
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Impact of electrolyte impurities and SEI composition on battery safety†
Florian
Baakes
 ,
Daniel
Witt
and
Ulrike
Krewer
*
,
Daniel
Witt
and
Ulrike
Krewer
*
Institute for Applied Materials – Electrochemical Technologies, Karlsruhe Institute of Technology, Adenauerring 20b, 76131 Karlsruhe, Germany. E-mail: ulrike.krewer@kit.edu
First published on 3rd November 2023
Abstract
Li-ion batteries have a potential risk of thermal runaway. Current safety evaluations in academia and industry rely on experiments or semi-empirical simulations. This limits the understanding of processes leading to or occurring during thermal runaway and how chemical species and impurities can impact them. The limited (quantitative) understanding in turn hinders a holistic safety assessment and optimisation of countermeasures through design or operation. The here presented thermal-runaway model contains a detailed degradation reaction network, which allows the impact of chemical species and impurities on thermal runaway to be studied. We set a particular focus on water impurities and solid-electrolyte interphase (SEI) properties, as both are known to impact life-time of batteries. SEI composition and thickness change during ageing, which is shown here to impact battery safety significantly. The model can reproduce reported experimental behaviour: aged cells are more safe, as they start self-heating, i.e. heat production without an external heat source, at 15–20 °C higher temperatures than fresh cells. Our model suggests a thick inorganic and thus less reactive SEI as the underlying cause. Furthermore, we could show that extensive electrode drying to remove water impurities before building battery cells will not significantly improve safety characteristics. In contrast, electrodes not subjected to any drying procedure cause an earlier start of the self-heating phase, i.e. have a higher risk of thermal runaway. These insights into the sensitivity to thermal runaway allow robust methods to be tailored for its prevention, from controlling battery and SEI properties during production to adjusting safety assessment for effects of ageing.
1 Introduction
The increase in electromobility demands cost-effective and safe Li-ion battery materials.1 With ever-increasing energy densities, safety aspects in Li-ion batteries receive more and more attention from researchers.2 The most critical safety issue with Li-ion batteries is thermal runaway. This phenomenon of uncontrolled temperature increase in the battery starts with slow self-heating, i.e. heating without external heat sources, and can end in explosions, fires, and cell temperatures above 800 °C.3Especially the initial period of self-heating provides an opportunity to apply counteracting measures to prevent thermal runaway. Yet, designing effective countermeasures demands a thorough understanding of the processes triggering and occurring during this critical initial period. In a previous study, we could show that the self-heating phase is characterised by an intricate balance between endothermic and exothermic processes.4 The main exothermic reactions are related to the decomposition and reformation of the solid electrolyte interphase (SEI), a protective layer at the surface of carbon-based negative active material particles.4,5 Presently, not much is known about the effect of SEI properties and highly reactive impurities such as water on the self-heating behaviour of Li-ion batteries. This motivates us to conduct a detailed analysis in this publication.
Since its discovery in the late 1980s, the SEI is among the most investigated parts of Li-ion batteries.6–8 As a naturally formed surface layer on the carbon electrode, it originates from solvent and conductive salt reduction during the first charge–discharge cycle, i.e., during the so-called cell formation process. After formation, the SEI serves as an electric isolator in the ion-conducting phase. Stabilising the SEI and preventing its continuous growth due to ongoing SEI reactions is crucial for reaching long battery life. Tremendous efforts have been undertaken to learn more about its composition, structure, and effects on performance.7–9 In a recent study, Liu et al.10 combined electrochemical measurements, an electrochemical quartz crystal microbalance, atomic force microscopy, X-ray photoelectron spectroscopy, and online electrochemical mass spectrometry to estimate the composition and thickness of the SEI after formation. With this combined approach, they were the first to show that the initially formed organic SEI component lithium ethylene dicarbonate (LEDC) is partially oxidised to form Li2O and several gases. However, the influence on battery performance was not investigated in their study. Son et al.11 compared the influence of the SEI-forming additives fluoroethylene carbonate (FEC) and vinylene carbonate (VC) on battery performance. They found that FEC could enable better fast-charging capabilities and long-term capacity retention. This is assumed to be connected to a higher resistance for the cell with VC, which is caused by a more polymeric SEI stemming from VC as an additive. The higher resistances on the electrode surface lead to higher overpotentials at elevated currents, eventually resulting in lithium plating during charging. As the SEI is a nanometer-thin surface layer, experimental investigations are very complex,12 and quantitative analysis has not yet been possible. Thus, a plethora of simulation methods have been used to tackle this problem.13
In a simulation study using a kinetic Monte-Carlo (kMC) approach connected to a Newman-type battery cell model, Röder et al.14 showed that higher currents during initial SEI formation may lead to a gradient in the SEI thickness along the electrode thickness. With a holistic model-based analysis of experimental electrochemical impedance spectroscopy data and C-rate tests, we could relate the ageing behaviour of Li-ion cells, among other factors, to an evolution of SEI thickness and interfacial properties.15 The model contained a Newman-type cell model extended with an SEI. In a recent study Esmaeilpour et al.16 used a kMC approach with over 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 simulation conditions to find that the most probable way of SEI formation is a sequence of formation, dissolution and finally, aggregation. Furthermore, approaches using kMC methods in combination with computational chemistry support long-time existing hypotheses about the composition and morphology of the SEI.17–19 These studies illustrate how simulations can be used as powerful tools to unravel, supplement or support existing theories on SEI growth and functionality.
000 simulation conditions to find that the most probable way of SEI formation is a sequence of formation, dissolution and finally, aggregation. Furthermore, approaches using kMC methods in combination with computational chemistry support long-time existing hypotheses about the composition and morphology of the SEI.17–19 These studies illustrate how simulations can be used as powerful tools to unravel, supplement or support existing theories on SEI growth and functionality.
It was already 20 years ago when the first experiments by Dahn et al. connected the self-heating behaviour of Li-ion cells to SEI decomposition.5 In experimental studies of aged cells, an increase in the self-heating temperature of about 15–20 °C had been suggested to stem from a thicker and more inorganic SEI.20–23 However, neither detailed experimental nor simulation-based studies have so far been performed on the influence of SEI composition or thickness on battery safety, especially during the self-heating phase. Thus, this publication aims to tackle this open challenge.
H2O is a known impurity during battery manufacturing.24 Hygroscopy of electrodes is the most often referenced origin of water in cells.25,26 Several studies showed the detrimental effect of water on battery performance.26–28 However, also positive impacts of H2O addition for Li-metal batteries have been reported.29 LiF is formed from the reaction of H2O with LiPF6, and it is a stable and highly conducting SEI component leading to better performance. In contrast, worse performance is usually connected to formation of the phosphorous decomposition products PF5 and POF3. These reactions are known to occur at room temperature and are inherently linked to battery ageing. Weber et al.30 investigated the ageing of an electrolyte mixture stored at 95 °C and identified 12 different organo-phosphoric decomposition products. They suggested that a detailed analysis of the formation of these products during ageing could be used to identify “ageing stages” for LiPF6-based electrolyte composition. Stich et al.31 investigated the kinetics of these decomposition reactions by purposely contaminating an electrolyte solution with 1000 ppm water and measuring the concentrations of H2O, HF and HPO2F2. They found that hydrolysis is not following a simple rate law and thus developed a kinetic model to describe their experiments. Huttner et al.32 studied the influence of different drying strategies for water removal on battery performance. They found that extensive drying can decrease performance. This is due to the extreme conditions the battery materials are subjected to during drying. In general, H2O contamination and follow–up reactions might not immediately influence battery performance, as Zheng et al. showed.28 They found that after 100 cycles, the capacity retention for batteries contaminated with H2O and without contamination was in the same range, with around 95% remaining capacity for water-free and 90% remaining capacity for water-containing batteries. However, after 300 cycles, increased H2O content drastically reduced capacity retention. The results show 90% remaining capacity for water-free batteries and 55% for water-contaminated ones. The water-containing battery had 14 mg water added to an 18650 battery. Despite these efforts to qualitatively and quantitatively correlate H2O contamination and battery performance, we have not found any reports in the literature on tests to investigate the effect of water on battery safety, especially during the crucial self-heating phase. This contribution will shed light on the extent to which H2O contamination influences battery safety.
To address both the sensitivity of water impurities and of SEI composition to thermal self-heating and thermal runaway, we use a component-based Li-ion battery degradation model. This approach allows us to assess the impact of each participating chemical species on the progression towards thermal runaway at open circuit potential. Our comprehensive model encompasses 12 decomposition reactions and 20 participating species, which will be parameterised through two separate experiments. First, for the conductive salt decomposition, the experiments conducted by Stich et al.31 will be used. The complete set of reactions is then parameterised against an accelerated rate calorimetry (ARC) measurement.33 The thus parameterised model is then used to conduct in-depth case studies on the effect of LEDC content in the SEI, the SEI thickness, and the H2O contamination on self-heating and thermal runaway. Eventually, we present a broader parameter study that combines all three effects, examining potential interdependencies between them and illustrating dominant processes and properties.
2 Methods and study cases
This section outlines the underlying reaction network. Furthermore, it provides a detailed description of the model with its assumptions, and the experiments employed for model parameterisation and validation. Eventually, the case study as well as the initial conditions of the reference case are introduced.2.1 Reaction network
The reaction network of SEI and electrolyte degradation forms the core of this comprehensive case study as it allows for the traceback of thermal behaviour to single reactions, reaction interactions and species. The subsequent section provides a thorough overview of the postulated reactions, citing the relevant literature and discussing explicit and implicit interactions. Table 1 summarises the reactions, including the abbreviations used in this work, their reaction enthalpies and the prospective temperature range of a notable start of each reaction.| Name | Abbr. | Equation | Δr![[H with combining low line]](https://www.rsc.org/images/entities/b_i_char_0048_0332.gif) /kJ mol−1 /kJ mol−1 |
T Start/°C |
|---|---|---|---|---|
| a * slow process during cell formation; ** process considered indirectly. | ||||
| Conductive salt decomposition29 | CSD | LiPF6 ⇌ LiF + PF5 |

|
25*/60–80 |
| PF5 decomposition29 | PFD | PF5 + H2O ⇌ 2HF + POF3 |

|
25*/60–80 |
| POF3 decomposition29 | POFD | POF3 + H2O → HF + HPO2F2 |

|
25*/60–80 |
| Organic SEI production30,31 | OSP | 2LiC6 + 2C3H4O3 (EC) → (CH2OCO2Li)2 + C2H4 + 2C6 |

|
25**/80–120 |
| Inorganic SEI production30,31 | ISP | 2LiC6 + C3H4O3 (EC) → Li2CO3 + C2H4 + 2C6 |

|
25**/80–120 |
| LiOH production20 | LSP | LiC6 + H2O → LiOH + 0.5H2 + C6 |

|
25**/80–120 |
| Organic SEI decomposition5,31 | OSD | (CH2OCO2Li)2 → Li2CO3 + C2H4 + CO2 + 0.5O2 |

|
60–120 |
| Inorganic SEI decomposition32 | ISD | Li2CO3 + 2HF → 2LiF + H2O + CO2 |

|
25 |
| LiOH decomposition33 | LSD | LiC6 + LiOH → Li2O + 0.5H2 + C6 |
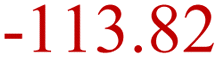
|
100–120 |
| Cathode decomposition34 | CD |
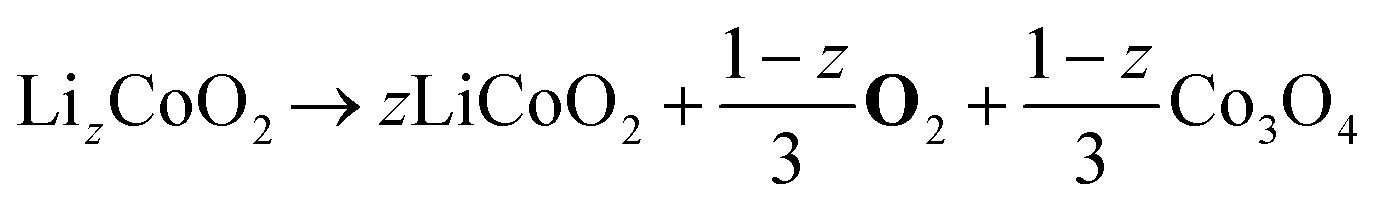
|

|
150–220 |
| EMC combustion34 | EMCD | 3.5O2 + C4H8O3 (EMC) → 4CO2 + 4H2O |

|
180–350 |
| EC combustion34 | ECD | 2.5O2 + C3H4O3 (EC) → 3CO2 + 2H2O |

|
180–350 |
Considering thermal battery decomposition, the first reactions that occur during heating of the cells at notable rates are the conductive salt decomposition (CSD) and the subsequent decomposition to POF3 (PFD) and HPO2F2 (POFD). They are reported to occur at elevated temperatures around 60 °C to 80 °C. They also happen at room temperature, but only slowly and in the range of days, much longer than during thermal abuse, where reactions occur within hours or minutes. Following the publication of Stich et al.,31 we consider the decomposition of LiPF6 to PF5 and LiF (CSD) and the subsequent decomposition of PF5 with H2O to POF3 and HF (PFD) as equilibrium reactions. In their work,31 PF5 directly reacts with two H2O to form three HF and HPO2F2. However, in a study by Solchenbach et al.,34 POF3, an intermediate, was detected by online electrochemical mass spectrometry. This detection implies that the lifespan of POF3 is sufficiently long to be considered significant and cannot be disregarded in our analysis. Thus, we consider the first decomposition of PF5 with H2O to form POF3 and HF (PFD) and the subsequent decomposition of POF3 with H2O to form HF and HPO2F2 (POFD) as individual reactions.
The decomposition of LiPF6 and the subsequent reactions of its decomposition products PF5 and POF3 initiate the release of HF, which causes the breakdown of Li2CO3 by reacting with HF to form LiF, H2O, and CO2 (ISD). Freiberg et al.35 reported that Li2CO3 coated on a carbon electrode reacted immediately with protons, and thus HF, to release CO2. From this, we conclude that the decomposition reaction of Li2CO3 with HF or H+ is not rate-limiting and that it will happen rapidly as soon as LiPF6 is decomposed and releases HF.
The decomposition of the organic SEI component LEDC (OSD) is reported to start somewhere in a wide temperature range from 60 °C to 120 °C.5 It is responsible for the transition into the self-heating phase of an ARC measurement.5,36
The decomposition of LiOH with lithium metal or Li intercalated in graphite to form H2 and Li2O (LSD) is frequently reported in the literature.25 However, we could not find any information on the temperature range for this reaction. Thus, we depend on a study of the decomposition of LiOH with LiH into Li2O and H2, which reported a decomposition temperature around 120 °C.37
The decomposition of SEI compounds, including Li2CO3, LEDC, and LiOH, eventually exposes the bare electrode surface. Given the solvent's instability against the anode's low potentials, subsequent SEI formation reactions occur at the electrode. These SEI-producing reactions (OSP, ISP, and LSP) are direct results of the preceding decomposition and are linked to the battery's self-heating.31
All reactions so far occur already below 130 °C and involve species in the anode or electrolyte. At significantly higher temperatures above 150 °C, self-heating is strongly accelerated by the decomposition of the cathode active material LiCoO2 (CD). The released O2, in turn, leads to the highly exothermic combustion of solvent molecules (ECD and EMCD),36 further accelerating thermal self-heating.
Fig. 1 illustrates the intricate interaction of the thermal degradation processes at the electrolyte and the SEI. The reactions are strongly connected via the participating species, leading to a complex interconnected reaction network, including a reaction cycle involving HF and H2O.
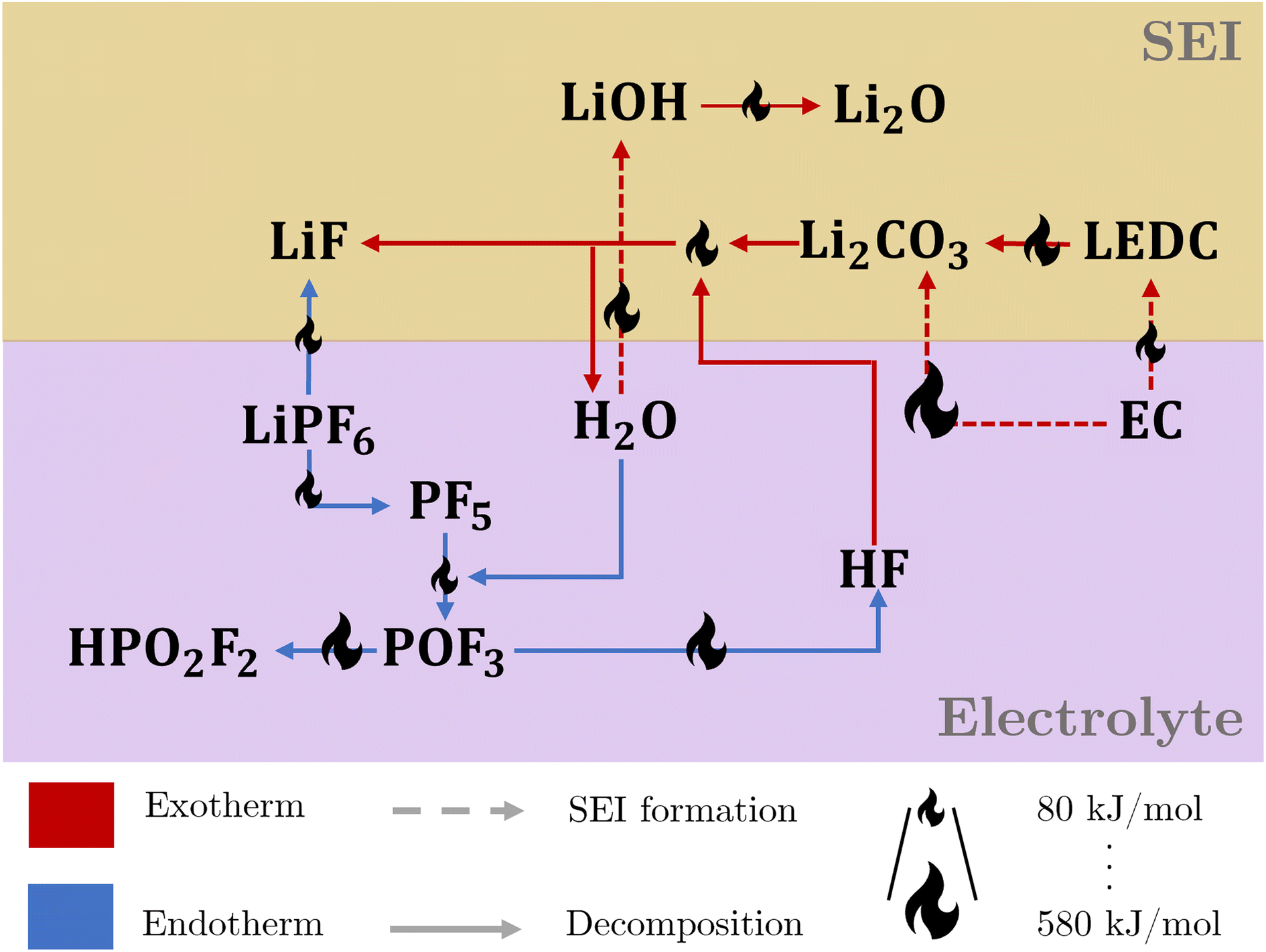 | ||
| Fig. 1 Reaction network for thermal composition of the SEI and electrolyte, covering the interaction between salt, solvent, and SEI. Red and blue arrows indicate exo- and endothermic reactions, respectively. The size of the flame icon on the arrows scales with the corresponding amount of consumed or released reaction heat. | ||
2.2 Mathematical model
This section presents the modelling approach, the modelled system and the underlying assumptions. Our previous publication4 provides a more detailed description of the basic model.| Description | Equation | No. |
|---|---|---|
| Species balance |
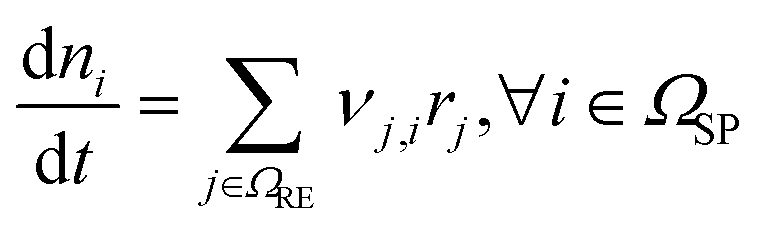
|
(1) |
| Reaction kinetics |
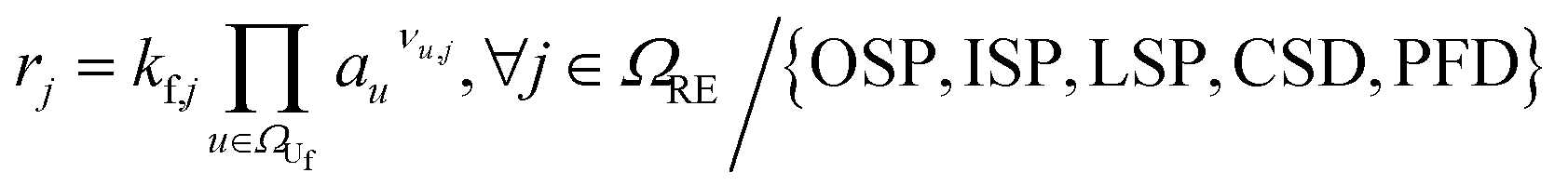
|
(2) |
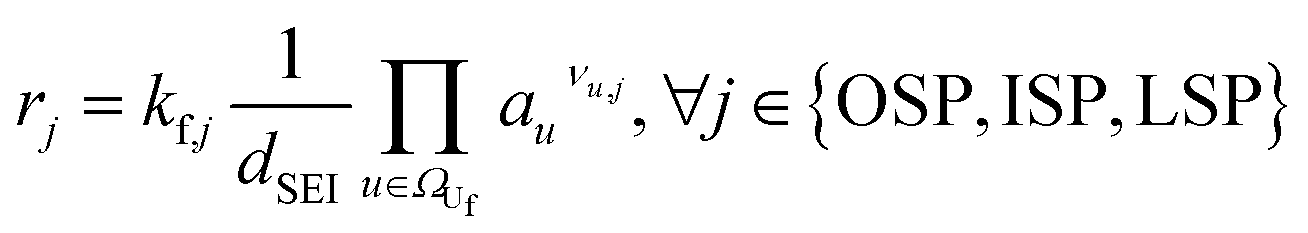
|
(3) | |
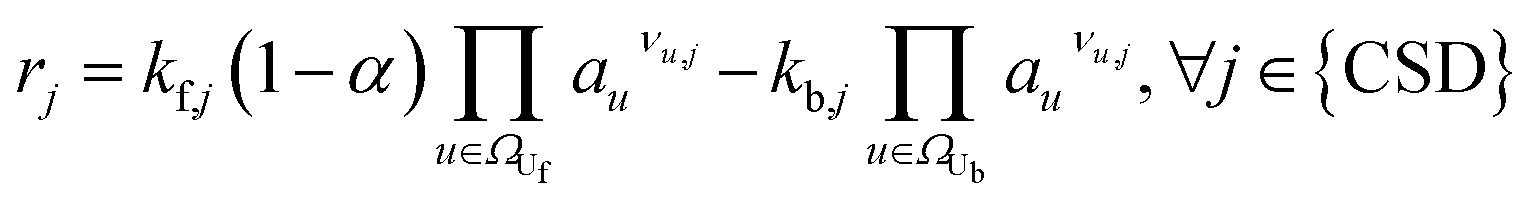
|
(4) | |
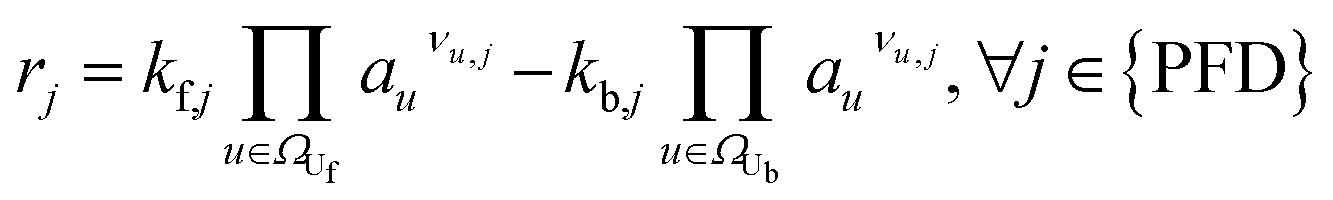
|
(5) | |
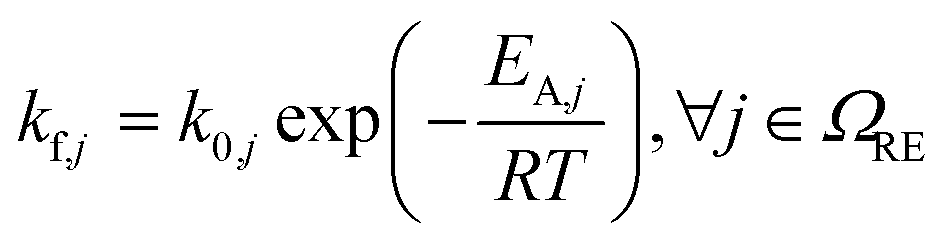
|
(6) | |
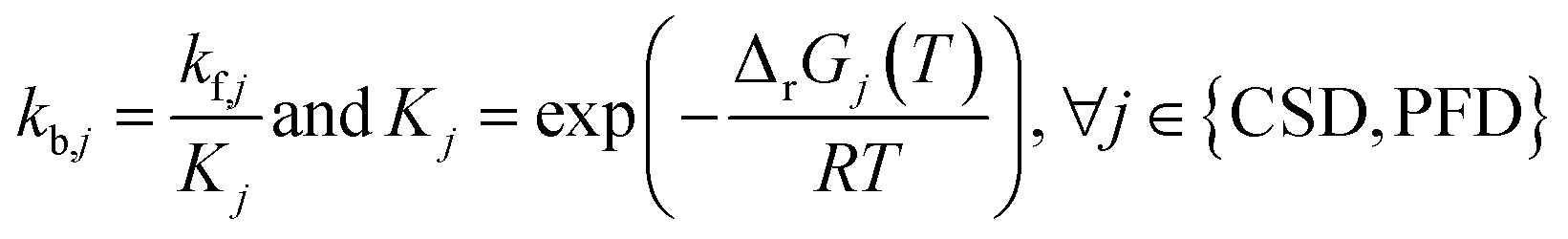
|
(7) and (8) | |
| ΔrGj(T) = ΔrHj(T) − TΔrSj(T), ∀j ∈ {CSD, PFD} | (9) | |
| Energy balance |
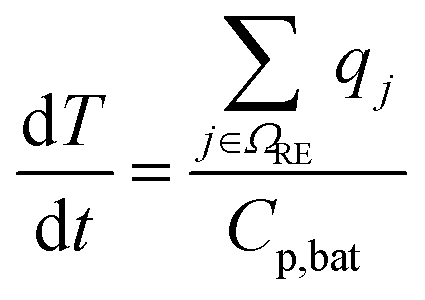
|
(10) |
• All reactants are ideally mixed, so no spatial discretisation is applied. Since the time constant for diffusion and thermal conductivity is on the order of seconds and the ARC measurement takes hours, we deem the assumption of a perfectly mixed system a sound approach.
• A pressure increase due to gas evolution does not impact liquid-phase reactions.
• All reactions except for LiPF6 and PF5 decomposition are irreversible.
• SEI formation reactions are modelled as chemical reactions.
2.3 Experiments and parameterisation
This study aims to reveal the influence of H2O impurities and SEI composition on battery safety at high temperatures. Kinetic parameters, k0 and EA, are obtained from two distinct experimental studies: Stich et al.31 focused on the decomposition of LiPF6 with H2O as an impurity, providing an isolated study of LiPF6, PF5 and POF3 decomposition. Meanwhile, Maleki et al.33 performed an ARC measurement encompassing all concurrent reactions. In the following, the experiments themselves are described, as well as our parameterisation procedure.Stich et al.31 used a 1:1 v:v EC:DEC 1 M LiPF6 electrolyte with an initial concentration of ≤ 15 ppm water and ≤ 50 ppm HF. They then added 1000 ppm of water. The concentrations of H2O, HF and HPO2F2 have been monitored for 15 days at room temperature by coulometric Karl Fischer titration, acid–base titration, and ion chromatography.
An ARC measurement by Maleki et al.33 is used to parameterise and study the transition from a safe battery at room temperature over its self-heating phase starting around 100 °C until the transition into rapid thermal runaway. The ARC measurements are conducted by inserting the battery into an oven-like apparatus. Following this, a linear temperature gradient is usually applied to reach a temperature where the first degradation reactions are expected to occur. In the experiments of Maleki et al.,33 this was 40 °C. Then, a heat–wait–seek (HWS) procedure is applied, where a 10 °C step-wise increase in temperature is conducted. Each step is followed by a period where the battery cell is equilibrating with the new temperature, and the temperature is monitored. No further heating is supplied when self-heating is detected during the seeking procedure. A detailed explanation of the underlying algorithm and the experiment used to parameterise our model is given in our previous publication.4 The cell used by Maleki et al.33 had a capacity of 550 mA h. The chemistry was reported to be a carbon-based anode, a LiCoO2 cathode, and polyvinylidene fluoride as the binder in both electrodes, and an ethylene carbonate (EC)/ethyl methyl carbonate (EMC) mixture with LiPF6 as the conductive salt. Since the original publication lacked some data crucial for simulation, the following assumptions have been made:
• The electrolyte consists of a 50:50 EC:EMC (v:v) mixture with 1200 mol m−3 LiPF6.
• The geometry of the cell is taken from a commercially available one that has a capacity and cell chemistry comparable to the cell used in the study by Maleki et al.44
• The electrode geometry is similar to other published electrodes of the same chemistry45,46
The kinetic parameters for the decomposition of LiPF6 and the subsequent reactions of PF5 and POF3, namely CSD, PFD, and POFD, have been parameterised such that two constraints are met. The first is that the experimental data31 of changes in the electrolyte at room temperature could be reproduced. The second is that the reactions notably accelerate within the reported temperature interval (see Table 1). For the decomposition of Li2CO3, the kinetic constants are set such that this reaction is not limiting.
For the other reactions, the kinetic parameters have been adjusted to meet two constraints: the first one is that the reactions occur within the reported temperature interval for each given reaction (Table 1). The second constraint is that the simulation can reproduce the experimental data from Maleki et al.33 Exceptions to this are the SEI forming reactions, OSP, ISP, and LSP. Given that they are, in nature, electrochemical reactions, they should notably occur even at room temperature with an unprotected electrode. Reported energy barriers for these reactions are very low, if not 0.17,18,47 The term introduced to account for the inhibition effect of the SEI (see eqn (3)) does not prevent notable reactions even at low temperatures with these low barriers. Therefore, the energy barriers are adjusted such that these reactions occur in a temperature range concurrently with the decomposition of the existing SEI.
The identified parameter values are given in the ESI.† Model equations and parameters were implemented in MATLAB, and the simulation was performed using the ode15s solver. All calculations have been performed with MATLAB Version 2022a,48 using an i7-9750H processor with 16 GB RAM. The average simulation time was six minutes.
2.4 Reference conditions and variation in water content and SEI properties
With their intricate internal decomposition mechanisms during formation, operation and thermal abuse, Li-ion batteries present a complexity that hinders the precise determination of the amount of species present at the beginning of a thermal decomposition study. Due to these restrictions, educated estimates for some components are necessary. For the sake of brevity, the procedures to arrive at the initial conditions will be shown in the ESI.† The values of interest are the conductive salt decomposition products, namely PF5, POF3, HPO2F2, HF, and H2O and the initial amount of the SEI components, Li2O, LEDC, LiOH, LiF, and Li2CO3, as well as the initial SEI thickness, see Table 3.| Parameter | Scenario | ||||||
|---|---|---|---|---|---|---|---|
| Reference | Thick SEI | Thin SEI | Organic SEI | Inorganic SEI | Wet electrode | Dry electrode | |
| d SEI/nm | 50 | 75 | 25 | 50 | 50 | 50 | 50 |
| ε LEDC/vol% | 45 | 45 | 45 | 90 | 0 | 46 | 45 |
| ε Li2CO3/vol% | 34 | 34 | 33 | 8.4 | 39 | 30 | 34 |
| ε LiF/vol% | 10.4 | 10.4 | 11.4 | 1 | 30.4 | 9 | 10.6 |
| ε Li2O/vol% | 10 | 10 | 10 | 0 | 30 | 10 | 10 |
| ε LiOH/vol% | 0.6 | 0.6 | 0.6 | 0.6 | 0.6 | 5 | 0.4 |
| C H2O/ppm | 260 | 260 | 260 | 260 | 260 | 505 | 168 |
| C HF/ppm | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| C PF3/ppm | 992 | 992 | 992 | 992 | 992 | 880 | 1017 |
| C POF3/ppm | 46 | 46 | 46 | 46 | 46 | 95 | 31 |
| C HPO2F2/ppm | 1306 | 1306 | 1306 | 1306 | 1306 | 5332 | 536 |
H2O impurities, SEI compositions and SEI thickness can be influenced during manufacturing processes such as electrode drying or formation, as well as through battery ageing. At present, it is unfeasible to model and incorporate all these processes. Nevertheless, we implicitly investigate the effects and variability of different manufacturing conditions on battery behaviour during thermal abuse by designing a case study that uses various combinations of manufacturing and ageing inspired initial values for water impurity, SEI composition and thickness (Table 3). The variation in H2O content is based on a study of different drying procedures by Huttner et al.32 The H2O content will change due to different water contents in the electrodes following Huttner's results for the undried “wet”, the medium dried, and the highly dried electrodes. The different values in the anode of 2422 ppm, 286 ppm, and 214 ppm translate into 5 vol%, 0.6 vol% and 0.4 vol% of LiOH content within the SEI. The reported values of 2644 ppm, 500 ppm and 464 ppm in the separator and 313 ppm, 156 ppm, and 63 ppm for the cathode translate into an H2O increment in the electrolyte of 930 ppm, 334 ppm, and 172 ppm, respectively.
In the early 2000s it was revealed that LEDC is the primary decomposition product of EC.49 A recent study by Wang et al.,50 however, questions the stability of LEDC and proposed that lithium ethylene mono-carbonate (LEMC) is the stable alternative. Following this, Xie et al.51 investigated the formation pathways of LEMC, including the decomposition of LEDC and found that all kinetically favourable pathways need water as a reactant. It is apparent that there is still a lively discussion on the exact composition of the SEI. Thus, we will consider a wide range of differing compositions and assume LEDC as the major organic compound: here, 90 vol%, 45 vol% and 0 vol% LEDC are investigated. We did not choose the pure LEDC content since LiOH has to be part of the SEI to account for water impurities. Furthermore, the complete lack of Li2CO3 would lead to an accumulation of HF according to the above-described behaviour, which we deem very improbable. Thus, the remaining volume percentages are Li2CO3.
The SEI thickness has been varied between 25 nm, 50 nm, and 75 nm, which are within reported values for SEI thicknesses.10,52,53
All variations undergo the same formation and conditioning procedure described in the ESI.† From this, the initial values listed in Table 3 are calculated.
An extensive parameter variation, including all possible 27 variations (3 × 3 × 3), has been simulated to investigate the interdependence. The additional 18 initial values, apart from the 9 already listed in Table 3, can be found in the ESI.†
The initial and maximum temperatures are 25 °C and 220 °C, respectively. The pressure is constant at p = 101325 Pa.
The initial conditions for all modelled species and their physical parameters and data on the electrode structure can be found in the ESI.†
3 Results and discussion
The upcoming section evaluates the propagation and characteristics of the thermal runaway during thermal abuse of the given Li-ion battery for the various manufacturing and ageing scenarios. First, sensitivities of ARC measurements to water content, SEI composition and SEI thickness are identified and quantified. This is followed by an in-depth analysis of this behaviour and the underlying causes based on the progression of reactions, their interplay, and their contribution to the temperature evolution during the abuse test. Particular focus is given to the transition from the heat–wait–seek to the exothermal self-heating mode, as this is the crucial point determining a battery's safety range. Then we elucidate the effects of different water contents and initial SEI properties on the state of the battery and occurring reactions and their consequences in an ARC measurement.3.1 Impact of the SEI state and H2O on temperature evolution
In Fig. 2, the simulated temperature evolution is displayed for a variation in (a) SEI composition, (b) SEI thickness, and (c) H2O content. The preheating procedure (1 °C min−1) can be observed up to 40 °C. From then on, the ARC switches to the heat–wait–seek phase and performs 10 °C heating steps, including wait-and-seek periods. This is repeated until the first self-heating is detected. This happens for the reference scenario (black line) at 108 °C and 7.8 h, marked by SH-II. Then, the ARC switches to the self-heating mode and starts to follow the cell temperature. For the reference scenario, self-heating at this stage is not sustained, and after a period of 1 h at 110 °C, an additional heating step is performed. At 119 °C and 10.1 h, marked by SH-III, another self-heating of the cell leads to switching to the exothermic mode of the ARC again. The temperature monotonically increases throughout the subsequent 13 h until the thermal runaway is triggered at 174 °C and 23 h marked by TR.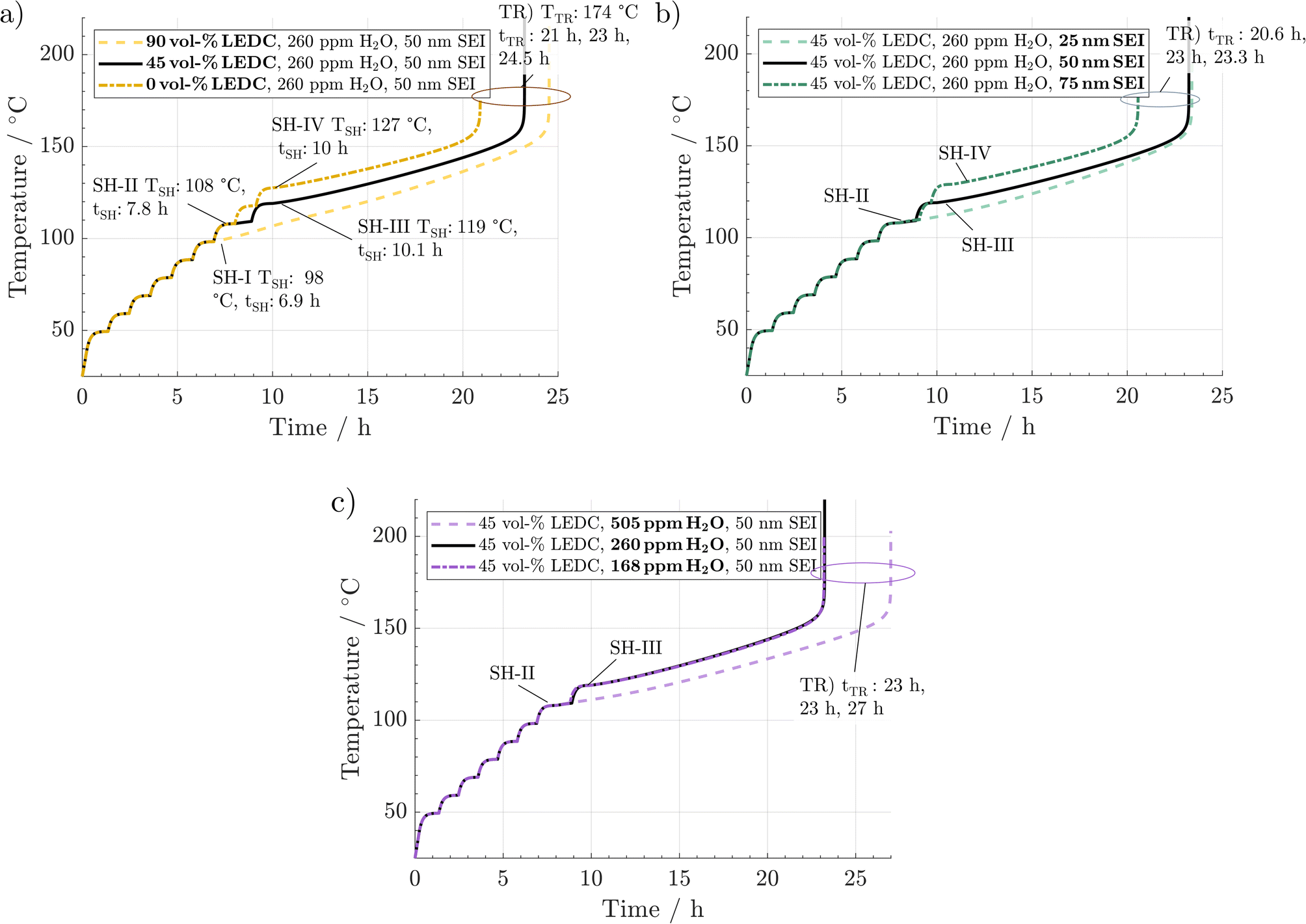 | ||
| Fig. 2 ARC simulations with variations of (a) SEI composition, (b) SEI thickness, and (c) water content originating from the electrodes. The solid black line refers to the reference case in all subfigures. The lighter dashed line refers to the scenario featuring the lower self-heating temperature TSH. The dash-dotted darker line indicates the scenario with the higher self-heating temperature. Characteristic self-heating temperatures, self-heating times, tSH, and runaway times, tTR, are indicated in the graphs. | ||
Fig. 2a shows the effect of variations in SEI composition at 90 vol% LEDC (dashed line) and 0 vol% (dash-dotted line). No significant difference between the scenarios can be observed during the preheating phase and in the first six heating steps. At 98 °C and 6.9 h, marked by SH-I, significant self-heating is observed for the 90 vol% LEDC-case, which is 11 °C earlier compared to the reference case. Temperature increases monotonically without a further heating step, and the cell reaches the thermal runaway at 173 °C and 24.5 h. For the inorganic SEI without LEDC, one additional heating step compared to the reference case is needed, with self-heating starting only at 127 °C and 10 h. This makes a difference of +9 °C compared to the reference case. Yet, the cell goes faster into the thermal runaway. It can be concluded that SEI composition has a notable effect on thermal safety of a battery, with cells with more LEDC, e.g. due to less ageing, being more likely to enter a thermal event, leading finally to thermal runaway.
In Fig. 2b, the effect of SEI thickness on thermal runaway is presented. The behaviour during the preheating and first heating steps shows no deviation from that of the reference case. The thin SEI enters one heating step earlier into the self-heating phase than the reference case. Self-heating progresses and causes a rapid thermal runaway at 174 °C and 23.3 h. In contrast, the thick SEI case reaches the self-heating only after a further heating step, at around 127 °C and 10 h, and enters the final runaway phase earlier, at 174 °C and 20.6 h. According to this analysis, SEI thickness also significantly impacts battery safety, with a thicker SEI leading less quickly to self-heating. It should be noted that the results presented here are specific to the investigated system of a graphite anode combined with EC/EMC 1.2 M LiPF6 liquid electrolyte. For example, in the case of Li metal and all solid state batteries, a decrease in safety with increasing SEI thickness was found.54,55
Finally Fig. 2c shows the impact of H2O impurities. As in the variations before, no difference is observed until 108 °C and 7.8 h. Then, the cell with high H2O amounts enters one heating-step earlier into the sustained self-heating phase. The thermal runaway is reached at 174 °C and 27 h. In contrast, the curves for low and medium water amounts are identical. Thus, high amounts of H2O seem to be detrimental to thermal safety of Li-ion batteries.
From the above-described results, we conclude that all three parameters significantly impact thermal safety. The most apparent difference between all cases is the change in self-heating temperature and the time from there until the thermal runaway is reached. For a better comparison, these key parameters are summarised in Fig. 3. Here, a clear trend can be observed. A lower self-heating temperature corresponds to a longer time until the cell reaches the thermal runaway eventually. These opposing trends of earlier self-heating but later thermal runaway pose a fundamental question as to how to produce inherently safer batteries: while higher self-heating temperatures can be interpreted as safer, a shorter time until reaching the thermal runaway, and with this, a virtually unstoppable thermal event could be considered unsafe. Manufacturers must perform a risk analysis to make a good trade-off for these safety-critical parameters. While discussing the case study, including 27 variations, in Section 3.3, we offer an alternative metric to include both characteristics in the safety assessment of Li-ion batteries. Note that the temperature for the start of the rapid thermal runaway is identical in all cases. Similarly, as reported in the literature, this is almost exclusively caused by the onset of cathode active material decomposition. In our study, the cathode material was not varied. Thus, this behaviour was expected.
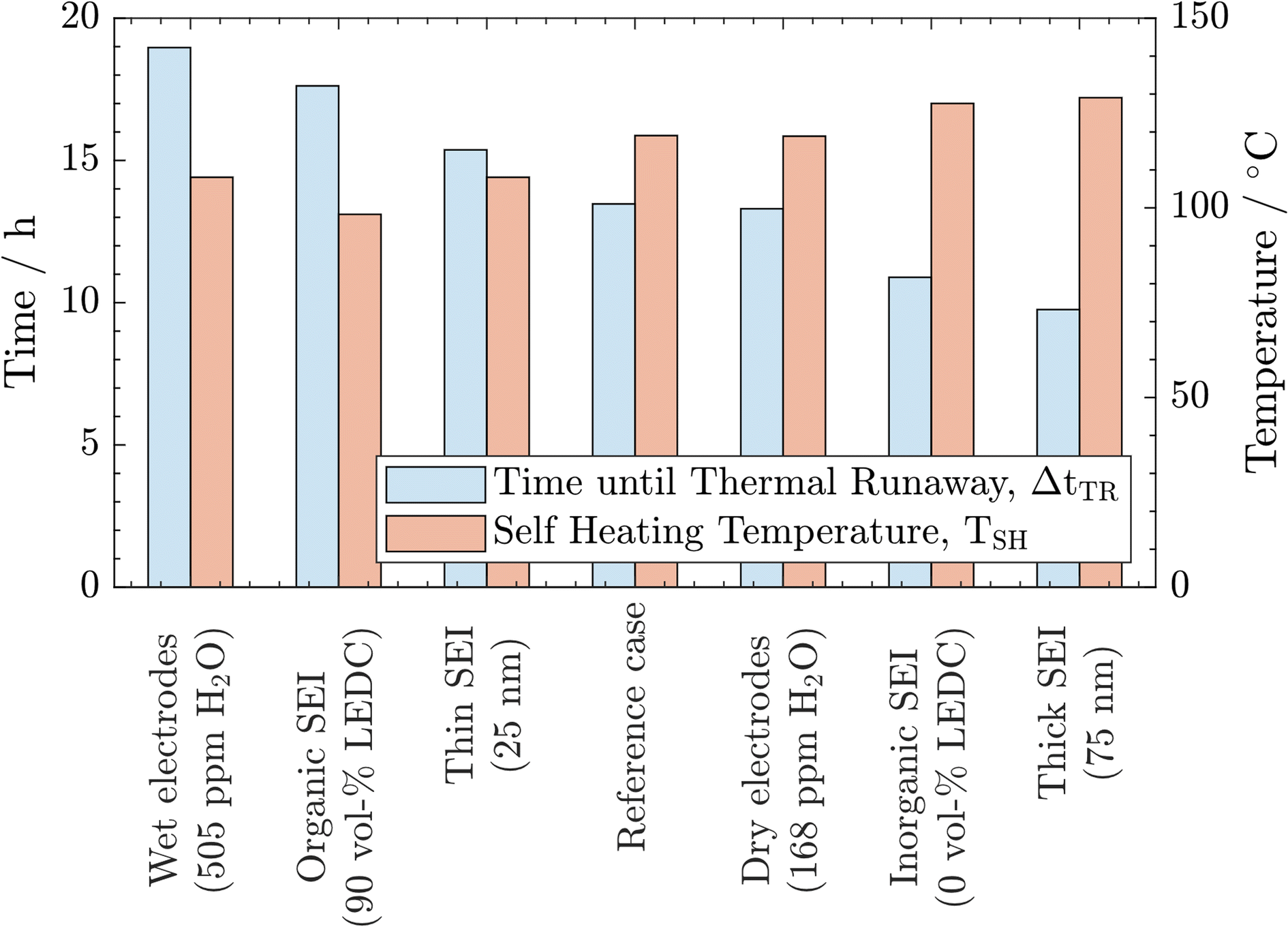 | ||
| Fig. 3 Comparison of self-heating temperature TSH and time until the thermal runaway is reached after the first exothermic phase ΔtTR as key indicators for all cases. | ||
In the following section, a deeper analysis of processes during the thermal abuse tests is performed to reveal the origin of the manufacturing- and ageing-specific differences in safety behaviour.
3.2 Analysis of processes during thermal abuse
To understand the main reactions impacting the self-heating and their sensitivity to the SEI state and water impurities, we here analyse the progression of reactions, related species concentrations and produced or consumed heat during the thermal misuse. For better readability, the figures display only those reactions that are substantially influenced by SEI composition or water content. For the same reason, all SEI-forming reactions, namely OSP, ISP, and LSP, are summed up. For individual contributions, see the ESI.† Analysis starts at 40 °C, where the first degradation reactions are observed (Fig. 4): the initial decomposition of the conductive salt is triggered by a shift of the equilibrium towards the decomposition product with increasing temperature. Furthermore, the decomposition of PF5 (PFD) and POF3 (POFD) starts at 60 °C, peaks around 75 °C, and ends for the first time around 110 °C (SH-II). The HF released from these reactions initiates the decomposition of Li2CO3 (ISD), which, thus, happens simultaneously to PFD and POFD. Note that the concentration of Li2CO3 is only slightly declining due to the only small amounts of H2O, and thus reactant HF, whereas it is produced in significant rates also from LEDC decomposition. Decomposition of PF5, POF3 and Li2CO3 happen simultaneously, and the ratio of produced heat by Li2CO3 decomposition vs. the consumed heat by PF5 and POF3 decomposition is always below 1 and decreases with temperature. Thus, the reactions caused by salt decomposition products, i.e. PFD, POFD, and ISD, act as heat sinks. The cause of the extinction of these reactions is the depletion of the necessary reactant H2O. As the reaction rates and, therefore, their interplay are strongly dependent on water availability, we continue the discussion when analysing the wet and dry case scenarios. Reactions connected to salt decomposition are complemented by the exothermic decomposition of LEDC with notable reaction rates occurring above 80 °C. Eventually, by increased decomposition of LEDC and already decreasing endothermic heat due to H2O depletion, the first self-heating starts at 108 °C (SH-II). This phase is, however, not self-sustaining because the exothermic LEDC decomposition slows down when much of the available LEDC has been consumed, so that a further heating step is required to trigger a thermal event. The transition to continuous self-heating of the battery at 118 °C (SH-III) is caused by the almost complete decomposition of LiPF6, which reduces the endothermic heat to nearly 0. The self-heating phase until the rapid thermal runaway is dominated by the re-formation of the organic SEI, i.e. LEDC, and the inorganic SEI, i.e. Li2CO3, and further decomposition of LEDC. The declining concentration of LiOH denotes its decomposition to Li2O (LSD) around 120 °C. The amount of LiOH introduced into the system by H2O in the anode after drying is too small to produce notable amounts of exothermic heat. The thermal runaway is eventually set in motion by the decomposition of the cathode active material (CSD) starting around 150 °C. The thus produced O2 triggers the subsequent solvent combustion, first of EC (ECD) and then of EMC (EMCD); see the ESI.† The combustion product H2O, in turn, triggers an exponential increase in PF5 (PFD) and POF3 (POFD) decomposition reactions; despite being endothermic, they cannot compensate for the strongly exothermic reactions. Simultaneously, the exothermic decomposition of Li2CO3 is also re-initiated by HF produced from the PF5 and POF3 decomposition. Finally, it should be noted that above 130 °C, the LEDC concentration is kept at around zero, as any generated LEDC is directly consumed. In contrast, the LiF concentration increases until all LiPF6 is decomposed and then stays constant. The SEI is thus completely inorganic at higher temperatures. Concentrations of HF, LiPF6 and Li2O are either 0 (HF) or can be deduced from LiF (LiPF6) or LiOH (Li2O) progressions. For the sake of clarity, they are shown in the ESI.†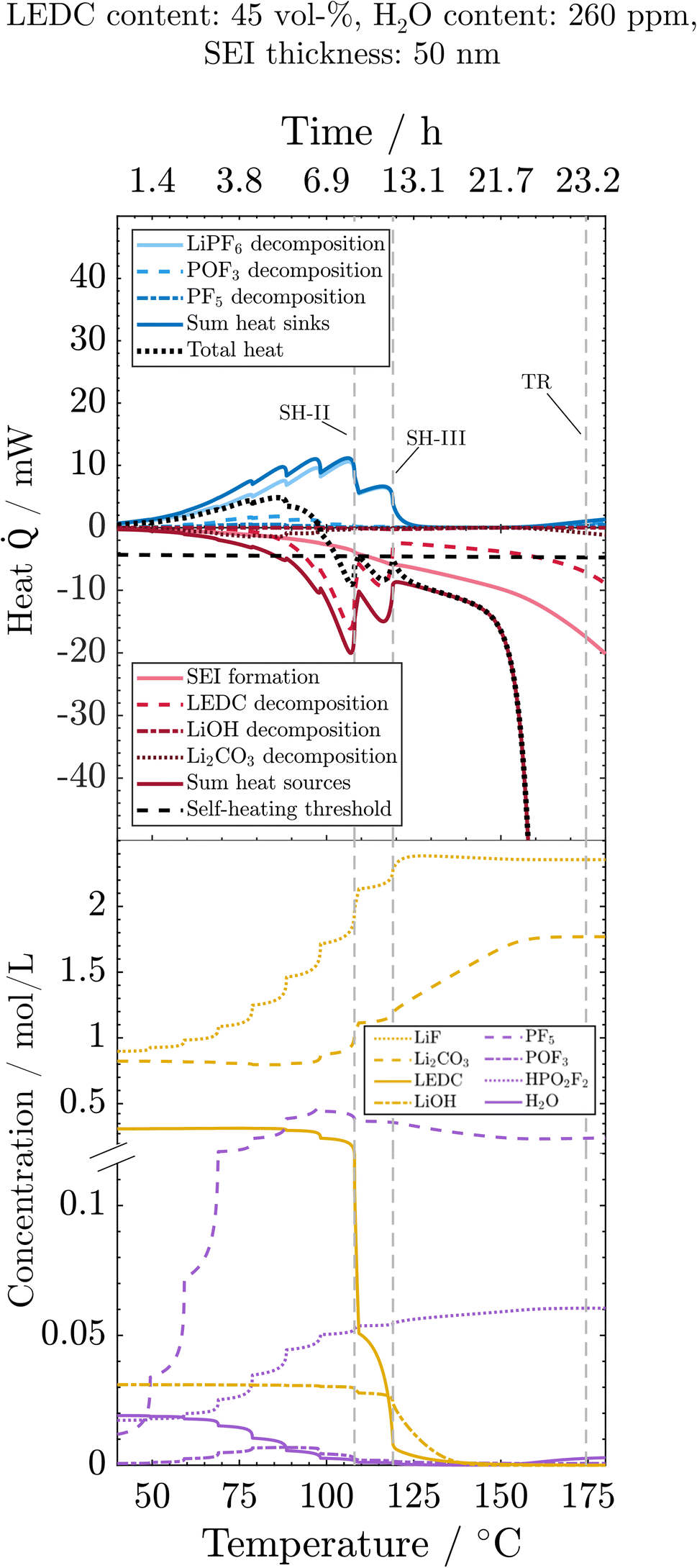 | ||
| Fig. 4 Processes during the ARC test for the reference case: evolution of heat sinks (blue) and heat sources (red) and related concentrations, as well as the total heat (black) and the self-heating threshold (horizontal dashed line). Vertical dashed lines indicate events marked in Fig. 2. The y-axis break is at 0.12 mol m−3. | ||
Having understood the process interplay for the reference case, we now evaluate how their dependence on SEI composition and thickness, and water content can explain the observed change in thermal self-heating and thermal runaway behaviour.
As SEI composition impacts mainly the SEI formation and SEI decomposition reactions, their corresponding heat is presented in Fig. 5a. Li2CO3 decomposition is only marginally affected by changes in the SEI composition and is thus displayed together with other reactions in the ESI.†
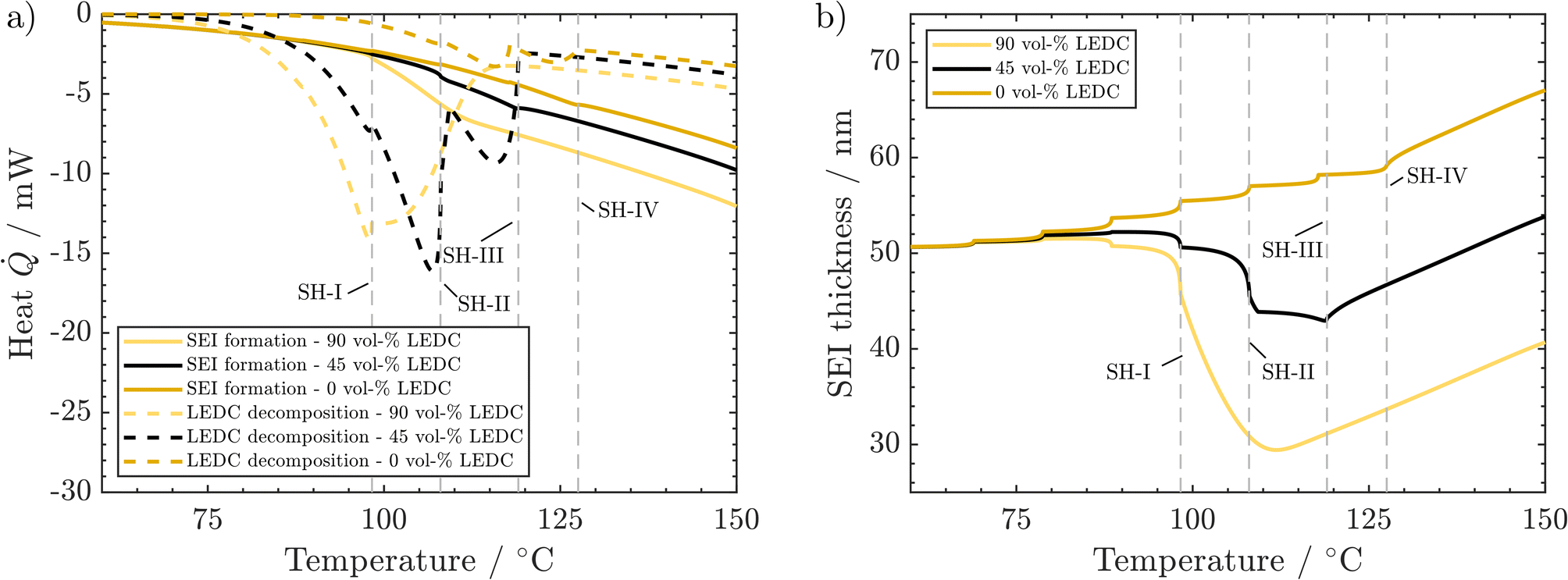 | ||
| Fig. 5 Effect of SEI composition on behaviour during the ARC test: (a) produced heat from LEDC decomposition and SEI formation. (b) Corresponding changes in SEI thickness. Conditions: 260 ppm water, 50 nm thick SEI. | ||
A higher LEDC volume fraction leads to notably earlier on-set of LEDC decomposition. This leads to the earlier self-heating on-set at 98 °C (SH-I) for the 90 vol% LEDC case compared to the reference case (45 vol% LEDC). For 90% LEDC, heat production from this reaction decreases much slower after reaching self-heating because there is still a significant amount of LEDC in the SEI (see the ESI†). Together with the higher SEI formation reaction, this can explain why the 90 vol% LEDC case needs no further heating step to proceed to thermal runaway. The large fraction of LEDC in the initial SEI and its rapid but not complete consumption lead to a significant drop in SEI thickness to 60% before SEI formation sets in and rebuilds the SEI. The decline is much more significant than for the reference case, where ca. 80% of the SEI, mostly inorganic, remains and is subsequently rebuilt. For the inorganic case, i.e. 0 vol% LEDC in the initial SEI, the missing exothermic heat from the decomposition of the initial LEDC and the slow exothermic SEI formation lead to additional heating steps. At around 128 °C, marked by SH-IV, eventually, self-heating of the battery sets in due to higher exothermic SEI formation rates at this temperature. As no LEDC is present in the initial SEI in this case, and as Li2CO3 decomposition is negligible and compensated by its production, SEI thickness increases monotonously. In all three cases, during proceeding self-heating >120 °C, the heat from LEDC decomposition is smaller than that from SEI formation. It can be concluded that LEDC content in the SEI is strongly impacting thermal safety, as the self-heating onset is strongly impacted by LEDC decomposition and formation rates, and thus by LEDC availability.
We now discuss the impact of initial SEI thickness on heat evolution from LEDC decomposition and reformation (Fig. 6a) and on the resulting changes in SEI thickness (Fig. 6b). From Fig. 2b, we know that a thicker initial SEI leads to a higher self-heating temperature. This is counterintuitive as more SEI, i.e., more LEDC, means more reactants for low-temperature decomposition. Indeed, more SEI leads to more LEDC decomposition (Fig. 6a); yet, during the seek period, LEDC decomposition heat decreases to similar values for all SEI thicknesses, whereas, for the thin SEI, exothermic SEI formation is almost double that of the thicker SEIs. The thickness-dependent SEI formation rate, eqn (3), is the key to explain why a thin SEI still leads to lower self-heating temperatures. The thinner the SEI is, the higher the formation rate and thus the heat produced from SEI formation reactions. SEI thickness can therefore be seen as a beneficial property of the SEI to prevent further formation and early thermal runaway. Our findings are also in good agreement with ARC tests of cycling-aged cells in the temperature range of 35–45 °C from the literature by Feng et al.,23 Feinauer et al.,22 Börner et al.,20 and Waldmann et al.21 All studies independently found that these ageing procedures lead to an increase in the on-set temperature of self-heating by 15–20 °C when compared to their fresh reference. Our study now delivers an explanation for this increase: when cells age, the SEI becomes thicker and more inorganic; both effects have been shown here to lead to a delayed self-heating temperature. Röder et al.56 in contrast found a lowered self-heating temperature for calendaric-aged cells at 60 °C. Our model may explain this behaviour also: either the SEI had a much more total amount of LEDC, probably dissolved also in the electrolyte, or the LEDC had reacted at 60 °C and left a thin, less inhibiting SEI.
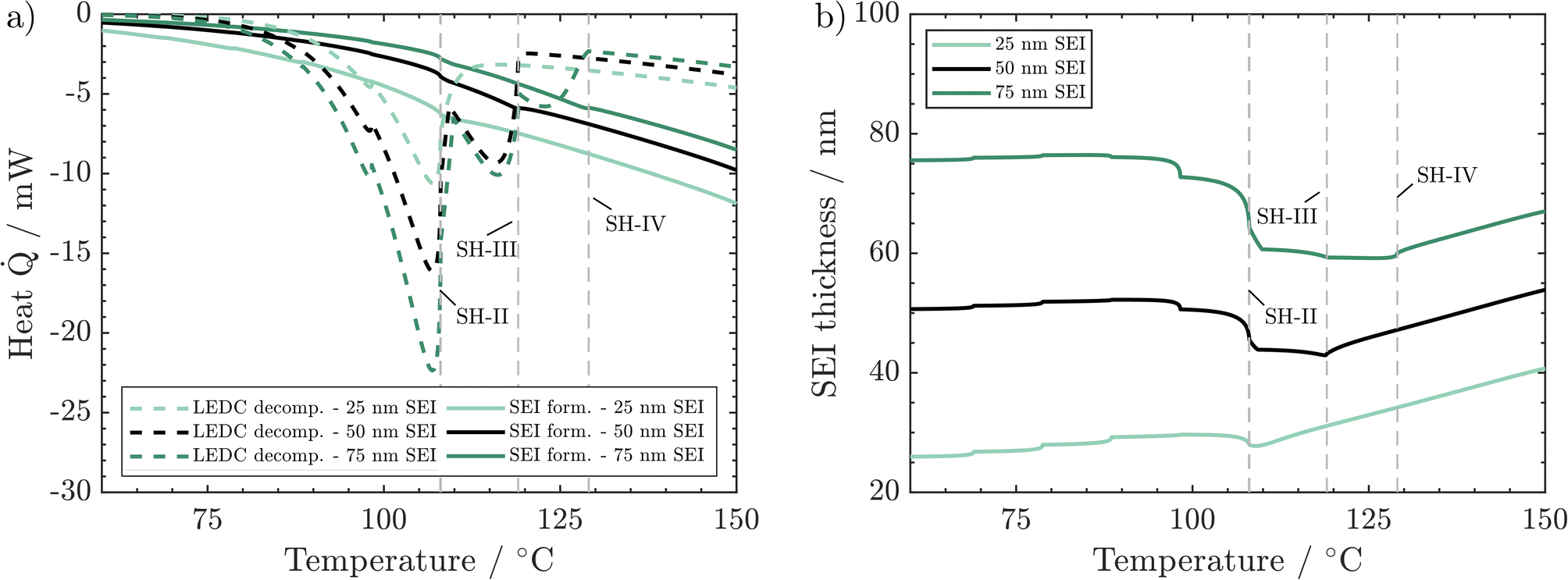 | ||
| Fig. 6 Effect of SEI thickness on behaviour during the ARC test: (a) produced heat from LEDC decomposition and SEI formation. (b) Corresponding changes in SEI thickness. Conditions: 260 ppm water, 45 vol% LEDC. | ||
Finally, we analyse the effect of H2O content deeper to understand why low and medium water impurities lead to the same temperature evolution while high water content leads to earlier self-heating. PF5, POF3 and Li2CO3 decomposition reactions (PFD, POFD, and ISD) happen almost simultaneously. Their added values are endothermic and increase with water content (Fig. 7a). It can also be observed that the higher the water content, the earlier the on-set of these reactions. In the case of dry and reference H2O concentrations, the released heats are almost identical and very small. From ca. 80 °C onwards, in the wet electrode case, most of the endothermic heat is still small and released in a temperature range where no exothermic counterpart exists. With increasing impurity concentrations, the temperature gradient after sustained self-heating changes from 4.4 °C h−1 (dry case) to 4.65 °C h−1 (medium dried case) to 4.1 °C h−1 (wet case). These changes are connected to the increased concentration of LiOH, which decomposes exothermically, and the corresponding decrease in self-heating temperature (wet case). First, when moving from the dry case to the medium dry case, the temperature gradient only increases slightly which can be explained only by a small increase in LiOH content from 0.4 vol% to 0.6 vol%. However, in the wet case the temperature gradient decreases, which is counterintuitive at first but is also connected to the higher LiOH content of 5 vol%. The significantly higher concentration of LiOH in the wet electrode case leads to notable LiOH decomposition rates and exothermic heat in the self-heating critical temperature frame of 100–130 °C. This leads to a transition into self-heating one temperature step earlier. The lower temperature in turn leads to a slower progression of all occurring reactions and, thus, a lower temperature gradient. This points to an important characteristic of an ARC measurement. As discrete temperature steps are used, more produced heat can either increase the temperature gradient (dry → medium dry), when self-heating starts during the same time step. Or it leads to a lower self-heating temperature, with a subsequent lower temperature gradient. Thus, it is important to discuss both characteristics together. In order to illustrate these impacts, the water variation case was simulated using a 2.5 °C temperature step instead of the 10 °C used before (see the ESI†). Even though the wet case also transitions into self-heating one step earlier, the gradients, 2.5210 °C h−1 (dry), 2.5459 °C h−1 (medium) and 2.6975 °C h−1 (wet), now better correspond to the produced heat as the temperatures are closer together.
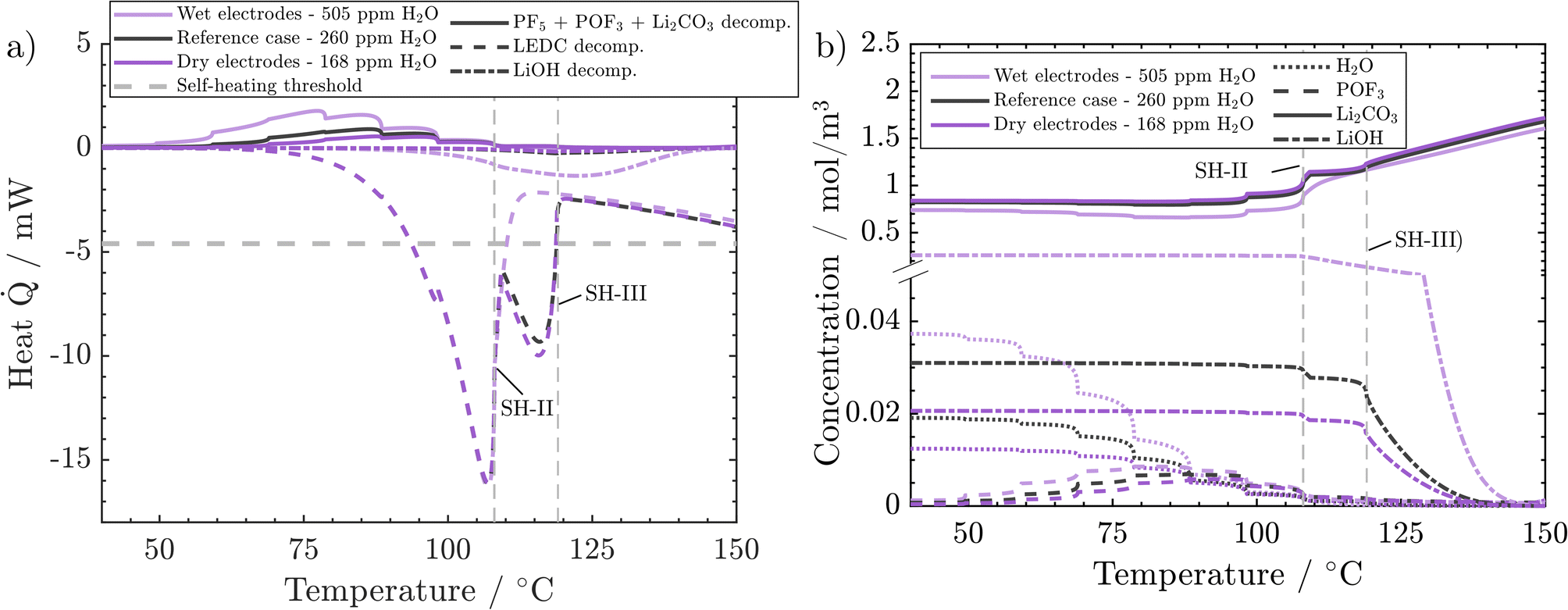 | ||
| Fig. 7 Effect of H2O impurities on behaviour during the ARC test: (a) combined endothermic heat of decomposition of PF5, POF3, and Li2CO3, vs. exothermic heat of LEDC and LiOH decomposition. (b) Concentrations of H2O, POF3, Li2CO3, and LiOH. Conditions: 45 vol% LEDC, 50 nm SEI. | ||
In conclusion, H2O impurities only play a significant role in battery safety when the electrodes have not been dried properly. The impact is negligible as soon as even a medium-intense drying procedure (260 ppm residual H2O) is applied. Also, further drying (168 ppm residual H2O) does not bring any noticeable benefit, because already at medium rates, LiOH decomposition rates are too low to give a significant heat contribution to tilt the balance towards sustained heating. As such, at least under the analysed circumstances and battery chemistry, extensive drying is unnecessary for a safer performance. This result aligns with the experimental findings of Huttner et al.,32 who found a negative impact on performance metrics for intense drying procedures. Two more points should be accounted for in a holistic analysis of battery safety, which may be followed in further studies: the acids produced from H2O, such as HPO2F2, are present in higher concentrations, i.e., 0.175 mol m−3 in the wet case compared to 0.078 mol m−3 in the reference case. The acids were reported to lead to increased dissolution of transition metals from the cathode, which will then promote SEI decomposition.57 Eventually, even though the energetic impact of these reactions is small, all are gassing reactions and will impact cell pressure.
3.3 Impact of variation in the SEI and H2O content
So far, we have analysed how the thermal safety behaviour changes when varying a single variable, SEI thickness, composition or water content. In reality, multiple factors change due to ageing or different manufacturing processes. In the following, the impact of the cross-influence of the three variables is analysed and trends and generalisations are deduced.
Fig. 8a shows the temperature gradient in the region between self-heating on-set and thermal runaway,  , over the inverse self-heating temperature,
, over the inverse self-heating temperature,  , for all 27 variations. Here, a high temperature gradient refers to a fast heating rate and, thus, a lower time to intervene. A low self-heating temperature indicates a lower resistance of the battery against thermal abuse. Thus, both characteristics can be considered indicators of battery safety and have therefore been chosen for this comparison. Cases that are located in the lower left corner of the figure can be considered rather safe, because it represents high self-heating temperatures and a low self-heating rate, whereas, the cases closer to the upper right show the opposite characteristics and, thus, can be considered rather critical. There are cases in all four quadrants of the figure, so no general correlation between the cases with fast self-heating and self-heating temperature can be found. The graph shows that the highly critical variations almost exclusively include high LEDC contents and a thin to medium thick SEI. The safe region on the other hand embodies almost exclusively all thick SEI variations with a large proportion of the inorganic cases. We can also observe that the thin SEI cases tend to have high temperature gradients probably due to the faster reformation at higher temperature. Higher LEDC content correlates with lower self-heating temperatures, as LEDC decomposition already occurs at low temperatures, whereas Li2CO3 decomposition is less strong at these temperatures. Variations of H2O are scattered over the whole figure, which indicates that its influence is not as significant and straight-forward as the SEI properties even for high contaminations. This confirms the lower sensitivity to H2O content than to SEI thickness and LEDC content.
, for all 27 variations. Here, a high temperature gradient refers to a fast heating rate and, thus, a lower time to intervene. A low self-heating temperature indicates a lower resistance of the battery against thermal abuse. Thus, both characteristics can be considered indicators of battery safety and have therefore been chosen for this comparison. Cases that are located in the lower left corner of the figure can be considered rather safe, because it represents high self-heating temperatures and a low self-heating rate, whereas, the cases closer to the upper right show the opposite characteristics and, thus, can be considered rather critical. There are cases in all four quadrants of the figure, so no general correlation between the cases with fast self-heating and self-heating temperature can be found. The graph shows that the highly critical variations almost exclusively include high LEDC contents and a thin to medium thick SEI. The safe region on the other hand embodies almost exclusively all thick SEI variations with a large proportion of the inorganic cases. We can also observe that the thin SEI cases tend to have high temperature gradients probably due to the faster reformation at higher temperature. Higher LEDC content correlates with lower self-heating temperatures, as LEDC decomposition already occurs at low temperatures, whereas Li2CO3 decomposition is less strong at these temperatures. Variations of H2O are scattered over the whole figure, which indicates that its influence is not as significant and straight-forward as the SEI properties even for high contaminations. This confirms the lower sensitivity to H2O content than to SEI thickness and LEDC content.
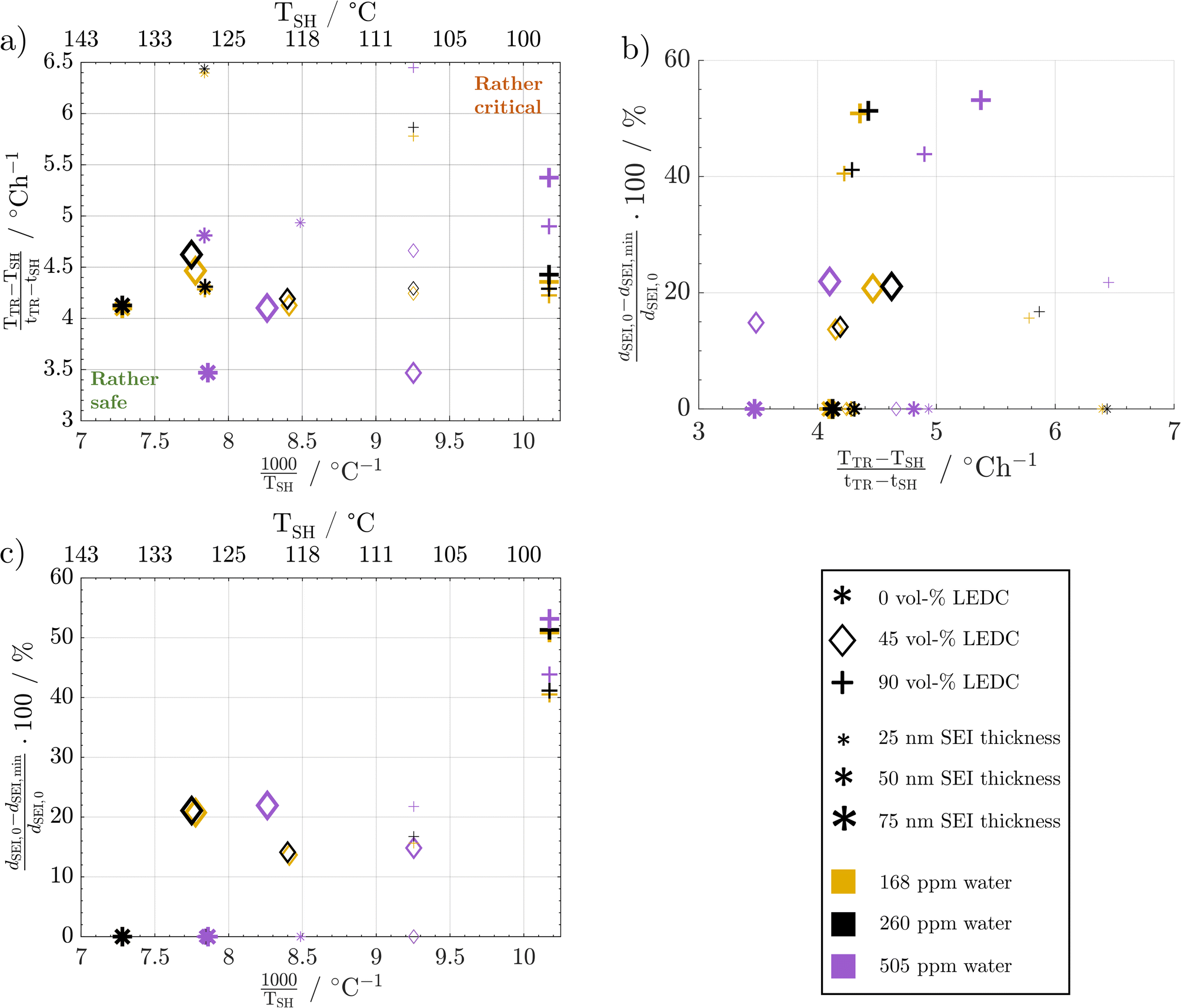 | ||
| Fig. 8 Impact of changes in water content and SEI properties on: (a) temperature gradient over the inverse self-heating temperature scaled by 1000, (b) the percentage of SEI thickness reduction over the temperature gradient, and (c) the percentage of SEI thickness reduction over the inverse self-heating temperature scaled by 1000. | ||
The complex water effects warrant further analysis. For cells with thick inorganic SEIs (large stars, Fig. 8a) the wet case (violet) has a lower self-heating temperature and a lower gradient compared to the dry and medium dry case. This means that it enters the self-heating phase earlier but then heats up slower, i.e. one metric gets worse and the other gets better. In contrast, for the high LEDC contents (+) the higher water contamination value does not lead to lower self-heating temperature and the temperature gradient shows the opposite behaviour to the inorganic case: it increases. The root cause lies in the onset of LiOH decomposition. Only in cases of high water contamination is the LiOH amount high enough to significantly impact the temperature progression, which we showed in Fig. 7a. The onset of this exothermic decomposition is around >115 °C, which is higher than the self-heating temperature of all high LEDC cases. This means that the LEDC cases are already in sustained self-heating when the LiOH decomposes; thus, water content does not impact the self-heating temperature for high LEDC content cases, whereas for all cases that have not transitioned to sustained self-heating before LiOH decomposes, the heat from LiOH decomposition impacts the self-heating temperature: the higher the water content and thus LiOH concentration, the more LiOH decomposition heat, thus the lower the self-heating temperature. The observed lower temperature gradient for the wet, inorganic and reference LEDC concentration cases is directly correlated with the lower temperature itself. The lower the temperature the slower all reactions progress, and thus, the less heat is produced.
A low SEI thickness has been shown to accelerate the exothermic SEI reformation; thus, we analyse for the different cases how much the SEI is reduced and how this correlates to the temperature gradient (Fig. 8b) and the self-heating temperature (Fig. 8c). The high LEDC content cases (+) exhibit the highest decline in SEI thickness compared to the medium and inorganic cases. The low thicknesses for high LEDC contents in turn accelerate the reformation and, thus, contribute to higher temperature gradients. The high concentration of LEDC also leads to a low self-heating temperature. The pure inorganic cases, represented by stars, and most of the thin SEI cases, represented by small symbols, do not show any thickness reduction. Inorganic SEI cases do not show a decrease in SEI thickness because the decomposition of Li2CO3 is not substantial enough to compensate for the SEI formation rates even for the thick SEI cases. Therefore, no decrease in SEI thickness can be observed. For the thin SEI cases, the thickness does not decrease substantially because the thin SEI accelerates SEI reformation, which counteracts SEI decomposition reactions. Most of the thin SEI and the inorganic SEI cases show a correlation between the temperature gradient, the self-heating temperature and the SEI thickness: the thinner the SEI the higher the temperature gradient and the lower the self-heating temperature. For the high LEDC content and thin SEI cases, the high amount of LEDC decomposes before substantial reformation starts. Thus, they show a slight SEI thickness reduction. Due to their anyway thin SEI thickness they also show the highest temperature gradient among all organic cases. Comparing the thin SEI cases with the medium thick SEI cases it shows that the medium thick SEI cases exhibit lower temperature gradients and higher self-heating temperatures. This is explained by the accelerated reformation rates for the thin SEI cases, shereas, the comparison of the medium thick SEI and thick SEI cases reveals that the thick SEI cases have a higher temperature gradient. This is explained by the higher self-heating temperature of the thick SEI cases. Since they transition at higher temperatures into the self-heating phase, the reactions occur faster and more heat is produced.
From these extensive variations and their impact on thermal safety behaviour, it can be concluded that the SEI thickness and LEDC content are the dominant effects in terms of battery safety. A safer battery has an inorganic, thick SEI. Safety decreases with increasing LEDC content and reducing SEI thickness. Besides these two, H2O impurities only play a role when severe contamination is present and generally contribute less than the SEI properties. The effect of water is also more challenging to address. High contamination affects the self-heating temperature and the temperature gradient differently depending on the composition of the SEI.
4 Conclusion
This study has elucidated the impact of the initial SEI state and water impurities on the thermal safety behaviour of Li-ion batteries with EC:EMC 1.2 M LiPF6 and a graphite anode, including when and why self-heating occurs and the subsequent progression to rapid thermal runaway. Initial concentrations of SEI components, impurities and conductive salt decomposition products were rigorously derived from assessing manufacturing, production and ageing effects.Dominant detrimental effects are high LEDC concentrations and a thin SEI, such as those found in rather fresh cells. Here, a high LEDC content could be connected to an earlier onset of self-heating. In contrast, a thinner SEI relates to faster SEI reformation and thus to a higher temperature gradient. The experimentally observed increase in self-heating temperature for aged cells20–23 is thus attributed to an ageing-induced change from a foremost organic SEI to an inorganic SEI and a thicker SEI, which delays exothermic SEI reformation processes. The impact of H2O impurities on battery safety is found to be marginal as long as a moderate drying procedure is applied to the electrodes during manufacturing. Thus, we could show that extreme electrode drying does not benefit battery safety. However, high H2O contamination during production should be avoided as this will have a substantial negative impact. Here, the effect of high contamination was found to depend on the SEI composition. For an inorganic and mixed SEI, the contamination will reduce the self-heating temperature due to decomposition of LiOH. High LEDC content cases, on the other hand, exhibit a higher temperature gradient, because here the self-heating already starts before LiOH decomposition sets in.
The insights gained here contribute significantly to understanding and controlling Li-ion battery behaviour during thermal abuse. The trends for impact of water as an electrolyte impurity, the complexities of SEI properties, and their combined battery safety have been shown, and they cross influence each other. The presented degradation reactions and kinetics are suitable for integration into full cell models to evaluate the impact of local hotspots and heat removal, and thus to reveal battery runaway and propagation on cell and pack levels. The studies may be further extended to include the effects of different active materials and electrolytes, as reactions, reactivity and mechanical stability may change. Different experimental behaviours were reported here, especially for the highly reactive Li metal and solid state electrolytes. Also of special importance is the interaction of the cathode with water impurities and of metal dissolution on the reaction network and the thermal safety behaviour.
Abbreviations
| a | Activity (—) |
| α | Dissociation degree (—) |
| C p,bat | Battery heat capacity (J K−1) |
| d SEI | SEI thickness (m) |
| E A | Activation energy (J mol−1) |
| ΔrG | Gibbs free energy of reaction (J mol−1) |
| ΔrHj | Enthalpy of reaction (J mol−1) |
| k f/b | Frequency factor forward/backward reaction (mol s−1, (mol m) s−1) |
| K | Equilibrium constant (—) |
| ν | Stoichiometric coefficient (—) |
| n | Molar amount (mol) |
| r | Reaction rate (mol s−1) |
| R | Molar gas constant (J (mol−1 K−1)) |
| ΔrS | Entropy of reaction (J (mol−1 K−1)) |
| t | Time (h) |
| t SH | Self-heating time (h) |
| t TR | Thermal runaway time (h) |
| T | Temperature (K, °C) |
| T SH | Self-heating temperature (K, °C) |
| T TR | Thermal runaway temperature (K, °C) |
Data availability
Source data are provided with this paper in the KITopen repository under https://doi.org/10.35097/1804.Author contributions
Florian Baakes: conceptualisation, methodology, software, writing –original draft, validation, investigation, visualisation. Daniel Witt: conceptualisation, writing – review & editing. Ulrike Krewer: conceptualisation, writing – review & editing, supervision, funding acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the German Federal Ministry for Economic Affairs and Climate Action through funding of the project “SimDural – Simulation-based Safety Analysis of an Uncontrolled Thermal Runaway in Aged Battery Cells” (16BZF325C) and the Friedrich and Elisabeth Boysen Foundation (BOY-174).References
- K. Turcheniuk, D. Bondarev, G. G. Amatucci and G. Yushin, Mater. Today, 2021, 42, 57 CrossRef CAS.
- J. Liu, J. Li and J. Wang, J. Energy Storage, 2021, 35, 102253 CrossRef.
- X. Liu, D. Ren, H. Hsu, X. Feng, G.-L. Xu, M. Zhuang, H. Gao, L. Lu, X. Han, Z. Chu, J. Li, X. He, K. Amine and M. Ouyang, Joule, 2018, 2, 2047 CrossRef CAS.
- F. Baakes, M. Lüthe, M. Gerasimov, V. Laue, F. Röder, P. B. Balbuena and U. Krewer, J. Power Sources, 2022, 522, 230881 CrossRef CAS.
- M. N. Richard and J. R. Dahn, J. Electrochem. Soc., 1999, 146, 2078 CrossRef CAS.
- E. Peled, J. Electrochem. Soc., 1979, 126, 2047 CrossRef CAS.
- E. Peled and S. Menkin, J. Electrochem. Soc., 2017, 164, A1703–A1719 CrossRef CAS.
- B. Horstmann, J. Shi, R. Amine, M. Werres, X. He, H. Jia, F. Hausen, I. Cekic-Laskovic, S. Wiemers-Meyer, J. Lopez, D. Galvez-Aranda, F. Baakes, D. Bresser, C.-C. Su, Y. Xu, W. Xu, P. Jakes, R.-A. Eichel, E. Figgemeier, U. Krewer, J. M. Seminario, P. B. Balbuena, C. Wang, S. Passerini, Y. Shao-Horn, M. Winter, K. Amine, R. Kostecki and A. Latz, Energy Environ. Sci., 2021, 14, 5289 RSC.
- X. He, D. Bresser, S. Passerini, F. Baakes, U. Krewer, J. Lopez, C. T. Mallia, Y. Shao-Horn, I. Cekic-Laskovic, S. Wiemers-Meyer, F. A. Soto, V. Ponce, J. M. Seminario, P. B. Balbuena, H. Jia, W. Xu, Y. Xu, C. Wang, B. Horstmann, R. Amine, C.-C. Su, J. Shi, K. Amine, M. Winter, A. Latz and R. Kostecki, Nat. Rev. Mater., 2021, 6, 1036 CrossRef CAS.
- T. Liu, L. Lin, X. Bi, L. Tian, K. Yang, J. Liu, M. Li, Z. Chen, J. Lu, K. Amine, K. Xu and F. Pan, Nat. Nanotechnol., 2019, 14, 50 CrossRef CAS PubMed.
- H. B. Son, M.-Y. Jeong, J.-G. Han, K. Kim, K. H. Kim, K.-M. Jeong and N.-S. Choi, J. Power Sources, 2018, 400, 147 CrossRef CAS.
- H. Adenusi, G. A. Chass, S. Passerini, K. V. Tian and G. Chen, Adv. Energy Mater., 2023, 13, 2203307 CrossRef CAS.
- A. Wang, S. Kadam, H. Li, S. Shi and Y. Qi, npj Comput. Mater., 2018, 4, 15 CrossRef.
- F. Röder, V. Laue and U. Krewer, Batteries Supercaps, 2019, 2, 248 CrossRef.
- D. Witt, F. Röder and U. Krewer, Batteries Supercaps, 2022, 5, e20220006 Search PubMed.
- M. Esmaeilpour, S. Jana, H. Li, M. Soleymanibrojeni and W. Wenzel, Adv. Energy Mater., 2023, 13(14), 2203966 CrossRef CAS.
- M. Gerasimov, F. A. Soto, J. Wagner, F. Baakes, N. Guo, F. Ospina-Acevedo, F. Röder, P. B. Balbuena and U. Krewer, J. Phys. Chem. C, 2023, 127(10), 4872–4886, DOI:10.1021/acs.jpcc.2c05898.
- J. Wagner, D. Kuai, M. Gerasimov, F. Röder, P. Balbuena and U. Krewer, Nat. Commun., 2023, 14, 6823 CrossRef PubMed.
- E. W. C. Spotte-Smith, R. L. Kam, D. Barter, X. Xie, T. Hou, S. Dwaraknath, S. M. Blau and K. A. Persson, ACS Energy Lett., 2022, 7, 1446 CrossRef CAS.
- M. Börner, A. Friesen, M. Grützke, Y. P. Stenzel, G. Brunklaus, J. Haetge, S. Nowak, F. M. Schappacher and M. Winter, J. Power Sources, 2017, 342, 382 CrossRef.
- T. Waldmann, J. B. Quinn, K. Richter, M. Kasper, A. Tost, A. Klein and M. Wohlfahrt-Mehrens, J. Electrochem. Soc., 2017, 164, A3154–A3162 CrossRef CAS.
- M. Feinauer, A. A. Abd-El-Latif, P. Sichler, A. Aracil Regalado, M. Wohlfahrt-Mehrens and T. Waldmann, J. Power Sources, 2023, 570, 233046 CrossRef CAS.
- X. Feng, S. Zheng, D. Ren, X. He, L. Wang, H. Cui, X. Liu, C. Jin, F. Zhang, C. Xu, H. Hsu, S. Gao, T. Chen, Y. Li, T. Wang, H. Wang, M. Li and M. Ouyang, Appl. Energy, 2019, 246, 53 CrossRef CAS.
- A. Kwade, W. Haselrieder, R. Leithoff, A. Modlinger, F. Dietrich and K. Droeder, Nat. Energy, 2018, 3, 290 CrossRef.
- D. Aurbach, I. Weissman, A. Zaban and P. Dan, Electrochim. Acta, 1999, 45, 1135 CrossRef CAS.
- U. Langklotz, M. Schneider and A. Michaelis, J. Ceram. Sci. Technol., 2013, 4(2), 69–76 Search PubMed.
- T. Osaka, T. Momma, T. Tajima and Y. Matsumoto, J. Electrochem. Soc., 1995, 142, 1057 CrossRef CAS.
- L.-Q. Zheng, S.-J. Li, H.-J. Lin, Y.-Y. Miao, L. Zhu and Z.-J. Zhang, Russ. J. Electrochem., 2014, 50, 904 CrossRef CAS.
- R. M. Kasse, N. R. Geise, J. S. Ko, J. Nelson Weker, H.-G. Steinrück and M. F. Toney, J. Mater. Chem. A, 2020, 8, 16960 RSC.
- W. Weber, V. Kraft, M. Grützke, R. Wagner, M. Winter and S. Nowak, J. Chromatogr. A, 2015, 1394, 128 CrossRef CAS PubMed.
- M. Stich, M. Göttlinger, M. Kurniawan, U. Schmidt and A. Bund, J. Phys. Chem. C, 2018, 122, 8836 CrossRef CAS.
- F. Huttner, W. Haselrieder and A. Kwade, Energy Technol., 2020, 8, 1900245 CrossRef CAS.
- H. Maleki, G. Deng, A. Anani and J. Howard, J. Electrochem. Soc., 1999, 146, 3224 CrossRef CAS.
- S. Solchenbach, M. Metzger, M. Egawa, H. Beyer and H. A. Gasteiger, J. Electrochem. Soc., 2018, 165(13), A3022–A3028 CrossRef CAS.
- A. T. Freiberg, J. Sicklinger, S. Solchenbach and H. A. Gasteiger, Electrochim. Acta, 2020, 346, 136271 CrossRef CAS.
- R. Spotnitz and J. Franklin, J. Power Sources, 2003, 113, 81 CrossRef CAS.
- W. D. Machin and F. C. Tompkins, Trans. Faraday Soc., 1966, 62, 2205–2218 RSC.
- H. Shin, J. Park, S. Han, A. M. Sastry and W. Lu, J. Power Sources, 2015, 277, 169 CrossRef CAS.
- D. Bedrov, O. Borodin and J. B. Hooper, J. Phys. Chem. C, 2017, 121, 16098 CrossRef CAS.
- H. Ye, S. Gui, Z. Wang, J. Chen, Q. Liu, X. Zhang, P. Jia, Y. Tang, T. Yang, C. Du, L. Geng, H. Li, Q. Dai, Y. Tang, L. Zhang, H. Yang and J. Huang, ACS Appl. Mater. Interfaces, 2021, 13, 44479 CrossRef CAS PubMed.
- C. Stetson, Y. Yin, C.-S. Jiang, S. C. DeCaluwe, M. Al-Jassim, N. R. Neale, C. Ban and A. Burrell, ACS Energy Lett., 2019, 4, 2770 CrossRef CAS.
- P. Atkins, and J. de Paula, Thermodynamics and Kinetics, W.H. Freeman and Co, New York, 2005 Search PubMed.
- H. Maleki and J. N. Howard, J. Power Sources, 2004, 137, 117 CrossRef CAS.
- Renata Batteries Datasheet, 2019, ICP622540PMT Search PubMed.
- A. M. Colclasure and R. J. Kee, Electrochim. Acta, 2010, 55, 8960 CrossRef CAS.
- B. Rieger, S. Schlueter, S. V. Erhard, J. Schmalz, G. Reinhart and A. Jossen, J. Energy Storage, 2016, 6, 213 CrossRef.
- Y. Wang, S. Nakamura, M. Ue and P. B. Balbuena, J. Am. Chem. Soc., 2001, 123, 11708 CrossRef CAS PubMed.
- MATLAB, Version 9.12.0.1956245 (R2022a), The MathWorks Inc, Natick, Massachusetts, 2022 Search PubMed.
- G. V. Zhuang, K. Xu, H. Yang, T. R. Jow and P. N. Ross, J. Phys. Chem. B, 2005, 109, 17567 CrossRef CAS PubMed.
- L. Wang, A. Menakath, F. Han, Y. Wang, P. Y. Zavalij, K. J. Gaskell, O. Borodin, D. Iuga, S. P. Brown, C. Wang, K. Xu and B. W. Eichhorn, Nat. Chem., 2019, 11, 789 CrossRef CAS PubMed.
- X. Xie, E. W. Clark Spotte-Smith, M. Wen, H. D. Patel, S. M. Blau and K. A. Persson, J. Am. Chem. Soc., 2021, 143, 13245 CrossRef CAS PubMed.
- M. Nie, D. Chalasani, D. P. Abraham, Y. Chen, A. Bose and B. L. Lucht, J. Phys. Chem. C, 2013, 117, 1257 CrossRef CAS.
- F. A. Soto, Y. Ma, J. M. La Martinez de Hoz, J. M. Seminario and P. B. Balbuena, Chem. Mater., 2015, 27, 7990 CrossRef CAS.
- B. Lu, D. Cheng, B. Sreenarayanan, W. Li, B. Bhamwala, W. Bao and Y. S. Meng, ACS Energy Lett., 2023, 8, 3230 CrossRef CAS.
- B. S. Vishnugopi, M. T. Hasan, H. Zhou and P. P. Mukherjee, ACS Energy Lett., 2023, 8, 398 CrossRef CAS.
- P. Röder, B. Stiaszny, J. C. Ziegler, N. Baba, P. Lagaly and H.-D. Wiemhöfer, J. Power Sources, 2014, 268, 315 CrossRef.
- C. Jayawardana, N. Rodrigo, B. Parimalam and B. L. Lucht, ACS Energy Lett., 2021, 6, 3788 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc04186g |
| This journal is © The Royal Society of Chemistry 2023 |