
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFrom non-conductive MOF to proton-conducting metal-HOFs: a new class of reversible transformations induced by solvent-free mechanochemistry†
Magdalena
Lupa-Myszkowska
 ab,
Marcin
Oszajca
a and
Dariusz
Matoga
*a
ab,
Marcin
Oszajca
a and
Dariusz
Matoga
*a
aFaculty of Chemistry, Jagiellonian University, Gronostajowa 2, 30-387 Kraków, Poland. E-mail: dariusz.matoga@uj.edu.pl
bDoctoral School of Exact and Natural Sciences, Jagiellonian University, ul. prof. S. Łojasiewicza 11, 30-348 Kraków, Poland
First published on 27th November 2023
Abstract
Proton-conducting materials play an important role as solid electrolytes in electrochemical devices for energy storage and conversion, including proton exchange membrane fuel cells. Metal–organic frameworks (MOFs), covalent-organic frameworks (COFs) and more recently hydrogen-bonded organic frameworks (HOFs) have emerged as useful crystalline platforms for proton transport that provide high conductivity and enable insight into conduction pathways. Here, we present two new HOFs with high conductivity, reaching 2 × 10−2 S cm−1 at 60 °C and 75% relative humidity, obtained in reactions that represent a new class of reversible transformations of solids. The reactions are induced by solvent-free mechanochemistry and involve breaking of coordination linkages in a MOF and formation of extended hydrogen-bonded networks of metal-HOFs (MHOFs). This unprecedented class of MOF-to-MHOF transformations has been demonstrated using a non-conductive MOF (JUK-1) and formamidinium or methylammonium thiocyanates as solid reactants. Structural details of the solid-state reactions are revealed by powder X-ray diffraction and Rietveld refinements for the MHOF products. None of the attempts using conventional methods were successful in obtaining the MHOFs, emphasizing a unique role of mechanochemical stimuli in the reactivity of supramolecular polymer solids, including crystalline MOFs and HOFs. The reversible nature of non-covalent interactions in such materials may be utilized for the development of healable polymer systems.
Introduction
Hydrogen-bonded organic frameworks (HOFs) represent a novel class of crystalline, functional porous structures that self-assemble through intermolecular hydrogen-bonding interactions from a multitude of organic building blocks or metal–organic building blocks,1,2 the latter case leading to a subclass of metal hydrogen-bonded organic frameworks (MHOFs).3 Whereas a number of H-bonding motifs were successfully used for constructing porous HOFs,4 the nature of relatively weak, reversible and flexible hydrogen bonds often entails the presence of polymorphs, which complicates synthesis, characterization and reproducibility.5–7 Despite these challenges, however, well-defined HOFs were recently demonstrated in a variety of applications,8 such as gas adsorption,9–11 sensing,12–16 catalysis,17–20 biomedicine,21,22 and proton conduction.23–27 Proton-conducting materials are of tremendous importance for renewable energy technologies, including fuel cells and proton exchange membrane-based devices. The inherent well-defined H-bonded networks, porosity, and tunability of HOFs make them excellent solid-state candidates for efficient water-mediated high superprotonic conduction.28,29 In other related open framework materials such as metal–organic frameworks (MOFs) and covalent organic frameworks (COFs), in addition to proper backbone functionalization, high proton conductivity has been achieved by introducing pore-filling Brønsted acid centres (molecules or ions), but examples of such HOFs remain uncommon.29–35Chemical reactions induced by mechanical force, have emerged as a promising and sustainable method for developing new materials.36–43 This new approach in synthetic chemistry has shown tremendous potential in revolutionizing traditional synthesis methods with energy-efficient, simple, low-temperature, and highly economical processes.44–51 Mechanochemical reactions can be performed in solvent-free conditions, decreasing environmental impact and providing access to unique reaction pathways that are often inaccessible by conventional methods.30,34,35,52,53 In particular, future perspectives of mechanochemistry in the synthesis and fine-tuning of functional HOFs, including proton-conducting ones, are also promising.54 Additionally, mechanochemistry offers the capability to access elusive polymorphs and increase the reproducibility of HOFs production, responding to one of the crucial challenges facing the field.7
In this work, we provide an accessible strategy for postsynthetic conversion of a metal–organic framework (MOF) into metal hydrogen-bonded organic frameworks (MHOFs) with the simultaneous introduction of proton conduction by solvent-free mechanochemistry. We demonstrate that grinding the layered metal–organic framework JUK-1 ({[Mn2(ina)4(H2O)2]·2EtOH}n)55 with methylammonium (MA) or formamidinium (FA) thiocyanate leads to the formation of highly proton-conducting metal hydrogen-bonded organic frameworks. In comparison with our previous research covering the inter-MOF transformation from JUK-1 to JUK-2 that contains ammonium (AM) cations ({(AM)2[Mn(ina)2(NCS)2]}n·xH2O),30,52 the mechanochemical reactions carried out in this work lead to the cleavage of coordination bonds between COO− and manganese, and the stabilization of the resulting framework by hydrogen bonding. The crystal structures of the MHOFs, determined by applying Rietveld refinement, as well as proton conductive properties of the MHOFs, elucidated by electrochemical impedance spectroscopy (EIS), are compared and discussed. A postsynthetic modification using ionic compounds demonstrates for the first time the capability to obtain metal hydrogen-bonded organic frameworks from a metal–organic framework by mechanochemical approach. Reverse incorporations of metal ions into suspensions of HOF solids that led to the formation of MOFs or 1D coordination polymers have been recently reported in the literature.56,57
Results and discussion
Postsynthetic modification and physicochemical characterization
Postsynthetic modifications of a layered JUK-1 with methylammonium (MA) or formamidinium (FA) thiocyanates were performed, resulting in two HOFs with coordinated NCS− ligands and extra-framework cations as proton carriers: (MA)2[Mn(ina)2(NCS)2(H2O)2] and (FA)2[Mn(ina)2(NCS)2(H2O)2], denoted Mn-HOF-MA and Mn-HOF-FA, respectively. Both syntheses involved fast solvent-free grinding of JUK-1 with an appropriate thiocyanate salt (see the ESI for details†). The formation of new crystalline Mn-HOFs phases can be detected by powder X-ray diffraction (PXRD) and infrared (IR) spectroscopy. For both reactions, stoichiometry (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2; metal:thiocyanate ratio) and the evidence of thiocyanate coordination are provided by IR band shift of the strong ν(CN) band from 2059 for the initial FASCN to 2098 cm−1 (Mn-HOF-FA), whereas for Mn-HOF-MA the band at 2045 cm−1 (for the initial MASCN) shifts to 2078 cm−1 (Fig. 1 and S2–S9, ESI†). The reactions carried out at higher stoichiometries of thiocyanate salts result in mixtures of uncoordinated thiocyanates and Mn-HOFs (observable by the appearance of extra X-ray reflections of MASCN/FASCN or extra ν(CN) IR bands). On the other hand, the use of lower stoichiometric ratios leads to mixtures of unreacted JUK-1 and Mn-HOFs due to a deficiency of salts.
:2; metal:thiocyanate ratio) and the evidence of thiocyanate coordination are provided by IR band shift of the strong ν(CN) band from 2059 for the initial FASCN to 2098 cm−1 (Mn-HOF-FA), whereas for Mn-HOF-MA the band at 2045 cm−1 (for the initial MASCN) shifts to 2078 cm−1 (Fig. 1 and S2–S9, ESI†). The reactions carried out at higher stoichiometries of thiocyanate salts result in mixtures of uncoordinated thiocyanates and Mn-HOFs (observable by the appearance of extra X-ray reflections of MASCN/FASCN or extra ν(CN) IR bands). On the other hand, the use of lower stoichiometric ratios leads to mixtures of unreacted JUK-1 and Mn-HOFs due to a deficiency of salts.
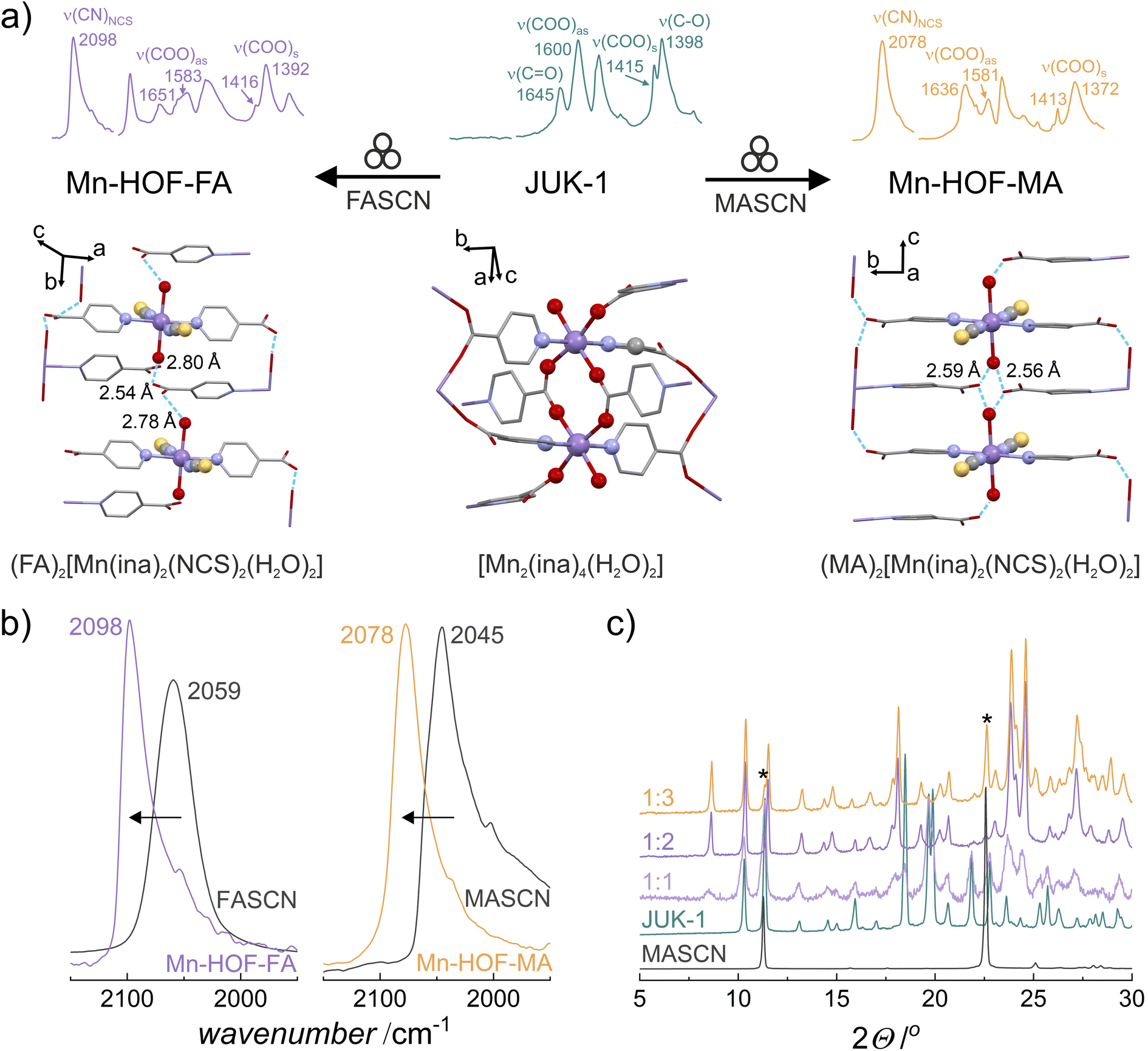 | ||
| Fig. 1 Solid-state reaction of JUK-1 with CH(NH2)2SCN (FASCN) or CH3NH3SCN (MASCN): (a) general reaction scheme for the mechanochemical synthesis of Mn-HOFs. IR spectra in the 2150–1300 cm−1 range with characteristic wavenumbers in cm−1 are given for comparison – top. Structural details of molecular rearrangement inside the coordination region, cations omitted for clarity (H atoms omitted, Mn – purple, O – red, N – blue, S – yellow, C – grey) – bottom. (b) FT-IR spectra of Mn-HOFs with characteristic band shift of the strong ν(CN) band. Numbers indicate wavenumbers (in cm−1) of the CN stretches. (c) PXRD patterns of mixtures after grinding JUK-1 with various amounts of MASCN; 1:1, 1:2 (Mn-HOF-MA) and 1:3 (given as JUK-1 to MASCN stoichiometric ratio), characteristic reflections of MASCN are labeled with “*”. | ||
For verification of potential Mn-HOFs formation in solution, JUK-1 was immersed in saturated solutions of FASCN or MASCN in ethanol – the IR measurements showed the absence of CN bands (see the ESI for details, Fig. S10†). Additionally, the de novo synthetic trials to obtain Mn-HOFs were also performed (see the ESI for details, Fig. S11 and S12†). None of these attempts succeeded in obtaining modified frameworks, confirming the necessity of mechanochemical grinding. Significantly, the solid-state modifications are fully reversible, with both Mn-HOF-FA and Mn-HOF-MA returning to JUK-1 after immersion in ethanol (see the ESI for details, Fig. S13†).
Structural details of solid-state reactions
Using X-ray diffraction on polycrystalline samples followed by the Rietveld refinement allowed the structural elucidation of Mn-HOF-MA and Mn-HOF-FA (Fig. S15 and S16, ESI†). The solid-state reaction of JUK-1 with appropriate thiocyanate salts includes the coordination of two thiocyanates and two labile aqua ligands per one manganese centre (with accompanying conversion of dinuclear Mn2 to mononuclear Mn nodes) as well as the breaking of coordination bonds of the COO− ions with manganese, and stabilization of the resulting discrete complexes via hydrogen bonding (Fig. 1 and 2). Considering the formation of Mn-HOF-MA, a comparable, yet significantly different situation occurred in our previous work when NH4SCN was used instead of MASCN and a new coordination polymer (JUK-2) was formed. Here, in contrast, as a result of 2D MOF to 3D MHOF structural transformation (Fig. S20, ESI†), isonicotinates are no longer coordination linkers and together with new aqua ligands become involved in strong hydrogen bonds forming a new extended network (donor–acceptor distances for O–H⋯O are approx. 2.6 Å; Fig. S18 and Table S3†). Additionally, extraframework CH3NH3+ ions are stabilized by hydrogen bonds with thiocyanate (N–H⋯S) and carboxylate (N–H⋯O), in which donor–acceptor distances are approx. 3.5 Å and 2.6 Å, respectively (Fig. S19†). Per analogy with JUK-2, the Mn-HOF-MA system can also be viewed as consisting of hydrogen-bonded layers (based on O–H⋯O bonds) that are further stabilized by the presence of MA cations that are involved in hydrogen bonding with adjacent layers (via N–H⋯S and N–H⋯O bonds) with the inter-layer separation of 10.5 Å. In contrast, despite the same type of hydrogen bonds, such a clear hierarchical distinction between intra- and inter-hydrogen-bonded layers cannot be made for Mn-HOF-FA, in which these hydrogen bonds interconnect FA cations and complex [Mn(ina)2(NCS)2(H2O)2]2− ions making up for 3D MHOF. Carboxylates engage with aqua ligands from adjacent complexes with donor–acceptor distances (O–H⋯O) of approx. 2.5, 2.8 and 2.9 Å (Fig. S22 and S23 and Table S5, ESI†), and additionally, the extraframework CH(NH2)2+ ions are stabilized by hydrogen bonds with thiocyanates (N–H⋯S, D⋯A distances 3.2 Å and 3.4 Å) and carboxylates (N–H⋯O, D⋯A distances 2.7 Å and 2.9 Å).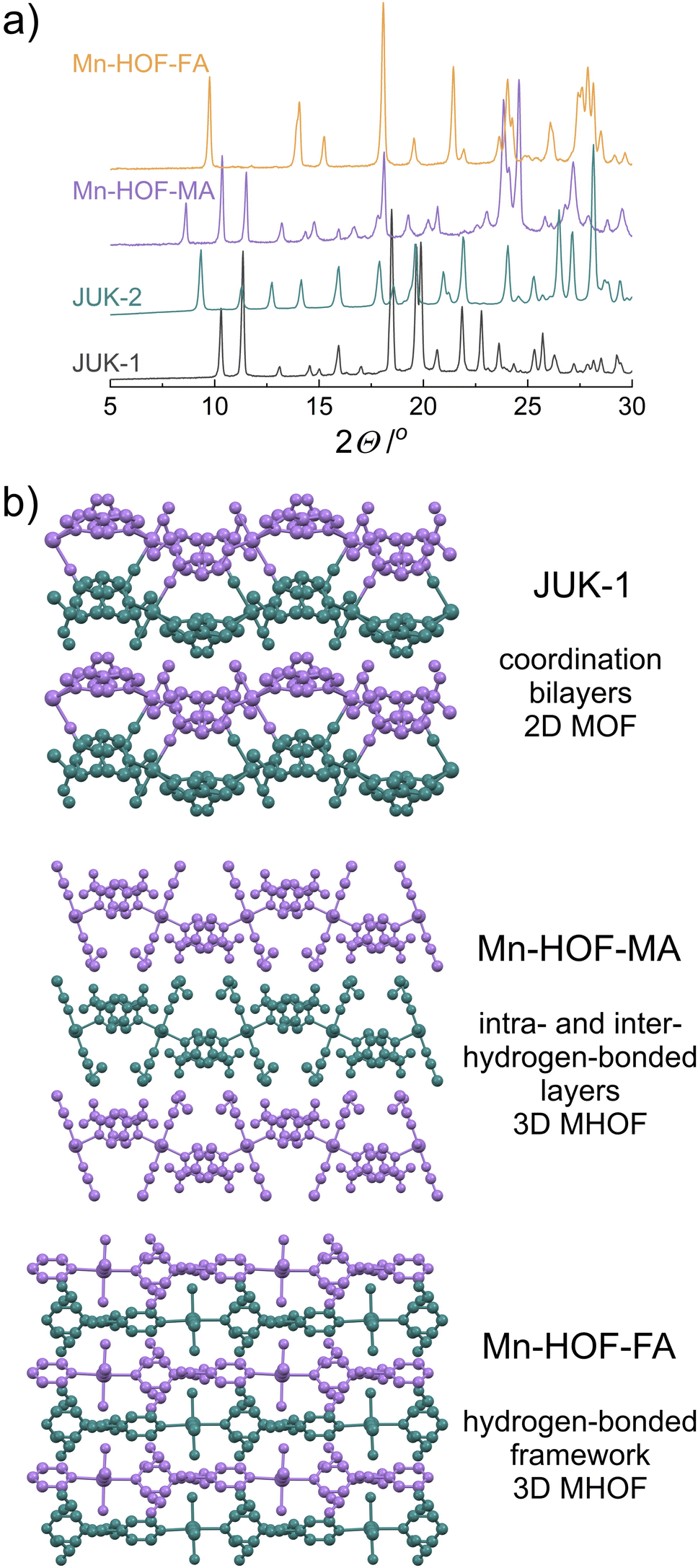 | ||
| Fig. 2 Structural transformation between JUK-1 and Mn-HOFs. (a) PXRD patterns for JUK-1 and Mn-HOFs, compared with related JUK-2. (b) Stacked coordination bilayers in the initial JUK-1 (top), and the resulting: stacked hydrogen-bonded layers in Mn-HOF-MA (middle) and 3D hydrogen-bonded network in Mn-HOF-FA (bottom), all packing views along the c axes. The layers in JUK-1 and Mn-HOF-MA are stabilized by interlayer hydrogen bonds. | ||
The confirmation of the structural transformations associated with thiocyanate complexation is also provided by IR spectra (Fig. 1), showing not only a clear shift of the CN band of used salts but also a change in the vibrations of carboxylates. Strong hydrogen bonds in Mn-HOFs significantly affect the oscillations of carboxylates, and despite the absence of coordination bonds between COO− and manganese, the bands corresponding to asymmetric and symmetric vibrations are observable at 1636 and 1581 cm−1 – ν(COO)as and at 1413 and 1372 cm−1 – ν(COO)s for Mn-HOF-MA and at 1651 and 1583 cm−1 – ν(COO)as and at 1416 and 1392 cm−1 – ν(COO)s for Mn-HOF-FA (Fig. S9, ESI†).
Proton conductive properties
Our previous work has shown the effectiveness of introducing appropriate thiocyanate salts (NH4SCN, MASCN and FASCN) to induce proton conduction in various porous frameworks. Successful modifications of both rigid CPO-27 3D MOFs and flexible JUK-1 2D MOFs resulted in high proton conductivity of CPO-27-NCS3,4 and JUK-2.30 Here we show an unprecedented effect of mechanochemical stoichiometric modification in the solid state of a flexible metal–organic framework to obtain proton-conducting metal hydrogen-bonded organic frameworks. For comparison, electrochemical impedance spectroscopy (EIS) measurements were performed for pelletized samples of Mn-HOFs and JUK-2 at variable temperatures (from 25 to 60 °C) and different relative humidity (RH) values (from 30 to 75%) over the frequency range of 4 Hz to 5 MHz (Fig. 3, S27–S30 and Table S6, ESI†). The highest proton conductivity, reaching 2 × 10−2 S cm−1 at 60 °C and 75% RH was observed for Mn-HOF-FA. Due to deliquescent nature, the EIS measurements of Mn-HOF-MA and JUK-2 were not available at 75% RH, as indicated by water adsorption studies. At lower 60% RH, these materials show conductivity of the same value, reaching nearly 10−3 S cm−1 at 60 °C. In line with earlier work, it was also found that relative humidity strongly affects conductivity values, which indicates that ions are the primary carriers in the charge transport observed. Representative equivalent circuit modelling carried out for AC impedance plots at 30% RH and various temperatures for Mn-HOF-FA indicates the presence of bulk resistance, grain boundary resistance and electrode/electrolyte interface in the studied sample (Fig. 3).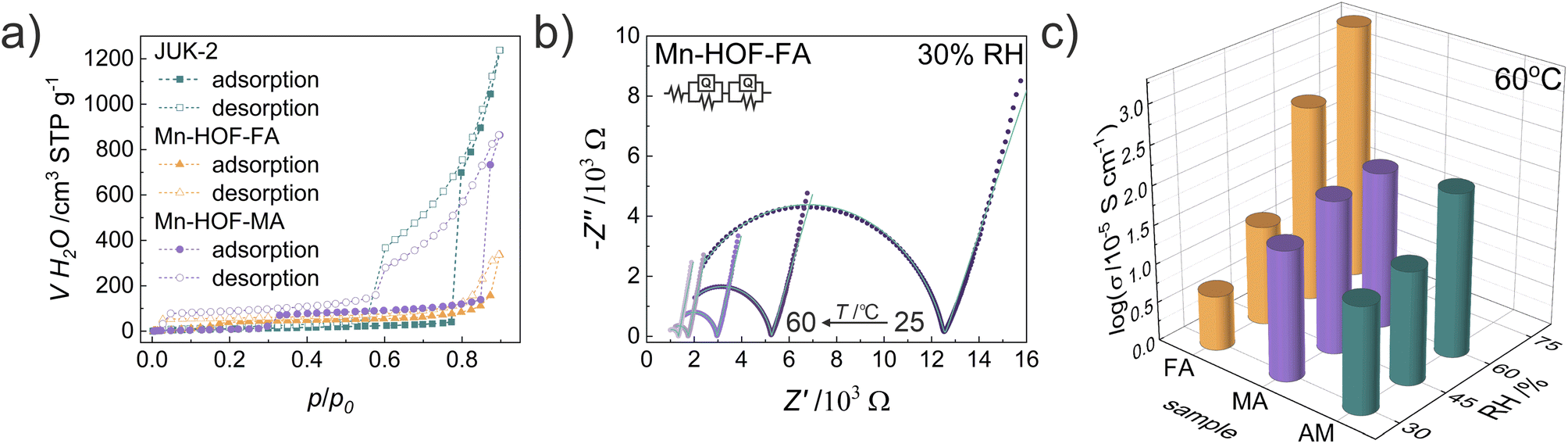 | ||
| Fig. 3 Humidity-dependent proton conduction for Mn-HOFs, compared with JUK-2. (a) Water vapour adsorption at 293 K. (b) Representative temperature-dependent AC impedance plots for Mn-HOF-FA at 30% RH with equivalent circuit used for fitting. (c) Proton conductivities at 60 °C and different RH values (Mn-HOF-FA, Mn-HOF-MA and JUK-2 abbreviated FA, MA and AM, respectively). | ||
The Arrhenius plots interpretation provided activation energies (Ea) for proton transport in Mn-HOFs. The activation energies mainly indicate a humidity-independent vehicle mechanism involving diffusion of CH(NH2)2+ and CH3NH3+ (Fig. S31, ESI†). At lower temperatures (25–30 °C), Ea is higher, between 1.4 and 3.5 eV, while in the range of 40–60 °C it is generally below 1 eV. These two different slopes suggest the occurrence of MHOF phase transitions under pressure, temperature and humidity applied during the EIS measurements, which is likely, given the fact that the MHOFs are based on flexible low-energy hydrogen bonds. The tendency of Mn-HOFs to undergo phase transitions is also clearly pronounced in their variable-temperature PXRD patterns (Fig. S25 and S26, ESI†). PXRD patterns recorded for both Mn-HOFs directly after EIS measurements at 60 °C at 60–75% RH also show the presence of new reflections (Fig. S33 and S34, ESI†), which on the other hand practically disappear after the water vapor adsorption–desorption cycle is performed on the samples at room temperature (Fig. S44, ESI†). This behaviour indicates the return to initial crystalline phases. The comparison of IR spectra of the as-synthesized Mn-HOFs with those after the EIS measurements at elevated temperature and humidity show no changes which indicates that the observed changes in the PXRD patterns are not associated with any coordination bond rearrangements. The water vapor adsorption measurements recorded for the samples after the impedance measurements at elevated temperature and humidity are identical with those observed for the as-synthesized samples (Fig. S43, ESI†). Additionally, SEM-EDX imaging also confirm the stability of these materials after grinding and proton conductivity measurements, with the retainment of crystallite morphology and homogeneous dispersion of elements (Fig. S45 and S46, ESI†). Similarly, under the measurement conditions (i.e., 25–60 °C), the Mn-HOFs are thermally stable, and no crystal phase transitions are observed in this temperature range (Fig. S24–S26, ESI†) under ambient pressure and humidity.
Conclusions
In summary, our work demonstrates a new class of reversible transformations of solids that involve the decomposition of metal–organic frameworks into metal hydrogen-bonded frameworks. Noteworthy, the breaking of extended network of coordination bonds and formation of extended network of hydrogen bonds is induced by ecological and economical solvent-free mechanochemistry and it leads to functionalization of the system. Our approach to functionalization through disintegration of coordination linkages can be applied to other flexible and reactive systems based on coordination bonds. Moreover, the reversibility of non-covalent interactions in framework materials, such as MOFs and HOFs, can be utilized in the future for the development of new healable polymer systems as well as switchable and responsive devices.Data availability
Crystallographic data for Mn-HOF-FA and Mn-HOF-MA has been deposited at the CCDC under 2288811 and 2288812, respectively, and can be obtained from https://www.ccdc.cam.ac.uk/structures/. The datasets supporting this article have been uploaded as part of the ESI.†Author contributions
Magdalena Lupa-Myszkowska: investigation, formal analysis, visualization, writing – original draft preparation. Marcin Oszajca: investigation (crystal structure determination and refinement). Dariusz Matoga: conceptualization, formal analysis, writing – original draft preparation, writing – review and editing, supervision, project administration, funding acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The National Science Centre (NCN, Poland) is gratefully acknowledged for the financial support (Grant no. 2019/35/B/ST5/01067).Notes and references
- I. Hisaki, C. Xin, K. Takahashi and T. Nakamura, Angew. Chem., Int. Ed., 2019, 58, 11160–11170 CrossRef CAS PubMed
.
- L. Chen, B. Zhang, L. Chen, H. Liu, Y. Hu and S. Qiao, Mater. Adv., 2022, 3, 3680–3708 RSC
- R.-B. Lin, Y. He, P. Li, H. Wang, W. Zhou and B. Chen, Chem. Soc. Rev., 2019, 48, 1362–1389 RSC
- B. Wang, R.-B. Lin, Z. Zhang, S. Xiang and B. Chen, J. Am. Chem. Soc., 2020, 142, 14399–14416 CrossRef CAS PubMed
- C. A. Zentner, H. W. H. Lai, J. T. Greenfield, R. A. Wiscons, M. Zeller, C. F. Campana, O. Talu, S. A. FitzGerald and J. L. C. Rowsell, Chem. Commun., 2015, 51, 11642–11645 RSC
- D. V. Horváth, T. Holczbauer, L. Bereczki, R. Palkó, N. V. May, T. Soós and P. Bombicz, CrystEngComm, 2018, 20, 1779–1782 RSC
- T. Stolar, J. Alić, I. Lončarić, M. Etter, D. Jung, O. K. Farha, I. Đilović, E. Meštrović and K. Užarević, CrystEngComm, 2022, 24, 6505–6511 RSC
- R.-B. Lin and B. Chen, Chem, 2022, 8, 2114–2135 CAS
- H. Wang, B. Li, H. Wu, T.-L. Hu, Z. Yao, W. Zhou, S. Xiang and B. Chen, J. Am. Chem. Soc., 2015, 137, 9963–9970 CrossRef CAS PubMed
- S. Nandi, D. Chakraborty and R. Vaidhyanathan, Chem. Commun., 2016, 52, 7249–7252 RSC
- J. Liang, S. Xing, P. Brandt, A. Nuhnen, C. Schlüsener, Y. Sun and C. Janiak, J. Mater. Chem. A, 2020, 8, 19799–19804 RSC
- Z. Sun, Y. Li, L. Chen, X. Jing and Z. Xie, Cryst. Growth Des., 2015, 15, 542–545 CrossRef CAS
- H. Wang, Z. Bao, H. Wu, R.-B. Lin, W. Zhou, T.-L. Hu, B. Li, J. C.-G. Zhao and B. Chen, Chem. Commun., 2017, 53, 11150–11153 RSC
- S. Cai, H. Shi, Z. Zhang, X. Wang, H. Ma, N. Gan, Q. Wu, Z. Cheng, K. Ling, M. Gu, C. Ma, L. Gu, Z. An and W. Huang, Angew. Chem., Int. Ed., 2018, 57, 4005–4009 CrossRef CAS PubMed
- Y. Wang, D. Liu, J. Yin, Y. Shang, J. Du, Z. Kang, R. Wang, Y. Chen, D. Sun and J. Jiang, Chem. Commun., 2020, 56, 703–706 RSC
- B. Wang, R. He, L.-H. Xie, Z.-J. Lin, X. Zhang, J. Wang, H. Huang, Z. Zhang, K. S. Schanze, J. Zhang, S. Xiang and B. Chen, J. Am. Chem. Soc., 2020, 142, 12478–12485 CrossRef CAS PubMed
- Y. Tang, M. Yuan, B. Jiang, Y. Xiao, Y. Fu, S. Chen, Z. Deng, Q. Pan, C. Tian and H. Fu, J. Mater. Chem. A, 2017, 5, 21979–21985 RSC
- B. Han, H. Wang, C. Wang, H. Wu, W. Zhou, B. Chen and J. Jiang, J. Am. Chem. Soc., 2019, 141, 8737–8740 CrossRef CAS PubMed
- W. Gong, D. Chu, H. Jiang, X. Chen, Y. Cui and Y. Liu, Nat. Commun., 2019, 10, 600 CrossRef CAS PubMed
- H. Coskun, A. Aljabour, P. Luna, H. Sun, N. Nishiumi, T. Yoshida, G. Koller, M. G. Ramsey, T. Greunz, D. Stifter, M. Strobel, S. Hild, A. W. Hassel, N. S. Sariciftci, E. H. Sargent and P. Stadler, Adv. Mater., 2020, 32, 1902177 CrossRef CAS PubMed
- Q. Yin, P. Zhao, R.-J. Sa, G.-C. Chen, J. Lü, T.-F. Liu and R. Cao, Angew. Chem., Int. Ed., 2018, 57, 7691–7696 CrossRef CAS PubMed
- W. Liang, F. Carraro, M. B. Solomon, S. G. Bell, H. Amenitsch, C. J. Sumby, N. G. White, P. Falcaro and C. J. Doonan, J. Am. Chem. Soc., 2019, 141, 14298–14305 CrossRef CAS PubMed
- A. Karmakar, R. Illathvalappil, B. Anothumakkool, A. Sen, P. Samanta, A. V. Desai, S. Kurungot and S. K. Ghosh, Angew. Chem., Int. Ed., 2016, 55, 10667–10671 CrossRef CAS PubMed
- G. Xing, T. Yan, S. Das, T. Ben and S. Qiu, Angew. Chem., Int. Ed., 2018, 57, 5345–5349 CrossRef CAS PubMed
- S. Chand, S. C. Pal, A. Pal, Y. Ye, Q. Lin, Z. Zhang, S. Xiang and M. C. Das, Chem.–Eur. J., 2019, 25, 1691–1695 CrossRef CAS PubMed
- Y. Wang, M. Zhang, Q. Yang, J. Yin, D. Liu, Y. Shang, Z. Kang, R. Wang, D. Sun and J. Jiang, Chem. Commun., 2020, 56, 15529–15532 RSC
- S. C. Pal, D. Mukherjee, R. Sahoo, S. Mondal and M. C. Das, ACS Energy Lett., 2021, 6, 4431–4453 CrossRef CAS
- Y. Qin, T. Gao, W.-P. Xie, Z. Li and G. Li, ACS Appl. Mater. Interfaces, 2019, 11, 31018–31027 CrossRef CAS PubMed
- Y. Wang, J. Yin, D. Liu, C. Gao, Z. Kang, R. Wang, D. Sun and J. Jiang, J. Mater. Chem. A, 2021, 9, 2683–2688 RSC
- D. Matoga, M. Oszajca and M. Molenda, Chem. Commun., 2015, 51, 7637–7640 RSC
- F.-M. Zhang, L.-Z. Dong, J.-S. Qin, W. Guan, J. Liu, S.-L. Li, M. Lu, Y.-Q. Lan, Z.-M. Su and H.-C. Zhou, J. Am. Chem. Soc., 2017, 139, 6183–6189 CrossRef CAS PubMed
- M. K. Sarango-Ramírez, D.-W. Lim, D. I. Kolokolov, A. E. Khudozhitkov, A. G. Stepanov and H. Kitagawa, J. Am. Chem. Soc., 2020, 142, 6861–6865 CrossRef PubMed
- S. Chen, Y. Wu, Y. Zhang, W. Zhang, Y. Fu, W. Huang, T. Yan and H. Ma, J. Mater. Chem. A, 2020, 8, 13702–13709 RSC
- M. Lupa, P. Kozyra, G. Jajko and D. Matoga, ACS Appl. Mater. Interfaces, 2021, 13, 29820–29826 CrossRef CAS PubMed
- M. Lupa, P. Kozyra and D. Matoga, ACS Appl. Energy Mater., 2023, 6, 9118–9123 CrossRef CAS
- A. Krusenbaum, S. Grätz, G. T. Tigineh, L. Borchardt and J. G. Kim, Chem. Soc. Rev., 2022, 51, 2873–2905 RSC
- F. Cuccu, L. De Luca, F. Delogu, E. Colacino, N. Solin, R. Mocci and A. Porcheddu, ChemSusChem, 2022, 15, e202200362 CrossRef CAS PubMed
- D. Virieux, F. Delogu, A. Porcheddu, F. García and E. Colacino, J. Org. Chem., 2021, 86, 13885–13894 CrossRef CAS PubMed
- J. G. Hernández and C. Bolm, J. Org. Chem., 2017, 82, 4007–4019 CrossRef PubMed
- C. Bolm and J. G. Hernández, Angew. Chem., Int. Ed., 2019, 58, 3285–3299 CrossRef CAS PubMed
-
F. Leon and F. Garcia, in Comprehensive Coordination Chemistry III, Elsevier, 2021, pp. 620–679 Search PubMed
- S. Hwang, S. Grätz and L. Borchardt, Chem. Commun., 2022, 58, 1661–1671 RSC
- D. Tan and F. García, Chem. Soc. Rev., 2019, 48, 2274–2292 RSC
- C. Mottillo and T. Friščić, Molecules, 2017, 22, 144 CrossRef PubMed
- J.-L. Do and T. Friščić, ACS Cent. Sci., 2017, 3, 13–19 CrossRef CAS PubMed
- K. Roztocki, D. Jędrzejowski, M. Hodorowicz, I. Senkovska, S. Kaskel and D. Matoga, Cryst. Growth Des., 2018, 18, 488–497 CrossRef CAS
- T. Friščić, C. Mottillo and H. M. Titi, Angew. Chem., 2020, 132, 1030–1041 CrossRef
- E. Colacino, V. Isoni, D. Crawford and F. García, Trends Chem., 2021, 3, 335–339 CrossRef CAS
- X. Liu, Y. Li, L. Zeng, X. Li, N. Chen, S. Bai, H. He, Q. Wang and C. Zhang, Adv. Mater., 2022, 2108327 CrossRef CAS
- O. Galant, G. Cerfeda, A. S. McCalmont, S. L. James, A. Porcheddu, F. Delogu, D. E. Crawford, E. Colacino and S. Spatari, ACS Sustainable Chem. Eng., 2022, 10, 1430–1439 CrossRef CAS
- D. Jędrzejowski, M. Ryndak, J. J. Zakrzewski, M. Hodorowicz, S. Chorazy and D. Matoga, ACS Appl. Mater. Interfaces, 2023, 15, 25661–25670 CrossRef PubMed
- D. Matoga, K. Roztocki, M. Wilke, F. Emmerling, M. Oszajca, M. Fitta and M. Bałanda, CrystEngComm, 2017, 19, 2987–2995 RSC
- W. Qin, D. Si, Q. Yin, X. Gao, Q. Huang, Y. Feng, L. Xie, S. Zhang, X. Huang, T. Liu and R. Cao, Angew. Chem., Int. Ed., 2022, 61, e202202089 CrossRef CAS PubMed
- I. Akhmetova, M. Rautenberg, C. Das, B. Bhattacharya and F. Emmerling, ACS Omega, 2023, 8, 16687–16693 CrossRef CAS PubMed
- D. Matoga, B. Gil, W. Nitek, A. D. Todd and C. W. Bielawski, CrystEngComm, 2014, 16, 4959 RSC
- P. Meng, A. Brock, Y. Xu, C. Han, S. Chen, C. Yan, J. McMurtrie and J. Xu, J. Am. Chem. Soc., 2020, 142, 479–486 CrossRef CAS PubMed
- Z.-X. Cai, Y. Xia, Y. Ito, M. Ohtani, H. Sakamoto, A. Ito, Y. Bai, Z.-L. Wang, Y. Yamauchi and T. Fujita, ACS Nano, 2022, 16, 20851–20864 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, synthetic procedures, FTIR spectra, PXRD patterns, TGA data, proton conduction data, gas sorption and crystallographic details. CCDC 2288811 and 2288812. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc04401g |
| This journal is © The Royal Society of Chemistry 2023 |