and spectroscopic analysis, we have revealed that halogens can diffuse within the crystal structure at elevated temperatures, acting as “repairmen” to rectify oxidation and significantly reducing interface defects within the QDs. Under the guidance of this protocol, InP core/shell QDs were synthesized by a hydrofluoric acid-free method with a full width at half-maximum of 37.0 nm and an absolute quantum yield of 86%. To further underscore the generality of this strategy, we successfully applied it to CdSe core/shell QDs as well. These findings provide fundamental insights into interface defect engineering and contribute to the advancement of innovative solutions for semiconductor nanomaterials.
Introduction
Colloidal quantum dots (QDs) have garnered significant interest in diverse markets, including electronics and photovoltaics, due to their adjustable light absorption,1–3 high color purity,4–7 controllable electronic transport, and solution processability.8–10 These QDs are usually prepared in a core–shell geometry, featuring excellent stability and efficient radiation.11–15 One critical aspect that significantly affects their optoelectronic properties is the presence of trap states within the band gap, which lead to the localization of charge carriers.16,17 These trap states are commonly associated with surface and interface defects.18–23 Achieving precise control over trap states in core–shell QDs remains a formidable challenge, yet addressing it would lead to significant advancements in their diverse applications.24–26A considerable amount of ongoing work in this field aims to uncover valuable insights that will advance our understanding of trap states passivation in core–shell QDs. To achieve QDs with nearly unity photoluminescence quantum yield (PLQY), surface oxides were eliminated using fluorinating agents prior to shell growth.27–29 Furthermore, numerous studies have highlighted surface ligand engineering to enhance PLQY.30–33 Alongside surface defects, interface defects inevitably arise during the preparation of core–shell QDs and significantly impact their performance.34,35 Meanwhile, exploring universal strategies to address the interface defects of QDs remains an open challenge.12,18
Herein, we present a highly versatile strategy for effectively addressing both surface and interface defects in core–shell QDs through a simple post-synthesis treatment. First, we examined the role of halide ions in the post-treatment, and uncovered that halide ions not only adhere to the surface through ligand coordination but also gradually permeate into the interior of the QDs at elevated temperatures. This unique behavior enables halide ions to bind the cations on the cores, effectively mitigating core oxidation during synthesis and significantly reducing interface defects within the QDs. To demonstrate the generality of this approach, we successfully applied it to both InP and CdSe core/shell QDs. The post-treatment InP core–multishell QDs exhibit an impressive full width at half-maximum (FWHM) of 37.0 nm and an absolute PLQY of 86%.
Results and discussion
Optical properties of zinc halide post-treated InP core–multishell QDs
The synthetic procedure for the fabrication of InP core–multishell QDs is depicted in Fig. 1a. This scheme involves hot injection to product InP cores, followed by cores purification, subsequent shell growth, and post-treatment processes. The InP core exhibits a well-defined first exciton absorption peak, displaying a VD (valley depth) value of 0.43 (Fig. S1a†). The VD value is defined as VD = 1 − (Absmin/Absmax), where Absmax and Absmin represent absorbance at the first maximum and minimum inflection points, respectively. A higher VD value indicates better size uniformity.27 The size distribution of InP cores was improved through the growth of a ZnSe shell, as evidenced by the increase in the VD values from 0.43 to 0.53. To relieve strain between the ZnSe and ZnS layers, a ZnSeS gradient alloy shell is employed. Prior to ZnSeS growth, the residual indium precursors were effectively eliminated through the purification of the InP/ZnSethin QDs, contributing to the superior photoluminescence (PL) properties of the InP core–multishell QDs.36 After growing a thick ZnSeS, the FWHM of the QDs decreased to 36.5 nm, and the VD value reached 0.58 due to uniform shell growth over the QDs. The PLQY of InP/ZnSe/ZnSeS increases from 38% to 53% compared to InP/ZnSethin due to the thicker shell layer that can effectively passivate the surface defect states. Finally, a ZnS shell was coated as the outermost layer to provide effective quantum confinement of excitons within the emissive InP cores. This resulted in a further increase of PLQY to 73%, with FWHM reaching 36.9 nm and a slight decrease in the VD value to 0.54.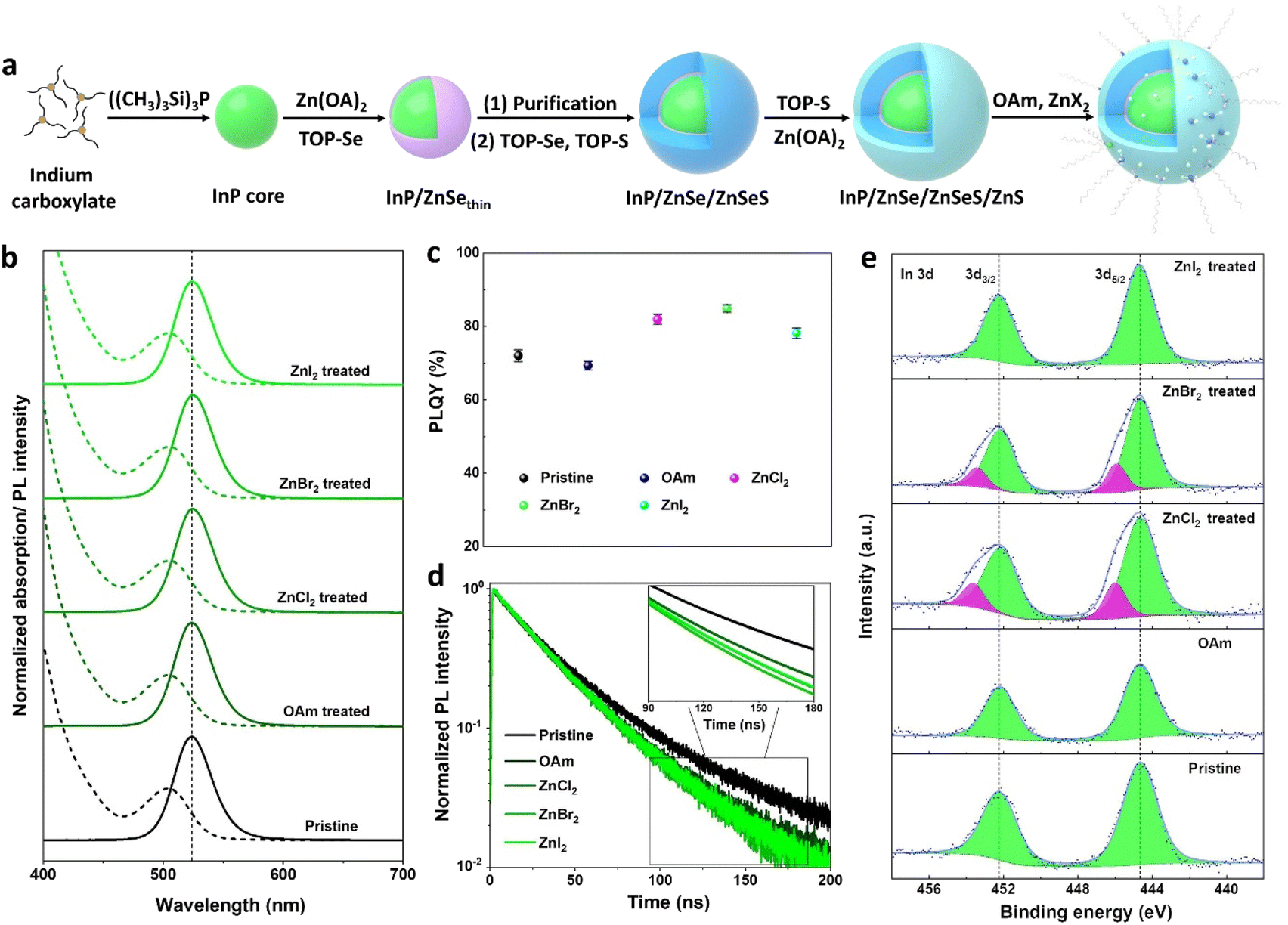 | ||
| Fig. 1 .Optical properties of InP core–multishell QDs post-treated with zinc halides. (a) Schematic of the synthesis procedure. (b) Normalized PL spectra (solid lines) and optical absorption (dashed lines), (c) PLQY values and (d) PL decay curves of pristine (black line) and post-treated (green lines) InP core–multishell QDs. The inset depicts lifetime fitting curves between 90 and 180 ns. (e) High-resolution XPS spectra of In 3d element. | ||
In previous reports on the synthesis of InP based QDs, zinc halides (ZnX2) were widely considered as Z-type ligands that effectively passivated the surface anionic trap states of QDs.16,37,38 Here, zinc halides (ZnX2) were adopted as post-treatment agents to improve the optical properties of InP/ZnSe/ZnSeS/ZnS QDs. Typically, the zinc halide treatment agents (0.2 M) were prepared by dissolving 2 mmol ZnCl2, ZnBr2, and ZnI2 into 10 ml mixed solvent (VOAm![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :VODE = 2:13) of OAm and ODE, respectively. The use of mixed solvents is to reduce the negative impact of excessive OAm on the performance of QDs during high-temperature post-processing. The crude solution of core–shell QDs was heated to 300 °C, and zinc halide treatment agents were injected at 4 ml h−1 for 30 min and maintained at this temperature for another 30 min. After treatment, QDs were purified and dispersed in toluene for further characterization.
:VODE = 2:13) of OAm and ODE, respectively. The use of mixed solvents is to reduce the negative impact of excessive OAm on the performance of QDs during high-temperature post-processing. The crude solution of core–shell QDs was heated to 300 °C, and zinc halide treatment agents were injected at 4 ml h−1 for 30 min and maintained at this temperature for another 30 min. After treatment, QDs were purified and dispersed in toluene for further characterization.
The X-ray diffractogram (XRD, Fig. S1d†) confirms the zinc blende structure of the QDs, which remains unchanged after treatment. Transmission electron microscopy (TEM, Fig. S2†) demonstrates the excellent size uniformity of InP core–multishell QDs, and the particle size and distribution remain consistent between OAm and ZnX2 treated samples. Therefore, the slight red shift observed in the PL peaks of ZnBr2 and ZnCl2 treated QDs (Fig. 1b) can be reasonably attributed to electron delocalization resulting from the binding of highly electronegative halogen ligands.39 As expected, the PLQY values of the QDs treated with ZnX2 show a notable improvement (Fig. 1c), as summarized in Table S1.† Specifically, the PLQY of the ZnBr2 treated sample increases from 73% to 86% compared to the pristine QDs. To exclude the contribution of OAm solvents to the enhancement of PLQY, a control treatment using an equivalent amount of OAm but without zinc halides was conducted. Interestingly, OAm exhibits a detrimental impact on the PLQY enhancement, which can be attributed to the replacement of the relatively stronger-binding oleic acid ligands with the weaker-binding OAm ligands, resulting in a decrease in PLQY.33 To clarify whether zinc in zinc halides plays a key role in improving PLQY, another control treatment experiment was performed. 0.1 M OAm halides were prepared by dissolving 1 mmol of OAm-Cl, OAm-Br, and OAm-I each in 10 ml of ODE. Using the same treatment process as above, the crude solution of multishell QDs was heated with OAm halides at 300 °C for 60 min, and all treated samples show a similar improvement in PLQY as the treatment with Zn halides (Fig. S3†). This means that the halide ions are the main contributors to the improved PLQY.
To elucidate the origin of higher PLQY values in halides treated QDs, the time-resolved photoluminescence (TRPL) spectra were examined (Fig. 1d). The TRPL decay curves were fitted using a double exponential function, and the fitting parameters are listed in Table S1.† Typically, the shorter component (τ1) of the fluorescence lifetime is associated with band-edge transition emission, while the longer component (τ2) is linked to defect-related emission.40 It is observed that the band-edge transition portions (τ1%) significantly increase in ZnX2 treated QDs. Specifically, the band-edge transition portion of ZnBr2 treated QDs improved from 50.1% to 79.2% compared with untreated QDs. This evident improvement is ascribed to the passivation of trap states of the QDs after ZnX2 treatment. Besides, the average fluorescence lifetimes of ZnX2 treated QDs were significantly shortened compared with untreated QDs (Table S1†). Specifically, the average lifetime of the ZnBr2 treated sample dropped from 47.4 ns to 37.1 ns relative to the untreated samples. We attribute the observed shorter lifetime of post-treatment samples to the incorporation of more Zn2+ into the InP lattice, resulting in a faster hole trapping rate.41 In addition, the average lifetime of ZnX2 post-treated samples is shorter than that of OAm post-treated samples, which also confirms this conjecture. This is because adding an additional zinc source will accelerate the entry of Zn2+ into the InP lattice, resulting in a shorter fluorescence lifetime.
The chemical states of InP core–multishell QDs were investigated by XPS measurements (Fig. 1e). It is noteworthy that in addition to the normal In 3d peaks at 444.6 eV identified as the chemical states of InP,39 new chemical states appear in the ZnBr2 and ZnCl2 treated samples at 446.0 eV. These new chemical states in the In 3d spectra are identified as the coordination of In with halogens (i.e., InBrx and InClx),39 and the In 3d XPS spectra of InP QDs (Fig. S3c†) treated with OAm halides also exhibit the same shift towards higher binding energy. The relatively inconspicuous In 3d peak shift from the ZnI2 treated sample can be attributed to the weaker binding strength or more obvious steric effects of iodine ions. Furthermore, the Cl 2p, Br 3d, and I 3d XPS spectra confirm the presence of Cl, Br, and I ions in the InP QDs (Fig. S4†).
The diffusion of halogens into InP/ZnSe/ZnSeS/ZnS QDs
The diffusion of halogens in perovskite crystals has been widely reported,42,43 but rarely reported in II–VI and III–V semiconductors. In our case, to determine whether the halogens are only bonded to the surface of QDs or also diffused into the ZnS shell, the ZnBr2-treated InP core–multishell QDs were layer-by-layer etched by wet chemical processes, with detailed procedures provided in the Methods section of the ESI.† InP core–multishell QDs were gradually etched to reach the surface of the InP core after five rounds of etching, as shown in the size distribution histograms of TEM (Fig. 2d). This was further confirmed by measuring the change of In:Zn element ratios by ICP analysis with the etching cycles (Fig. 2e). The In:Zn ratios rapidly increased after five etching cycles, indicating the etching reached the InP cores. Ion chromatography was employed to quantitatively analyze the content of Br ions in the etching supernatant of InP QDs, which varied with etching cycles (Fig. 2f) and could reflect the distribution of halide ions within the QDs to some extent. During the sixth etching, the InP core was completely etched, and the Br ion content was almost zero. Therefore, it is reasonable to speculate that the Br ions did not diffuse into the InP core. This phenomenon can be attributed to the higher diffusion activation energy required for halogen diffusion in the InP lattice.
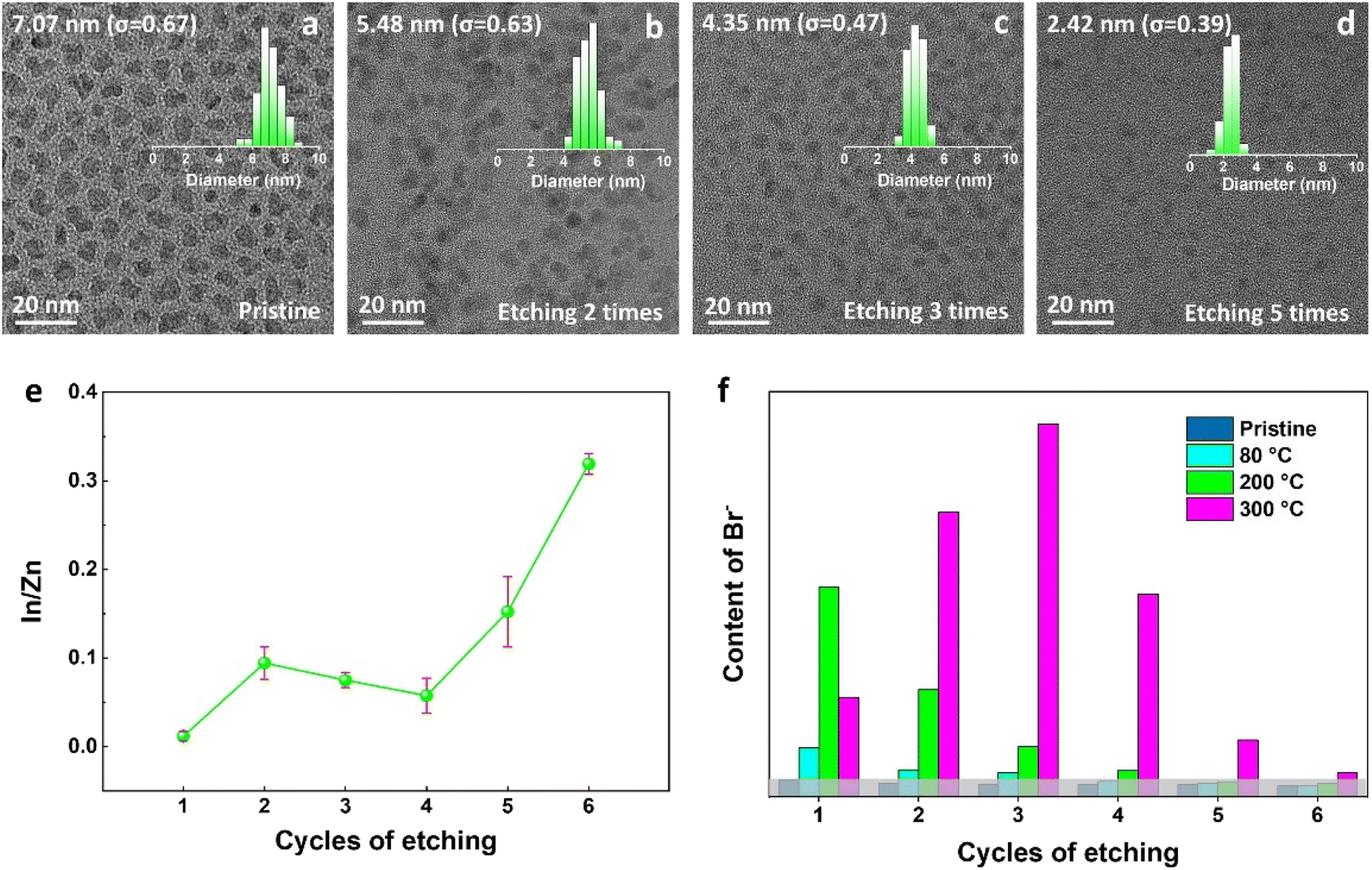 | ||
| Fig. 2 .Chemical etching of pristine and ZnBr2-treated InP core–multishell QDs. (a–d) TEM images and size distribution histograms (top-right) of QDs after various etching times. (e) Changes in the In:Zn element ratios and (f) variations in the Br− content of InP QDs processed at different temperatures as a function of etching cycles. The grey background represents the noise level. | ||
Surprisingly, based on the samples processed at 300 °C, Br ions are not only localized on the surface of multi-shell QDs but also distributed throughout all the shells, as shown in Fig. 2f. It is also found that the distribution of Br ions exhibits temperature-dependent features. This phenomenon can be explained as follows: firstly, an activation barrier must be surmounted for the ZnX2 ligands to attach to the QDs' surface,16 which could explain the significantly higher concentration of Br ions on the QDs' surface when treated at 200 °C compared to 80 °C. Secondly, under much higher post-treat temperatures, e.g. at 300 °C, the highest concentration of Br ions occurs in the third etch instead of the first one, which we attribute to the gradual diffusion of Br ions into the QD shells after overcoming the specific diffusion barrier,44 resulting in a relative decrease in the surface coordinated halide ions. Therefore, we can conclude that halogens can diffuse inside QDs and exhibit temperature-dependent characteristics.
Fig. 3a illustrates the temperature-dependent diffusion mechanism of halide atoms within InP core–multishell QDs. At higher temperatures, an increased number of vacancies occurs, facilitating the diffusion of halide atoms.45,46 The temperature dependence of PLQY values in InP QDs treated with ZnBr2 is also evident (Fig. 3b), with QDs post-treated at higher temperatures displaying higher PLQY values. We attribute this to a greater number of halogens entering the interior of QDs for defect repair at higher temperatures. This finding was further supported by the TRPL measurements, indicating that QDs prepared at higher post-treatment temperatures exhibit more band-edge emission components (Fig. 3d). The FWHM of ZnBr2-treated InP QDs, processed at different temperatures, exhibits varying changes with post-treatment time. Specifically, QDs post-treated at 300 °C demonstrate a smaller FWHM compared to those treated at 160 °C and 240 °C (Fig. 3f), which can be attributed to the higher temperature favoring the formation of better chemical homogeneity within the QDs.47
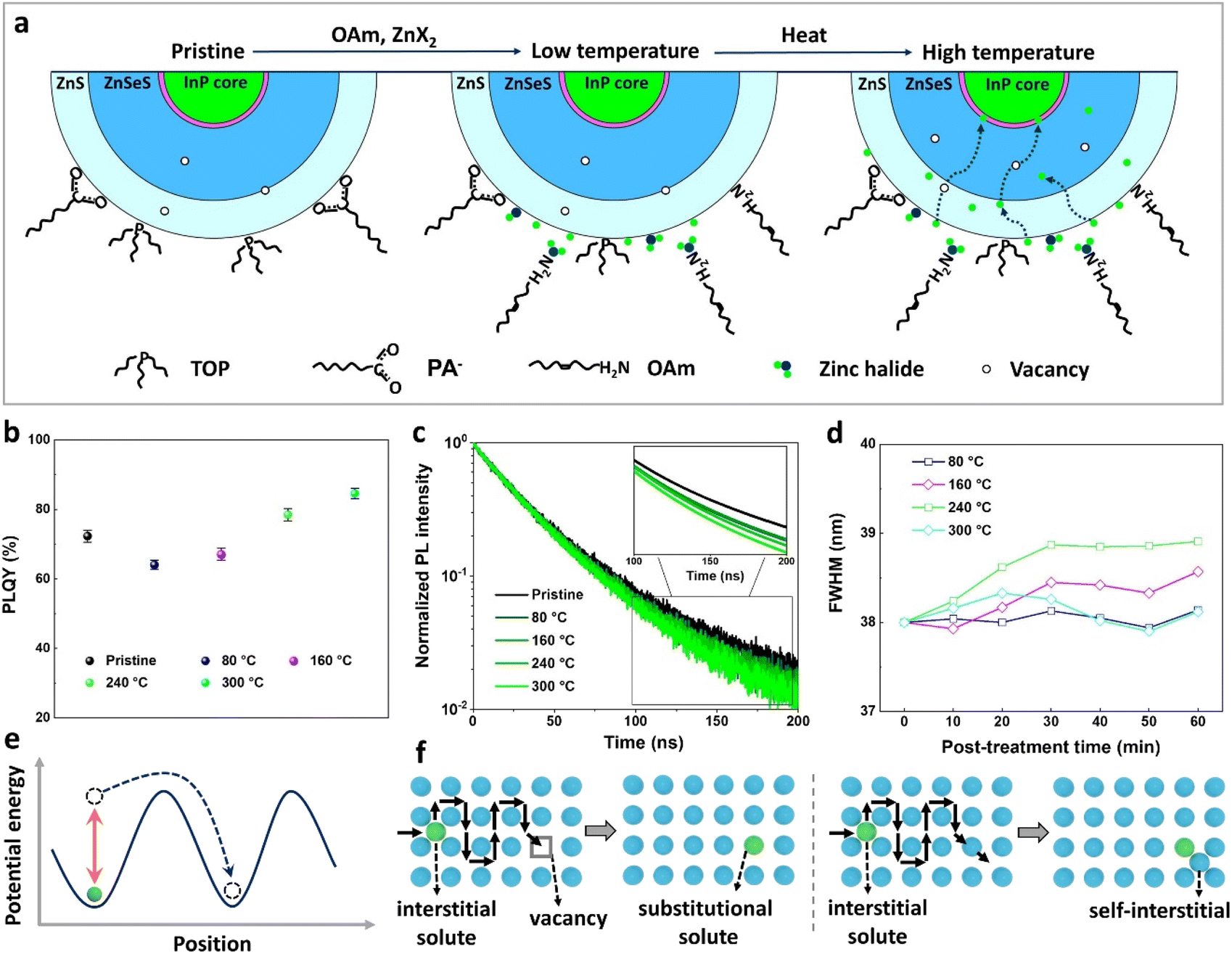 | ||
| Fig. 3 .The temperature dependent diffusion of halide ions. (a) Schematic illustration of halide ion diffusion in InP core–multishell QDs upon low and high temperature annealing. (b) PLQY and (c) PL decay curves of ZnBr2-treated InP QDs varying with temperature. The inset depicts lifetime fitting curves between 100 and 200 ns. (d) The FWHM of ZnBr2-treated InP QDs processed at different temperatures changes with post-treatment time. (e) Schematic diagram of temperature-driven atomic diffusion. (f) The simplified 2D model of interstitial–substitutional exchange mechanisms for foreign atom diffusion. Left: Dissociative mechanism. Right: Kick-out mechanism. | ||
The diffusion of ions within a crystal involves overcoming potential barriers, which are influenced by the internal lattice environment of the ions as well as external potential gradient forces. Interactions between ions and surrounding atoms create potential wells that bind the ions. Various gradient forces, including concentration gradients, electric fields, and thermal potential gradients, drive ions diffusion. At a given temperature, the mobility and diffusion coefficient of ions depend on factors such as the height of the potential barriers from the equilibrium position, the frequency of ion jumps, and the average distance of these jumps. As temperature increases, the ions become more active, facilitating their transition across the barrier to another equilibrium position, as illustrated in Fig. 3e. The temperature dependence of diffusion can be explained as Arrhenius formula48
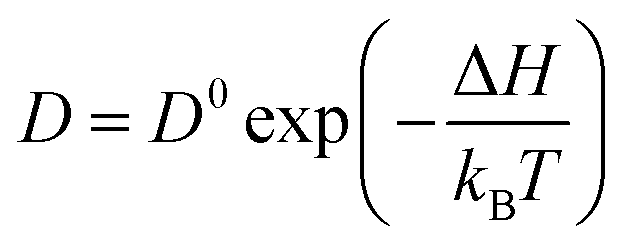 | (1) |
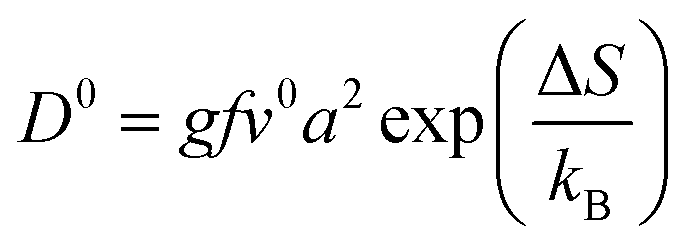 | (2) |
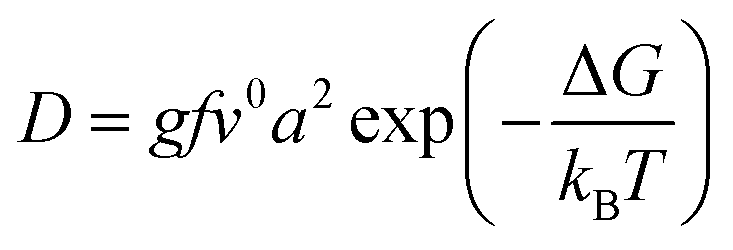 | (3) |
ZnS and ZnSe zinc blende lattices are open crystal structures with sufficient space for diffusion of interstitial species.48 And the ionic radii of chloride (Cl−, 181 pm) and bromide (Br−, 196 pm) are similar to those of selenium (Se2−, 198 pm) and sulfur (S2−, 184 pm). For this reason, we speculate that Cl− and Br− diffuse within the zinc blende lattice of ZnS and ZnSe mainly through interstitial diffusion and interstitial–substitutional exchange diffusion mechanisms, and the hybrid solutes (Cl−, Br−) change over to more soluble substitutional states via the dissociative or kick-out mechanisms (Fig. 3f).
The removal of internal oxidation defects by halogens
The discovery of the temperature dependence of halide ion diffusion in zinc blende lattices has enabled us to devise an internal trap state repair scheme for core–multishell QDs featuring a defect-free crystal structure. Previous reports have demonstrated that stacking faults in ZnTeSe/ZnSe/ZnS QDs can be effectively eliminated by treating them with both HF and ZnCl2 during shell growth.49 In a designed experiment, we have shown that the post-treatment of halides could also effectively remove the internal oxidation defects (Fig. S7†).The removal of internal oxidation defects by halogens was further confirmed in CdSe/ZnS and InP/ZnSeS core/shell QDs prepared from purposely oxidized cores. CdSe and InP QDs were first treated with dibenzoyl peroxide to prepare oxidized cores, followed by purification, shell growth, and finally post-treatment with OAm halides (Fig. 4a). The optical absorption and normalized PL spectra of these QDs are shown in Fig. S8.† The PLQYs of CdSe/ZnS and InP/ZnSeS with oxidized cores were poor, at 32% and 21%, respectively, which increased to 42% and 33%, respectively, after OAm-Br post-treatment (Fig. 4b). Further analysis of intrinsic factors that contribute to the enhancement of PLQY has been conducted by XRD measurements. The CdO/ZnO mixed oxide peaks are observed in the X-ray diffractogram (Fig. 4c) of oxidized CdSe/ZnS QDs,50 which are obviously weakened after being treated with OAM-Br. In high-resolution XPS spectra (Fig. 4d and e), the pristine samples prepared without BPO treatment exhibited the In 3d5/2 peak at 444.6 eV and Cd 3d5/2 center at 405.0 eV, corresponding to the chemical states of InP and CdSe, respectively. After BPO treatment, the Cd 3d peaks shifted to lower binding energy, while the In 3d peaks exhibited a slight shift towards higher binding energy, indicating the generation of Cd–O and In–O species, which is consistent with what has been previously reported.51,52 It is noteworthy that both the binding energy of Cd 3d and In 3d shifted towards higher binding energy after OAm–Br treatment, suggesting the formation of Cd–Br and In–Br compounds.39,53 Therefore, it is reasonable to speculate that the PLQY enhancement is associated with the removal of internal oxidation defects.
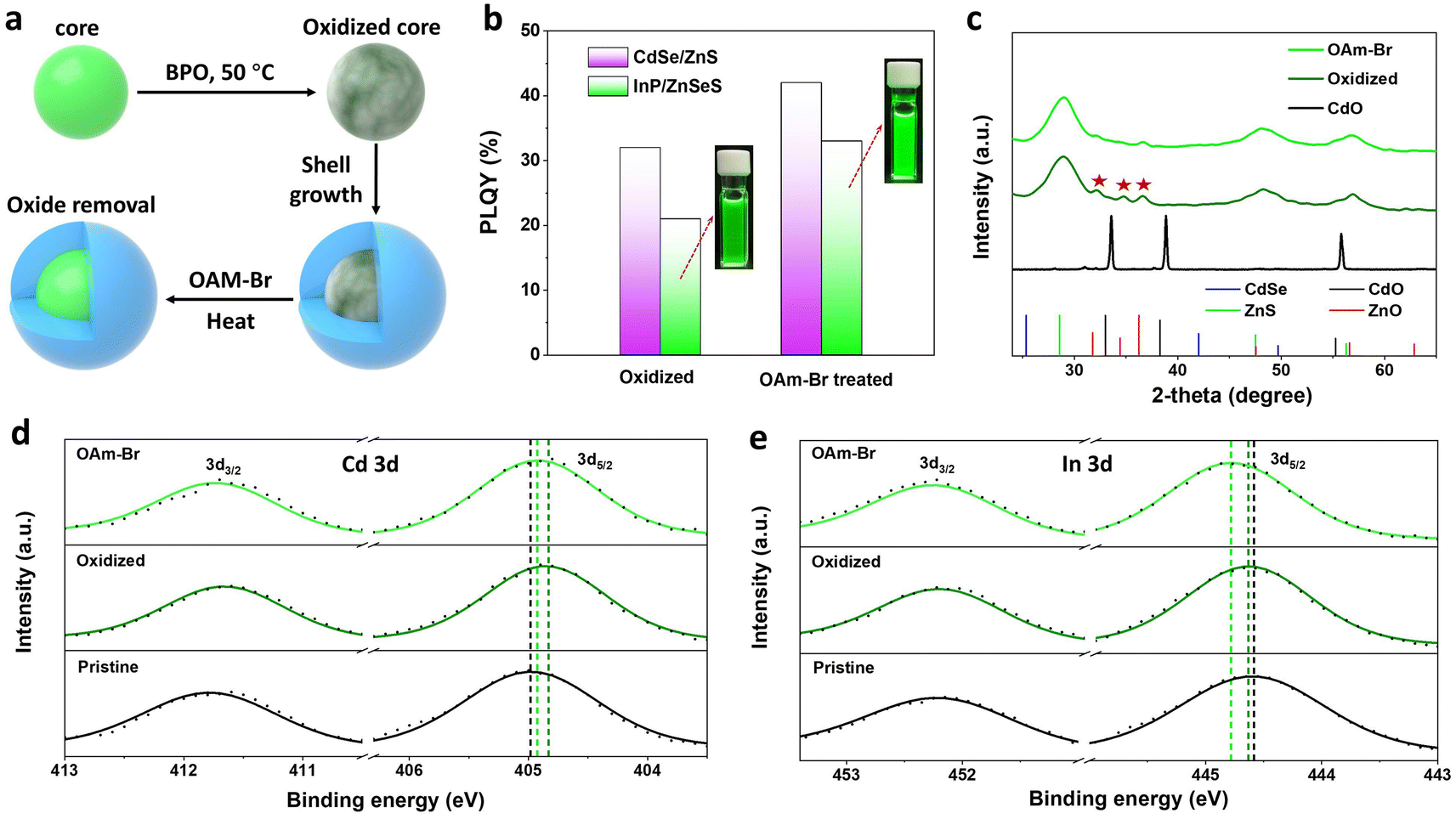 | ||
| Fig. 4 .Optical properties of QDs treated with OAm halides. (a) Schematic diagram of BPO treatment of QD cores, subsequent shell growth and post-treatment synthesis. (b) PLQY of InP/ZnSeS and CdSe/ZnS. Both types of QDs possess oxidized cores and were prepared without any post-treatment or with OAm-Br post-treatment. The insets show photographs of InP/ZnSeS QDs under UV excitation at 365 nm. (c) X-ray diffractogram of CdO (black line) and post-treated (green lines) CdSe/ZnS QDs. The corresponding high-resolution XPS spectra of (d) Cd 3d and (e) In 3d elements. | ||
Conclusions
In summary, we have shed light on the microcosmic mechanisms of QD passivation using halide agents for post-treatment. Meticulous chemical etching demonstrates the potential diffusion of halide ions in ZnS and ZnSe zinc blende lattices, and the diffusion processes have temperature-dependent features. Multilateral XPS analysis elucidated the molecular mechanisms of halogen passivation by conversion of oxide species to metal halides. Based on this profound understanding of the halide passivation mechanisms, facile post-treatment schemes were devised for core–multishell QDs. Following the guidelines, uniform and defect-free InP core–multishell QDs were prepared with a PLQY of 86%. This article provides new insights into the interactions between halogens and nanocrystals, which will help to deepen the understanding of defect passivation and develop novel synthetic strategies for other nanomaterials.Data availability
All the experimental data were provided in the main text and/or ESI.†Author contributions
C. Y. and L. L. prepared the methodology and original draft. The data curation and formal analysis were performed by C. Y., M. H., X. L., M. L., Q. W., L. K., and Q. Z. L. L., L. K., and Z. Q. were involved in the funding acquisition, resources and supervision.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Macau Science and Technology Development Fund (FDCT-0005/2022/AKP); Jiangxi Key Research and Development Program (20223BBH80W01); Joint Funds of the National Natural Science Foundation of China (No. U21A20320); the National Natural Science Foundation of China (22175113, 42007125, 22205136); and the Start-up Funding for Scientific Research from Macau University of Science and Technology (SSF-22-001-MIMSE).References
- R. Yadav, Y. Kwon, C. Rivaux, C. Saint-Pierre, W. L. Ling and P. Reiss, J. Am. Chem. Soc., 2023, 145, 5970–5981 CrossRef CAS PubMed.
- Y. Deng, F. Peng, Y. Lu, X. Zhu, W. Jin, J. Qiu, J. Dong, Y. Hao, D. Di, Y. Gao, T. Sun, M. Zhang, F. Liu, L. Wang, L. Ying, F. Huang and Y. Jin, Nat. Photon., 2022, 16, 505–511 CrossRef CAS.
- C. Zhang, W. Li and L. Li, Angew. Chem., Int. Ed., 2021, 60, 7488–7501 CrossRef CAS PubMed.
- M. Liu, Q. Wan, H. Wang, F. Carulli, X. Sun, W. Zheng, L. Kong, Q. Zhang, C. Zhang, Q. Zhang, S. Brovelli and L. Li, Nat. Photon., 2021, 15, 379–385 CrossRef CAS.
- C. Zhang, Q. Wan, L. K. Ono, Y. Liu, W. Zheng, Q. Zhang, M. Liu, L. Kong, L. Li and Y. Qi, ACS Energy Lett., 2021, 6, 3545–3554 CrossRef CAS.
- Q. Zhang, S. Liu, M. He, W. Zheng, Q. Wan, M. Liu, X. Liao, W. Zhan, C. Yuan, J. Liu, H. Xie, X. Guo, L. Kong and L. Li, Angew. Chem., Int. Ed., 2022, 61, e202205463 CrossRef CAS PubMed.
- Y. Shu, X. Lin, H. Qin, Z. Hu, Y. Jin and X. Peng, Angew. Chem., Int. Ed., 2020, 59, 22312–22323 CrossRef CAS PubMed.
- F. P. Garcia de Arquer, D. V. Talapin, V. I. Klimov, Y. Arakawa, M. Bayer and E. H. Sargent, Science, 2021, 373, eaaz8541 CrossRef CAS PubMed.
- Y.-S. Park, J. Roh, B. T. Diroll, R. D. Schaller and V. I. Klimov, Nat. Rev. Mater., 2021, 6, 382–401 CrossRef CAS.
- M. He, Q. Zhang, F. Carulli, A. Erroi, W. Wei, L. Kong, C. Yuan, Q. Wan, M. Liu, X. Liao, W. Zhan, L. Han, X. Guo, S. Brovelli and L. Li, ACS Energy Lett., 2022, 8, 151–158 CrossRef.
- H. Moon, C. Lee, W. Lee, J. Kim and H. Chae, Adv. Mater., 2019, 31, e1804294 CrossRef PubMed.
- Y. Choi, D. Hahm, W. K. Bae and J. Lim, Nat. Commun., 2023, 14, 43 CrossRef CAS PubMed.
- N. Razgoniaeva, P. Moroz, M. Yang, D. S. Budkina, H. Eckard, M. Augspurger, D. Khon, A. N. Tarnovsky and M. Zamkov, J. Am. Chem. Soc., 2017, 139, 7815–7822 CrossRef CAS PubMed.
- D. Zhu, H. Bahmani Jalali, G. Saleh, F. Di Stasio, M. Prato, N. Polykarpou, A. Othonos, S. Christodoulou, Y. P. Ivanov, G. Divitini, I. Infante, L. De Trizio and L. Manna, Adv. Mater., 2023, 35, 2303621 CrossRef CAS PubMed.
- B. G. Jeong, J. H. Chang, D. Hahm, S. Rhee, M. Park, S. Lee, Y. Kim, D. Shin, J. W. Park, C. Lee, D. C. Lee, K. Park, E. Hwang and W. K. Bae, Nat. Mater., 2021, 21, 246–252 CrossRef PubMed.
- N. Kirkwood, J. O. V. Monchen, R. W. Crisp, G. Grimaldi, H. A. C. Bergstein, I. du Fosse, W. van der Stam, I. Infante and A. J. Houtepen, J. Am. Chem. Soc., 2018, 140, 15712–15723 CrossRef CAS PubMed.
- H. J. Lee, S. Im, D. Jung, K. Kim, J. A. Chae, J. Lim, J. W. Park, D. Shin, K. Char, B. G. Jeong, J. S. Park, E. Hwang, D. C. Lee, Y. S. Park, H. J. Song, J. H. Chang and W. K. Bae, Nat. Commun., 2023, 14, 3779 CrossRef PubMed.
- H. Van Avermaet, P. Schiettecatte, S. Hinz, L. Giordano, F. Ferrari, C. Nayral, F. Delpech, J. Maultzsch, H. Lange and Z. Hens, ACS Nano, 2022, 16, 9701–9712 CrossRef CAS PubMed.
- A. M. Dennis, M. R. Buck, F. Wang, N. F. Hartmann, S. Majumder, J. L. Casson, J. D. Watt, S. K. Doorn, H. Htoon, M. Sykora and J. A. Hollingsworth, Adv. Funct. Mater., 2019, 29, 1809111 CrossRef.
- R. F. Ubbink, G. Almeida, H. Iziyi, I. du Fossé, R. Verkleij, S. Ganapathy, E. R. H. van Eck and A. J. Houtepen, Chem. Mater., 2022, 34, 10093–10103 CrossRef CAS PubMed.
- Z. Li, L. Kong, S. Huang and L. Li, Angew. Chem., Int. Ed., 2017, 56, 8134–8138 CrossRef CAS PubMed.
- K. Kim, D. Yoo, H. Choi, S. Tamang, J. H. Ko, S. Kim, Y. H. Kim and S. Jeong, Angew. Chem., Int. Ed., 2016, 55, 3714–3718 CrossRef CAS PubMed.
- Q. Zhang, M. He, Q. Wan, W. Zheng, M. Liu, C. Zhang, X. Liao, W. Zhan, L. Kong, X. Guo and L. Li, Chem. Sci., 2022, 13, 3719–3727 RSC.
- K. E. Hughes, J. L. Stein, M. R. Friedfeld, B. M. Cossairt and D. R. Gamelin, ACS Nano, 2019, 13, 14198–14207 CrossRef CAS PubMed.
- L. Wang, X. Qian, Y. Ren, H. Lei, X. Hu, D. Chen, J. Li and X. Peng, Chem. Mater., 2022, 34, 8297–8305 CrossRef CAS.
- P. Xia, B. Sun, M. Biondi, J. Xu, O. Atan, M. Imran, Y. Hassan, Y. Liu, J. M. Pina, A. M. Najarian, L. Grater, K. Bertens, L. K. Sagar, H. Anwar, M. J. Choi, Y. Zhang, M. Hasham, F. P. Garcia de Arquer, S. Hoogland, M. W. B. Wilson and E. H. Sargent, Adv. Mater., 2023, 35, e2301842 CrossRef PubMed.
- Y. H. Won, O. Cho, T. Kim, D. Y. Chung, T. Kim, H. Chung, H. Jang, J. Lee, D. Kim and E. Jang, Nature, 2019, 575, 634–638 CrossRef CAS PubMed.
- H. Li, W. Zhang, Y. Bian, T. K. Ahn, H. Shen and B. Ji, Nano Lett., 2022, 22, 4067–4073 CrossRef CAS PubMed.
- M. Imran, W. Paritmongkol, H. A. Mills, Y. Hassan, T. Zhu, Y. K. Wang, Y. Liu, H. Wan, S. M. Park, E. Jung, J. Tam, Q. Lyu, G. F. Cotella, P. Ijaz, P. Chun and S. Hoogland, Adv. Mater., 2023, 2303528 CrossRef PubMed.
- Y. Bai, M. Hao, S. Ding, P. Chen and L. Wang, Adv. Mater., 2022, 34, e2105958 CrossRef PubMed.
- S. M. Click and S. J. Rosenthal, Chem. Mater., 2023, 35, 822–836 CrossRef CAS.
- F. W. Eagle, N. Park, M. Cash and B. M. Cossairt, ACS Energy Lett., 2021, 6, 977–984 CrossRef CAS.
- K. C. Dümbgen, J. Leemans, V. De Roo, M. Minjauw, C. Detavernier and Z. Hens, Chem. Mater., 2023, 35, 1037–1046 CrossRef.
- M. D. Tessier, E. A. Baquero, D. Dupont, V. Grigel, E. Bladt, S. Bals, Y. Coppel, Z. Hens, C. Nayral and F. Delpech, Chem. Mater., 2018, 30, 6877–6883 CrossRef CAS.
- J. L. Stein, W. M. Holden, A. Venkatesh, M. E. Mundy, A. J. Rossini, G. T. Seidler and B. M. Cossairt, Chem. Mater., 2018, 30, 6377–6388 CrossRef CAS.
- Y. Li, X. Hou, X. Dai, Z. Yao, L. Lv, Y. Jin and X. Peng, J. Am. Chem. Soc., 2019, 141, 6448–6452 CrossRef CAS PubMed.
- D. A. Taylor, J. A. Teku, S. Cho, W.-S. Chae, S.-J. Jeong and J.-S. Lee, Chem. Mater., 2021, 33, 4399–4407 CrossRef CAS.
- J. J. Calvin, J. K. Swabeck, A. B. Sedlak, Y. Kim, E. Jang and A. P. Alivisatos, J. Am. Chem. Soc., 2020, 142, 18897–18906 CrossRef CAS PubMed.
- T. G. Kim, D. Zherebetskyy, Y. Bekenstein, M. H. Oh, L. W. Wang, E. Jang and A. P. Alivisatos, ACS Nano, 2018, 12, 11529–11540 CrossRef CAS PubMed.
- X. Duan, J. Ma, W. Zhang, P. Liu, H. Liu, J. Hao, K. Wang, L. Samuelson and X. W. Sun, ACS Appl. Mater. Interfaces, 2023, 15, 1619–1628 CrossRef CAS PubMed.
- W. Yang, Y. Yang, A. L. Kaledin, S. He, T. Jin, J. R. McBride and T. Lian, Chem. Sci., 2020, 11, 5779–5789 RSC.
- K. Domanski, B. Roose, T. Matsui, M. Saliba, S.-H. Turren-Cruz, J.-P. Correa-Baena, C. R. Carmona, G. Richardson, J. M. Foster, F. De Angelis, J. M. Ball, A. Petrozza, N. Mine, M. K. Nazeeruddin, W. Tress, M. Grätzel, U. Steiner, A. Hagfeldt and A. Abate, Energy Environ. Sci., 2017, 10, 604–613 RSC.
- W. Zhan, M. Liu, Q. Wan, M. He, Q. Zhang, X. Liao, C. Yuan, L. Kong, Y. Wang, B. Sun, S. Brovelli and L. Li, Small, 2023, 19, 2304829 CrossRef CAS PubMed.
- J. Chen, L. Liang, S. Tan, S. Xi, C. H. Lin, T. Wu, Q. He and X. Liu, Nano Lett., 2023, 23, 7221–7227 CrossRef CAS PubMed.
- B. Tuck, Atomic diffusion in III-V semiconductors, Adam Hilger, 1988 Search PubMed.
- A. Seeger, Atomic Defects in Metals and Semiconductors, ed. J. Treusch, Springer Berlin, Heidelberg, 1976 Search PubMed.
- E. M. Janke, N. E. Williams, C. She, D. Zherebetskyy, M. H. Hudson, L. Wang, D. J. Gosztola, R. D. Schaller, B. Lee, C. Sun, G. S. Engel and D. V. Talapin, J. Am. Chem. Soc., 2018, 140, 15791–15803 CrossRef CAS PubMed.
- H. Mehrer, Diffusion in solids : fundamentals, methods, materials, diffusion-controlled processes, World Publishing Corporation, 2007 Search PubMed.
- T. Kim, K. H. Kim, S. Kim, S. M. Choi, H. Jang, H. K. Seo, H. Lee, D. Y. Chung and E. Jang, Nature, 2020, 586, 385–389 CrossRef CAS PubMed.
- N. A. Eom, T. S. Kim, Y. H. Choa, W. B. Kim and B. S. Kim, J. Nanosci. Nanotechnol., 2014, 14, 8024–8027 CrossRef CAS PubMed.
- L. Huang, Z. Li, C. Zhang, L. Kong, B. Wang, S. Huang, V. Sharma, H. Ma, Q. Yuan, Y. Liu, G. Shen, K. Wu and L. Li, Chem. Sci., 2019, 10, 6683–6688 RSC.
- Y. C. Pu, H. C. Fan, J. C. Chang, Y. H. Chen and S. W. Tseng, J. Phys. Chem. Lett., 2021, 12, 7194–7200 CrossRef CAS PubMed.
- W. van der Stam, I. du Fosse, G. Grimaldi, J. O. V. Monchen, N. Kirkwood and A. J. Houtepen, Chem. Mater., 2018, 30, 8052–8061 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc04136k |
| This journal is © The Royal Society of Chemistry 2023 |