
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Halogen bonding with carbon: directional assembly of non-derivatised aromatic carbon systems into robust supramolecular ladder architectures†
Jogirdas
Vainauskas
 ab,
Tristan H.
Borchers
ab,
Mihails
Arhangelskis
c,
Laura J.
McCormick McPherson
d,
Toni S.
Spilfogel
b,
Ehsan
Hamzehpoor
b,
Filip
Topić
b,
Simon J.
Coles
d,
Dmytro F.
Perepichka
b,
Christopher J.
Barrett
b and
Tomislav
Friščić
*ab
ab,
Tristan H.
Borchers
ab,
Mihails
Arhangelskis
c,
Laura J.
McCormick McPherson
d,
Toni S.
Spilfogel
b,
Ehsan
Hamzehpoor
b,
Filip
Topić
b,
Simon J.
Coles
d,
Dmytro F.
Perepichka
b,
Christopher J.
Barrett
b and
Tomislav
Friščić
*ab
aSchool of Chemistry, University of Birmingham, Edgbaston, Birmingham B15 2TT, UK. E-mail: t.friscic@bham.ac.uk
bDepartment of Chemistry, McGill University, 801 Sherbrooke St. W., H3A 0B8 Montreal, Canada
cFaculty of Chemistry, University of Warsaw, 1 Pasteura Street, Warsaw 02-093, Poland
dEPSRC National Crystallography Service, School of Chemistry, University of Southampton, Highfield, Southampton, UK
First published on 24th October 2023
Abstract
Carbon, although the central element in organic chemistry, has been traditionally neglected as a target for directional supramolecular interactions. The design of supramolecular structures involving carbon-rich molecules, such as arene hydrocarbons, has been limited almost exclusively to non-directional π-stacking, or derivatisation with heteroatoms to introduce molecular assembly recognition sites. As a result, the predictable assembly of non-derivatised, carbon-only π-systems using directional non-covalent interactions remains an unsolved fundamental challenge of solid-state supramolecular chemistry. Here, we propose and validate a different paradigm for the reliable assembly of carbon-only aromatic systems into predictable supramolecular architectures: not through non-directional π-stacking, but via specific and directional halogen bonding. We present a systematic experimental, theoretical and database study of halogen bonds to carbon-only π-systems (C–I⋯πC bonds), focusing on the synthesis and structural analysis of cocrystals with diversely-sized and -shaped non-derivatised arenes, from one-ring (benzene) to 15-ring (dicoronylene) polycyclic atomatic hydrocarbons (PAHs), and fullerene C60, along with theoretical calculations and a systematic analysis of the Cambridge Structural Database. This study establishes C–I⋯πC bonds as directional interactions to arrange planar and curved carbon-only aromatic systems into predictable supramolecular motifs. In >90% of herein presented structures, the C–I⋯πC bonds to PAHs lead to a general ladder motif, in which the arenes act as the rungs and halogen bond donors as the rails, establishing a unique example of a supramolecular synthon based on carbon-only molecules. Besides fundamental importance in the solid-state and supramolecular chemistry of arenes, this synthon enables access to materials with exciting properties based on simple, non-derivatised aromatic systems, as seen from large red and blue shifts in solid-state luminescence and room-temperature phosphorescence upon cocrystallisation.
Introduction
Polycyclic aromatic hydrocarbons (PAHs) are an extensive class of carbon-based molecules, estimated to hold 10–15% of all carbon in the known universe.1–3 PAH-based molecules are ubiquitous in the design of organic electronics4,5 and organic light emitting diodes (OLEDs).6,7 While the arrangement of PAH units in solids is of critical importance for their optical and electronic properties,8 controlling the assembly of PAHs in crystalline materials is a persistent challenge of organic solid-state chemistry.9 In most cases, guiding the arrangement of PAH units in the solid state requires derivatisation, either to introduce sterically demanding groups that modify molecular packing,10 or to introduce recognition sites for the formation of multi-component crystals (cocrystals) by directional interactions such as hydrogen (HB)11–13 or halogen bonding (XB).14Such derivatisation strategies are, however, not applicable to the assembly of pristine, non-derivatised PAHs. Indeed, while carbon is central to organic chemistry, it is rarely considered a target of supramolecular recognition. Directional interactions such as HB15 and XB16–18 are generally regarded as insufficiently robust to enable predictable, directional assembly of carbon-only π-systems. As a result, approaches to molecular recognition, and in particular to cocrystallisation, of PAHs have traditionally been limited to non-directional face-to-face π–π stacking.19 This presents the design of supramolecular architectures based on directional assembly of carbon-only systems as an unsolved fundamental problem of solid-state supramolecular chemistry, and of the supramolecular chemistry of carbon in general.20–22
Halogen bonding has emerged as a versatile directional interaction in the crystalline solid state, offering access to a wider range of acceptor atoms than seen for hydrogen bonding.23 Scattered structural reports show that halogen bonds can form between electron-deficient iodine atoms as donors and flat, electron-rich π-systems, such as anilines, as acceptors.24–34 While cocrystals exhibiting individual XBs to carbon have been reported or theoretically studied, the possibility of using such interactions for the predictable assembly of supramolecular architectures based on PAHs has not been established.35–42 Recently,43 we observed that cocrystals of azulene with XB donors 1,4-diiodotetrafluorobenzene (tfib) or trans-octafluoro-4,4′-diiodoazobenzene (ofiab) (Fig. 1b and c) exhibit a ladder-like halogen-bonded motif involving aromatic carbon, in which the PAHs act as the rungs and the XB donors as rails, identical to the motif seen in a cocrystal of naphthalene30 with 14tfib. This unexpected observation of the same C–I⋯πC ladder-like motif in three different cocrystals of naphthalene and azulene suggested an unexpected role for XB as a unique, possibly general tool for directional assembly of non-derivatised PAHs – contrasting the traditionally relied upon non-directional π-stacking.
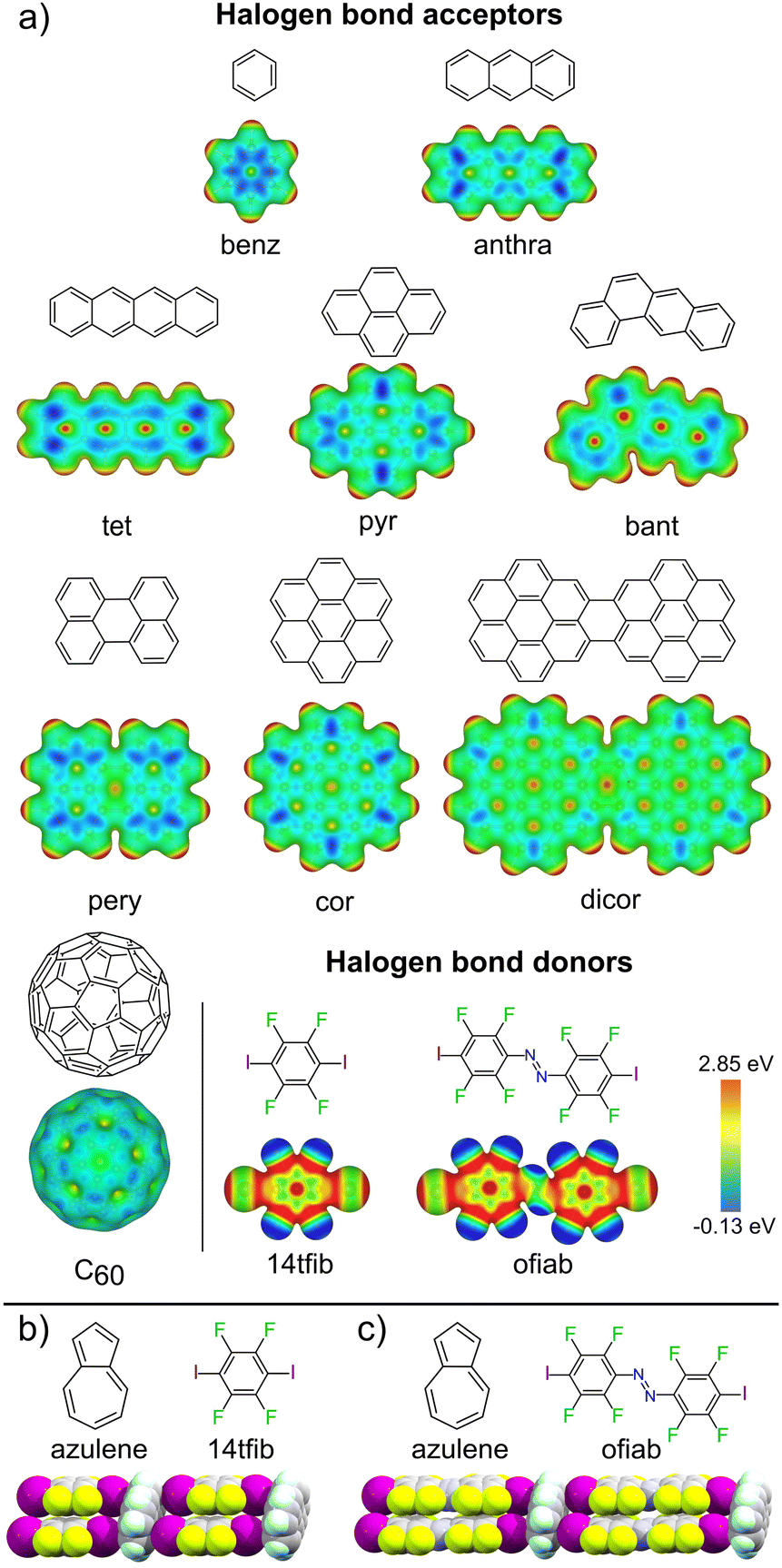 | ||
| Fig. 1 (a) Halogen bond acceptor and donor molecules explored in this work, along with corresponding electrostatic surface potential (ESP) maps, with isosurfaces plotted at 0.01 a.u. Fragments of the halogen-bonded C–I⋯πC supramolecular ladder seen in the cocrystals of: (b) (azulene)(14tfib)2 and (c) (azulene)(ofiab)2 along with corresponding molecular diagrams.43 | ||
We now provide an extensive and systematic study that demonstrates halogen bonding as a long-overlooked, reliable tool for the directional assembly of non-derivatised carbon-only aromatic systems. Our study, addressing diversely-shaped and -sized PAHs, as well as fullerene C60 (Fig. 1a), reveals the persistent formation of cocrystals based on a highly reproducible44–46 supramolecular ladder-like motif (Fig. 1b, c, and 2) of C–I⋯πC halogen bonds. The robustness of this motif presents it as a unique example of a supramolecular synthon47 for the assembly of exclusively carbon-based aromatic systems through directional interactions, independent of molecular size, shape, or presence of π⋯π or C–H⋯π contacts. The presented set of cocrystals involving different combinations of 9 XB acceptors and 2 donors, along with theoretical calculations and a systematic analysis of the Cambridge Structural Database (CSD),48 establishes a different paradigm for the crystal engineering based on aromatic carbon: not via surface-based π-stacking, but by directional halogen bonding. The potential of this approach is evident by luminescence studies of select cocrystals based on pyrene, coronene and perylene, demonstrating extensive modification of emission properties due to halogen bond-directed assembly.
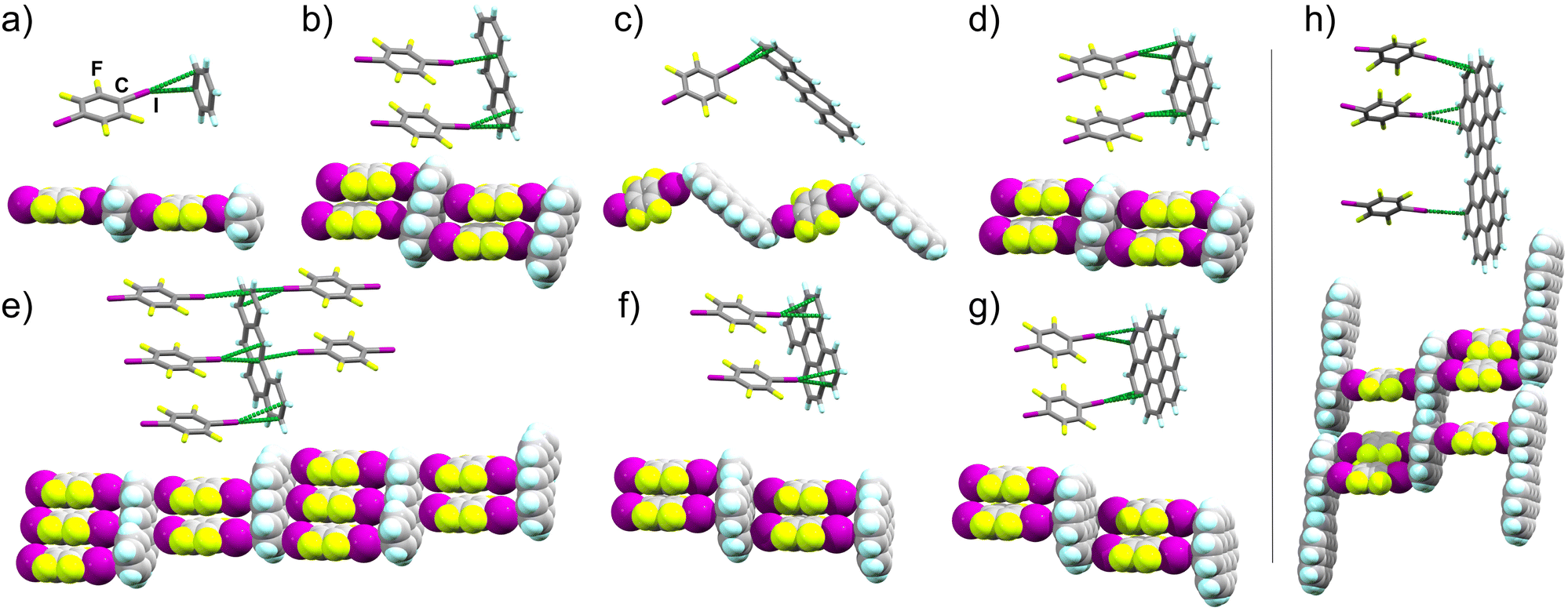 | ||
| Fig. 2 Stick model representation of halogen-bonded fragments in explored 14tfib cocrystals, with extended motifs shown using space-filling models: (a) (benz)(14tfib) (CCDC code 2240199), (b) (anthra)(14tfib)2 (CCDC code 2249511, also CSD JEJXOP),55 (c) (tet)(14tfib) (CCDC code 2240203), (d) (pyr)(14tfib)2 (CCDC code 2240200), (e) (bant)2(14tfib)5 (CCDC code 2281269), (f) (pery)(14tfib)2 (CCDC code 2240202), (g) (cor)(14tfib)2 (CCDC code 2240204), (h) (dicor)(14tfib)3 (CCDC code 2240207). The I⋯Cπ intermolecular distances shorter than the sum of van der Waals radii for iodine and carbon atoms (3.68 Å) are shown as green dotted lines.47 | ||
Results and discussion
Linear arenes (acenes)
Our first target was cocrystallisation of 14tfib with benzene (benz), the smallest possible arene that could be addressed in this study. Dissolving 14tfib in benz followed by slow evaporation produced large, colorless plate-like crystals. Removal of the crystals from the crystallisation vessel at room temperature, however, resulted in immediate degradation, evident by a loss of crystal transparency. Cooling the sample in dry ice enabled successful handling of the crystal and collection of X-ray single crystal diffraction data at 180 K (see ESI, Table S1†). Structure determination revealed that the crystals exhibit the composition (benz)(14tfib), based on one-dimensional (1D) supramolecular chains of alternating benz and 14tfib molecules, held by C–I⋯πC halogen bonds between 14tfib iodine atoms and each face of benz molecules (Fig. 2a). Specifically, each 14tfib molecule forms multiple short I⋯πC halogen-bonding contacts to the adjacent benz molecules, with intermolecular I⋯C distances (dI⋯C) of 3.48(2) Å and 3.51(2) Å, with the corresponding C–I⋯C angles (∠C–I⋯C) of 165° and 166°, respectively (Table 1, also ESI Table S2.†). The dI⋯C separations are shorter than the expected sum of van der Waals radii (3.68 Å) for carbon and iodine.49The simplicity of components makes it surprising that (benz)(14tfib) was not reported previously. The formation of halogen-bonded cocrystals with benz, however, is not without precedent, as 1D motifs similar to that in (benz)(14tfib) have been found with Br2 (ref. 50) and 1,4-diiodotetrachlorobenzene as XB donors.51 While the dI⋯C distances in (benz)(14tfib) are ∼5% shorter than the sum of corresponding van der Waals radii, the shortest distance between the iodine atom of a 14tfib donor and a benz acceptor molecule is to the centroid of a carbon–carbon bond (3.42(1) Å). This is consistent with previously noted optimal geometry for halogen bonding to π-systems,36,37 and with the calculated electrostatic surface potential (ESP) surface of benz (Fig. 1a), showing that the areas of most negative ESP, expected to be beneficial for halogen bonding, are located on the arene π-bonds.
Bulk analysis of (benz)(14tfib) was hindered by rapid decomposition in air. Eventually, the formation of bulk cocrystal in a slurry of 14tfib in excess benz was confirmed by powder X-ray diffraction (PXRD) analysis of a sample under a plastic-wrap cover (see ESI, Fig. S1†).52 The (benz)(14tfib) structure confirms that the simplest aromatic hydrocarbon can be used as an acceptor in forming C–I⋯πC chains (Fig. 2a) reminiscent of those in cocrystals of naphthalene and azulene with 14tfib or ofiab.30,43
Next, we targeted anthracene (anthra) and tetracene (tet) as XB acceptors. For anthra, a cocrystal of composition (anthra)(14tfib)2 was obtained by mechanochemical liquid-assisted grinding (LAG),53,54 using nitromethane as a liquid additive (ESI, Fig. S2†).55 Diffraction-quality single crystals of (anthra)(14tfib)2 were obtained by dissolving the mechanochemically made material in hot CH2Cl2, rapidly cooling in an ice bath for ca. 10 seconds, followed by slow evaporation. Structure analysis revealed that (anthra)(14tfib)2 is based on the ladder-like motif (Fig. 2b) with each face of the arene forming two C–I⋯πC bonds with 14tfib molecules. The dI⋯C distances ranged from 3.427(9) Å to 3.583(7) Å, with ∠C–I⋯C angles in the range 157° to 173°. The distance between the iodine atom and the centroid of the relevant carbon–carbon bond was even shorter, at 3.366(5) Å, and no other short contacts were observed that would indicate C–H⋯π hydrogen bonding or π⋯π stacking between anthra molecules. The structure of (anthra)(14tfib)2 was also recently reported by Azzali et al. and our experimental data fully agrees with it.55
In the case of tet, the cocrystal (tet)(14tfib) was obtained in the form of diffraction-quality single crystals by slow evaporation of a solution of tet and 14tfib in 1,2,4-trichlorobenzene. Unlike the cocrystal containing anthra, the (tet)(14tfib) cocrystal does not exhibit the ladder-like motif, but instead presents a zig-zag architecture of alternating tet and 14tfib molecules interconnected via short, linear C–I⋯πC contacts (Fig. 2c, Table 1 and ESI Table S2†). The individual I⋯C distances are again shorter than the sum of van der Waals radii for C and I atoms, at 3.410(6) Å and 3.509(7) Å, with respective ∠C–I⋯C angles of 163° and 174°. The corresponding distance of the 14tfib iodine atom to the centroid of the relevant C–C bond on tet is again even shorter, at 3.387(5) Å, supporting the targeting of the π-system on the PAH. While tet is the only PAH in our study that did not produce the expected supramolecular ladder architecture, the (tet)(14tfib) structure confirms the general reliability of C–I⋯πC halogen bonds for PAH cocrystallisation.
Non-linear arenes
In order to explore the possibility of using XB for the assembly of arenes beyond the acene family, we targeted cocrystallisation with pyrene (pyr), benzanthracene (bant), perylene (pery) and coronene (cor), constituted of four, five and six fused ring systems. Mechanochemical screening in all cases produced new cocrystal phases, with compositions (pyr)(14tfib)2, (bant)2(14tfib)5, (pery)(14tfib)2 and (cor)(14tfib)2 (Fig. 2d–g). All four cocrystals were obtained in the form of diffraction-quality single crystals by crystallisation from CH2Cl2, and structural analysis revealed in each case the ladder motif analogous to the one seen with naphthalene,30 azulene,43 and anthra, with each face of the PAH participating in at least two linear C–I⋯πC halogen bonds. The halogen-bonded distances were in each cocrystal comparable, in the range from 3.4 Å to 3.6 Å, with ∠C–I⋯C angles in the range from 156° to 174° (see Table 1 and ESI Table S2†). Each cocrystal was also obtained mechanochemically as a bulk powder, exhibiting PXRD patterns consistent with herein determined crystal structures (see ESI†). The stability and composition of each cocrystal was validated by differential scanning calorimetry and thermogravimetric analysis (DSC and TGA, respectively, see ESI†). The DSC analysis revealed that the cocrystals were generally stable up to ca. 110 °C, and upon decomposition exhibited a weight loss step consistent with a loss of 14tfib. Structures of (pyr)(14tfib)2, (bant)2(14tfib)5, (pery)(14tfib)2, (cor)(14tfib)2 and (dicor)(14tfib)3 (Fig. 2d–h) further highlight the robustness of the supramolecular ladder motif based on C–I⋯πC XB interactions, even with expanded aromatic systems as XB acceptors.In the case of pyr, two other cocrystals with 14tfib have previously been reported, with different compositions: (pyr)(14tfib)29 and (pyr)4(14tfib).56 Both of these previously reported stoichiomorphs contain 14tfib molecules involved in either extended (for (pyr)(14tfib) cocrystal) or discrete (for (pyr)4(14tfib) cocrystal) arene-perfluoroarene π-stacking motifs with pyr molecules. The 14tfib molecules in these two prior cocrystals also form C–I⋯C halogen bonds of ca. 3.5 Å to neighboring pyr units. It is, therefore, notable, that the (pyr)(14tfib)2 phase reported here presents C–I⋯πC halogen bonding as the dominant intermolecular interaction, without any notable herringbone C–H⋯π or arene-perfluoroarene π-stacking interactions seen in other stoichiomorphs (Fig. 2d, also see ESI Fig. S4†).29,56
Cocrystallisation of bant, an isomer of pyr and tet, with 14tfib resulted in a halogen-bonded cocrystal of composition (bant)2(14tfib)5. The cocrystal was found to again contain the anticipated ladder motif (Fig. 2e). The ladder motif in (bant)2(14tfib)5 is unique, however, with different faces of each bant unit alternatively taking part in either two or three C–I⋯πC halogen bonds.
Crystal structures of cocrystals (pery)(14tfib)2 and (cor)(14tfib)2 (Fig. 2f and g) are, to the best of our knowledge, the first examples of halogen bonding being used to form cocrystals with large, non-substituted PAHs comprising >4 aromatic rings. To further examine the ability of C–I⋯πC bonds to organise large arenes, we focused on dicoronylene (dicor), comprised of 15 aromatic rings (Fig. 1a). A sample of dicor was synthesised according to the reported procedure,57 by reaction of cor at 170 °C in a 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 by weight melt of AlCl3 and NaCl, followed by sublimation (see ESI†). Attempts to obtain cocrystals of 14tfib and dicor from common organic solvents were not successful due to poor arene solubility, with crystallisation eventually achieved from molten 14tfib at 110 °C. Single crystal X-ray analysis of the ruby-red crystals revealed a cocrystal structure with composition (dicor)(14tfib)3, exhibiting the C–I⋯πC ladder motif, but with each face of the arene now engaged in XBs with 3 separate 14tfib donors (Fig. 2h). The XB distances lie in the range from 3.424(7) Å to 3.560(9) Å, with corresponding distances to the centroids of carbon–carbon bonds being in the range 3.327(6) Å to 3.579(8) Å. The halogen bonds exhibit a linear geometry, with ∠C–I⋯C angles adopting values from 159–178°. Notably, the C–I⋯πC bonds form on the edges of each dicor molecule, consistent with the calculated arene ESP surface, which shows that the highest negative potential is localised on the molecular rim. This means that a large fraction of the dicor molecule surface is not likely to engage in halogen bonding, and could be accessible for other types of interactions. Indeed, the dicor molecules are also engaged in π–π stacking between adjacent halogen-bonded ladders.
:1 by weight melt of AlCl3 and NaCl, followed by sublimation (see ESI†). Attempts to obtain cocrystals of 14tfib and dicor from common organic solvents were not successful due to poor arene solubility, with crystallisation eventually achieved from molten 14tfib at 110 °C. Single crystal X-ray analysis of the ruby-red crystals revealed a cocrystal structure with composition (dicor)(14tfib)3, exhibiting the C–I⋯πC ladder motif, but with each face of the arene now engaged in XBs with 3 separate 14tfib donors (Fig. 2h). The XB distances lie in the range from 3.424(7) Å to 3.560(9) Å, with corresponding distances to the centroids of carbon–carbon bonds being in the range 3.327(6) Å to 3.579(8) Å. The halogen bonds exhibit a linear geometry, with ∠C–I⋯C angles adopting values from 159–178°. Notably, the C–I⋯πC bonds form on the edges of each dicor molecule, consistent with the calculated arene ESP surface, which shows that the highest negative potential is localised on the molecular rim. This means that a large fraction of the dicor molecule surface is not likely to engage in halogen bonding, and could be accessible for other types of interactions. Indeed, the dicor molecules are also engaged in π–π stacking between adjacent halogen-bonded ladders.
Of the three symmetrically distinct 14tfib molecules in the (dicor)(14tfib)3 structure, two also participate in F⋯F and C–H⋯F contacts58 with neighboring 14tfib and dicor molecules, while one 14tfib molecule also participates in F⋯I contacts.
The formation of this (dicor)(14tfib)3 shows that C–I⋯πC halogen bonds are sufficiently robust to enable the cocrystallisation of large PAHs – to the best of our knowledge, no cocrystal of a non-derivatised PAH of similar size to dicor has ever previously been reported. Attempts to produce a bulk sample of (dicor)(14tfib)3 have so far been unsuccessful.
Non-planar aromatic systems: C60
Next, we targeted a non-planar aromatic acceptor, buckminsterfullerene (C60). While previous work reported that C60 and 14tfib do not form a cocrystal,39 we found that milling of the two components produces a cocrystal of composition (C60)(14tfib)2. Dark-red block-shaped crystals of (C60)(14tfib)2 were subsequently obtained by slow evaporation of a toluene solution of the mechanochemically made material. Crystal structure analysis revealed that (C60)(14tfib)2 is based on interpenetrated square-grid topology (sql) nets of C–I⋯πC bonds (Fig. 3a) with dI⋯C separation of 3.54(1) Å and halogen bond angle of 163°. The sql-nets are formed by C60 molecules acting as four-fold nodes with 14tfib molecules as framework linkers (Fig. 3b and c). The structure also exhibits short C⋯F contacts between 14tfib and C60 molecules in adjacent nets (dF⋯C = 3.17(1) Å; ∠C–F⋯C = 102.8(4)°). Notably, nearest-neighbor C60 molecules do not exhibit any short mutual contacts, and at room temperature do not exhibit the rotational disorder commonly seen in C60-containing crystal structures.59–62 Analysis of (C60)(14tfib)2 by DSC and TGA reveals decomposition around 110 °C, evidenced by a single endothermic event in the DSC thermogram, accompanied by the loss of 50% of sample weight, consistent with the theoretical content of 14tfib (52.7%). | ||
| Fig. 3 Structural motifs in (C60)(14tfib)2 and cocrystals of anthra and pyr with ofiab: (a) the C–I⋯C contact between iodine and C60 in (C60)(14tfib)2 (CCDC code 2240201); (b) interpenetration of two sql-grids in (C60)(14tfib)2; (c) space-filling representation of three distinct (C60)(14tfib)2sql-grids; (d) fragment of the supramolecular ladder in (anthra)(ofiab)2 (CCDC code 2240205) and (e) in (pyr)(ofiab)2 (CCDC code 2240206). | ||
Replacement of the halogen bond donor
In order to further explore the generality of the supramolecular ladder motif formed with PAHs, we next attempted cocrystallisation of anthra and pyr with ofiab, a longer XB donor. In each case, milling of the XB donor and acceptor followed by PXRD analysis revealed a new cocrystal phase, with compositions (anthra)(ofiab)2 and (pyr)(ofiab)2. Crystallographic analysis of single crystals grown by recrystallisation of the mechanochemically-made material from CH2Cl2 revealed the expected C–I⋯πC ladder-like architectures in both cases (Fig. 3d, e, Table 1 and ESI Table S2†). Importantly, the measured I⋯C and I···centroid distances are not appreciably different from those in (anthra)(14tfib)2 and (pyr)(14tfib)2. Both (anthra)(ofiab)2 and (pyr)(ofiab)2 were found to melt around 160–170 °C, as established by DSC, with TGA showing that samples completely evaporate, in a single step, at temperatures around 200 °C.Periodic DFT calculations
The formation of herein reported cocrystals was also investigated by plane-wave periodic-density functional theory (DFT) calculations in CASTEP,63 using PBE64 functionals combined with Grimme D3 semiempirical dispersion correction.65 Periodic DFT was previously shown66 to be effective for understanding the thermodynamic stability of halogen-bonded cocrystals. The calculated cocrystal formation energies (Ecalc) with respect to individual crystalline components (Table 1) were found to be negative in all but one case, indicating that cocrystallisation was enthalpically favorable in general. The exception is (dicor)(14tfib)3, with Ecalc of +6.48 kJ mol−1 suggesting that its formation may be entropy driven. Vibrational contributions to the Free energies related to cocrystallisation were, however, not taken into account due to the high computational cost of periodic DFT phonon calculations.| Cocrystal | E calc/kJ mol−1 | E XB/kJ mol−1 | d(C–I⋯Cπ)/Å |
|---|---|---|---|
| a The full set of XB bonds and angles is provided in ESI Table S2. b Calculation based on the orthorhombic polymorph of benz (CSD code BENZEN). | |||
| (benz)(14tfib) | −10.4b | −16.43 | 3.48(2), 3.51(2) |
| (anthra)(14tfib)2 | −5.89 | −18.82 (−19.28, −18.36) | 3.427(9), 3.445(8), 3.583(7) |
| (tet)(14tfib) | −2.27 | −13.76 | 3.410(6), 3.509(7) |
| (pyr)(14tfib)2 | −5.90 | −18.86 (−18.63, −19.10) | 3.515(8), 3.56(1), 3.50(1), 3.50(1) |
| (bant)2(14tfib)5 | −5.54 | −19.48 (−19.13, −20.29, −18.30, −17.45, −22.25) | 3.50(2), 3.64(2), 3.50(1), 3.46(1), 3.51(3), 3.39(2), 3.48(2), 3.40(2), 3.55(1) |
| (pery)(14tfib)2 | −8.46 | −20.45 (−20.45, −20.45) | 3.509(9), 3.430(8), 3.482(9), 3.51(1) |
| (cor)(14tfib)2 | −5.93 | −20.45 (−20.45, −20.45) | 3.45(1), 3.46(2), 3.46(1), 3.51(2) |
| (dicor)(14tfib)3 | 6.48 | −20.12 (−19.54, −20.29, −20.55) | 3.440(8), 3.424(7), 3.368(8), 3.38(1), 3.560(9) |
| (anthra)(ofiab)2 | −2.91 | −19.66 (−19.30, −20.31) | 3.62(2), 3.43(1), 3.55(2), 3.63(2) |
| (pyr)(ofiab)2 | −0.17 | −20.33 (−20.00, −20.66) | 3.476(7), 3.394(7), 3.586(7), 3.448(6) |
| (C60)(14tfib)2 | −13.95 | −15.15 | 3.54(1) |
The energies associated with individual halogen bonds (EXB) were obtained by subtracting the total energies of individual component molecules from the total energy of each distinct XB donor–acceptor dimer unit found in the cocrystal structure. The EXB values were highly consistent across all explored XB acceptors, with all dimer interaction energies being exothermic and falling within the range of −13 kJ mol−1 to −21 kJ mol−1.
Analysis of the Cambridge structural database (CSD)
Our systematic experimental study reveals the robust supramolecular synthon based on C–I⋯πC halogen bonds for the directional assembly of carbon-only aromatic systems of various shapes and sizes. It is highly surprising that this expansive potential for directional arene assembly by C–I⋯πC bonds has not previously been noted, leading us to conduct an in-depth search of the Cambridge Structural Database (CSD) to probe for the appearance and geometry of such interactions involving the iodine atom of a C–I fragment as a donor and a carbon atom of a 5-, 6- or a 7-membered ring, a C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C, or a C
C, or a C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C moiety, as the acceptor. As XBs are expected to be shorter than the sum of the van der Waals radii of interacting atoms, the search was limited to I⋯C distances up to 4.68 Å, which is ca. 1 Å longer than the sum of van der Waals radii for carbon (1.7 Å) and iodine (1.98 Å).49
C moiety, as the acceptor. As XBs are expected to be shorter than the sum of the van der Waals radii of interacting atoms, the search was limited to I⋯C distances up to 4.68 Å, which is ca. 1 Å longer than the sum of van der Waals radii for carbon (1.7 Å) and iodine (1.98 Å).49
The CSD searches revealed that short contacts between iodine and 6-membered rings are the most prevalent, representing ca. 71% of the total short contacts examined, followed by contacts to CC bonds (13%), CC bonds (13%), while contacts involving 5- and 7-membered rings represented ca. 2% and 1% of all identified short contacts (Fig. 4a). The CSD searches indicated two geometry-distinct regimes of C–I⋯πC interactions, dependent on the I⋯Cπ distance. Specifically, there was no particular geometrical preference for longer I⋯C separations. However, shorter I⋯C distances were generally associated with a preference for linear C–I⋯C contacts, with angles in the range of ca. 140–180°. Such behavior was particularly pronounced for CC bonds and 6-membered rings as acceptors, where the majority of I⋯C distances below ∼3.7 Å were associated with a C–I⋯C angle of 140° or higher (Fig. 4b and c). Such preference for linear geometries at lower I⋯C distances is consistent with XBs,14d reinforcing the herein proposed role of halogen bonds as an overlooked, yet consistent, strategy for directional assembly of carbon-based aromatic system, and suggest the possible existence of other C–I⋯πC driven supramolecular synthons. In addition to structures identified in the CSD, during preparation of this manuscript two crystal structures were reported67 that strongly reinforce the view of XBs as reliable interactions to form supramolecular architectures based on PAHs (Fig. 4d and e). These structures are based on phenanthrene and on chrysene, an isomer of tet and bant, and exhibit the C–I⋯πC ladder motif with PAH as the XB acceptor and 14tfib as the donor.
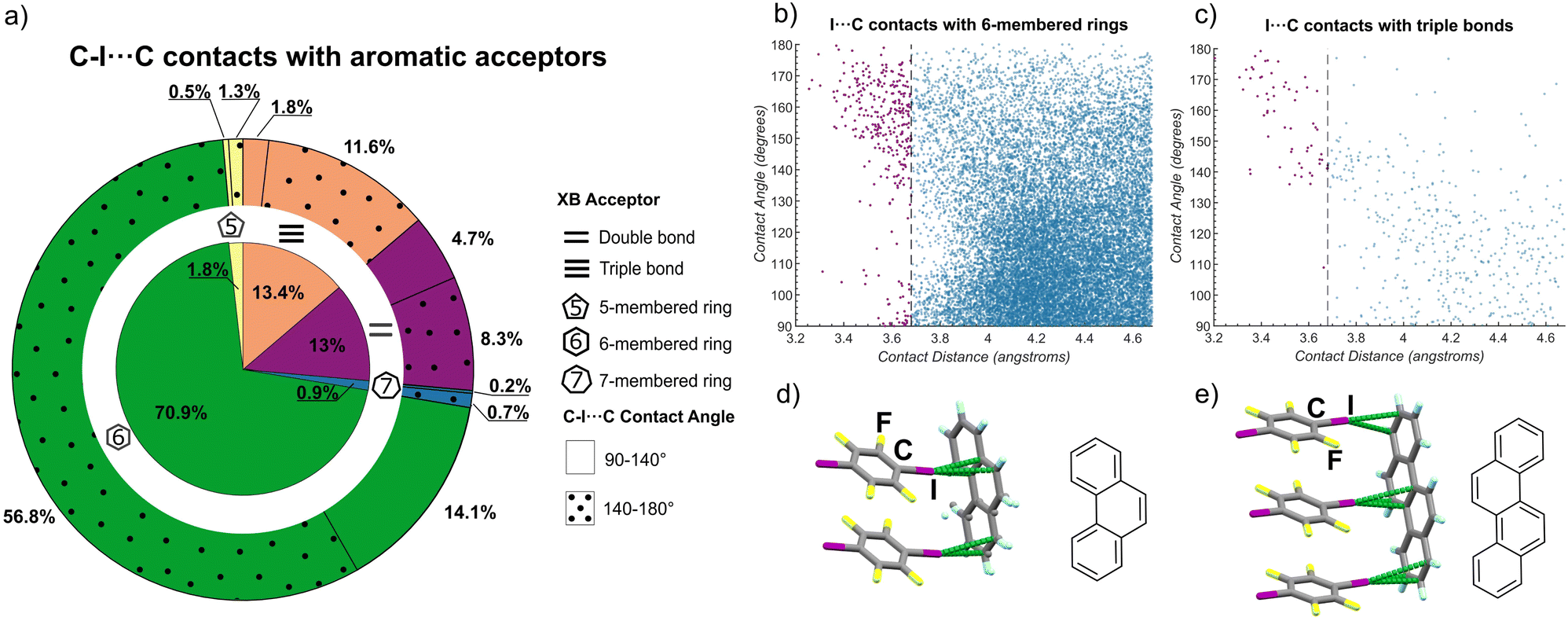 | ||
| Fig. 4 Outcomes of CSD searches for short C–I⋯C contacts to alkene, alkyne, 5-, 6- and 7-membered ring acceptors based on aromatic carbon and selected representative structures. (a) Distribution of the I⋯C contact lengths shorter than 4.68 Å across different acceptor types. For each XB acceptor type, the fraction of contacts with a linear geometry (C–I⋯C angle in the range from 140° to 180°) corresponding to halogen bonding, is indicated on the outer circle by a dotted pattern. Plots of the distribution of lengths and angles for C–I⋯C contacts to: (b) 6-membered ring and (c) alkyne (CC) acceptors. To guide the eye, contacts shorter than the van der Waals limit of 3.68 Å are shown in purple. Plots of the distribution of C–I⋯C contact lengths and angles for 5- and 7-membered ring, as well as alkene (CC) acceptors, are given in the ESI.† Fragment of the crystal structures67 of: (d) (phenanthrene)(14tfib)2 and (e) (chrysene)(14tfib)3, illustrating the appearance of the C–I⋯πC ladder motif. | ||
Luminescence properties
Organic materials with emissive triplet states and long emission lifetimes (phosphorescence) are being sought for applications in OLEDs,68 biological imaging,69 anticounterfeiting70 and more.71 However, purely organic systems rarely exhibit phosphorescence at room temperature, due to the spin-forbidden nature of the singlet-to-triplet transition.72–74 Recent work shows the critical roles of intermolecular interactions and crystal packing on organic room temperature phosphorescence efficiency.74–76 In this context, cocrystallisation of PAHs offers a promising platform for achieving organic phosphorescent materials, as the solid-state arrangement of aromatic units directly influences diverse optoelectronic properties, such as excitation/emission wavelengths, and lifetime kinetics.77,78 Halogen bonded arene cocrystals have been reported to exhibit phosphorescence decays on the scale of several milliseconds,27–32,42,67,77 with an isolated case of decays >200 ms.77b Recently, Abe et al. reported luminescence properties of a (phenanthrene)(14tfib)2 cocrystal, and found that partial replacement (up to 25%) of phenanthrene with pyr leads to an increase of the phosphorescence quantum yield from ca. 6 to >20%.67 Further replacement of phenanthrene with pyr (up to 50%), however, led to a drop of phosphorescence efficiency. Notably, a cocrystal of composition (pyr)(14tfib)2, in which all phenanthrene would be replaced by pyr, was not reported – but (pyr)(14tfib) showed a low luminescence quantum yield <1% and short lifetime of 50 μs67 (500 μs29).Having herein synthesised the previously missing cocrystal of composition (pyr)(14tfib)2, we explored its photoluminescence properties, along with those of the heavier congeners (pery)(14tfib)2 and (cor)(14tfib)2 (Fig. 5). The cocrystals of cor, pyr, or pery with 14tfib all exhibited strong luminescence shifts when compared to the pristine solid PAH. The previously not accessible (pyr)(14tfib)2 exhibited a remarkable bathochromic shift of almost 200 nm, with the maximum emission wavelength (λmax) at 678 nm (red), compared to 498 nm (blue) for solid pyr (Fig. 5a and b). The red-shifted λmax and the emission life-time (10.4 μs) are similar to the previously reported behavior of (pyr)(14tfib)29,67 and suggest the phosphorescent nature of the emission. A similar behavior was displayed by (cor)(14tfib)2 cocrystals: a significant ∼150 nm red shift of λmax (from 513 nm in pristine solid cor to 660 nm in the cocrystal, Fig. 5c and d) and very long emission life-time (4.2 ms). Notably, the emission of (cor)(14tfib)2 extends in the NIR region, beyond 750 nm, which is typically difficult to achieve because of the energy-gap law.78
 | ||
| Fig. 5 Change in luminescence spectra upon the formation of halogen-bonded cocrystals with aromatic XB acceptors. (a) Solid-state luminescence spectra for pyr (blue) and (pyr)(14tfib)2 (red). (b) A sample of (top) pyr and (bottom) (pyr)(14tfib)2 excited with 365 nm LED light. (c) Solid-state luminescence spectra for cor (green) and (cor)(14tfib)2 (red). (d) A sample of (top) cor and (bottom) (cor)(14tfib)2 excited with 365 nm LED light. (e) Solid-state luminescence spectra for α-pery and (pery)(14tfib)2. (f) A sample of (top) α-pery and (bottom) (pery)(14tfib)2 excited with 365 nm LED light. | ||
In a sharp contrast to the red-shifted emission (pyr)(14tfib)2 and (cor)(14tfib)2 cocrystals, the (pery)(14tfib)2 cocrystals exhibited a strong ∼100 nm hypsochromic shift of emission, from λmax = 607 nm (orange) in pristine pery solid (α-pery polymorph) to 521 nm (green) of the cocrystal (Fig. 5e and f). Such blue-shifting behavior is unusual, as cocrystals exhibiting C–I⋯πC interactions generally exhibit red-shifted emission compared to the pure emissive component solid.79 This behavior of (pery)(14tfib)2 is reminiscent of the J-aggregate emission of the β-pery crystals80 in which the pery molecules are arranged in off-diagonal interaggregate interactions, similar to (pery)(14tfib)2 (see ESI, Fig. S36†). Such J-aggregated states are known to enhance the fluorescence emission radiative rates, outcompeting intersystem crossing rates from populating triplet excitons.81 Indeed, the emission lifetime observed in (pery)(14tfib)2 was <0.62 ns (see ESI, Fig. S35†). The observed blue shift of (pery)(14tfib)2 cocrystals (as well as β-pery crystals) compared to α-pery is explained by disruption of the excimer emission resulting from the π-dimer packing motif of the latter structure.
Consequently, the incorporation of the C–I⋯πC interactions in co-crystals of the PAHs can enable their room-temperature phosphorescence (RTP) through the heavy atom effect, and is commonly manifested in the red-shifted emission of cocrystals compared to pure PAH. However, the RTP properties also depend on a balance of inter-system crossing vs. fluorescence transition which varies with the molecular and crystal structure. Thus, the fast fluorescence transition in (pery)(14tfib)2 brought about by the specific packing of PAH molecules (J-aggregation) prevents the observation of RTP.
Conclusions
In summary, we have presented an extensive and systematic experimental, theoretical, and database study that establishes halogen bonding to aromatic carbon systems as a powerful, but historically overlooked approach for the design of supramolecular architectures based on carbon as the molecular recognition site. This work coalesces and advances scattered reports of halogen bonds to π-systems, and demonstrates not only that C–I⋯πC halogen bonding can be reliably used as a directional interaction for the synthesis of cocrystals of differently-sized and -shaped carbon-only aromatic systems, but also establishes the supramolecular ladder structure based on C–I⋯πC halogen bonds as a unique supramolecular synthon generally applicable to non-substituted carbon systems.In particular, C–I⋯πC halogen bonding has been effective in cocrystallisation of all 9 explored aromatic carbon molecules, from benzene (one aromatic ring) to dicoronylene (15 fused aromatic rings), resulting in altogether 11 cocrystals. Focusing on PAHs only (i.e., excluding benzene and C60), C–I⋯πC halogen bonding led to the formation of the expected supramolecular ladder motif in 8 out of 9 cases, with PAH units acting as the rungs and halogen bond donor molecules as the rails. The reliability of a self-assembly motif can be evaluated through Aakeröy's concept of supramolecular yield.44–46 From that perspective, the synthesis of C–I⋯πC ladder motifs based on non-substituted arene hydrocarbons is highly reliable, with an 89% supramolecular yield. If one includes in this overview the recently reported cocrystals of naphthalene,30 azulene (two cocrystals),43 phenanthrene and chrysene,67 which were all found to exhibit the C–I⋯πC ladder motif, the supramolecular yield rises to 13 out of 14 cases, i.e. 93%.
The high reliability of this supramolecular cocrystal-forming reaction is surprising when considering that traditionally the only reliable way to achieve cocrystallisation of arenes was through π-stacking. The C–I⋯πC halogen-bonded ladder now suggests a route for crystal engineers to rationally use non-substituted PAHs as building blocks in designing new carbon-based supramolecular architectures, different from π-stacked arrays.‡ The C–I⋯πC halogen bonds underlying the ladder motif often coincide with the sites of greatest negative ESP on the arene, and a single side of a six-membered carbon ring is never observed to participate in more than one halogen bond. These observations establish a different element of design for the assembly of supramolecular architectures from non-substituted arenes, based on the directional interaction of halogen bonding, rather than the traditionally used and non-directional π-stacking. The ability to generate new structures based on simple PAH components is of significance in the context of materials with new properties. That is illustrated by very large red or blue shifts (in the range of ca. 100 nm to 200 nm), as well as room-temperature phosphorescence achieved for PAHs engaged in the supramolecular C–I⋯πC ladder structures. In particular, the phosphorescence lifetime of >4 ms observed for the coronene-based cocrystal provides further incentive to explore the design of fully organic phosphorescent systems based on halogen bonding to aromatic system.
Data availability
Data supporting this manuscript has been provided as ESI.† Selected data relevant to PXRD analysis, fluorescence emission spectroscopy, and theoretical modelling, is also available from the data repository Zenodo (DOI: https://doi.org/10.5281/zenodo.10064165). Crystallographic data in CIF format has been deposited with the Cambridge Structural Database (CCDC 2240199–2240207, 2249511 and 2281269).Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the support of McGill University, Natural Sciences and Engineering Research Council of Canada (NSERC) Discovery Grant (RGPIN-2017-06467; RGPIN-2018-06500); John C. Polanyi Award (JCP 562908-2022), the Government of Canada for Tier-1 Canada Research Chair Program (TF), the National Science Center of Poland (NCN) OPUS Grant (2020/37/B/ST5/02638), the Leverhulme Trust (Leverhulme International Professorship, LIP-2021-011) and the University of Birmingham. We thank the UK Engineering and Physical Sciences Research Council for funding the National Crystallography Service (EP/W02098X/1). Fonds de recherche du Québec – Nature et technologies (FRQNT) is acknowledged for a doctoral scholarship (EH).Notes and references
- E. Peeters and J. Cami, in Encyclopedia of Astrobiology, ed. M. Gargaud, R. Amils, J. C. Quintanilla, H. J. Cleaves, W. M. Irvine, D. L. Pinti and M. Viso, Springer Berlin Heidelberg, Berlin, Heidelberg, 2011, ch. 1250, pp. 1307–1321 Search PubMed.
- E. Peeters, C. Mackie, A. Candian and A. G. G. M. Tielens, Acc. Chem. Res., 2021, 54, 1921–1933 CrossRef CAS PubMed.
- C. S. Hansen, E. Peeters, J. Cami and T. W. Schmidt, Commun. Chem., 2022, 5, 94 CrossRef PubMed.
- C. Aumaitre and J. F. Morin, Chem. Rec., 2019, 19, 1142–1154 CrossRef CAS PubMed.
- J. Zaumseil and H. Sirringhaus, Chem. Rev., 2007, 107, 1296–1323 CrossRef CAS PubMed.
- J. H. Lee, C. H. Chen, P. H. Lee, H. Y. Lin, M. K. Leung, T. L. Chiu and C. F. Lin, J. Mater. Chem. C, 2019, 7, 5874–5888 RSC.
- N. Liang, Y. K. Zhao, Y. Z. Wu, C. R. Zhang and M. Shao, Appl. Phys. Lett., 2021, 119, 053301 CrossRef CAS.
- X. Yang, L. Lan, X. Pan, X. Liu, Y. Song, X. Yang, Q. Dong, L. Li, P. Naumov and H. Zhang, Nat. Commun., 2022, 13, 7874 CrossRef CAS PubMed.
- (a) P. P. Yu, Y. G. Zhen, H. L. Dong and W. P. Hu, Chem, 2019, 5, 2814–2853 CrossRef CAS; (b) P. Vishweshwar, J. A. McMahon, J. A. Bis and M. J. Zaworotko, J. Pharm. Sci., 2006, 95, 499–516 CrossRef CAS PubMed; (c) G. R. Desiraju, J. Am. Chem. Soc., 2013, 135, 9952–9967 CrossRef CAS PubMed; (d) M. K. Corpinot and D.-K. Bučar, Cryst. Growth Des., 2019, 19, 1426–1453 CrossRef CAS; (e) D. Braga, F. Grepioni and G. R. Desiraju, Chem. Rev., 1998, 98, 1375–1406 CrossRef CAS PubMed; (f) L. Sun, W. Zhu, X. Zhang, L. Li, H. Dong and W. Hu, J. Am. Chem. Soc., 2021, 143, 19243–19256 CrossRef CAS PubMed.
- (a) J. E. Anthony, Chem. Rev., 2006, 106, 5028–5048 CrossRef CAS PubMed; (b) B. Strehmel, A. M. Sarker, J. H. Malpert, V. Strehmel, H. Seifert and D. C. Neckers, J. Am. Chem. Soc., 1999, 121, 1226–1236 CrossRef CAS; (c) Z. Chen, P. Müller and T. M. Swager, Org. Lett., 2006, 8, 273–276 CrossRef CAS PubMed; (d) S. E. Wheeler, J. Am. Chem. Soc., 2011, 133, 10262–10274 CrossRef CAS PubMed.
- P. Gupta, D. P. Karothu, E. Ahmed, P. Naumov and N. K. Nath, Angew. Chem., Int. Ed., 2018, 57, 8498–8502 CrossRef CAS PubMed.
- S. P. Yelgaonkar, G. Campillo-Alvarado and L. R. MacGillivray, J. Am. Chem. Soc., 2020, 142, 20772–20777 CrossRef CAS PubMed.
- G. R. Krishna, R. Devarapalli, G. Lal and C. M. Reddy, J. Am. Chem. Soc., 2016, 138, 13561–13567 CrossRef CAS PubMed.
- (a) C. B. Aakeröy, M. Baldrighi, J. Desper, P. Metrangolo and G. Resnati, Chem.–Eur. J., 2013, 19, 16240–16247 CrossRef PubMed; (b) C. Gilday, S. W. Robinson, T. A. Barendt, M. J. Langton, B. R. Mullaney and P. D. Beer, Chem. Rev., 2015, 115, 7118–7195 CrossRef PubMed; (c) J. Teyssandier, K. S. Mali and S. De Feyter, ChemistryOpen, 2020, 9, 225–241 CrossRef CAS PubMed; (d) G. Cavallo, P. Metrangolo, R. Milani, T. Pilati, A. Priimagi, G. Resnati and G. Terraneo, Chem. Rev., 2016, 116, 2478–2601 CrossRef CAS PubMed; (e) G. R. Desiraju, P. S. Ho, L. Kloo, A. C. Legon, R. Marquardt, P. Metrangolo, P. Politzer, G. Resnati and K. Rissanen, Pure Appl. Chem., 2013, 85, 1711–1713 CrossRef CAS; (f) N. Juneja, N. M. Shapiro, D. K. Unruh, E. Bosch, R. H. Groeneman and K. M. Hutchins, Angew. Chem., Int. Ed., 2022, 61, e202202708 CrossRef CAS PubMed; (g) M. A. Sinnwell and L. R. MacGillivray, Angew. Chem., Int. Ed., 2016, 55, 3477–3480 CrossRef CAS PubMed; (h) R. Gutzler, C. Fu, A. Dadvand, Y. Hua, J. M. MacLeod, F. Rosei and D. F. Perepichka, Nanoscale, 2012, 4, 5965–5971 RSC.
- (a) G. Ferguson, J. F. Gallagher, C. Glidewell and C. M. Zakaria, Acta Crystallogr., Sect. C: Struct. Chem., 1994, 50, 70–73 CrossRef; (b) M. Nishio, Phys. Chem. Chem. Phys., 2011, 13, 13873–13900 RSC; (c) J. L. Atwood, F. Hamada, K. D. Robinson, G. W. Orr and R. L. Vincent, Nature, 1991, 349, 683–684 CrossRef CAS; (d) K. Fucke, K. M. Anderson, M. H. Filby, M. Henry, J. Wright, S. A. Mason, M. J. Gutmann, L. J. Barbour, C. Oliver, A. W. Coleman, J. L. Atwood, J. A. Howard and J. W. Steed, Chem.–Eur. J., 2011, 17, 10259–10271 CrossRef CAS PubMed; (e) F. H. Allen, V. J. Hoy, J. A. K. Howard, V. R. Thalladi, G. R. Desiraju, C. C. Wilson and G. J. McIntyre, J. Am. Chem. Soc., 1997, 119, 3477–3480 CrossRef CAS.
- U. Geiser, S. K. Kumar, B. M. Savall, S. S. Harried, K. D. Carlson, P. R. Mobley, H. H. Wang, J. M. Williams and R. E. Botto, Chem. Mater., 1992, 4, 1077–1082 CrossRef CAS.
- N. R. Goud, O. Bolton, E. C. Burgess and A. J. Matzger, Cryst. Growth Des., 2016, 16, 1765–1771 CrossRef CAS.
- D. Cao, M. Hong, A. K. Blackburn, Z. Liu, J. M. Holcroft and J. F. Stoddart, Chem. Sci., 2014, 5, 4242–4248 RSC.
- (a) H. J. Cho, S. W. Kim, S. Kim, S. Lee, J. Lee, Y. Cho, Y. Lee, T.-W. Lee, H.-J. Shin and C. Song, J. Mater. Chem. C, 2020, 8, 17289–17296 RSC; (b) G. W. Coates, A. R. Dunn, L. M. Henling, J. W. Ziller, E. B. Lobkovsky and R. H. Grubbs, J. Am. Chem. Soc., 1998, 120, 3641–3649 CrossRef CAS; (c) A. Friedrich, I. E. Collings, K. F. Dziubek, S. Fanetti, K. Radacki, J. Ruiz-Fuertes, J. Pellicer-Porres, M. Hanfland, D. Sieh, R. Bini, S. J. Clark and T. B. Marder, J. Am. Chem. Soc., 2020, 142, 18907–18923 CrossRef CAS PubMed; (d) J. C. Collings, K. P. Roscoe, E. G. Robins, A. S. Batsanov, L. M. Stimson, J. A. K. Howard, S. J. Clark and T. B. Marder, New J. Chem., 2002, 26, 1740–1746 RSC; (e) L. Wang, J. Deng, M. Jiang, C. Zhen, F. Li, S. Li, S. Bai, X. Zhang and W. Zhu, J. Mater. Chem. A, 2023, 11, 11235–11244 RSC; (f) B. L. Hu and Q. Zhang, Chem. Rec., 2021, 21, 116–132 CrossRef CAS PubMed; (g) T. Faisal, K. M. Solntsev, T. Kahs, N. Saleh, P. Commins, J. Whelan, S. Mohamed and P. Naumov, Energy Fuels, 2021, 35, 8742–8755 CrossRef CAS; (h) Z. M. Saeed, B. Dhokale, A. F. Shunnar, W. M. Awad, H. H. Hernandez, P. Naumov and S. Mohamed, Cryst. Growth Des., 2021, 21, 4151–4161 CrossRef CAS; (i) G. Raj, Y. Kikkawa, L. Catalano, R. Pasricha, Y. Norikane and P. Naumov, Chemnanomat, 2020, 6, 68–72 CrossRef CAS; (j) T. Schillmöller, R. Herbst-Irmer and D. Stalke, Adv. Opt. Mater., 2021, 9, 2001814 CrossRef; (k) N. G. Petrov, P. Chartier, T. Maris and J. D. Wuest, J. Am. Chem. Soc., 2022, 144, 556–572 CrossRef CAS PubMed; (l) K. Biradha and R. Santra, Chem. Soc. Rev., 2013, 42, 950–967 RSC; (m) M. C. Gerthoffer, B. Xu, S. Wu, J. Cox, S. Huss, S. M. Oburn, S. A. Lopez, V. H. Crespi, J. V. Badding and E. Elacqua, Polym. Chem., 2022, 13, 1359–1368 RSC; (n) A. J. Oyer, J.-M. Y. Carrillo, C. C. Hire, H. C. Schniepp, A. D. Asandei, A. V. Dobrynin and D. H. Adamson, J. Am. Chem. Soc., 2012, 134, 5018–5021 CrossRef CAS PubMed; (o) Y. Xu and M. von Delius, Angew. Chem., Int. Ed., 2020, 59, 559–573 CrossRef CAS PubMed; (p) M. Bartkowski and S. Giordani, Nanoscale, 2020, 12, 9352–9358 RSC.
- L. R. MacGillivray, CrystEngComm, 2004, 6, 77–78 RSC.
- C. A. Gunawardana and C. B. Aakeröy, Chem. Commun., 2018, 54, 14047–14060 RSC.
- A. K. Nangia and G. R. Desiraju, Angew. Chem., Int. Ed., 2019, 58, 4100–4107 CrossRef CAS PubMed.
- (a) D. Cinčić, T. Friščić and W. Jones, J. Am. Chem. Soc., 2008, 130, 7524–7525 CrossRef PubMed; (b) D. Cinčić, T. Friščić and W. Jones, CrystEngComm, 2011, 13, 3224–3231 RSC; (c) Y. Xu, J. Huang, B. Gabidullin and D. L. Bryce, Chem. Commun., 2018, 54, 11041–11043 RSC; (d) A. A. Eliseeva, D. M. Ivanov, A. V. Rozhkov, I. V. Ananyev, A. Frontera and V. Y. Kukushkin, JACS Au, 2021, 1, 354–361 CrossRef CAS PubMed; (e) A. Bauza, D. Quinonero, A. Frontera and P. M. Deya, Phys. Chem. Chem. Phys., 2011, 13, 20371–20379 RSC; (f) E. A. Katlenok, A. V. Rozhkov, O. V. Levin, M. Haukka, M. L. Kuznetsov and V. Y. Kukushkin, Cryst. Growth Des., 2021, 21, 1159–1177 CrossRef CAS; (g) I. Benito, R. M. Gomila and A. Frontera, CrystEngComm, 2022, 24, 4440–4446 RSC; (h) K. Lisac, F. Topić, M. Arhangelskis, S. Cepic, P. A. Julien, C. W. Nickels, A. J. Morris, T. Friščić and D. Cinčić, Nat. Commun., 2019, 10, 61 CrossRef CAS PubMed; (i) P. J. Costa, Phys. Sci. Rev., 2017, 2 Search PubMed; (j) T. H. Borchers, F. Topić, J. C. Christopherson, O. S. Bushuyev, J. Vainauskas, H. M. Titi, T. Friščić and C. J. Barrett, Nat. Chem., 2022, 14, 574–581 CrossRef CAS PubMed.
- X. H. Ding, Y. Z. Chang, C. J. Ou, J. Y. Lin, L. H. Xie and W. Huang, Natl. Sci. Rev., 2020, 7, 1906–1932 CrossRef CAS PubMed.
- S. A. Sharber, W. J. Mullin and S. W. Thomas, Chem. Mater., 2021, 33, 6640–6661 CrossRef CAS.
- R. Liantonio, S. Luzzati, P. Metrangolo, T. Pilati and G. Resnati, Tetrahedron, 2002, 58, 4023–4029 CrossRef CAS.
- W. Z. Wang, Y. Zhang and W. J. Jin, Coord. Chem. Rev., 2020, 404, 213107 CrossRef CAS.
- L. L. Li, Z. F. Liu, W. X. Wu and W. J. Jin, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2018, 74, 610–617 CrossRef CAS.
- Q. J. Shen, H. Q. Wei, W. S. Zou, H. L. Sun and W. J. Jin, CrystEngComm, 2012, 14, 1010–1015 RSC.
- Q. J. Shen, X. Pang, X. R. Zhao, H. Y. Gao, H.-L. Sun and W. J. Jin, CrystEngComm, 2012, 14, 5027–5034 RSC.
- H. Y. Gao, X. R. Zhao, H. Wang, X. Pang and W. J. Jin, Cryst. Growth Des., 2012, 12, 4377–4387 CrossRef CAS.
- H. Y. Gao, Q. J. Shen, X. R. Zhao, X. Q. Yan, X. Pang and W. J. Jin, J. Mater. Chem., 2012, 22, 5336–5343 RSC.
- L. Li, H. Wang, W. Wang and W. J. Jin, CrystEngComm, 2017, 19, 5058–5067 RSC.
- L. Li, W. X. Wu, Z. F. Liu and W. J. Jin, New J. Chem., 2018, 42, 10633–10641 RSC.
- S. J. Ang, A. M. Mak, M. B. Sullivan and M. W. Wong, Phys. Chem. Chem. Phys., 2018, 20, 8685–8694 RSC.
- A. Forni, S. Pieraccini, S. Rendine, F. Gabas and M. Sironi, ChemPhysChem, 2012, 13, 4224–4234 CrossRef CAS PubMed.
- D. Y. Kim, J. M. L. Madridejos, M. Ha, J. H. Kim, D. C. Yang, C. Baig and K. S. Kim, Chem. Commun., 2017, 53, 6140–6143 RSC.
- T. J. Mooibroek and P. Gamez, CrystEngComm, 2013, 15, 1802–1805 RSC.
- Y. Zhang, J. G. Wang, X. Sun, Q. Liu, W. Wang and Y. B. Wang, ChemPlusChem, 2018, 83, 470–477 CrossRef CAS PubMed.
- A. S. Mikherdov, A. S. Novikov, V. P. Boyarskiy and V. Y. Kukushkin, Nat. Commun., 2020, 11, 2921 CrossRef CAS PubMed.
- A. S. Mikherdov, R. A. Popov, A. S. Smirnov, A. A. Eliseeva, A. S. Novikov, V. P. Boyarskiy, R. M. Gomila, A. Frontera, V. Y. Kukushkin and N. A. Bokach, Cryst. Growth Des., 2022, 22, 6079–6087 CrossRef CAS; A. S. Smirnov, E. A. Katlenok, A. S. Mikherdov, M. A. Kryukova, N. A. Bokach and V. Y. Kukushkin, Int. J. Mol. Sci., 2023, 24, 13324 CrossRef PubMed.
- (a) S. d'Agostino, F. Grepioni, D. Braga and B. Ventura, Cryst. Growth Des., 2015, 15, 2039–2045 CrossRef; (b) G. Lapadula, N. Judaš, T. Friščić and W. Jones, Chem.–Eur. J., 2010, 16, 7400–7403 CrossRef CAS PubMed; (c) S. Y. Oh, C. W. Nickels, F. Garcia, W. Jones and T. Friščić, CrystEngComm, 2012, 14, 6110–6114 RSC.
- J. Vainauskas, F. Topić, O. S. Bushuyev, C. J. Barrett and T. Friščić, Chem. Commun., 2020, 56, 15145–15148 RSC.
- C. B. Aakeröy, A. M. Beatty and B. A. Helfrich, J. Am. Chem. Soc., 2002, 124, 14425–14432 CrossRef PubMed.
- C. B. Aakeröy, A. M. Beatty and B. A. Helfrich, Angew Chem. Int. Ed. Engl., 2001, 40, 3240–3242 CrossRef.
- C. B. Aakeröy and D. J. Salmon, CrystEngComm, 2005, 7, 439–448 RSC.
- G. R. Desiraju, Angew Chem. Int. Ed. Engl., 1995, 34, 2311–2327 CrossRef CAS.
- C. R. Groom, I. J. Bruno, M. P. Lightfoot and S. C. Ward, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2016, 72, 171–179 CrossRef CAS PubMed.
- M. Mantina, A. C. Chamberlin, R. Valero, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. A, 2009, 113, 5806–5812 CrossRef CAS PubMed.
- O. Hassel, K. O. Strømme, H. Haraldsen, A. Grönvall, B. Zaar and E. Diczfalusy, Acta Chem. Scand., 1958, 12, 1146 CrossRef CAS.
- E. Bosch, IUCrData, 2019, 4, x190993 CrossRef CAS.
- I. Huskić, J. C. Christopherson, K. Užarević and T. Friščić, Chem. Commun., 2016, 52, 5120–5123 RSC.
- T. Friščić, S. L. Childs, S. A. A. Rizvi and W. Jones, CrystEngComm, 2009, 11, 418–426 RSC.
- T. Friščić, A. V. Trask, W. Jones and W. D. Motherwell, Angew Chem. Int. Ed. Engl., 2006, 45, 7546–7550 CrossRef PubMed.
- A. Azzali, S. d'Agostino, M. Capacci, F. Spinelli, B. Ventura and F. Grepioni, CrystEngComm, 2022, 24, 5748–5756 RSC.
- H. Jain, D. Sutradhar, S. Roy and G. R. Desiraju, Angew Chem. Int. Ed. Engl., 2021, 60, 12841–12846 CrossRef CAS PubMed.
- M. Zander and W. Franke, Chem. Ber./Recl., 1958, 91, 2794–2797 CrossRef CAS.
- Z. Mu, L. Shu, H. Fuchs, M. Mayor and L. Chi, J. Am. Chem. Soc., 2008, 130, 10840–10841 CrossRef CAS PubMed.
- Y. Quo, N. Karasawa and W. A. Goddard, Nature, 1991, 351, 464–467 CrossRef.
- D. André, A. Dworkin, H. Szwarc, R. Céolin, V. Agafonov, C. Fabre, A. Rassat, L. Straver, P. Bernier and A. Zahab, Mol. Phys., 1992, 76, 1311–1317 CrossRef.
- A. Giri and P. E. Hopkins, J. Phys. Chem. Lett., 2017, 8, 2153–2157 CrossRef CAS PubMed.
- T. Matsuno, Y. Nakai, S. Sato, Y. Maniwa and H. Isobe, Nat. Commun., 2018, 9, 1907 CrossRef PubMed.
- S. J. Clark, M. D. Segall, C. J. Pickard, P. J. Hasnip, M. J. Probert, K. Refson and M. C. Payne, Z. Kristallogr. Cryst. Mater., 2005, 220, 567–570 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- S. Ehrlich, J. Moellmann, W. Reckien, T. Bredow and S. Grimme, ChemPhysChem, 2011, 12, 3414–3420 CrossRef CAS PubMed.
- (a) K. Lisac, V. Nemec, F. Topić, M. Arhangelskis, P. Hindle, R. Tran, I. Huskić, A. J. Morris, T. Friščić and D. Cinčić, Cryst. Growth Des., 2018, 18, 2387–2396 CrossRef CAS; (b) F. Topić, K. Lisac, M. Arhangelskis, K. Rissanen, D. Cinčić and T. Friščić, Chem. Commun., 2019, 55, 14066–14069 RSC; (c) M. Arhangelskis, F. Topić, P. Hindle, R. Tran, A. J. Morris, D. Cinčić and T. Friščić, Chem. Commun., 2020, 56, 8293–8296 RSC; (d) L. Kumar, K. Leko, V. Nemec, D. Trzybinski, N. Bregovic, D. Cinčić and M. Arhangelskis, Chem. Sci., 2023, 14, 3140–3146 RSC.
- A. Abe, K. Goushi, M. Mamada and C. Adachi, Adv. Mater., 2023, e2211160 CrossRef PubMed.
- G. Zhan, Z. Liu, Z. Bian and C. Huang, Front. Chem., 2019, 7, 305 CrossRef CAS PubMed.
- J. Yang, X. Zhen, B. Wang, X. Gao, Z. Ren, J. Wang, Y. Xie, J. Li, Q. Peng, K. Pu and Z. Li, Nat. Commun., 2018, 9, 840 CrossRef PubMed.
- Y. Su, S. Z. F. Phua, Y. Li, X. Zhou, D. Jana, G. Liu, W. Q. Lim, W. K. Ong, C. Yang and Y. Zhao, Sci. Adv., 2018, 4, eaas9732 CrossRef PubMed.
- W. J. Zhao, Z. K. He and B. Z. Tang, Nat. Rev. Mater., 2020, 5, 869–885 CrossRef CAS.
- Kenry, C. Chen and B. Liu, Nat. Commun., 2019, 10, 2111 CrossRef CAS PubMed.
- E. Hamzehpoor, C. Ruchlin, Y. Tao, C. H. Liu, H. M. Titi and D. F. Perepichka, Nat. Chem., 2023, 15, 83–90 CrossRef CAS PubMed.
- Z. An, C. Zheng, Y. Tao, R. Chen, H. Shi, T. Chen, Z. Wang, H. Li, R. Deng, X. Liu and W. Huang, Nat. Mater., 2015, 14, 685–690 CrossRef CAS PubMed.
- J. Yang, X. Zhen, B. Wang, X. Gao, Z. Ren, J. Wang, Y. Xie, J. Li, Q. Peng, K. Pu and Z. Li, Nat. Commun., 2018, 9, 840 CrossRef PubMed.
- E. Hamzehpoor and D. F. Perepichka, Angew. Chem., Int. Ed., 2020, 59, 9977–9981 CrossRef CAS PubMed.
- (a) H. Wang, R. X. Hu, X. Pang, H. Y. Gao and W. J. Jin, CrystEngComm, 2014, 16, 7942–7948 RSC; (b) S. d'Agostino, F. Spinelli, P. Taddei, B. Ventura and F. Grepioni, Cryst. Growth Des., 2018, 19, 336–346 CrossRef; (c) M. Singh, K. Liu, S. L. Qu, H. L. Ma, H. F. Shi, Z. F. An and W. Huang, Adv. Opt. Mater., 2021, 9, 2002197 CrossRef CAS.
- Y. C. Wei, S. F. Wang, Y. Hu, L. S. Liao, D. G. Chen, K. H. Chang, C. W. Wang, S. H. Liu, W. H. Chan, J. L. Liao, W. Y. Hung, T. H. Wang, P. T. Chen, H. F. Hsu, Y. Chi and P. T. Chou, Nat. Photonics, 2020, 14, 570–577 CrossRef CAS.
- Y. Huang, Z. Wang, Z. Chen and Q. Zhang, Angew Chem. Int. Ed. Engl., 2019, 58, 9696–9711 CrossRef CAS PubMed.
- K. Sato and R. Katoh, Chem. Phys. Lett., 2019, 730, 312–315 CrossRef CAS.
- N. J. Hestand and F. C. Spano, Chem. Rev., 2018, 118, 7069–7163 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Details of experimental and computational procedures, as well as selected crystallographic, PXRD, FTIR-ATR, TGA/DSC, database mining, and fluorescence emission data. CCDC 2240199–2240207, 2249511 and 2281269. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc04191c |
| ‡ d'Agostino and co-workers42 have reported that the aromatic hydrocarbons stilbene and tolane form halogen-bonded cocrystals with 14tfib which, although they do not exhibit the C–I⋯πC ladder synthon, are based on a closely-related sheet architecture. |
| This journal is © The Royal Society of Chemistry 2023 |