
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNickel-catalysed asymmetric hydromonofluoromethylation of 1,3-enynes for enantioselective construction of monofluoromethyl-tethered chiral allenes†
Ying
Zhang‡
a,
Jimin
Yang‡
b,
Yu-Long
Ruan‡
a,
Ling
Liao
a,
Chuang
Ma
a,
Xiao-Song
Xue
 *bd and
Jin-Sheng
Yu
*ac
*bd and
Jin-Sheng
Yu
*ac
aState Key Laboratory of Molecular & Process Engineering, Shanghai Engineering Research Center of Molecular Therapeutics and New Drug Development, School of Chemistry and Molecular Engineering, East China Normal University, Shanghai 200062, China. E-mail: jsyu@chem.ecnu.edu.cn
bKey Laboratory of Organofluorine Chemistry, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, Shanghai 200032, China. E-mail: xuexs@sioc.ac.cn
cHubei Key Laboratory of Quality Control of Characteristic Fruits and Vegetables, Hubei Engineering University, Xiaogan, 432000, China
dSchool of Chemistry and Materials Science, Hangzhou Institute for Advanced Study, University of Chinese Academy of Sciences, Hangzhou 310024, China
First published on 23rd October 2023
Abstract
An unprecedented nickel-catalysed enantioselective hydromonofluoromethylation of 1,3-enynes is developed, allowing the diverse access to monofluoromethyl-tethered axially chiral allenes, including the challenging deuterated monofluoromethyl (CD2F)-tethered ones that are otherwise inaccessible. It represents the first asymmetric 1,4-hydrofunctionalization of 1,3-enynes using low-cost asymmetric nickel catalysis, thus opening a new avenue for the activation of 1,3-enynes in reaction development. The utility is further verified by its broad substrate scope, good functionality tolerance, mild conditions, and diversified product elaborations toward other valuable fluorinated structures. Mechanistic experiments and DFT calculations provide insights into the reaction mechanism and the origin of the enantioselectivity.
Introduction
The increasing demand for organofluorine compounds in the discovery of new drugs, agrochemicals, and functional materials has aroused extensive interest in developing new synthetic methods to access structurally diverse fluorine-containing molecules.1 In particular, monofluoromethyl (CH2F)-containing chiral compounds are valuable, because the introduction of a CH2F group as a bioisosteric substitute for methyl, hydroxymethyl and other functionalities can often result in profound changes in the chemical and biological properties of parent molecules (Scheme 1a).2 Consequently, significant progress has been made in the selective synthesis of CH2F-containing chiral molecules in the recent decade.3 While much attention has been paid to the synthesis and application of CH2F-tethered stereocenters, the construction of CH2F-tethered axial chirality remains an unconquered challenge (Scheme 1b).3a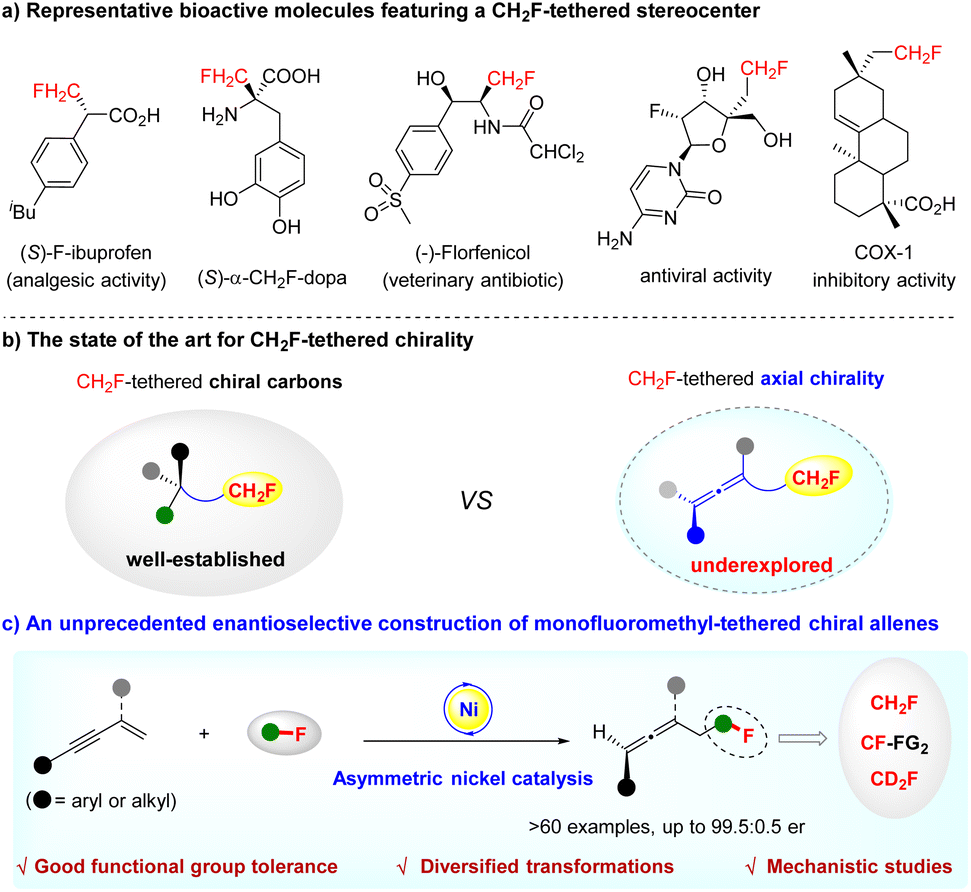 | ||
| Scheme 1 The state of the art for CH2F-tethered chirality and the current work. | ||
On the other hand, axial chirality represents a prominent structural motif in pharmaceuticals and bioactive natural products. Among them, chiral allenes are a particularly important type of axially chiral pharmacophore,4 as well as versatile building blocks in organic synthesis.5 The catalytic asymmetric synthesis of axially chiral allenes has accordingly attracted considerable attention over the past decade.6 Despite great achievements, the enantioselective construction of optically active fluorine-containing allenes is still largely underdeveloped.7 Therefore, the design and synthesis of diverse fluorinated chiral allenes, especially the challenging CH2F- or CD2F-tethered chiral allenes, represent a very important task.
Against this background, together with our research interest in selective fluoroalkylation of unsaturated hydrocarbons,8 we envisioned whether it would be possible to realize the access of CH2F-tethered chiral allenes via a regio- and enantio-selective hydromonofluoromethylation of 1,3-enynes9 by asymmetric nickel catalysis10 (Scheme 1c). Such research is, however, confronted with the following challenges: (i) the feasibility of a nickel catalyst to activate 1,3-enynes—nickel-catalysed hydrofunctionalization of 1,3-enynes has not yet been explored; (ii) not knowing how a nickel species initiates the reaction, and effects that control the enantioselectivity; (iii) the quest for a readily convertible fluoroalkylation reagent.
Herein, we successfully implemented this challenging transformation. We develop an unprecedented nickel-catalysed regio- and enantio-selective hydromonofluoromethylation of 1,3-enynes with fluorobis(phenylsulfonyl)methane (FBSM)11 that proves to be a robust monofluoromethyl reagent for installing a CH2F-tethered stereocenter.3a,12 It provides a facile and efficient route to axially chiral allenes featuring a CH2F or CD2F moiety that cannot be accessed by known methods (Scheme 1c). Notably, this constitutes the first application of an earth-abundant and low-cost redox-neutral asymmetric nickel catalysis for enabling the asymmetric 1,4-hydrofunctionalization of 1,3-enynes.
Results and discussion
Optimization of the reaction conditions
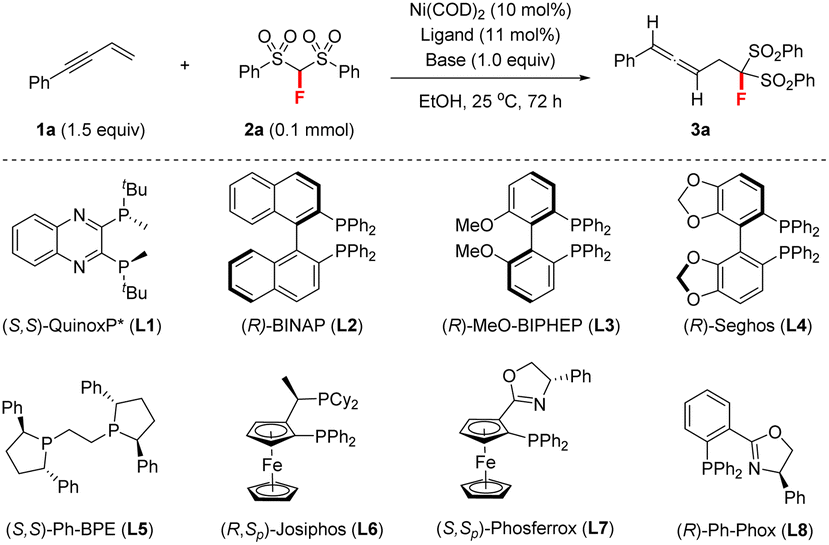
We initiated this study by investigating different chiral ligands for the model reaction of but-3-en-1-yn-1-ylbenzene 1a and FBSM 2a in EtOH at room temperature (rt) under Ni(COD)2 (10 mol%) catalysis, as shown in Table 1. P-Chiral ligand (S,S)-QuinoxP* (L1, 10 mol%) was first examined under base-free conditions; however, only a trace amount of the desired fluorine-containing allene 3a was detected, accompanied by the remainder of both starting materials (entry 1). We then proceeded to add a base, to enable the reaction. To our delight, we found that upon the addition of Et3N (1.0 equiv.) the reaction proceeded smoothly, affording allene 3a in 48% yield with 94![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :6 er (entry 2). Encouraged by this positive result, we then explored the performance of other chiral bisphosphine ligands and P,N-based PHOX ligands in the presence of Et3N (entries 3–9). The use of axially chiral (R)-MeO-BIPHEP (L3) greatly improved the reactivity and afforded 3a in 68% yield with 94.5:5.5 er (entry 4). Subsequently, an investigation into the solvent effect revealed that almost no product was observed when using a non-protonic solvent such as toluene or THF (entries 10 and 11). EtOH remained the best solvent (entry 4), although the use of other alcoholic solvents could give a similar reaction outcome (entries 12 and 13). Furthermore, organic bases, including DIPEA, DABCO, and quinuclidine, were investigated (entries 14–16). DABCO proved to be the optimal base in terms of reactivity and enantioselectivity (entry 15). Finally, we found that reducing the loading of DABCO or chiral nickel catalyst significantly decreased the yield of 3a, albeit without loss of enantioselectivity (entries 17–20).
:6 er (entry 2). Encouraged by this positive result, we then explored the performance of other chiral bisphosphine ligands and P,N-based PHOX ligands in the presence of Et3N (entries 3–9). The use of axially chiral (R)-MeO-BIPHEP (L3) greatly improved the reactivity and afforded 3a in 68% yield with 94.5:5.5 er (entry 4). Subsequently, an investigation into the solvent effect revealed that almost no product was observed when using a non-protonic solvent such as toluene or THF (entries 10 and 11). EtOH remained the best solvent (entry 4), although the use of other alcoholic solvents could give a similar reaction outcome (entries 12 and 13). Furthermore, organic bases, including DIPEA, DABCO, and quinuclidine, were investigated (entries 14–16). DABCO proved to be the optimal base in terms of reactivity and enantioselectivity (entry 15). Finally, we found that reducing the loading of DABCO or chiral nickel catalyst significantly decreased the yield of 3a, albeit without loss of enantioselectivity (entries 17–20).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Ligand | Base | Solvent | Yieldb (%) | erc |
| a Conditions: 1a (0.15 mmol), 2a (0.10 mmol), Ni(COD)2 (10 mol%), ligand (11 mol%), and base (1.0 equiv.), at 25 °C for 72 h in the indicated solvent (1.0 mL), unless otherwise noted. b Determined by 1H NMR analysis of the crude product using 1,3,5-trimethoxybenzene as the internal standard. c Determined by chiral HPLC analysis. d DABCO (50 mol%). e DABCO (20 mol%). f Ni(COD)2 (5 mol%) and L3 (5.5 mol%) was used. g Ni(COD)2 (1 mol%) and L3 (1.1 mol%) was used. | |||||
| 1 | L1 | — | EtOH | Trace | — |
| 2 | L1 | Et3N | EtOH | 48 | 94:6 |
| 3 | L2 | Et3N | EtOH | 57 | 86:14 |
| 4 | L3 | Et3N | EtOH | 68 | 94.5:5.5 |
| 5 | L4 | Et3N | EtOH | 40 | 94:6 |
| 6 | L5 | Et3N | EtOH | 38 | 95:5 |
| 7 | L6 | Et3N | EtOH | 15 | 78:22 |
| 8 | L7 | Et3N | EtOH | 26 | 93:7 |
| 9 | L8 | Et3N | EtOH | 12 | 81:19 |
| 10 | L3 | Et3N | Toluene | nr | — |
| 11 | L3 | Et3N | THF | nr | — |
| 12 | L3 | Et3N | MeOH | 68 | 93.5:6.5 |
| 13 | L3 | Et3N | iPrOH | 62 | 95.5:4.5 |
| 14 | L3 | DIPEA | EtOH | 73 | 94.5:5.5 |
| 15 | L3 | DABCO | EtOH | 93 | 96:4 |
| 16 | L3 | Quinuclidine | EtOH | 21 | 95:5 |
| 17d | L3 | DABCO | EtOH | 81 | 95:5 |
| 18e | L3 | DABCO | EtOH | 52 | 95:5 |
| 19f | L3 | DABCO | EtOH | 49 | 96:4 |
| 20g | L3 | DABCO | EtOH | 16 | 95:5 |
Evaluation of substrate scope
Having established the optimized reaction conditions, we then explored the scope of hydromonofluoromethylation between 1,3-enynes 1 and FBSM 2. The effect of the substituents on the aromatic ring of 1,3-enynes 1 was evaluated (see Scheme 2). 1,3-Enynes with electron-withdrawing and electron-donating groups were all suitable substrates, affording the corresponding products 3b–3q with up to 97:3 er. Notably, this transformation tolerated various functionalities on the phenyl ring of 1,3-enynes, such as halide (3b–3e), CF3 (3f), cyano (3g), ester (3h), formyl (3i), acetyl (3j), hydroxymethyl (3p), and diacetone fructose (3q). Slightly lower enantiomeric ratios were obtained for 2-naphthyl- and 3-thienyl-substituted 1,3-enynes (3r and 3s). The absolute configuration of 3s was determined to be (R) by its X–ray diffraction (XRD) analysis. Subsequently, (R) was assigned to all other disubstituted allenes 3 by analogy. Aliphatic 1,3-enynes with a cyclohexyl or phenylethyl group also afforded the targets 3t and 3u with moderate to high enantioselectivities, when using (S)-difluorphos (L9) or Josiphos SL-J009-1 (L10) as the ligand. Additionally, differently substituted FBSM 2 materials were compatible with the reaction conditions, giving rise to products 3v–3x in 82–98% yields with 95.5:4.5–96:4 er. Remarkably, 1,3-enynes featuring a methyl group at the 2-position were also tolerated.13 The corresponding fluorine-containing trisubstituted allenes 3aa–3ac were obtained with up to 94:6 er under the action of a 20 mol% (S,S)-BenzP* (L11) decorated chiral nickel catalyst at 60 °C. XRD analysis revealed that the absolute configuration of 3ac was (R) and that of products 3aa and 3ab was assigned by analogy.
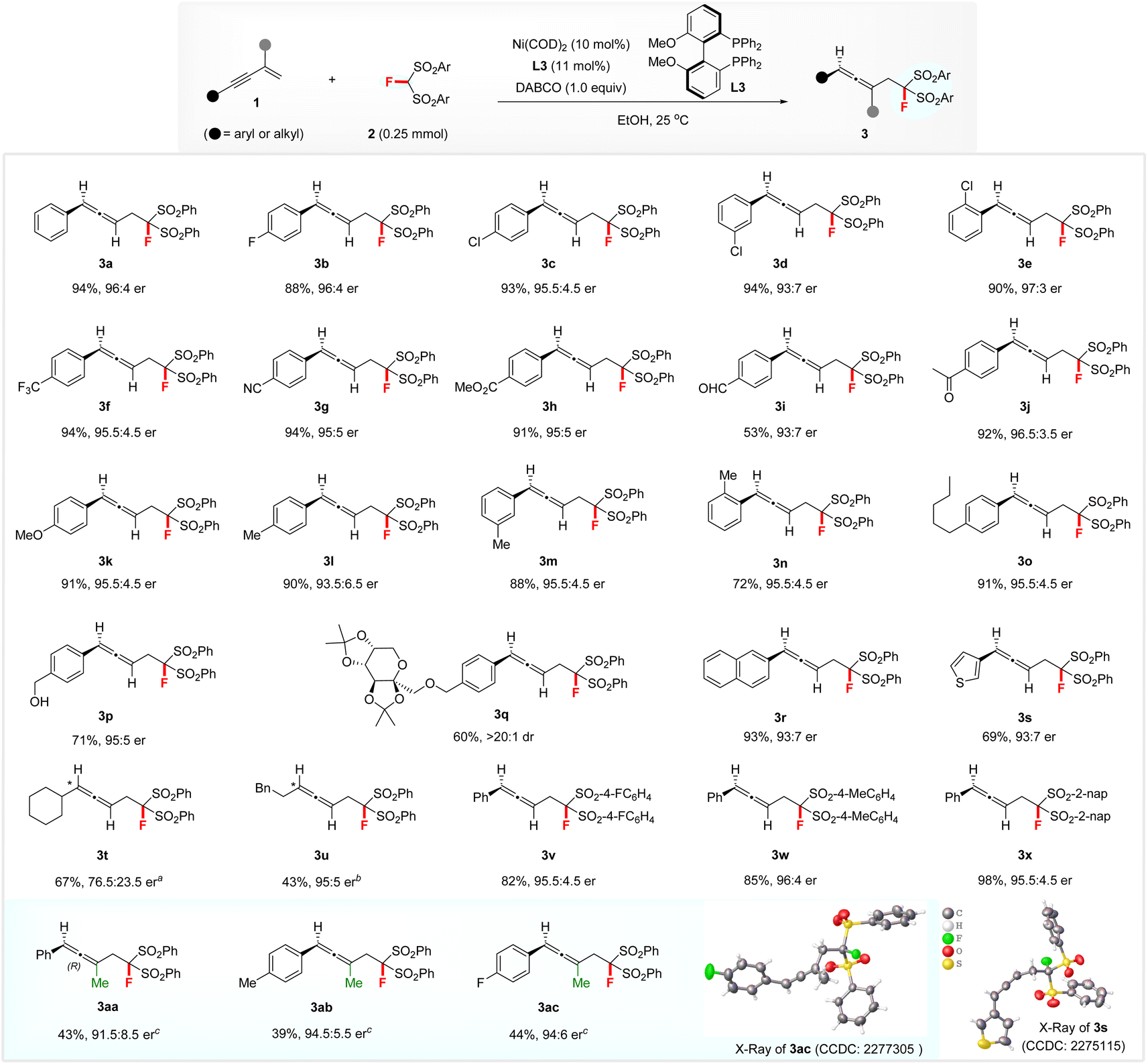 | ||
| Scheme 2 Scope of hydromonofluoromethylation of 1,3-enynes 1 with FBSM 2. Reaction conditions: 1 (0.375 mmol), 2 (0.25 mmol), Ni(COD)2 (10 mol%), L3 (11 mol%), and DABCO (1.0 equiv.), at 25 °C in EtOH (2.5 mL), unless otherwise noted. Yields of isolated products are reported. The er was determined by chiral HPLC analysis. a(S)-Difluorphos (L9) was used instead of L3, at 40 °C. bJosiphos SL-J009-1 (L10) was used instead of L3, at 40 °C. c(S,S)-BenzP* (L11) was used instead of L3, at 60 °C. | ||
To illustrate the generality of this nickel-catalysed enantioselective hydrofluoromethylation process, we next investigated the reaction of 1,3-enynes 1 with diethyl fluoromalonate 4 for the construction of functionalized chiral allenes featuring a fluorine atom and two convertible ester groups14 (Scheme 3). Gratifyingly, a variety of aryl 1,3-enynes 1 were amenable to the reaction under the above standard conditions, affording a variety of functionalized chiral allenes 5a–5u in good to excellent yields with 95:5–98:2 er. The position and nature of the substituents on the aryl group have no obvious influence on enantioselectivity and reactivity. The diacetone fructose-derived aryl 1,3-enyne appeared to also be tolerated, affording the product 5q in 94% yield with >20:1 dr. Both 2-naphthyl- and 1-naphthyl-substituted 1,3-enynes afforded the corresponding allenes 5s in 96% yield with 95:5 er and 5t in 83% yield with 95:5 er, respectively. Heteroaromatic 3-thienyl 1,3-enyne also delivered allene 5u with 95.5:4.5 er. In addition, cyclohexyl-substituted aliphatic 1,3-enyne was a viable substrate under the optimized conditions, affording the 1,3-dialkyl allene 5v with 87.5:12.5 er when employing L9 instead of L3 at elevated temperature. Notably, 2-methyl 1,3-enynes also reacted smoothly with 4 in the presence of 20 mol% (S,S)-BenzP* (L11)/Ni(COD)2 at 60 °C, affording the functionalized trisubstituted allenes 5aa with 97:3 er and 5ab with 99:1 er.
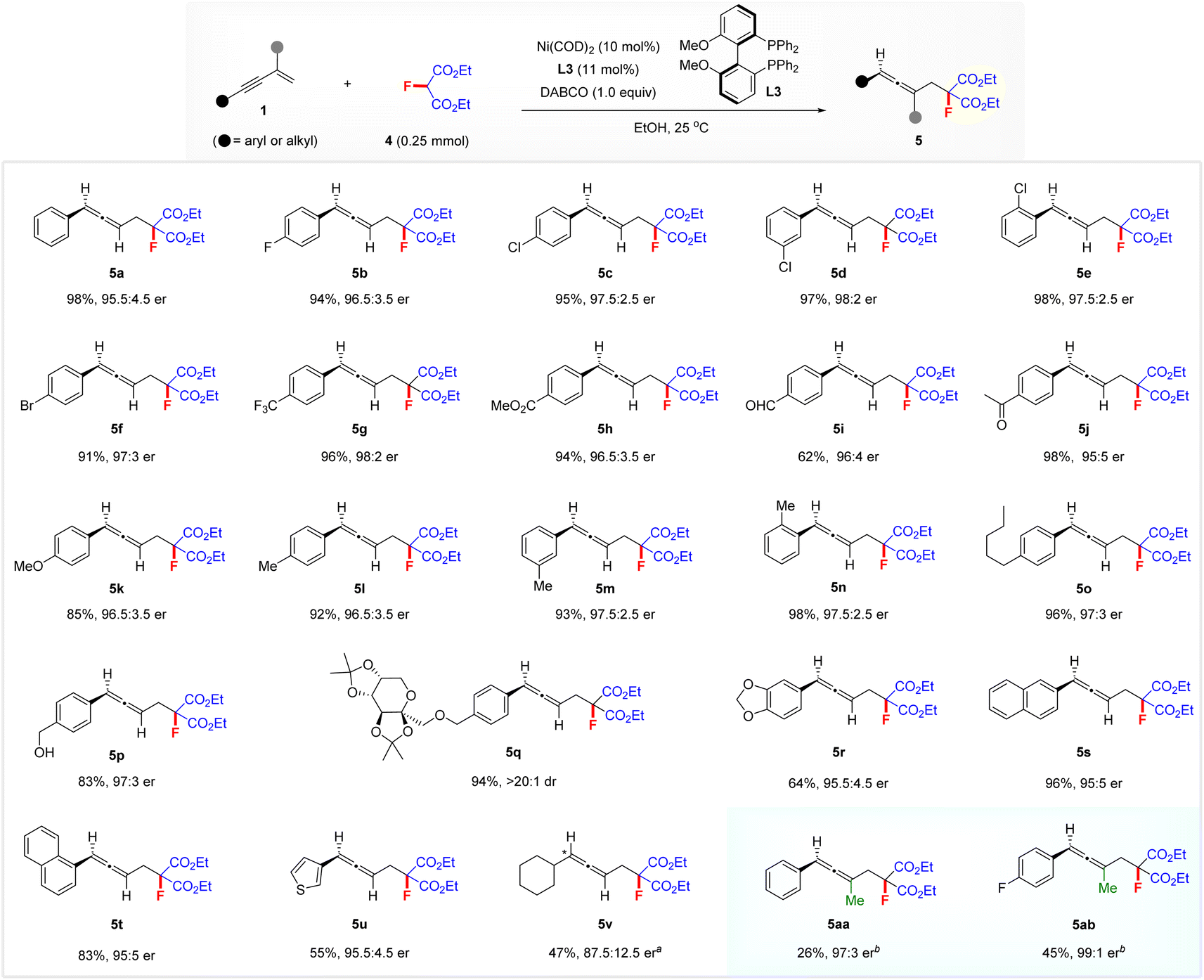 | ||
| Scheme 3 Scope of hydromonofluoroalkylation of 1,3-enynes 1 with diethyl fluoromalonate 4. Reaction conditions: 1 (0.375 mmol), 4 (0.25 mmol), Ni(COD)2 (10 mol%), L3 (11 mol%), and DABCO (1.0 equiv.), at 25 °C in EtOH (1.25 mL), unless otherwise noted. Yields of isolated products are reported. The er was determined by chiral HPLC analysis. aL9 was used instead of L3, at 40 °C. bL11 was used instead of L3, at 60 °C. | ||
Synthetic utility
The synthetic potential of this method was further highlighted by the direct synthesis of CH2F-or CD2F-tethered chiral allenes, gram-scale synthesis, and the diverse product transformations toward other fluorinated compounds, as depicted in Scheme 4. First, we directed our efforts toward the challenging CH2F- or CD2F-tethered chiral allenes by combining Ni-catalysed hydromonofluorobis(phenyl-sulfonyl)methylation with a sequential Mg-enabled reductive desulfonylation. Gratifyingly, treatment of the crude product of the hydrofluoroalkylation with Mg/MeOH smoothly afforded the desired CH2F-tethered chiral allenes 6a–6e with high to excellent enantioselectivity, while the corresponding CD2F-tethered chiral allenes D-6a–6e were obtained with up to 95.5:4.5 er when using CD3OD as the solvent in the desulfonylation step (Scheme 4a). Notably, the stereoselective installation of a CD2F group into the stereocenter is still a challenging task and remains underexplored, while the development of efficient protocols toward deuterated molecules is of current interest.15
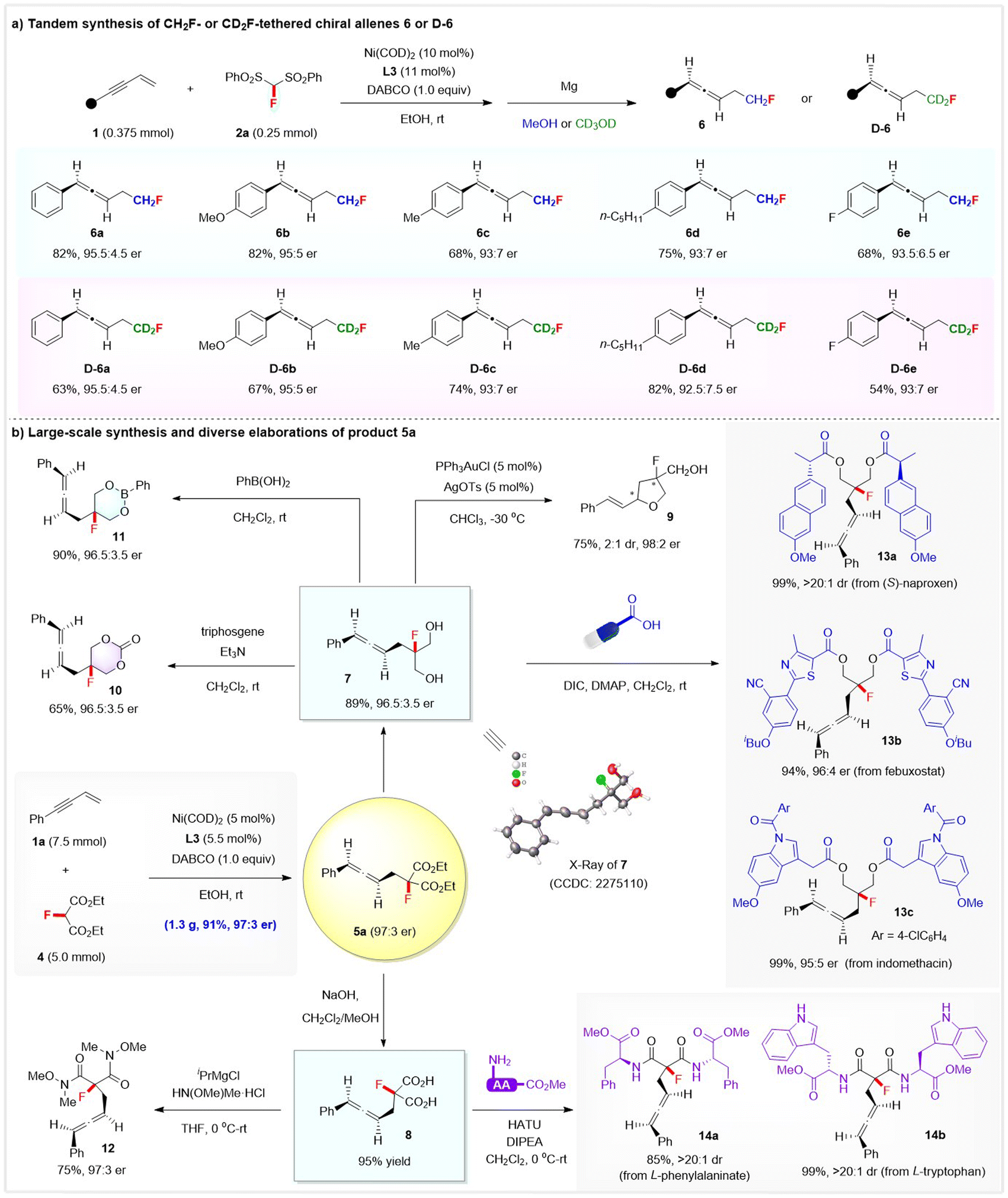 | ||
| Scheme 4 Synthetic utility. | ||
Furthermore, a large-scale reaction between 1a (7.5 mmol) and 4 (5.0 mmol) proceeded smoothly by simply using 5 mol% of chiral Ni complex, affording 1.3 g of allene 5a in 91% yield with 97:3 er. The product 5a was rich in functionality and useful as a versatile synthon to access other fluorine-containing molecules (Scheme 4b). The two ester functionalities in 5a were readily either reduced to form the chiral allene-tethered fluorinated diol 7 (89% yield and 96.5:3.5 er) or hydrolyzed to allene-substituted dicarboxylic acid 8 (95% yield). XRD analysis of diol 7 confirmed its absolute configuration to be (R), and thus the absolute configuration of the other allene products 5 could be assigned. Interestingly, allene diol 7 underwent a gold-catalysed cyclization via an axial-to-central chirality transfer process to deliver optically active fluorinated tetrahydrofuran 9 featuring 1,3-stereocenters—a key skeleton in many natural products and bioactive molecules such as the anti-HCV drug sofosbuvir. Chiral allene-containing fluorinated 1,3-dioxane-2-one 10 (65% yield and 96.5:3.5 er) or 1,3,2-dioxaborinane 11 (90% yield and 96.5:3.5 er) was readily obtained upon treatment of diol 7 with triphosgene or PhB(OH)2, respectively. Dicarboxylic acid 8 reacted well with HN(OMe)Me·HCl to afford the corresponding allene Weinreb amide 12 in 75% with 97:3 er. Moreover, enantioenriched allene diol 7 and acid 8 proved to be very useful synthons in the late-stage modification of drugs and amino acids, as exemplified by the diverse assembly of chiral allene-containing drug derivatives 13a–13c from (S)-naproxen, febuxostat, and indomethacin, as well as 14a–14b from methyl L-tryptophanate or L-phenylalaninate.
Mechanistic studies
To gain insight into the reaction mechanism, we first conducted a deuterium-labeling experiment (Scheme 5a). When performing the reaction of 1a and 2a in ethanol-d1 (EtOD) under standard conditions, we found that the deuterium label is confined to C1 in 3a (87% D-labeled); no deuterium incorporation was detected in the recovered enyne 1a (eqn (1)). Under the above conditions using EtOD, there was also no H–D scrambling in enyne 1a in the absence of 2a (eqn (2)). We therefore concluded that insertion of the alkyne into the nickel hydride (Ni–H) occurs selectively, and the protonation of 1a might not be accomplished by ethanol independently.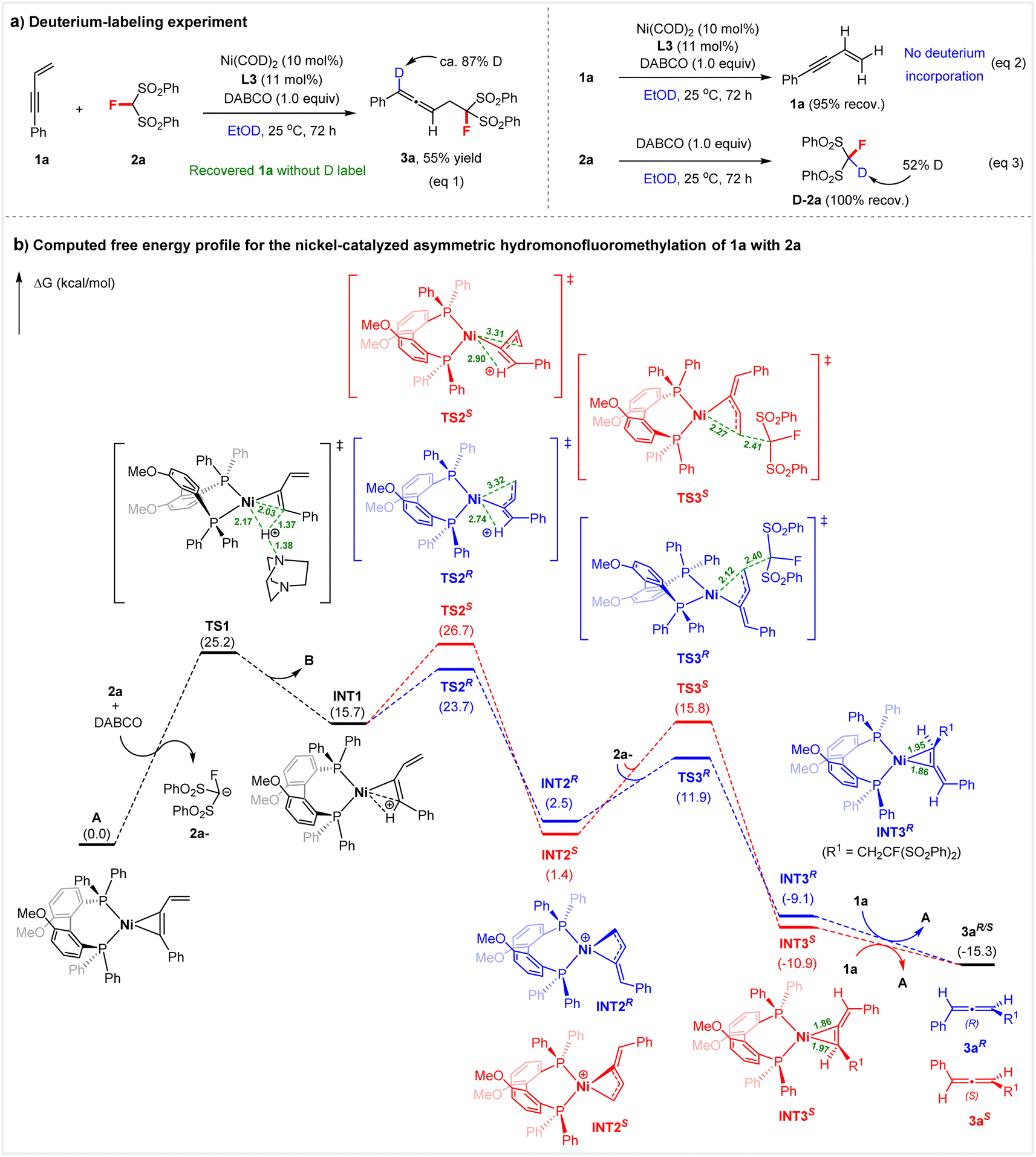 | ||
| Scheme 5 Mechanistic investigation. Free energy profile and selected geometric parameters of key species (distances in angstrom) at the PBE0-D3(BJ)/def2-TZVP/PCM(EtOH)//PBE0-D3(BJ)/def2-SVP level. | ||
Meanwhile, DFT calculations at the PBE0-D3(BJ)16a,b/def2-TZVP16c/PCM(EtOH)17//PBE0-D3(BJ)/def2-SVP16c level were performed to gain a better understanding of the mechanism and the origin of the enantioselectivity of the hydromonofluoromethylation of 1a with 2a (Scheme 5b). The catalytic cycle commences with the protonation of active species A (see Fig. S4† for more details on the generation of A from Ni(COD)2) by protonated DABCO (BH+), which is formed from DABCO (B) and 2a, giving η1-coordinated butadienyl–Ni complex INT1. This step needs to overcome a free energy barrier of 25.2 kcal mol−1. The observed H–D exchange of 2a with EtOD in the presence of DABCO supports the process (eqn (3), Scheme 5a).
On this basis, the mechanism for the generation of intermediate INT1via the activation of 1,3-enyne with the Ni complex is different from the traditional Pd–H species initiated enantioselective 1,3-enyne hydrofunctionalizations.9b,h Subsequently, isomerization of INT1 by rotation of the protonated enyne ligand occurs viaTS2 to give a more stable η3-butadienyl-Ni INT2, which is then attacked by the anion 2a− in an outer-sphere manner, to give the 3a ligated complex INT3. Finally, ligand exchange between 1a and 3a takes place to complete the catalytic cycle. The enantioselectivity-determining step is the isomerization of INT1, in which there are two directions for ligand rotation, corresponding to TS2S and TS2R, respectively. TS2S is 3.0 kcal mol−1 less stable than TS2R (26.7 kcal mol−1vs. 23.7 kcal mol−1). For the subsequent nucleophilic attack step, the R-conformation is more facile than the S-conformation (11.9 kcal mol−1 of TS3Rvs. 15.8 kcal mol−1 of TS3S). These results are consistent with experimental observations.
To shed more light on the origin of enantioselectivity, energy decomposition analysis18 towards TS2S and TS2R was carried out. In these transition states (TSs), the neutral Ni catalyst part is defined as fragment F1 and the protonated enyne moiety as fragment F2 (Scheme 6). The activation energy ΔE‡ of the transition states can be written as ΔE‡ = ΔEdef + ΔEint, where ΔEdef (deformation energy) is the energy difference that arises from structural changes toward the TS formation, and ΔEint (interaction energy) corresponds to the energy difference between the two fragments (F1 and F2) and the complex at the TS structure. The results indicate that although the interaction between the protonated enyne and Ni catalyst in TS2S is stronger than that in TS2R (ΔΔEint = −4.1 kcal mol−1), the deformation energy of TS2S is much higher than that of TS2R by 7.1 kcal mol−1, especially for the deformation of protonated enyne moiety F2, which governs the energy disparity between the two TSS. Besides, the smaller C1–C2–C3 angle in TS2S could also reflect the larger deformation of F2 (Scheme 6b). It is likely due to the repulsion between the phenyl groups of the chiral ligand and vinyl group of F2 with the nearest H⋯H distance of 2.29 Å in TS2S, while in TS2R the corresponding nearest H⋯H distance is significantly larger (2.46 Å). Such an effect could be the key factor causing the high enantioselectivity in this reaction.
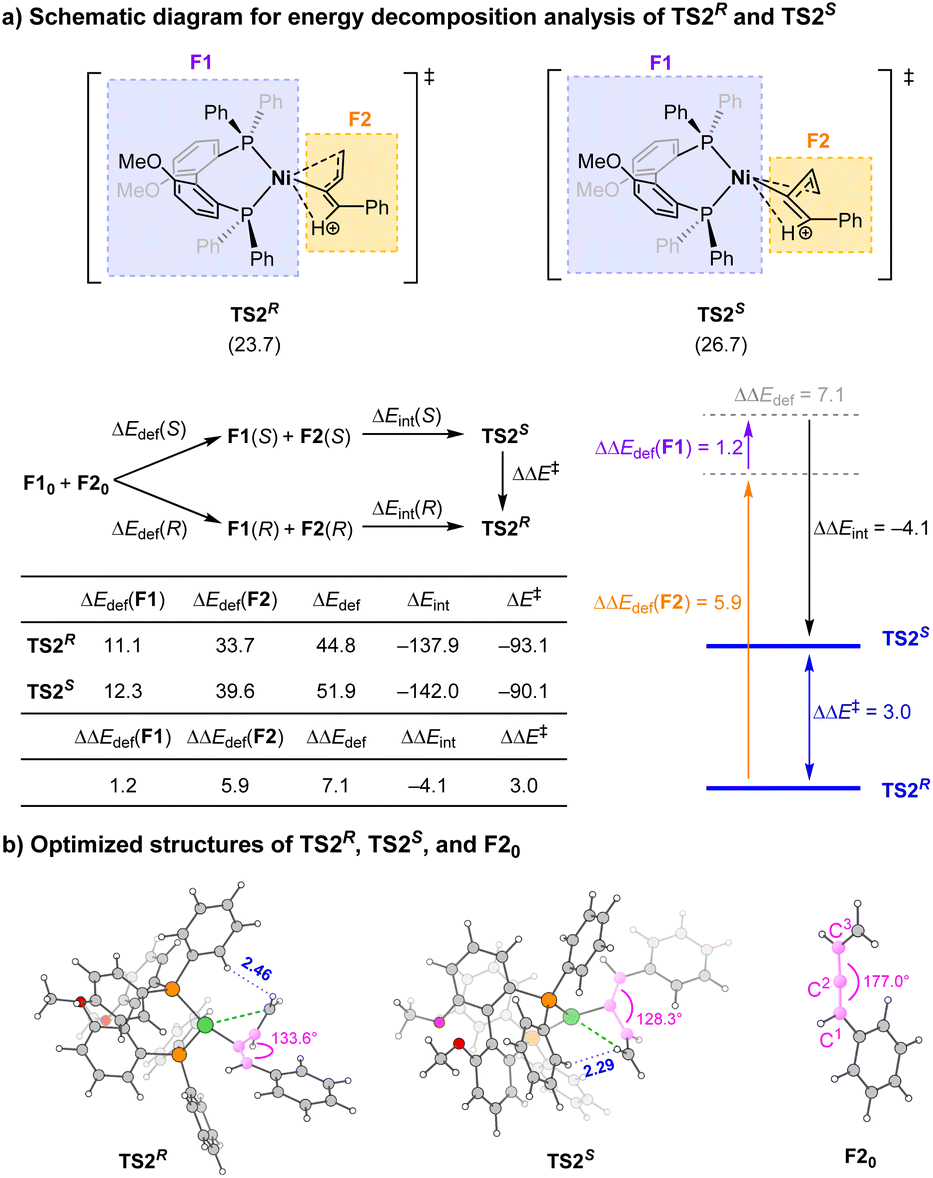 | ||
| Scheme 6 Energy decomposition analysis. Energy decomposition analysis of TS2R and TS2S at the PBE0-D3(BJ)/def2-TZVP/PCM(EtOH) level, and ΔΔEdef(F1) = ΔEdef(F1-S) − ΔEdef(F1-R) (energies in kcal mol−1); optimized structures and selected geometric parameters of TS2R, TS2S, and F20 (distances in angstrom). | ||
Conclusions
To summarize, we have established a regio- and enantio-selective hydromonofluoromethylation of 1,3-enynes with FBSM or diethyl fluoromalonate by redox-neutral asymmetric nickel catalysis, enabling us to access various monofluoromethylated chiral allenes, including the challenging CH2F- or CD2F-tethered ones. Moreover, the synthetic utility of this method is demonstrated by the following: the unprecedented preparation of CH2F- or CD2F-tethered chiral allenes, broad substrate scope, good functional group tolerance, and diverse product transformations to valuable functionalized fluorine-containing allenes or fluorinated tetrahydrofurans. The reaction mechanism and the origin of enantioselectivity were elucidated by DFT calculations and experimental studies. This also constitutes the first application of low-cost asymmetric nickel catalysis in activating 1,3-enynes for realizing asymmetric 1,4-hydrofunctionalizations, thus providing motivation for further developing enantioselective Ni-catalysed transformations of 1,3-enynes. The development of other regioselective hydrofluoroalkylation processes of 1,3-enynes by asymmetric nickel catalysis is currently ongoing in our laboratory.Data availability
All of the experimental data have been included in the ESI.† Crystallographic data can be obtained from the CCDC (2277305, 2275110 and 2275115).Author contributions
J.-S. Y. conceived the idea; Y. Z., Y.-L. R., L. L., and C. M. performed the experiments; J. Y. and X.-S. X. performed the DFT calculations; Y. Z. and Y.-L. R. collected and analyzed the data. J.-S. Y. and X.-S. X. directed the project and co-wrote the manuscript. All authors reviewed and edited the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Key Research and Development Program of China (2021YFC2102400), the National Natural Science Foundation of China (Grant No. 22171087 and 22122104, 22193012, and 21933004), the Shanghai Science and Technology Innovation Action Plan (21N41900500 and 20JC1416900), the Ministry of Education (PCSIRT) and the Fundamental Research Funds for the Central Universities. J.-S. Y. also acknowledges financial support from the “Zijiang Scholar Program” of East China Normal University (ECNU). We acknowledge Prof. Dr Jian Zhou at ECNU for his valuable discussion.Notes and references
-
(a) S. Purser, P. R. Moore, S. Swallow and V. Gouverneur, Chem. Soc. Rev., 2018, 37, 320–330 RSC
; (b) J. Wang, M. Sánchez-Roselló, J. Aceña, C. del Pozo, A. E. Sorochinsky, S. Fustero, V. A. Soloshonok and H. Liu, Chem. Rev., 2014, 114, 2432–2506 CrossRef CAS PubMed
-
(a) J. Kollonitsch, A. Patchett, S. Marburg, A. L. Maycock, L. M. Perkins, G. A. Doldouras, D. E. Duggan and S. D. Aster, Nature, 1978, 274, 906–908 CrossRef CAS PubMed
- For leading reviews:
(a) M. Reichel and K. Karaghiosoff, Angew. Chem., Int. Ed., 2020, 59, 12268–12281 CrossRef CAS PubMed
- A. Hoffmann-Röder and N. Krause, Angew. Chem., Int. Ed., 2004, 43, 1196–1216 CrossRef PubMed
-
(a) S. Ma, Acc. Chem. Res., 2009, 42, 1679–1688 CrossRef CAS PubMed
-
(a)
Axially Chiral Compounds: Asymmetric Synthesis and Applications, ed. B. Tan, Wiley-VCH GmbH, 2021, ch. 6, p. 141 Search PubMed
- Known examples for the construction of fluorinated chiral allenes:
(a) T. J. O'Connor, B. K. Mai, J. Nafie, P. Liu and F. D. Toste, J. Am. Chem. Soc., 2021, 143, 13759–13768 CrossRef PubMed
-
(a) J.-S. Yu, F.-M. Liao, W.-M. Gao, K. Liao, R.-L. Zuo and J. Zhou, Angew. Chem., Int. Ed., 2015, 54, 7381–7385 CrossRef CAS PubMed
- For typical examples on the asymmetric functionalization of 1,3-enynes:
(a) J.-W. Han, N. Tokunaga and T. Hayashi, J. Am. Chem. Soc., 2001, 123, 12915–12916 CrossRef CAS PubMed
-
(a) C. L. Drennan, Nat. Chem., 2010, 2, 900 CrossRef CAS PubMed
- For the seminal work regarding FBSM:
(a) T. Fukuzumi, N. Shibata, M. Sugiura, H. Yasui, S. Nakamura and T. Toru, Angew. Chem., Int. Ed., 2006, 45, 4973–4977 CrossRef CAS PubMed
- For a review:
(a) A.-N. R. Alba, X. Companyo and R. Rios, Chem. Soc. Rev., 2010, 39, 2018–2033 RSC
- We thank one referee for suggesting the examination of the result for 1-substituted 1,3-enynes. It was found that no desired product was observed when subjecting pent-3-en-1-yn-1-ylbenzene to the reaction with FBSM 2a under the standard conditions, accompanied by the recovery of 2a with >95%.
- P. Xu and Z. Huang, Nat. Chem., 2021, 13, 634–642 CrossRef CAS PubMed
-
(a) C. Schmidt, Nat. Biotechnol., 2017, 35, 493–494 CrossRef CAS PubMed
-
(a) C. Adamo and V. Barone, J. Chem. Phys., 1999, 110, 6158–6170 CrossRef CAS
-
(a) S. Miertuš, E. Scrocco and J. Tomasi, Chem. Phys., 1981, 55, 117–129 CrossRef
- F. M. Bickelhaupt and K. N. Houk, Angew. Chem., Int. Ed., 2017, 56, 10070–10086 CrossRef CAS PubMed
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2275110, 2275115 and 2277305. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc04474b |
| ‡ Y. Z., J. Y. and Y.-L. R. contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |