
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAn erythrocyte membrane-camouflaged fluorescent covalent organic framework for starving/nitric oxide/immunotherapy of triple-negative breast cancer†
Fang
Yuan
 ,
Cuiling
Zhang
*,
Xianzhu
Luo
,
Shasha
Cheng
,
Yingxin
Zhu
and
Yuezhong
Xian
*
,
Cuiling
Zhang
*,
Xianzhu
Luo
,
Shasha
Cheng
,
Yingxin
Zhu
and
Yuezhong
Xian
*
Shanghai Engineering Research Center of Molecular Therapeutics and New Drug Development, Department of Chemistry, School of Chemistry and Molecular Engineering, East China Normal University, Shanghai 200241, China. E-mail: clzhang@chem.ecnu.edu.cn; yzxian@chem.ecnu.edu.cn
First published on 28th July 2023
Abstract
It is a great challenge to effectively treat triple-negative breast cancer (TNBC) due to lack of therapeutic targets and drug resistance of systemic chemotherapy. Rational design of nanomedicine with good hemocompatibility is urgently desirable for combination therapy of TNBC. Herein, an erythrocyte membrane-camouflaged fluorescent covalent organic framework (COF) loaded with an NO donor (hydroxyurea, Hu), glucose oxidase (GOx) and cytosine–phosphate–guanine oligonucleotides (CPG) (COF@HGC) was developed for imaging-guided starving/nitric oxide (NO)/immunization synergistic treatment of TNBC. The substances of HGC are easily co-loaded onto the COF due to the ordered pore structure and large surface area. And a folic acid-modified erythrocyte membrane (FEM) is coated on the surface of COF@HGC to improve targeted therapy and haemocompatibility. When COF@HGC@FEM is internalized into tumor cells, hemoglobin (Hb) on FEM and GOx loaded on the COF can trigger cascade reactions to kill tumor cells due to the simultaneous production of NO and exhaustion of glucose. Meanwhile, the COF with excellent fluorescence properties can be used as a self-reporter for bioimaging. Furthermore, the CPG can reprogram tumor-associated macrophages from tumor-supportive phenotype to anti-tumor phenotype and enhance immunotherapy. Through the “three-in-one” strategy, the biomimetic nanoplatform can effectively inhibit tumor growth and reprogram the tumor immunosuppression microenvironment in the TNBC mouse model.
Introduction
As the most aggressive and heterogeneous subtype of breast cancer, triple-negative breast cancer (TNBC) lacks three kinds of hormone receptors including for progesterone, estrogen, and human epidermal growth factor receptor 2.1,2 Though the occurrence rate is only 15–20% among all breast cancers, TNBC patients have shown high metastasis and high mortality in recent years.3,4 Combination chemotherapy is a leading strategy for TNBC treatment in the clinic. However, it suffers from non-targeting, high systemic toxicity, rapid clearance with blood circulation and drug resistance.5,6 Nanomedicines have made significant progress in cancer therapy because of reducing multi-drug resistance, prolonging systemic blood circulation, and enhancing tumor infiltrate and accumulation.7,8 Various nano delivery systems (e.g. liposomes, mesoporous silica nanoparticles, polymer micelles, metal organic frameworks, etc.) have been developed for targeting delivery drugs to tumor sites.9–12 However, these nanocarriers are still limited by poor targeting ability and haemocompatibility, low drug-loading efficiency or potential heavy metal toxicity.7 Therefore, there is an urgent need to develop accurate and efficient nanomedicine for TNBC.As a class of crystalline porous networks, covalent organic frameworks (COFs) are constructed by pure organic building blocks, which exhibit highly tunable structures and well-customized functionalities.13 Introducing a luminous monomer into the COF skeleton will endow it with good fluorescence properties.14 COFs show high crystallinity and porosity, excellent drug-loading performance, multiple post-modification sites and good biocompatibility. These features are favorable to develop a multifunctional integration nanoplatform for synergistic tumor therapy.15–19 Liu et al. reported encapsulating DOX onto a COF via a one-pot method with a high drug-loading capacity of 32.1%.20 Gao et al. developed a COF nanoprobe for simultaneous survivin mRNA detection and cancer photodynamic therapy.21 Although COFs show great potential in biomedical fields, only a handful of studies on COF-based nanomedicine have been reported. The significant bottlenecks of COFs for therapeutics are their relatively large size and poor dispersibility in water.22 Great efforts have been made to reduce their size to the nanoscale and improve their dispersibility and stability in biological systems. Two main methods were utilized to achieve these goals, (1) the bottom-up method through solvent or surfactant-assisted homogeneous polymerization to form stable colloidal suspensions of COF nanoparticles and (2) the top-down method through convenient solvent-assisted exfoliation of the bulk COF.23–25 Although the above-mentioned strategies have contributed to the great progress in the preparation of nanoscale COFs, for in vivo therapy, the COFs still suffer from long-term stability in the physiological system, short systemic circulation time and rapid immune clearance by the mononuclear phagocyte system.26 The natural erythrocyte membrane (EM) camouflage method is a promising strategy to prevent COF π–π stacking and aggregation, prolong half-life circulation and avoid immune elimination because of the presence of CD47 protein on the EM, which can be recognized as signal regulatory protein α (SIRPα) and is accompanied by the release of a “don't eat me” signal.27,28 Therefore, the EM camouflage strategy might eliminate the obstacle to the application of COF in vivo diagnosis and therapy.
Gas therapy has emerged as a ‘green’ tumor therapy paradigm by using gasotransmitters to kill cancer cells.29 Among the gasotransmitters, NO is an essential biological messenger associated with various physiological and pathological processes, such as neuronal communication, stem-cell therapy, wound healing, etc.30,31 Generally, a low concentration of NO (∼1.0 nM) can regulate endothelial cell proliferation and migration, thus promoting vasodilatation, angiogenesis and vascular remodeling.32 Recently, Liu et al. reported the generation of NO through an enzyme-mediated reaction based on Hb existing on EM, which was coated on coacervate protocells for blood vessel vasodilation.33 The system can rapidly produce NO due to the cascade catalysis reaction of Hb (peroxidase activity) and glucose oxidase (GOx) by using glucose and hydroxyurea (Hu) as substrate, respectively. It has been reported that high concentrations of NO (1.0 μM–1.0 mM) show distinct anti-tumor efficiency through nitrosation of mitochondrial DNA and protein in cancerous cells.29,34,35 Although NO has been applied in anti-tumor research, it suffers from a slow release rate and insufficient treatment concentration. It is still a great challenge to construct an NO gas generation platform that can precisely release NO at a rapid rate and high dose at the tumor site.
Inspired by the advantages of fluorescent COF and enzyme-mediated NO production, we rationally designed and developed an integrated bioplatform for TNBC treatment. In this system, a folic acid (FA) modified EM (FEM) was coated on a fluorescent COF for targeting the delivery of NO donor (Hu), GOx and immune adjuvant cytosine–phosphate–guanine oligonucleotides (CPG) (Scheme 1). The introduction of specific ligands of FA on the EM could enhance its tumor-targeting capacity due to the over-expressed FA receptor (FR) on TNBC cells.36 Owing to the GOx loading on the COF, starvation therapy was realized by depleting glucose based on a GOx-directed catalytic reaction (stage I). Meanwhile, the loaded Hu was used as an NO donor, which can be further employed to promote NO-mediated gas therapy in the light of Hb-catalyzed reaction in the presence of H2O2 (stage II). We selected CPG (a Toll-like receptor 9 (TLR9) agonist) to endow the bioplatform with the function of immunotherapy (stage III). CPG has been identified to reprogram immunosuppressive tumor-supportive phenotype (M2)-like tumor-associated macrophages (TAMs) into immunostimulatory anti-tumor phenotype (M1)-like TAMs.37–40 Moreover, it can promote the secretion of proinflammatory cytokines, such as tumor necrosis factor alpha (TNF-α) and interleukin-12 (IL-12) to kill tumor cells and reduce the levels of anti-inflammatory cytokines such as IL-10 and transforming growth factor-β1 (TGF-β1).9 Thus, the nanoplatform with excellent fluorescence can boost imaging-guided TNBC therapy through the synergistic starving/NO/immunization treatment.
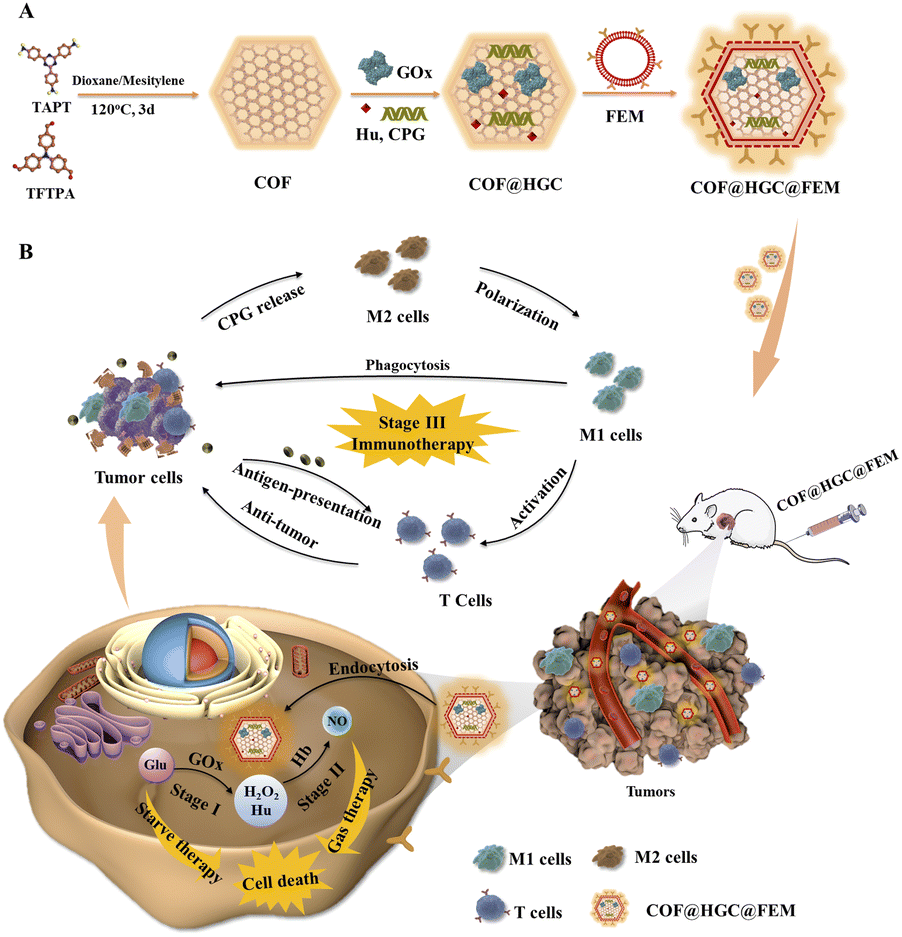 | ||
| Scheme 1 (A) Schematic illustration of the preparation of a biomimetic COF@HGC@FEM nanoplatform. (B) Synergistic treatment process for suppressing tumor growth through enzyme-directed cascade catalytic reaction and immunotherapy. | ||
Results and discussion
Synthesis and characterization of COF@HGC@FEM
The EM-derived fragments were extracted using the established method.33 The optical photos of erythrocyte and EM are shown in Fig. S1A and B.† The target FA molecules were modified on the surface of EM. It is well known that FR is over-expressed in various cancer cells, such as 4T1, MCF-7, and HeLa cells.39,41 Therefore, FA molecules are typically used for specific tumor targeting due to their high affinity toward FR. Benefiting from the fluidity of lipid bilayer membranes, the FEM can be easily generated by physically mixing DSPE-PEG-FA with EM at 37 °C for 0.5 h. Fluorescent DSPE-PEG-Cy5 was used to visualize the decoration. As shown in Fig. S1C,† the bright Cy5 red fluorescence can be observed under CLSM imaging, indicating DSPE-PEG-Cy5 has been successfully inserted into EM. The mass fraction of Hb on FEM lyophilized powder was determined to be 23.8% by using bHb as a standard (Fig. S2†). Simultaneously, the imine-linked COF nanosheets were synthesized and exfoliated through sonication according to our previous work.42 Hu, GOx and CPG were loaded on COF by electrostatic and hydrophobic interaction. The mixture of FEM fragments and COF@HGC suspension was treated with ultrasound to induce the encapsulation of FEM fragments on the surface of COF@HGC. A series of characterizations were conducted to verify the successful assembly of COF@HGC@FEM. As shown in Fig. 1A, the TEM image of the original COF shows irregular nanosheet morphology with an average size of about 184 nm. After the Hu, GOx and CPG decoration, the COF@HGC shows a rougher morphology compared with bare COF nanosheets. After further cloaked COF@HGC with FEM, a typical core–shell structure is observed and the thickness of the shell layer of FEM on COF@HGC@FEM is approximately 10 nm. The hydrodynamic radius of COF, COF@HGC, and COF@HGC@FEM is about 195, 214, and 239 nm, respectively (Fig. 1B). The embedding of Hu, GOx and CPG on COF leads to an about 19 nm increase in hydrodynamic diameter. FEM camouflage on COF@HGC results in an about 25 nm additional increase.26 As can be seen in Fig. 1C, the zeta potential of the pristine COF is about +20.9 mV due to the presence of amino groups. As for the COF@HGC, it is about −27.5 mV due to the immobilization of negatively charged GOx and CPG. The zeta potential of COF@HGC@FEM is about −21.1 mV which is close to that of FEM (−21.6 mV). This result can be interpreted by the charge screening effect due to FEM camouflage.43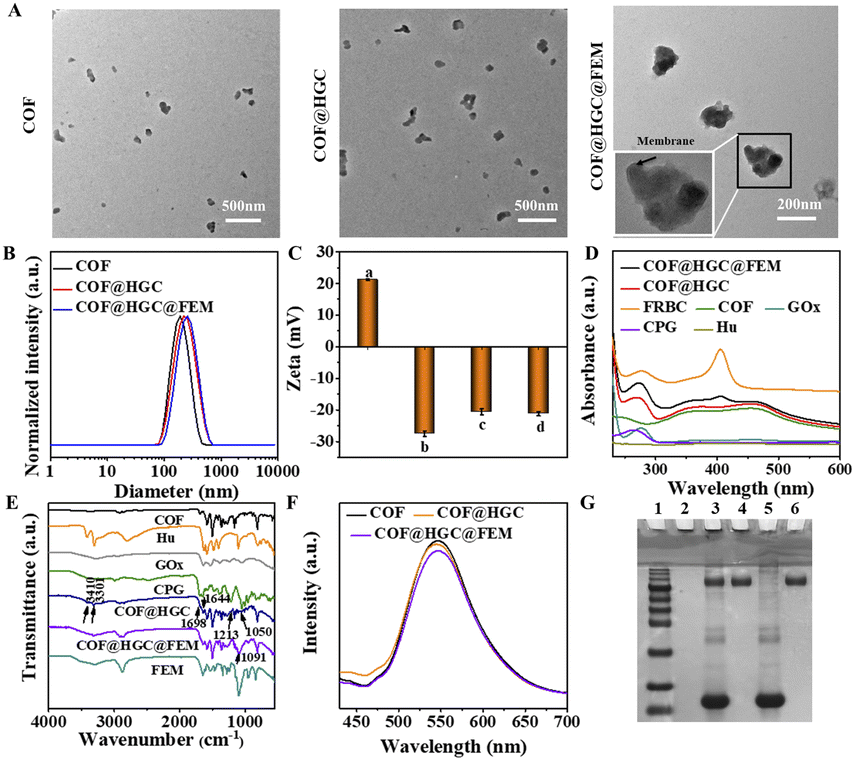 | ||
| Fig. 1 Characterization of COF@HGC@FEM. (A) TEM images of COF, COF@HGC, and COF@HGC@FEM, respectively. (B) Hydrodynamic size distribution of COF, COF@HGC and COF@HGC@FEM, respectively. (C) Zeta potentials of (a) COF, (b) COF@HGC, (c) COF@HGC@FEM and (d) FEM, respectively. (D) UV-vis and (E) FTIR spectra of COF@HGC@FEM, COF@HGC, COF, Hu, CPG, FEM and free GOx. (F) Fluorescence spectra of COF, COF@HGC and COF@HGC@FEM (λex = 405 nm). (G) SDS-PAGE protein analysis of (1) protein markers, (2) COF, (3) COF@HGC@FEM, (4) COF@HGC, (5) FEM, and (6) GOx, respectively. | ||
From the UV-vis spectra of COF@HGC@FEM shown in Fig. 1D, COFs exhibit two broad absorption bands at 350 and 470 nm, respectively. After Hu, GOx and CPG embedding, the absorption peak located at 270 nm can be observed which might result from the co-immobilization of CPG (ca. 260 nm) and GOx (ca. 280 nm). Notably, after FEM camouflage, a new peak appears at 410 nm, which belongs to Hb absorption originating from FEM.44 This suggests that FEM has been successfully enveloped on the surface of COF@HGC. The content of Hu was quantitatively analyzed through HPLC and the loading capacity was about 32.9%. Also, the loading capacities of GOx, CPG and Hb on COF@HGC@FEM obtained through the corresponding standard curves are about 17.6%, 2.3%, and 4.4%, respectively (Fig. S2–S4†).
Fig. 1E displays the FTIR spectra of the corresponding substances. The FTIR spectrum of COF exhibits a characteristic imine (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N) stretching vibration at 1621 cm−1. The bands at 3436 and 3348 cm−1 are assigned to the stretching vibrations of amino groups (N–H), and the band at 1680 cm−1 is assigned to the stretching vibration of carbonyl groups (CO) due to the –NH2 and –CHO groups on COF.42 After loading with Hu, GOx and CPG, new bands at 3410 and 3301 cm−1 (antisymmetric stretching of –NH2 from Hu),45 1698 cm−1 (CO from CPG), 1213 cm−1, 1250 cm−1 (antisymmetric stretching vibration of the PO from CPG),46 and 1644 cm−1 (amide I bond of GOx)47 can be observed, indicating the successful encapsulation of corresponding substances onto COF. After FEM wrapping, the peak of the phosphate group at 1091 cm−1 is enhanced, proving the successful construction of COF@HGC@FEM. Besides, the XRD patterns revealed that the COF maintained high crystallinity, and both the drug loading and FEM coating processes did not affect the crystallinity of the pristine COF (Fig. S5†). The Brunauer–Emmett–Teller (BET) surface area of COF was 1223 m2 g−1 with an average pore size of 1.9 nm for the COF (Fig. S6†). However, after the formation of COF@HGC@FEM, the BET surface area and pore size decreased to 129 m2 g−1 and 1.7 nm, respectively. The reduction confirmed the successful entrapment of drugs within the porous lattice.
N) stretching vibration at 1621 cm−1. The bands at 3436 and 3348 cm−1 are assigned to the stretching vibrations of amino groups (N–H), and the band at 1680 cm−1 is assigned to the stretching vibration of carbonyl groups (CO) due to the –NH2 and –CHO groups on COF.42 After loading with Hu, GOx and CPG, new bands at 3410 and 3301 cm−1 (antisymmetric stretching of –NH2 from Hu),45 1698 cm−1 (CO from CPG), 1213 cm−1, 1250 cm−1 (antisymmetric stretching vibration of the PO from CPG),46 and 1644 cm−1 (amide I bond of GOx)47 can be observed, indicating the successful encapsulation of corresponding substances onto COF. After FEM wrapping, the peak of the phosphate group at 1091 cm−1 is enhanced, proving the successful construction of COF@HGC@FEM. Besides, the XRD patterns revealed that the COF maintained high crystallinity, and both the drug loading and FEM coating processes did not affect the crystallinity of the pristine COF (Fig. S5†). The Brunauer–Emmett–Teller (BET) surface area of COF was 1223 m2 g−1 with an average pore size of 1.9 nm for the COF (Fig. S6†). However, after the formation of COF@HGC@FEM, the BET surface area and pore size decreased to 129 m2 g−1 and 1.7 nm, respectively. The reduction confirmed the successful entrapment of drugs within the porous lattice.
Moreover, it is worth noting that the biomimetic modification has no significant influence on the fluorescence intensity of the COF (Fig. 1F), which ensures further fluorescence imaging in vivo. The SDS-PAGE method was also used to further confirm the integrity of membrane proteins on COF@HGC@FEM by using FEM and GOx as controls. As shown in Fig. 1G, no band is observed for COF due to the absence of any proteins. As for the COF@HGC sample, one protein band similar to that of free GOx at 150 kDa can be observed. In the case of COF@HGC@FEM, except for the GOx band, it also displays bands identical to those of natural FEM. The results show that most pristine membrane proteins on FEM are well preserved after fabrication, and GOx is also successfully embedded into the FEM-cloaked nanocomposites. In the long-term stability test, the hydrodynamic diameter and PDI of COF@HGC@FEM show negligible changes after 14 days of storage in H2O, PBS, DMEM, and 10% FBS solution (Fig. S7A†). However, the hydrodynamic diameter of COF increases from approximately 195 to 260 nm in these media (Fig. S7B†). These results validate that the FEM cloak effectively enhances the long-term stability of COF under physiological conditions. The above results suggested that the FEM camouflaged biomimetic nanocarrier encapsulated with Hu, GOx and CPG has been successfully constructed and can be used for subsequent experiments.
We evaluated the drug release behaviors of COF@HGC@FEM in PBS with different pH values (5.5 and 7.4), which were selected to simulate the pH of lysosomes (5.5) and blood plasma (7.4). As shown in Fig. S8,† only 23.5%, 27.8%, and 26.4% of Hu, GOx and CPG were released from COF@HGC@FEM at pH 7.4 over 72 h. The results indicate that COF@HGC@FEM effectively prevents premature drug leakage during blood circulation and thus enhances drug availability to tumors. In contrast, at pH 5.5, 77.5%, 87.3%, and 86.2% of Hu, GOx and CPG were released within the same time. This phenomenon can be attributed to the protonation of amino groups in COF at pH 5.5, which led to a decrease in the electrostatic and hydrophobic interaction between drugs and COF, and subsequently triggered the release of drugs.
The catalytic behavior of COF@HGC@FEM
Due to the peroxidase-like activity of Hb on EM,48 we explore the COF@HGC@FEM as a nanocarrier to deliver enzymes and produce NO through cascade GOx- and Hb-directed enzymatic catalysis (Fig. 2A). The process included two steps: (1) GOx-directed catalytic generation of gluconic acid and H2O2 in the presence of glucose and oxygen; (2) peroxidase-like Hb-induced catalytic production of NO in the presence of H2O2 and Hu. At first, the catalytic ability of GOx embedded in COF was evaluated using a GOx/HRP/ABTS colorimetric assay. The produced H2O2 was monitored by the absorption of the ABTS-diradical product at 418 nm. The catalytic activity of free GOx, COF, COF@HGC and COF@HGC@FEM was tested and is shown in Fig. S9A.† COF shows no apparent catalytic activity, while COF@HGC and COF@HGC@FEM display comparable catalytic ability similar to that of free GOx. The peroxidase activity of FEM was also estimated by using the H2O2/ABTS colorimetric assay. The bHb was used as a control. As shown in Fig. S9B,† pure COF and COF@HGC show no apparent absorption at 418 nm. In contrast, COF@HGC@FEM displays a similar absorption relative to that of free bHb, indicating the peroxidase activity of FEM.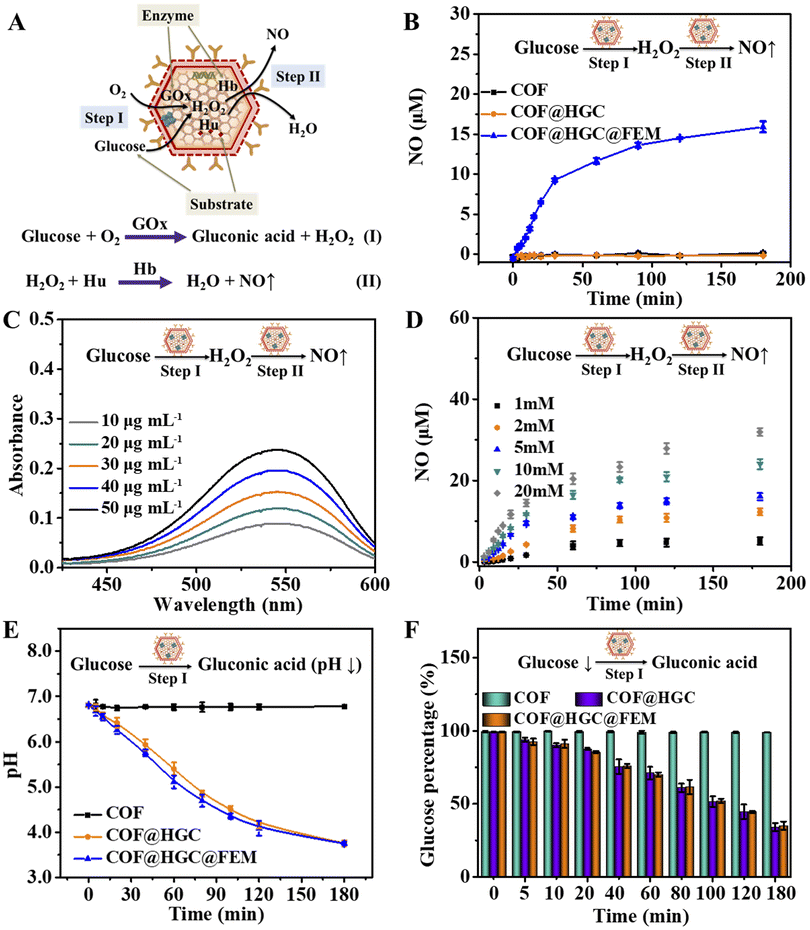 | ||
| Fig. 2 In vitro enzymatic activity of COF@HGC@FEM. (A) Schematic illustration of the cascade catalytic reactions driven by COF@HGC@FEM. (B) NO generation at different times in the system containing 5.0 mM glucose and 30 μg mL−1 COF, COF@HGC or COF@HGC@FEM, respectively. (C) Absorption spectra of Griess reagent triggered by cascade catalysis reaction at different concentrations (10, 20, 30, 40 and 50 μg mL−1) of COF@HGC@FEM in the presence of 5.0 mM glucose for 3 h. (D) Time-dependence of NO generation in the system containing 30 μg mL−1 COF@HGC@FEM and different concentrations (1, 2, 5, 10 and 20 mM) of glucose. Time-dependence of (E) pH variation and (F) glucose consumption in 30 μg mL−1 COF, COF@HGC or COF@HGC@FEM in the presence of 5.0 mM glucose. Data are presented as the mean ± SD (n = 3). | ||
Next, the generation of NO through cascade catalysis by COF@HGC@FEM was determined by Griess colorimetric assay. As shown in Fig. 2B, we cannot observe the NO generation for COF due to the absence of GOx and FEM simultaneously. For the COF@HGC system, there was no NO generation due to the absence of FEM wrapping, and the cascade catalysis could not occur. In COF@HGC@FEM suspension, a remarkable production of NO was observed with an initial rate of around 0.328 μM min−1. After 180 min reaction, the NO content reaches a plateau and the concentration is about 15.4 μM, implying the excellent enzyme catalytic activity of COF@HGC@FEM. Moreover, it can be seen from Fig. 2C that the catalytic capacity of COF@HGC@FEM is concentration-dependent over the range from 10 to 50 μg mL−1 which is similar to the enzyme-catalyzed reaction. Next, the effect of pH on the enzymatic cascade reaction was examined at pH 6.8 and 7.4, respectively. As shown in Fig. S10,† the Griess reagent absorption is enhanced at pH 6.8. This implies that the acidic TME might benefit glucose consumption and NO generation, thereby promoting starvation and NO gas therapy performance.
The amount of NO production at different glucose concentrations was measured to study the cascade catalytic kinetics. As shown in Fig. 2D, the NO amount is highly related to substrate concentration. Much more NO is produced with the elevation of substrate concentration. This is because more GOx can catalytically decompose more glucose to produce H2O2 and subsequently participate in the oxidation of Hu to yield NO by Hb on COF@HGC@FEM. The maximum reaction rate (Vmax) of COF@HGC@FEM toward glucose is about 1.5 μmol L−1 min−1 and the Michaelis–Menten constant (Km) is approximately 21.8 mM according to the Michaelis–Menten equation and Lineweaver–Burk fitting equation, 1/V = Km/Vmax(1/[S] + 1/Km), where [S] is the substrate concentration and V is the initial velocity.49 The data indicate the NO generation by COF@HGC@FEM is faster than that of the GOx-based NO cascade catalysis system (0.25 μmol L−1 min−1),33 which might be ascribed to the high loading efficiency of COF. Consideration of the intracellular glucose varied from 0.1 to 5.0 mM,50 hence, 5.0 mM glucose was used as the input substrate concentration in our system.
The generation of gluconic acid might lead to the down-regulation of pH. Thus, we tested the changes in pH with time. As depicted in Fig. 2E, the pH value of the system containing COF suspension and 5.0 mM glucose remains constant within 180 min. In contrast, for COF@HGC, the pH continuously decreases from 6.8 to 3.8 within 180 min because of the continuous production of gluconic acid. Coincidentally, for the COF@HGC@FEM system, a similar pH decline trend to that of COF@HGC is observed. The time-dependent glucose consumption was also monitored by a 3,5-dinitrosalicylic acid (DNS) method based on a colorimetric assay. As shown in Fig. 2F, the percentage of glucose declines to about 33% and 36% after 180 min in the presence of COF@HGC and COF@HGC@FEM. As for the COF system, the glucose concentration remains stable even after 180 min. The outcomes indicate that the immobilization of GOx on COF can effectively deplete glucose, which might be used to achieve tumor starvation therapy. As stated above, the biomimetic COF@HGC@FEM system shows the potential to kill tumor cells through glucose consumption and intracellular NO generation if it can be efficiently delivered to tumor tissue.
Cell targeting capacity
Before the targeting experiment, the cellular internalization of COF@HGC@FEM was investigated in TNBC cell lines (4T1 and MDA-MB-231 cells). It can be observed from Fig. S11† that the intracellular fluorescence intensity of COF@HGC@FEM is gradually enhanced with the extended culture time and reaches a maximum after 3 h in both cell lines. Hence, 3 h was selected as the experiment time. Next, the tumor targeting experiment of COF@HGC@FEM was operated on MCF-10A (human normal breast cells, FR−), 4T1 (FR+) and MDA-MB-231 (FR+) cell lines, respectively.39,51 As depicted in Fig. 3A, after being treated with COF@HGC@FEM or COF@HGC@EM for 3 h, the green emission of COF can be observed. Moreover, the COF@HGC@FEM treated 4T1 and MDA-MB-231 cells showed significantly high fluorescence intensity compared to MCF-10A cells. This result indicated that FA-modified EM possessed superior tumor-targeting ability to normal cells. Besides, the CLSM imaging indicates that the green fluorescence signal in 4T1 and MDA-MB-231 cells stained with COF@HGC@FEM is much stronger than that of the COF@HGC@EM group, confirming FA can promote the targeting efficiency. These data indicate that COF@HGC@FEM can be specific and efficiently internalized by 4T1 and MDA-MB-231 cells via FR-mediated endocytosis. This also shows that COF@HGC@FEM can be used as an excellent self-reporter for targeting delivery.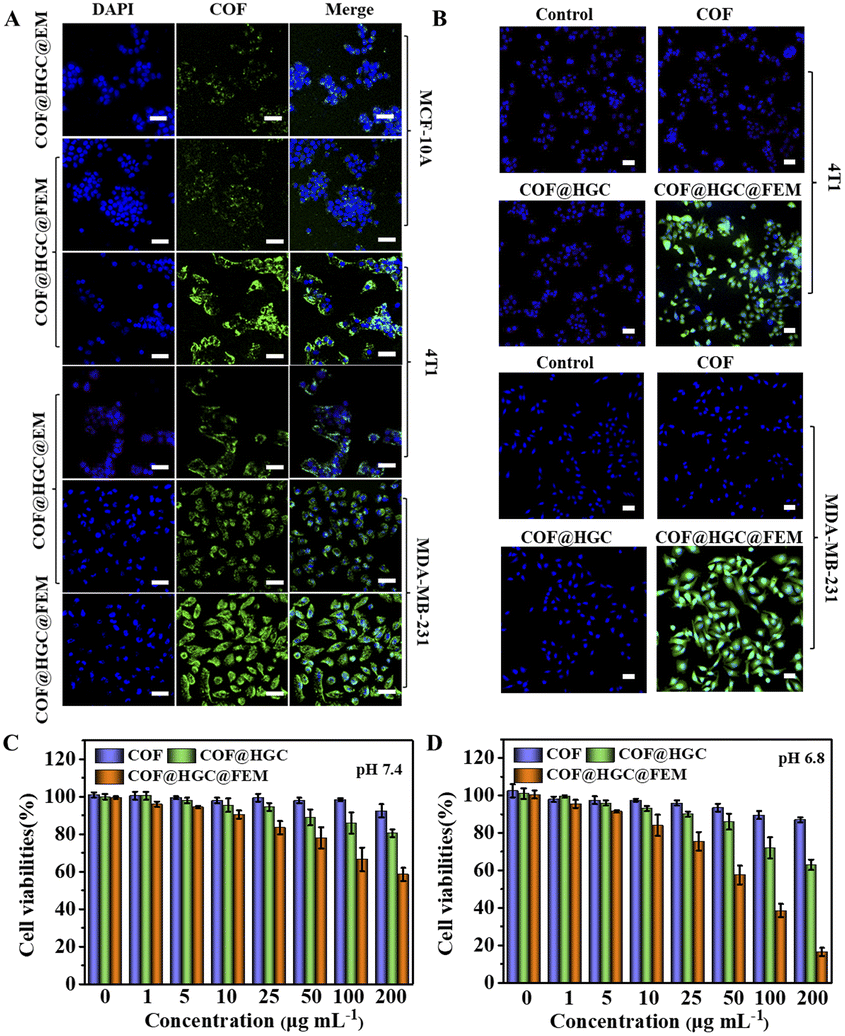 | ||
| Fig. 3 (A) CLSM images of MCF-10A, 4T1 and MDA-MB-231 cells after being incubated with COF@HGC@FEM or COF@HGC@EM for 3 h. Scale bar = 50 μm. (B) CLSM images of intracellular NO generation in 4T1 and MDA-MB-231 cells using DAF-FM DA as probe treated with COF, COF@HGC or COF@HGC@FEM for 3 h. Scale bar = 50 μm. Cell viability of 4T1 cells treated with different concentrations of COF, COF@HGC or COF@HGC@FEM at pH 7.4 (C) and 6.8 (D) for 24 h, respectively. Data are presented as the mean ± SD (n = 3). | ||
We further assessed intracellular NO generation using the specific NO probe of DAF-FM DA (Fig. 3B). The CLSM images indicate that the fluorescence signal of the NO probe after COF or COF@HGC treatment is nearly invisible, indicating no NO generation. Notably, the 4T1 and MDA-MB-231 cells incubated with COF@HGC@FEM display bright green fluorescence signals of the NO probe, implying a high level of NO generation under these conditions. To further validate the targeting capacity of COF@HGC@FEM towards tumor cells, a three-dimensional (3D) multicellular tumor spheroid (MTS) model was built to simulate the physiological condition of the solid tumor. The permeability of COF and COF@HGC@FEM into MTSs was examined after 12 h incubation. The fluorescence images for different layers of MTSs were captured by CLSM from top to bottom. As shown in Fig. S12,† for the COF treated group, the green fluorescence from the COF can be visualized at about 40 μm depth. With the z-axis distance increasing, the green fluorescence is only distributed in the margin areas of the layer, signifying the limited penetration depth of COF. However, for the COF@HGC@FEM group, strong green fluorescence can be observed throughout the whole MTS with a penetration depth of over 100 μm. This experiment further manifested the superior penetration ability of COF@HGC@FEM by specific FA targeting.
The cytotoxicity and in vitro therapy efficiency
The cytotoxicity was evaluated on MCF-10A, 4T1 and MDA-MB-231 cells by a 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide CCK-8 assay. It can be seen from Fig. S13† that COF@HGC@FEM showed negligible cytotoxicity on MCF-10A, indicating good biocompatibility to normal cells. Besides, Fig. 3C and D and S14A and B† showed that the 4T1 and MDA-MB-231 cells treated with COF show little side effect on cell viability. At pH 7.4, the treatment with COF@HGC (starvation therapy) or COF@HGC@FEM (synergistic therapy) displays certain cytotoxicity due to the endogenous H2O2 and a small amount of NO production. For the starvation therapy group at pH 6.8, about 37% and 35% of 4T1 and MDA-MB-231 cells are dead after incubation with COF@HGC at 200 μg mL−1 for 24 h. This implies that the COF@HGC has a moderate cell-killing ability on account of intracellular glucose consumption, which can inhibit the energy supply for cell proliferation. For the synergistic therapy group at pH 6.8, about 83% and 76% of 4T1 and MDA-MB-231 cells have been killed at the 200 μg mL−1 concentration due to the synergistic effect of GOx-induced starvation therapy and Hb-mediated NO gas therapy through cascade catalysis reaction. The results are in accordance with the enzymatic assay and identify the highly catalytic effect of COF@HGC@FEM in the weak acidic tumor microenvironment.The combined therapeutic efficacy was also visualized by live/dead cell fluorescence staining with calcein-AM/propidium iodide (PI). As depicted in Fig. 4A and B and S15A and B,† compared with the control group, the cells treated with COF present a similar green fluorescence signal of calcein-AM, implying that almost all cells are alive and have low cytotoxicity of COF nanosheets. In contrast, the cells treated with COF@HGC show some red fluorescence, indicating a moderate therapeutic efficiency due to cell starvation. Remarkably, the cancer cells treated with COF@HGC@FEM show bright red fluorescence, indicating the majority of cells have been killed. This directly reflects the excellent combined therapeutic performance. This result was further confirmed through flow cytometric apoptosis analysis. Fig. 4C shows that the apoptosis rates (including early and late apoptosis) of 4T1 cells were 1.94, 42.59 and 72.83% in COF, COF@HGC and COF@HGC@FEM groups, respectively, whereas it was 2.41% in the control group. The COF@HGC@FEM group showed the highest cell apoptosis ratio, which was 1.71-fold higher than that in the COF@HGC group (Fig. 4D). The abovementioned results suggest the biomimetic COF@HGC@FEM is favorable for antitumor application.
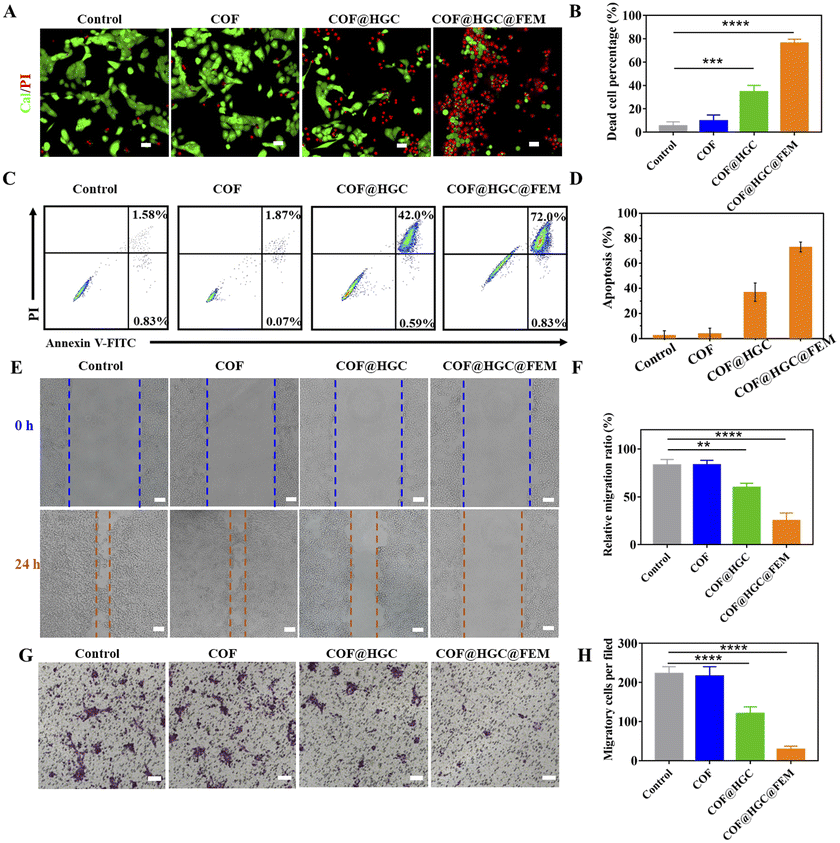 | ||
| Fig. 4 (A) CLSM images of 4T1 cells co-stained with calcein-AM and PI after being incubated with COF, COF@HGC or COF@HGC@FEM for 24 h, respectively. Scale bar = 50 μm. (B) Percentage of dead cells according to calcein-AM and PI staining. (C) Flow cytometric plots of 4T1 cell apoptosis after different treatments. (D) Quantitative analysis of 4T1 cell apoptosis from flow cytometry analysis. (E) Wound healing images and (F) corresponding wound migration ratio of 4T1 cells after being treated with COF, COF@HGC or COF@HGC@FEM for 24 h. Scale bar = 100 μm. (G) Microscopy images and (H) quantification of transwell migration number of 4T1 cells treated with COF, COF@HGC or COF@HGC@FEM for 24 h. Scale bar = 100 μm. Data are presented as the mean ± SD (n = 3). | ||
Considering that recurrence and metastasis are two critical factors affecting cancer curation efficacy, we further assessed the migration and invasion behavior of tumor cells via wound healing and transwell experiments. It can be seen from Fig. 4E and F and S15E and F† that the COF-treatment group presents a similar cell migration trend to that of the control group, manifesting the high migration feature of tumor cells and the low inhibition cell migration ability of COF. In contrast, the COF@HGC treated cells display a moderate migration ability, indicating the cell migration ability is suppressed to some extent due to the starvation therapy. After being treated with COF@HGC@FEM, the cells show the lowest migration rate, demonstrating that cell migration ability is almost inhibited due to the cellular energy supply being cut off and cytotoxicity arising from NO production. Next, the transwell test was employed to evaluate the invasion capacity of tumor cells. As shown in Fig. 4G and H and S15G and H,† the COF treatment cannot influence cell invasion, while the COF@HGC treatment inhibits cell invasion to a certain extent. Notably, treatment with COF@HGC@FEM almost completely inhibits cell invasion. All the above results highlight that COF@HGC@FEM is likely to be a promising combinative therapeutic platform for inhibiting the growth and migration of tumor cells.
COF@HGC@FEM induces repolarization of M2 to M1 in vitro
CPG loading was designed to modulate the tumor immunosuppression microenvironment by reprogramming M2 into M1 macrophages. We evaluated the macrophage state by morphology observation and specific phenotypic marker expression after different treatments to determine the feasibility. Specifically, M2 macrophages were obtained by stimulating RAW264.7 macrophages with interleukin 4 (IL-4). Then, we co-cultured M2 macrophages with pretreated 4T1 cells using a transwell system. 4T1 cells were seeded in the upper chamber and treated with COF, COF@HGC, or COF@HGC@FEM for 3 h, respectively. Afterward, the upper chamber was transferred to a 24-well plate with M2 macrophages seeded in the bottom chamber and co-cultured for another 24 h (Fig. 5A). The macrophages were stained with DIO and DAPI dye and their morphology was observed via CLSM. As shown in Fig. 5B, the cells treated with COF@HGC or COF@HGC@FEM group shows a round and flattened morphology, typical of M1 macrophages. As for the blank or COF treatment group, the elongated morphology reflects a representative of M2 macrophages.52 Next, the percentage of M1 and M2 macrophages was assessed using flow cytometry by labeling their specific phenotypic markers (M1 biomarker, CD80; M2 biomarker, CD206). As shown in Fig. 5C and D, compared to the control and COF-treated groups, the proportion of M2-type macrophages decreased to 39.3% in the COF@HGC group and 25.8% in the COF@HGC@FEM group. Conversely, the proportion of M1-type macrophages increased to 50.1% in the COF@HGC group and 63.1% in the COF@HGC@FEM group. These results demonstrate that nanocarriers containing CPG tended to transform the M2-type macrophage morphology to its M1-type counterparts through cytoskeleton rearrangement.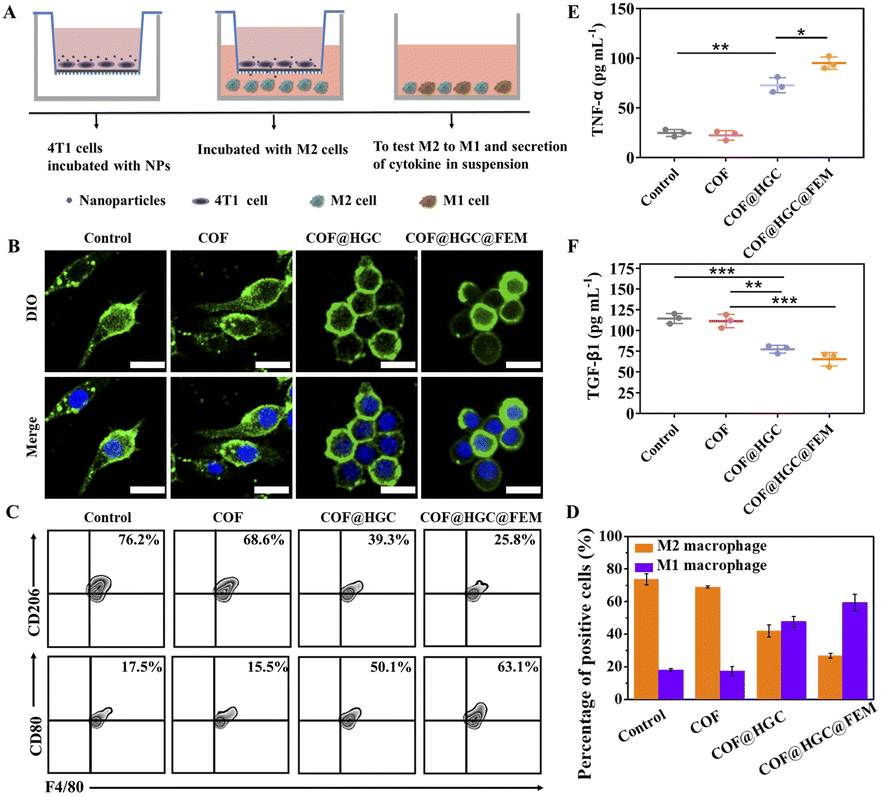 | ||
| Fig. 5 (A) Schematic illustration of co-culture 4T1 cells and M2 macrophages. (B) CLSM images of macrophages after being co-cultured with 4T1 cells treated with COF, COF@HGC, and COF@HGC@FEM, respectively. The cell nucleus (blue) and membrane (green) were stained with DAPI and DIO, respectively. Scale bar = 50 μm. (C) Flow cytometry plots of the M1 macrophage (F4/80+/CD80+) and M2 macrophage (F4/80+/CD206+) after various treatments. (D) Percentage of M1 and M2 macrophages according to flow cytometry analysis. (E) Cytokine levels of TNF-α and (F) TGF-β1 in cell supernatants detected by ELISA. Data are presented as the mean ± SD (n = 3). | ||
Macrophage polarization is often accompanied by cytokine secretion changes.53 The supernatant of the co-culture system was collected and detected by ELISA kits. Fig. 5E and F show that COF@HGC or COF@HGC@FEM can accelerate the pro-inflammatory cytokines secretion (TNF-α, M1 marker) and suppress anti-inflammatory cytokines secretion (TGF-β1, M2 marker). The COF group shows that the secretion of TNF-α and TGF-β1 is similar to that of the control due to the lack of CPG. In contrast, for COF@HGC and COF@HGC@FEM groups, the TNF-α level is significantly up-regulated and the TGF-β1 level is down-regulated. As such, these findings confirm the robust capability of COF@HGC@FEM for reduction of the M2 phenotype to the M1 phenotype to elevate the native immune ability and cancer immunotherapy efficiency.
In vivo fluorescence imaging
The successful synergistic therapy on TNBC cell models inspires us to evaluate the performance in vivo. To investigate the distribution of COF and COF@HGC@FEM in vivo, the 4T1 tumor-bearing mice were intravenously injected with nanocarriers and the fluorescence was monitored using an IVIS spectrum system at different times. As shown in Fig. 6A, for the administration of COF, the fluorescence signals in the tumor site gradually increase and reach their maximum after 24 h. After that, the fluorescence intensity is attenuated and nearly disappears after 48 h. Comparatively, for the COF@HGC@FEM treatment group, the signal in the tumor site can be observed after 2 h injection and reaches its maximum at 24 h. After 48 h, it still maintains a high level. It is worth noting that the fluorescence intensity of the COF@HGC@FEM group is stronger than that of the COF group at each time point. This implies that the encapsulation of FEM can extend its blood circulation time and enable highly specific targeting of tumor tissue because of FA. The results also indicate that COF@HGC@FEM can be used as an in vivo self-reporter for biodistribution. After post-injection for 24 h, these mice were euthanized, then the tumors and major organs were harvested for in vitro fluorescence imaging as shown in Fig. 6B. Compared with the COF group, the fluorescence signals in the liver and lung are reduced. In contrast, they are enhanced in the tumor site for the COF@HGC@FEM administration group (Fig. 6C and D). This also reveals the excellent tumor-targeting ability of COF@HGC@FEM. Furthermore, the pharmacokinetics of COF@HGC@FEM were explored by measuring the fluorescence intensity of COF@HGC@FEM in the bloodstream. As shown in Fig. S16,† the blood circulation curve can be well fitted by a two-component model, displaying a blood distribution half-life (t1/2α) of 0.027 h and a clearance half-life (t1/2β) of COF@HGC@FEM of 3.05 h, respectively.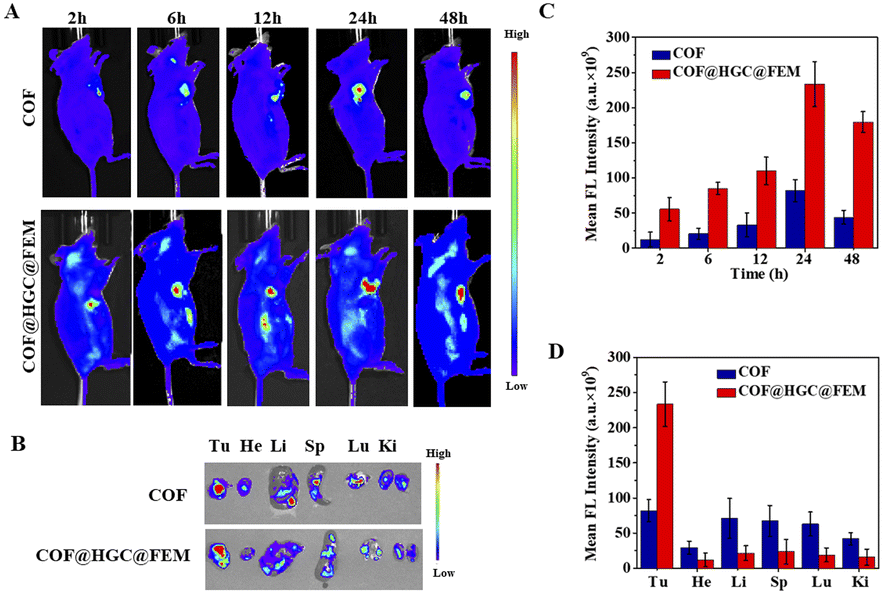 | ||
| Fig. 6 In vivo fluorescence imaging and biodistribution. (A) Fluorescence images of 4T1 tumor-bearing mice after post-injection of COF or COF@HGC@FEM at different time points. (B) Ex vivo fluorescence imaging of tumor tissues and major organs after 24 h post-injection. (C) The mean fluorescence intensity in tumor sites at different time points. (D) The mean fluorescence intensity in tumor sites and major organs after 24 h post-injection (tumor, Tu; heart, He; liver, Li; spleen, Sp; lung, Lu; kidney, Ki). Data are presented as the mean ± SD (n = 3). | ||
Before in vivo therapy, we evaluated the hemolytic performance of COF and COF@HGC@FEM. Generally, hemolysis ratios below 5% are considered as good hemocompatibility.54 As shown in Fig. S17A and B,† the hemolysis rate of COF increases with the up-regulated incubation concentration and reaches about 11% after incubation with 200 μg mL−1 COF for 8 h. As for the COF@HGC@FEM group, there is no obvious hemolysis (<5%) at 200 μg mL−1. It suggests the FEM coating can guarantee excellent biocompatibility and prolong the circulation time in vivo.
Synergistic antitumor treatment in vivo
The 4T1 tumor-bearing mice were divided into 6 groups randomly, and received the following treatment every two days with a therapy period of 2 weeks, (1) PBS (100 μL); (2) COF (100 μL, 1.0 mg mL−1); (3) CPG (100 μL, 23 μg mL−1); (4) COF@GOx (100 μL, 1.0 mg mL−1); (5) COF@HGC (100 μL, 1.0 mg mL−1); (6) COF@HGC@FEM (100 μL, 1.0 mg mL−1). The body weight and tumor size were monitored and recorded every two days. As illustrated in Fig. 7A–D, PBS and COF groups have little effect on tumor inhibition and the tumor grows rapidly. As a representative immune therapy, the CPG treatment group displays limited efficiency in inhibiting tumor growth due to its rapid elimination in vivo. For starvation therapy, the COF@GOx group exhibits moderate therapeutic ability with the merits of the EPR effect of the tumor region. As for the combination of starvation and immune therapy, the COF@HGC group shows better therapeutic ability and tumor ablation relative to COF@GOx groups. As expected, the mice injected with COF@HGC@FEM display the best therapy efficiency as evidenced by the smallest tumor volume and tumor weight (Fig. 7A and B). This was attributed to the synergetic therapy efficiency of the cascade reaction of GOx-mediated starvation therapy, Hb-mediated NO gas therapy and CPG-mediated immune therapy. Fig. 7D shows the representative tumor tissues of different treated groups at 14 d. The results also indicate the superiority of synergistic starvation/NO gas/CPG-mediated immunotherapy therapy. In addition, mice's body weights from all therapy groups show no significant changes, implying negligible side effects during the treatment process (Fig. 7C). Hematoxylin and eosin (H&E) and Ki-67 immunohistochemical stain experiments were used to evaluate the tissue apoptosis and proliferation, respectively. As shown in Fig. 7E, compared with other therapy groups, the tumor tissue treated with COF@HGC@FEM displays the worst damage due to tumor cells' severe necrosis and apoptosis. Furthermore, Ki-67 staining reveals that the COF@HGC@FEM group exhibits the lowest expression level of proliferative biomarker, suggesting that COF@HGC@FEM can strongly suppress cancer cell proliferation. Moreover, the H&E staining images of major organs from tumor-bearing mice reveal negligible pathological damage or abnormalities after different treatments (Fig. S18†). To further assess the biosecurity, the blood biochemical parameters of the mice were tested after therapy. As shown in Fig. S19,† liver function parameters (alanine transaminase (ALT) and aspartate aminotransferase (AST)) and kidney function parameters (blood urea nitrogen (BUN) and creatinine (CREA)) maintained normal levels in all treated groups, confirming no apparent hepatic or renal toxicity. Thus, the bioengineered therapeutic nanoplatform displays prominent biosafety to implement the cooperative anticancer treatment through starvation, NO gas and immune therapy.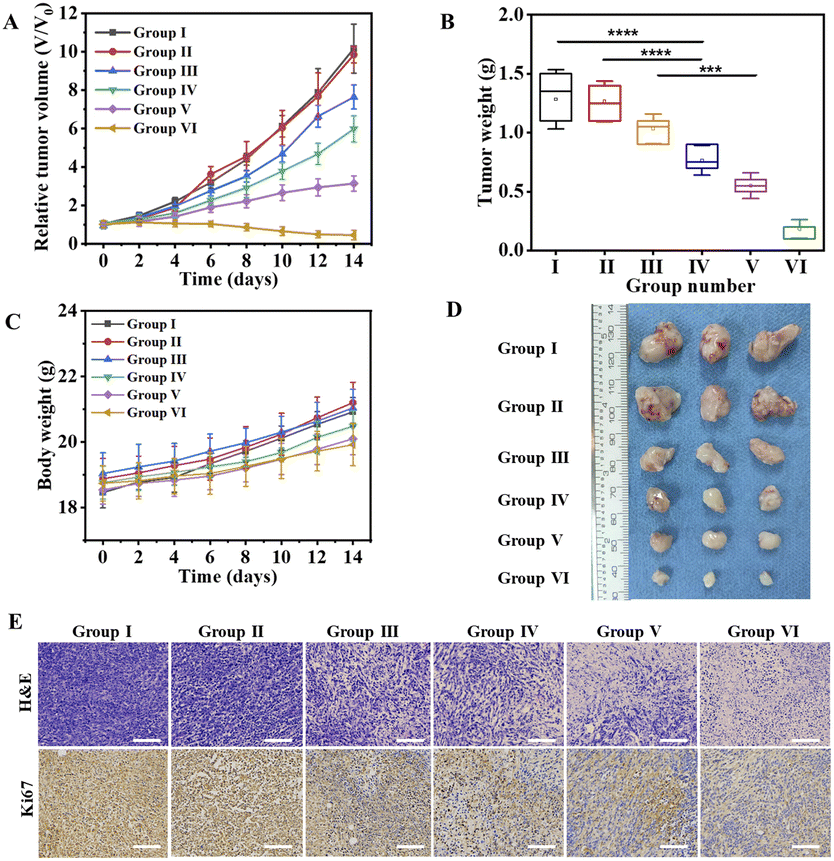 | ||
| Fig. 7 In vivo tumor inhibitory effects of COF@HGC@FEM. (A) Tumor growth curves during 14 days after receiving different treatments (Group I: PBS, Group II: COF, Group III: CPG, Group IV: COF@GOx, Group V: COF@HGC, Group VI: COF@HGC@FEM). (B) Relative tumor weight at the 14th day collected from mice after different treatments. (C) The body weight of mice recorded during 14 days after receiving different treatments. (D) Photographs of tumor excised from the mice of different groups at the 14th day. (E) Immunohistochemistry staining images of H&E and Ki-67 in tumor tissues after different treatments. Scale bar = 100 μm. Data are presented as the mean ± SD (n = 3). (*p < 0.05, **p < 0.01, ***p < 0.001). | ||
To evaluate the reversion of the TAM phenotype after various treatments in vivo, we assessed the number of M1 and M2 macrophages and the cytokine level in the tumor tissues. At first, the TAM phenotypes were classified by flow cytometry through the biomarker of M1 (CD11b+F4/80+CD80+) and M2 (CD11b+F4/80+CD206+) macrophages, respectively. The number of M1 macrophages is significantly increased, while the number of M2 macrophages is reduced dramatically after being treated with COF@HGC or COF@HGC@FEM (Fig. 8A). Fig. 8B shows the ratios of M1 to M2 after different treatments. The results reveal that most of the TAM phenotype is repolarized from M2 to M1 after being treated with substances containing CPG. As expected, the nanoplatform modified with FEM displays the highest proportion of M1-TAMs due to the excellent tumor-killing performance, which can accelerate the release of more CPG to simulate the immune effect. However, for the COF or COF@GOx group, the depletion of M2-TAMs is relatively weak, which might be related to the shortage of immunological CPG. While for the free CPG group, the immunotherapy efficiency is limited due to rapid systemic clearance and low cellular uptake of naked CpG.55 Furthermore, immunofluorescence staining also examined M1 and M2 macrophages in tumor tissue. As shown in Fig. 8C, the polarization of M2 to M1 macrophages is reflected by the enhancement of green fluorescence of the M1-related biomarker (CD80) and the reduction of red fluorescence of the M2-related biomarker (CD206). After injection of COF@HGC or COF@HGC@FEM, the number of CD206+ cells is significantly down-regulated and the number of CD80+ cells is up-regulated, which further identifies the effect of immunotherapy induced by CPG.
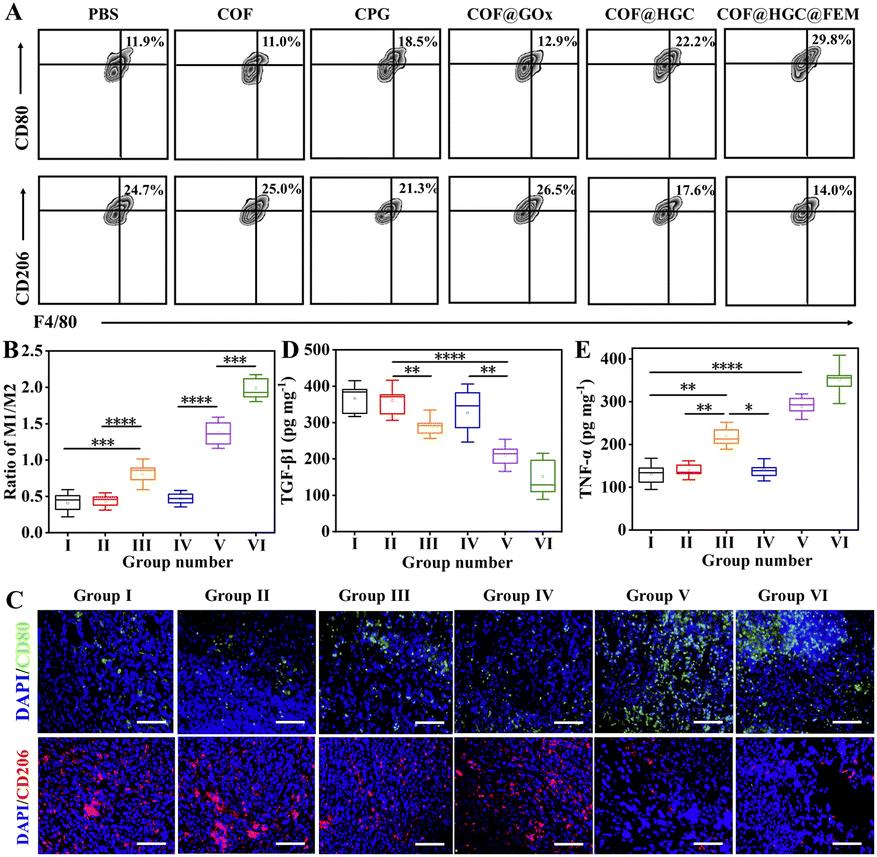 | ||
| Fig. 8 In vivo reprogramming of TAM and anti-tumor immune response induced by different treatments. (A) Flow cytometry analysis of the M1-macrophage (F4/80+CD11b+CD80+) and M2-macrophage (F4/80+CD11b+CD206+) in tumors isolated from the mice after various treatments. Data were gated on CD11b+ cells. (B) Ratios of M1 to M2 in tumors after different treatments according to flow cytometry analysis. (C) Representative immunofluorescence staining images of CD80 (green) and CD206 (red) in tumor tissues from different groups. Cell nuclei were labeled with DAPI. Scale bar = 100 μm. Cytokine levels of (D) TNF-α and (E) TGF-β1 in tumors after different treatments tested by ELISA. Data are presented as the mean ± SD (n = 3) (*p < 0.05, **p < 0.01, ***p < 0.001). | ||
Next, we investigated the secretion of pro-inflammatory cytokines and anti-inflammatory cytokines (TNF-α, TGF-β1) in tumors. As displayed in Fig. 8D and E, after administration with COF@HGC and COF@HGC@FEM, the level of TNF-α is increased while the level of TGF-β1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 43 is decreased compared with other groups. All these results suggest that the CPG-loaded platform possesses robust regulation capability in reprogramming TAM, excellent anti-tumor immune responses and a synergistic antitumor effect toward TNBC.
43 is decreased compared with other groups. All these results suggest that the CPG-loaded platform possesses robust regulation capability in reprogramming TAM, excellent anti-tumor immune responses and a synergistic antitumor effect toward TNBC.
Conclusions
In summary, a biomimetic multifunctional nanoplatform was successfully developed for the imaging and synergistic treatment of TNBC. Through FEM wrapping and HGC immobilization, the biomimetic COF@HGC@FEM can realize combination tumor treatment through starvation/NO gas/CPG-directed immunization therapy. Fluorescence signals from COF can be used as a self-reporter for bioimaging. The as-prepared COF@HGC@FEM nanoplatform shows an ingenious cascade catalytic reaction to cut off the energy supply of tumors and produce a high concentration of NO to suppress tumor cell proliferation. Taking advantage of FEM modification, COF@HGC@FEM can be specifically accumulated in the tumor site, further improving antitumor efficiency. Especially, CPG transported by COF@HGC@FEM can effectively repolarize TAM-associated M2 macrophages into M1 macrophages. Overall, both in vitro and in vivo experiments show that the COF@HGC@FEM exhibits a remarkable synergistic treatment efficacy for the inhibition of TNBC. Therefore, it is believed that this “three-in-one” therapeutic nanoplatform may open a new window to ablate breast tumors in the clinic.Ethical statement
All experimental procedures on mice were performed in compliance with the relevant laws and the Guidelines of the Animal Experimental Ethics Committee of East China Normal University. All mice experiments have been approved by the committee (No. m20210224).Data availability
All experimental supporting data and procedures are available in the ESI.†Author contributions
Fang Yuan: conceptualization, methodology, investigation, writing – original draft. Cuiling Zhang: supervision, conceptualization, writing – review & editing, funding acquisition. Xianzhu Luo: validation, investigation. Shasha Cheng: investigation, visualization. Yingxin Zhu: investigation. Yuezhong Xian: supervision, writing – review & editing, funding acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (22274054, 21974050 and 11727810), the Natural Science Foundation of Shanghai (20ZR1418000) and the Fundamental Research Funds for the Central Universities.Notes and references
- Y. Eralp, D. Derin, Y. Ozluk, E. Yavuz, N. Guney, P. Saip, M. Muslumanoglu, A. Igci, S. Kucucuk, M. Dincer, A. Aydiner and E. Topuz, Ann. Oncol., 2008, 19, 669–674 CrossRef CAS PubMed.
- S. Dawood, Drugs, 2010, 70, 2247–2258 CrossRef CAS PubMed.
- P. Sharma, Oncologist, 2016, 21, 1050–1062 CrossRef PubMed.
- G. Bianchini, J. M. Balko, I. A. Mayer, M. E. Sanders and L. Gianni, Nat. Rev. Clin. Oncol., 2016, 13, 674–690 CrossRef CAS PubMed.
- M. Zhao, X. Yang, H. Fu, C. Chen, Y. Zhang, Z. Wu, Y. Duan and Y. Sun, ACS Appl. Mater. Interfaces, 2021, 13, 32763–32779 CrossRef CAS PubMed.
- N. Verma, Y. Vinik, A. Saroha, N. U. Nair, E. Ruppin, G. Mills, T. Karn, V. Dubey, L. Khera, H. Raj, F. Maina and S. Lev, Sci. Adv., 2020, 6, eaba8968 CrossRef CAS PubMed.
- H. Cheng, R. F. Luo and Y. B. Wang, ACS Appl. Mater. Interfaces, 2020, 12, 41113–41126 CrossRef PubMed.
- M. M. Li, Y. S. Ning, J. L. Chen, X. C. Duan, N. Song, D. Ding, X. C. Su and Z. L. Yu, Nano Lett., 2019, 19, 7965–7976 CrossRef CAS PubMed.
- C. F. Deng, Q. Zhang, M. D. Jia, J. Zhao, X. Sun, T. Gong and Z. R. Zhang, Adv. Sci., 2019, 6, 1801868 CrossRef PubMed.
- K. Li, C. C. Lin, Y. He, L. Lu, K. Xu, B. L. Tao, Z. Xia, R. Zeng, Y. L. Mao, Z. Luo and K. Y. Cai, ACS Nano, 2020, 14, 14164–14180 CrossRef CAS PubMed.
- Z. Wang, B. Liu, Q. Q. Sun, L. L. Feng, F. He, P. A. Yang, S. L. Gai, Z. W. Quan and J. Lin, ACS Nano, 2021, 15, 12342–12357 CrossRef CAS PubMed.
- L. Wang, K. L. Ding, C. X. Zheng, H. F. Xiao, X. X. Liu, L. L. Sun, R. Omer, Q. H. Feng and Z. Z. Zhang, Nano Lett., 2020, 20, 6272–6280 CrossRef CAS PubMed.
- M. S. Lohse and T. Bein, Adv. Funct. Mater., 2018, 28, 1705553 CrossRef.
- C. Y. Liao and S. J. Liu, J. Mater. Chem. B, 2021, 9, 6116–6128 RSC.
- L. L. Yang, L. Zhang, S. C. Wan, S. Wang, Z. Z. Wu, Q. C. Yang, Y. Xiao, H. X. Deng and Z. J. Sun, Adv. Funct. Mater., 2021, 31, 2103056 CrossRef CAS.
- Q. Guan, L. L. Zhou, Y. A. Li, W. Y. Li, S. M. Wang, C. Song and Y. B. Dong, ACS Nano, 2019, 13, 13304–13316 CrossRef CAS PubMed.
- Q. Guan, L. L. Zhou, F. H. Lv, W. Y. Li, Y. A. Li and Y. B. Dong, Angew. Chem., Int. Ed., 2020, 59, 18042–18047 CrossRef CAS PubMed.
- Q. Guan, D. D. Fu, Y. A. Li, X. M. Kong, Z. Y. Wei, W. Y. Li, S. J. Zhang and Y. B. Dong, iScience, 2019, 14, 180 CrossRef CAS PubMed.
- S. J. Gan, X. N. Tong, Y. Zhang, J. H. Wu, Y. Q. Hu and A. H. Yuan, Adv. Funct. Mater., 2019, 29, 1902757 CrossRef CAS.
- S. N. Liu, C. L. Hu, Y. Liu, X. Y. Zhao, M. L. Pang and J. Lin, Chem.–Eur. J., 2019, 25, 4315–4319 CrossRef CAS PubMed.
- P. Gao, M. Wang, Y. Chen, W. Pan, P. Zhou, X. Wan, N. Li and B. Tang, Chem. Sci., 2020, 11, 6882–6888 RSC.
- K. Wang, Z. Zhang, L. Lin, K. Hao, J. Chen, H. Y. Tian and X. S. Chen, ACS Appl. Mater. Interfaces, 2019, 11, 39503–39512 CrossRef CAS PubMed.
- B. J. Smith, L. R. Parent, A. C. Overholts, P. A. Beaucage, R. P. Bisbey, A. D. Chavez, N. Hwang, C. Park, A. M. Evans, N. C. Gianneschi and W. R. Dichtel, ACS Cent. Sci., 2017, 3, 58–65 CrossRef CAS PubMed.
- S. Chandra, S. Kandambeth, B. P. Biswal, B. Lukose, S. M. Kunjir, M. Chaudhary, R. Babarao, T. Heine and R. Banerjee, J. Am. Chem. Soc., 2013, 135, 17853–17861 CrossRef CAS PubMed.
- I. Berlanga, M. L. Ruiz-Gonzalez, J. M. Gonzalez-Calbet, J. L. Fierro, R. Mas-Balleste and F. Zamora, Small, 2011, 7, 1207–1211 CrossRef CAS PubMed.
- A. Lin, Y. Liu, X. Zhu, X. Chen, J. Liu, Y. Zhou, X. Qin and J. Liu, ACS Nano, 2019, 13, 13965–13984 CrossRef CAS PubMed.
- P. A. Oldenborg, A. Zheleznyak, Y. F. Fang, C. F. Lagenaur, H. D. Gresham and F. P. Lindberg, Science, 2000, 288, 2051 CrossRef CAS PubMed.
- M. Z. Zou, W. L. Liu, F. Gao, X. F. Bai, H. S. Chen, X. Zeng and X. Z. Zhang, Adv. Mater., 2019, 31, 1904495 CrossRef CAS PubMed.
- W. Fan, B. C. Yung and X. Chen, Angew. Chem., Int. Ed., 2018, 57, 8383–8394 CrossRef CAS PubMed.
- Q. Yang, H. Yin, T. Xu, D. Zhu, J. Yin, Y. Chen, X. Yu, J. Gao, C. Zhang, Y. Chen and Y. Gao, Small, 2020, 16, 1906814 CrossRef CAS PubMed.
- Z. Yang, D. Gao, X. Q. Guo, L. Jin, J. J. Zheng, Y. Wang, S. J. Chen, X. W. Zheng, L. Zeng, M. Guo, X. C. Zhang and Z. M. Tian, ACS Nano, 2020, 14, 17442–17457 CrossRef CAS PubMed.
- T. Yang, A. N. Zelikin and R. Chandrawati, Small, 2020, 16, 1907635 CrossRef CAS PubMed.
- S. Liu, Y. Zhang, M. Li, L. Xiong, Z. Zhang, X. Yang, X. He, K. Wang, J. Liu and S. Mann, Nat. Chem., 2020, 12, 1165–1173 CrossRef CAS PubMed.
- N. Yang, F. Gong and L. Cheng, Chem. Sci., 2022, 13, 1883–1898 RSC.
- Y. Su, X. Zhang, L. Lei, B. Liu, S. Wu and J. Shen, ACS Appl. Mater. Interfaces, 2021, 13, 12960–12971 CrossRef CAS PubMed.
- P. S. Low, W. A. Henne and D. D. Doorneweerd, Acc. Chem. Res., 2008, 41, 120–129 CrossRef CAS PubMed.
- W. Ngamcherdtrakul, M. Reda, M. A. Nelson, R. Wang, H. Y. Zaidan, D. S. Bejan, N. H. Hoang, R. S. Lane, S. W. Luoh, S. A. Leachman, G. B. Mills, J. W. Gray, A. W. Lund and W. Yantasee, Adv. Mater., 2021, 33, 2100628 CrossRef CAS PubMed.
- Y. Cao, S. Ding, L. Zeng, J. Miao, K. Wang, G. Chen, C. Li, J. Zhou, X. W. Bian and G. Tian, ACS Appl. Mater. Interfaces, 2021, 13, 53504–53518 CrossRef CAS PubMed.
- L. Chen, L. L. Zhou, C. H. Wang, Y. Han, Y. L. Lu, J. Liu, X. C. Hu, T. M. Yao, Y. Lin, S. J. Liang, S. Shi and C. Y. Dong, Adv. Mater., 2019, 31, 1904997 CrossRef CAS PubMed.
- M. Sylvestre, C. A. Crane and S. H. Pun, Adv. Mater., 2020, 32, 1902007 CrossRef CAS PubMed.
- Y. Li, J. Lin, P. Wang, Q. Luo, H. Lin, Y. Zhang, Z. Hou, J. Liu and X. Liu, ACS Nano, 2019, 13, 12912–12928 CrossRef CAS PubMed.
- F. Yuan, Y. Kong, J. You, C. Zhang and Y. Xian, ACS Appl. Mater. Interfaces, 2021, 13, 51351–51361 CrossRef CAS PubMed.
- M. Li, H. Fang, Q. Liu, Y. Gai, L. Yuan, S. Wang, H. Li, Y. Hou, M. Gao and X. Lan, Biomater. Sci., 2020, 8, 1802–1814 RSC.
- P. Yuan, G. Dou, T. Liu, X. Guo, Y. Bai, D. Chu, S. Liu, X. Chen and Y. Jin, Biomaterials, 2021, 275, 120956 CrossRef CAS PubMed.
- Y. Tazhbayev, O. Mukashev, M. Burkeev and J. Kreuter, Pharmaceutics, 2019, 11, 410 CrossRef CAS PubMed.
- T. Rolim, J. Cancino and V. Zucolotto, Biomed. Microdevices, 2015, 17, 3 CrossRef PubMed.
- X. Jin, G. Li, T. Xu, L. Su, D. Yan and X. Zhang, Biosens. Bioelectron., 2022, 196, 113760 CrossRef CAS PubMed.
- D. Li, T. Chen, Y. Zhang, Y. Xu and H. Niu, Adv. Healthcare Mater., 2021, 10, 2100716 CrossRef CAS PubMed.
- Y. Xu, S. Y. Liu, L. Zeng, H. Ma, Y. Zhang, H. Yang, Y. Liu, S. Fang, J. Zhao, Y. Xu, C. R. Ashby Jr, Y. He, Z. Dai and Y. Pan, Adv. Mater., 2022, 34, 2204733 CrossRef CAS PubMed.
- L. Q. Zhang, F. Y. Su, S. Buizer, X. X. Kong, F. Lee, K. Day, Y. Q. Tian and D. R. Meldrum, Chem. Commun., 2014, 50, 6920–6922 RSC.
- E. Gholibegloo, T. Mortezazadeh, F. Salehian, H. Forootanfar, L. Firoozpour, A. Foroumadi, A. Ramazani and M. Khoobi, J. Colloid Interface Sci., 2019, 556, 128–139 CrossRef CAS PubMed.
- M. Chen, Y. Miao, K. Qian, X. Zhou, L. Guo, Y. Qiu, R. Wang, Y. Gan and X. Zhang, Nano Lett., 2021, 21, 6031–6041 CrossRef CAS PubMed.
- M. Chang, Z. Hou, D. Jin, J. Zhou, M. Wang, M. Wang, M. Shu, B. Ding, C. Li and J. Lin, Adv. Mater., 2020, 32, 2004647 CrossRef CAS PubMed.
- Y. T. Su, X. C. Zhang, G. H. Ren, Z. C. Zhang, Y. Liang, S. S. Wu and J. Shen, Chem. Eng. J., 2020, 400, 125949 CrossRef CAS.
- H. Chu, J. Zhao, Y. Mi, Z. Di and L. Li, Nat. Commun., 2019, 10, 2839 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc02022c |
| This journal is © The Royal Society of Chemistry 2023 |