
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The role of metal accessibility on carbon dioxide electroreduction in atomically precise nanoclusters†
Yingwei
Li‡
 ,
Grant J.
Stec‡
,
Agnes E.
Thorarinsdottir
,
Ryan D.
McGillicuddy
,
Shao-Liang
Zheng
and
Jarad A.
Mason
*
,
Grant J.
Stec‡
,
Agnes E.
Thorarinsdottir
,
Ryan D.
McGillicuddy
,
Shao-Liang
Zheng
and
Jarad A.
Mason
*
Department of Chemistry & Chemical Biology, Harvard University, 12 Oxford Street, Cambridge, Massachusetts 02138, USA. E-mail: mason@chemistry.harvard.edu
First published on 24th October 2023
Abstract
Atomically precise nanoclusters (NCs) can be designed with high faradaic efficiency for the electrochemical reduction of CO2 to CO (FECO) and provide useful model systems for studying the metal-catalysed CO2 reduction reaction (CO2RR). While size-dependent trends are commonly evoked, the effect of NC size on catalytic activity is often convoluted by other factors such as changes to surface structure, ligand density, and electronic structure, which makes it challenging to establish rigorous structure–property relationships. Herein, we report a detailed investigation of a series of NCs [AunAg46−n(C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CR)24Cl4(PPh3)2, Au24Ag20(CCR)24Cl2, and Au43(CCR)20/Au42Ag1(CCR)20] with similar sizes and core structures but different ligand packing densities to investigate how the number of accessible metal sites impacts CO2RR activity and selectivity. We develop a simple method to determine the number of CO2-accessible sites for a given NC then use this to probe relationships between surface accessibility and CO2RR performance for atomically precise NC catalysts. Specifically, the NCs with the highest number of accessible metal sites [Au43(CCR)20 and Au42Ag1(CCR)20] feature a FECO of >90% at –0.57 V vs. the reversible hydrogen electrode (RHE), while NCs with lower numbers of accessible metal sites have a reduced FECO. In addition, CO2RR studies performed on other Au–alkynyl NCs that span a wider range of sizes further support the relationship between FECO and the number of accessible metal sites, regardless of NC size. This work establishes a generalizable approach to evaluating the potential of atomically precise NCs for electrocatalysis.
CR)24Cl4(PPh3)2, Au24Ag20(CCR)24Cl2, and Au43(CCR)20/Au42Ag1(CCR)20] with similar sizes and core structures but different ligand packing densities to investigate how the number of accessible metal sites impacts CO2RR activity and selectivity. We develop a simple method to determine the number of CO2-accessible sites for a given NC then use this to probe relationships between surface accessibility and CO2RR performance for atomically precise NC catalysts. Specifically, the NCs with the highest number of accessible metal sites [Au43(CCR)20 and Au42Ag1(CCR)20] feature a FECO of >90% at –0.57 V vs. the reversible hydrogen electrode (RHE), while NCs with lower numbers of accessible metal sites have a reduced FECO. In addition, CO2RR studies performed on other Au–alkynyl NCs that span a wider range of sizes further support the relationship between FECO and the number of accessible metal sites, regardless of NC size. This work establishes a generalizable approach to evaluating the potential of atomically precise NCs for electrocatalysis.
Introduction
Heterogeneous catalysts composed of metal nanoparticles (NPs) dispersed on high-surface-area supports have been studied for more than a century,1–3 and these catalysts are of increasing interest for the electrochemical reduction of CO2 into chemical fuels and feedstocks.4–6 Gold- and silver-based NPs are particularly effective for the selective reduction of CO2 to CO.7 Though the effects of nanoparticle size,8–10 shape,11,12 and surface ligands13,14 on the CO2 reduction reaction (CO2RR) have been widely studied, the nonuniformity of metal NP catalysts is a long-standing challenge in the investigation of fundamental catalytic mechanisms.1,15 In particular, it is often difficult to identify the specific active sites that drive catalysis because of the wide distribution of local microenvironments in ligand-protected NPs that adopt varying sizes, shapes, and surface structures.16,17 For example, although functionalization with larger organic ligands has been shown to enhance the CO2RR activity of Au NPs,18,19 uncertainty over the exact arrangement of surface ligands makes it difficult to determine how bulky ligands impact selectivity and catalytic activity. Such molecular-level insight is, however, possible when atomically precise nanoclusters (NCs) are used as catalysts, since their uniformity allows the entire particle structure—including the ligand shell—to be resolved crystallographically.20–23Soon after the canonical Au25(SR)18 (SR = aryl or alkylthiolate) NC was first reported, it was shown to be effective for CO2RR, featuring a high faradaic efficiency for CO (FECO) at –1 V vs. the reversible hydrogen electrode (RHE).24 Though the CO2RR has since been studied for many other atomically precise Au NCs,25–28 there still remains much to be understood about how NC size, structure, and surface ligand identity influence catalytic activity and selectivity. For instance, relationships between NC size and CO2RR activity are challenging to identify because the ligand-to-metal ratio typically increases for smaller NCs,29–31 resulting in higher surface coverage. Changes to the arrangement of surface ligands and metal atoms—as well as the electronic structure of the NC—may also affect the outcome of catalytic reactions, further convoluting structure–property relationships.32 Indeed, differing trends have been reported for how NC size affects CO2RR activity. For example, in the series Au25(SR)18, Au38(SR)24, and Au144(SR)60 (SR = SC2H4Ph), CO2RR activity increases with increasing NC size,33 while other studies have found that the FECO of Au-SR NCs is not directly affected by NC size.34 Given the different size-dependent trends that have been observed for atomically precise NCs, the number of active sites is often a better predictor of catalytic behavior but is difficult to manipulate in a predictable fashion.
In an effort to decouple the role of metal active sites from NC size, structure, and ligand type, we designed a series of alkynyl-protected atomically precise Au/Ag NCs with similar sizes and core structures but different degrees of surface ligand coverage. We investigated the CO2RR performance of these NCs and developed a convenient computational method to quantitatively evaluate the accessibility of potential catalytically active sites. Critically, the use of acetylene-based ligands—bearing one rotatable bond—simplifies the conformational landscape at the metal–ligand interface, thereby clarifying the effect of ligand modification on surface coverage and the number of accessible metal sites. In particular, we found that the number and accessibility of surface metal sites is directly correlated to experimental CO2RR activity.
Results and discussion
Synthesis and characterization of alkynyl-protected Au/Ag NCs
We recently reported the isostructural alkynyl-protected NCs Au43(CCtBu)20 (Au43) and Au42Ag1(CCtBu)20 (Au42Ag1), which are synthesized by reducing an oligomeric AuI–CCtBu or AuI/AgI–CCtBu precursor with borane tert-butylamine then purifying via thin layer chromatography (Fig. S1A†).35 With a nearly identical NC core but an increased density of alkynyl surface ligands, we also selected the previously reported Au24Ag20(CCPhtBu)24Cl2 NC (Au24Ag20) for comparison (Fig. S1B†).36,37 To complete a series of NCs with similar sizes and varying surface ligand densities, we also targeted an Au/Ag–alkynyl NC with an even denser organic shell. This was achieved by introducing a bulky triphenylphosphine (PPh3) ligand through the “hydride-mediated conversion” method.38,39 Specifically, [Au9(PPh3)8]3+ (ref. 40) was reduced with NaBH4 to furnish a hydride-doped [HAu9(PPh3)8]2+ cluster, which was then reacted with CH3COOAg, meta-substituted phenylacetylene ligands, and triethylamine to yield NCs with a composition of AunAg46−n(CCPh–m–X)24Cl4(PPh3)2 (AunAg46−n, n = 16–19, X = H, F, CH3) (Fig. 1, see ESI† for experimental details). Though attempts to grow single crystals of AunAg46−n(CCPh)24Cl4(PPh3)2 suitable for structure determination were unsuccessful, crystal structures were successfully determined for AunAg46−n with X = F and CH3 (see Fig. 2). The successful crystallization of AunAg46−n with meta-substituted phenylacetylene ligands can be attributed to the additional interparticle C–H⋯π, or C–F⋯π interactions between the meta functional groups on one NC and the phenyl rings of ligands on another NC (Fig. S2 and S3†).41,42
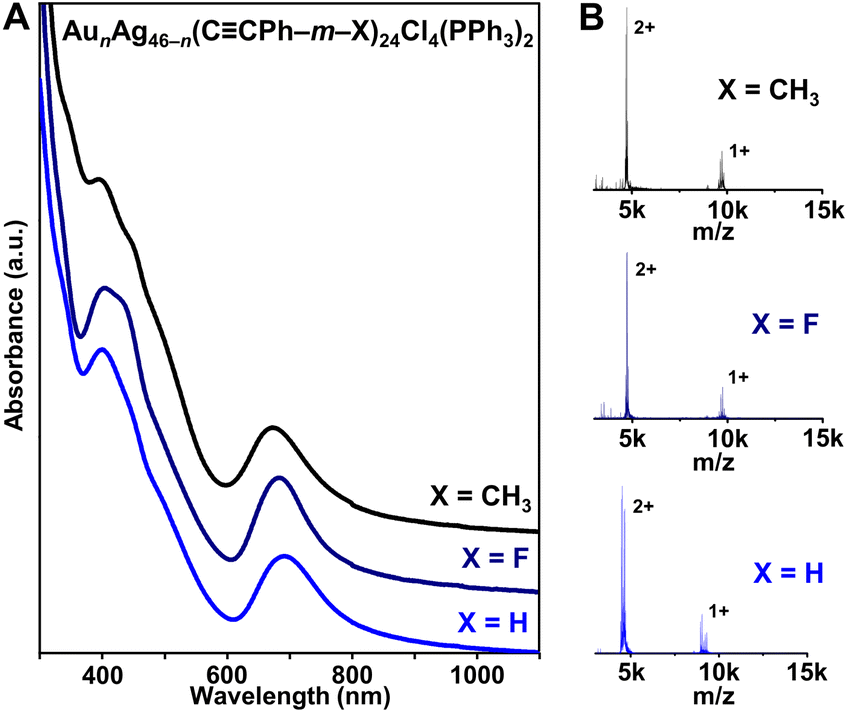 | ||
| Fig. 1 (A) UV-vis-NIR absorption spectra and (B) ESI-MS of AunAg46−n(CCPh–m–X)24Cl4(PPh3)2 (n = 16–19, X = H, F, or CH3). | ||
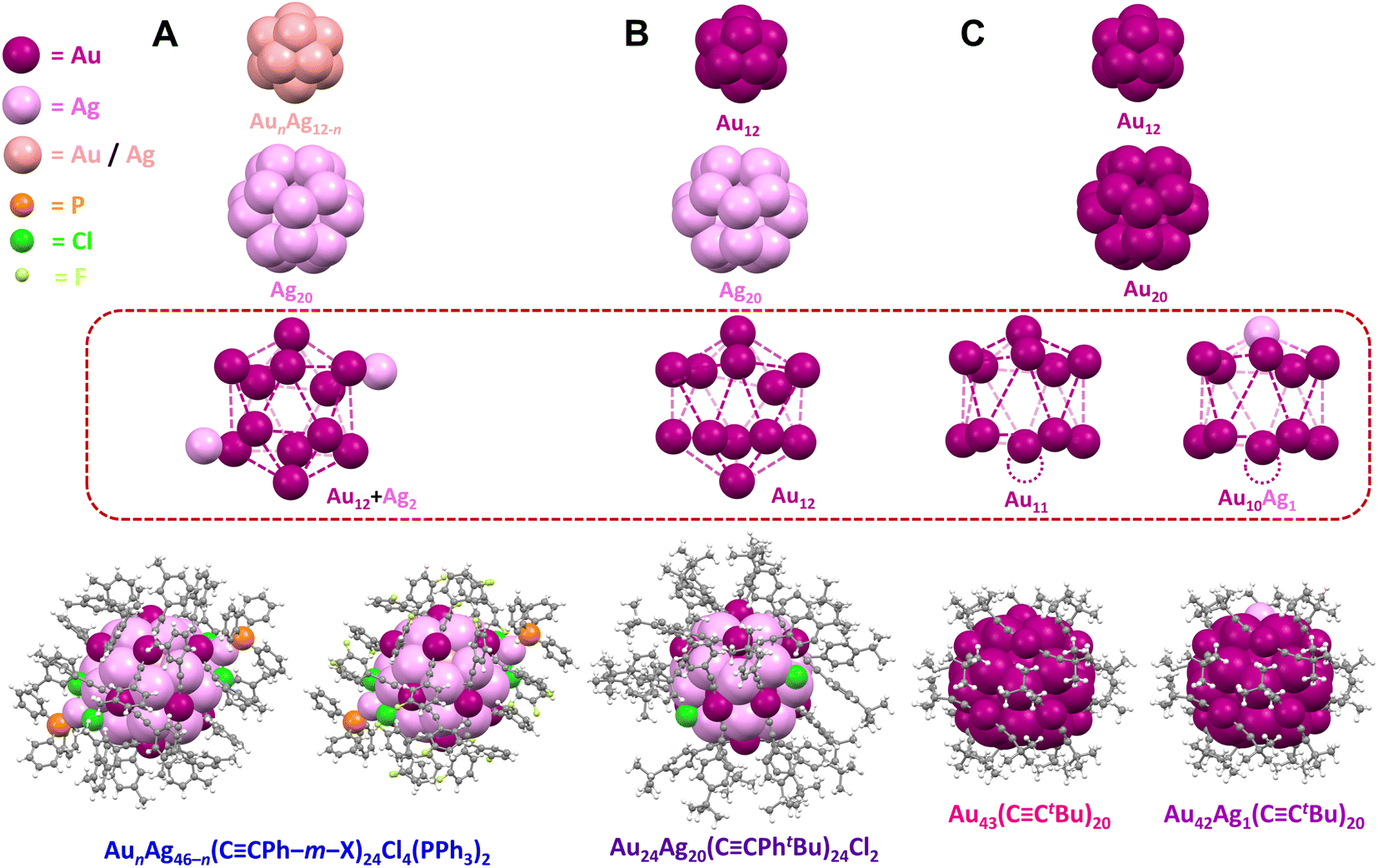 | ||
| Fig. 2 Structures of (A) AunAg46−n(CCPh–m–X)24Cl4(PPh3)2 (n = 16–19, X = F or CH3), (B) Au24Ag20(CCPhtBu)24Cl2, and (C) Au43(CCtBu)20 and Au42Ag1(CCtBu)20, Color code: magenta = Au; violet = Ag; light pink = Au/Ag; orange = P; green = Cl; light green = F; grey = C; white = H. The vacant surface sites in Au43(CCtBu)20 and Au42Ag1(CCtBu)20 are indicated with a dashed circle. | ||
The solution-phase UV-vis absorption spectra of all three AunAg46−n NCs with different protecting ligands exhibit sharp absorption peaks centered near 400 and 690 nm (Fig. 1A). Moreover, electrospray ionization mass spectrometry (ESI-MS) analysis shows 2+ and 1+ ion peaks for the NCs (Fig. 1B). Combined with the X-ray crystallography data, the 2+ and 1+ charges are attributed to ionization in ESI-MS—not the native charge states of the NCs—and the peaks correspond to NCs that have lost one or two PPh3 ligands, which has been commonly observed for similar atomically precise NCs (Fig. S4–S6†).43,44 The ESI-MS spectrum of AunAg46−n(CCPh–m–F)24Cl4(PPh3)2 contains peaks corresponding to n = 16, 17, and 18 (Fig. S5†), consistent with the crystallographically refined composition of Au17.67Ag28.33(CCPh–m–F)24Cl4(PPh3)2 (Tables S1 and S2†). When HCCPh–m–CH3 was used instead, three peaks corresponding to n = 17, 18, and 19 were found, which is consistent with the higher refined Au![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Ag ratio in Au19Ag27(CCPh–m–CH3)24Cl4(PPh3)2 (Tables S3 and S4†). The small inconsistency between MS and X-ray crystallography data has also been observed for other heterometallic NCs, which are known to be dynamic—and prone to rearrangement—in solution.45,46 Regardless, the similar UV-vis and ESI-MS spectra for AunAg46−n(CCPh)24Cl4(PPh3)2 and AunAg46−n(CCPh–m–X)24Cl4(PPh3)2 (X = F, CH3) NCs—along with the fact that the same synthesis conditions were used—suggest that the three NCs are isostructural. Note that small differences in absorption features near 400 nm and shifts in MS can be attributed to the different alkynyl ligands used.
:Ag ratio in Au19Ag27(CCPh–m–CH3)24Cl4(PPh3)2 (Tables S3 and S4†). The small inconsistency between MS and X-ray crystallography data has also been observed for other heterometallic NCs, which are known to be dynamic—and prone to rearrangement—in solution.45,46 Regardless, the similar UV-vis and ESI-MS spectra for AunAg46−n(CCPh)24Cl4(PPh3)2 and AunAg46−n(CCPh–m–X)24Cl4(PPh3)2 (X = F, CH3) NCs—along with the fact that the same synthesis conditions were used—suggest that the three NCs are isostructural. Note that small differences in absorption features near 400 nm and shifts in MS can be attributed to the different alkynyl ligands used.
The series of five atomically precise Au/Ag NCs exhibit similar core structures. Specifically, AunAg46−n has an icosahedral AunAg12−n kernel (n = 4–7 with Au and Ag randomly distributed), a dodecahedral Ag20 inner shell, and an icosahedral Au12 outer shell with two additional Ag atoms on the surface (Fig. 2A). Each of the additional Ag atoms on the surface of AunAg46−n is bonded to two chloride ligands, one Au atom in the outer shell, and one PPh3 ligand. Four chloride ligands are necessary for AunAg46−n to adopt a closed-shell superatomic electronic configuration (46 − 24 − 4 = 18 e–) similar to Au24Ag20 with two chloride ligands (24 + 20 − 24 − 2 = 18 e–). The structure of Au24Ag20 also consists of an icosahedral Au12 kernel, a dodecahedral Ag20 inner shell, and an icosahedral Au12 outer shell (Fig. 2B), while Au43/Au42Ag1 has an icosahedral Au12 kernel, a dodecahedral Au20 inner shell, and an incomplete icosahedral Au11 or Au10Ag1 outer shell with a single vacant surface site (Fig. 2C).
Adding or removing a single metal atom to or from the surface of NCs has provided insight into the optical and electronic properties of thiolate-capped metal NCs.47,48 The successful synthesis of our series of NCs with the same core (M46, M44, and M43; M = Au and/or Ag) represents—to the best of our knowledge— the first demonstration of atom-by-atom evolution for alkynyl-stabilized metal NCs structure (Fig. 2). Moreover, the linear directionality of alkynyl ligands in alkynyl-stabilized NCs offer advantages for catalytic studies since the NC/electrolyte interface is dominated by Au–CC bonds with similar local arrangements. Note that for AunAg46−n and Au24Ag20, Ag atoms are located in the inner dodecahedral Ag12 shell and/or the icosahedral AunAg12−n kernel which are not accessible to substrates interacting with the surface of the NC (Fig. 2, highlighted by the dashed red box). Importantly, the surface ligand density varies systematically across the series: 24 alkynyl, 4 chloride, and 2 phosphine ligands for AunAg46−n, 24 alkynyl and 2 chloride ligands for Au24Ag20, and only 20 alkynyl ligands for Au43 and Au42Ag1. Thus, this series of atomically precise NCs provides a powerful platform to study relationships between metal site accessibility and CO2RR activity.
Evaluation of CO2RR activity and selectivity
To investigate their efficacy for CO2RR catalysis, synthesized atomically precise NCs were mixed with carbon black (20 wt% NC loading) and deposited on a carbon paper electrode. For comparison, a carbon electrode was also prepared with spherical Au–SC2H4Ph NPs (Au–S NPs) with an average diameter of 3.1 ± 0.4 nm (Fig. S1C†) that were mixed with carbon black at the same mass loading. To provide an additional comparison, an 85 nm thick bulk gold layer was also deposited on one carbon paper electrode (referenced as Au layer) by electron beam deposition. Linear sweep voltammetry (LSV) was then performed for each electrode in CO2-saturated 0.5 M KHCO3 solution. For the atomically precise NCs, the current density (jtotal) was found to increase in the order of AunAg46−n < Au24Ag20 < Au42Ag1 < Au43 (Fig. 3A). Larger sized Au–S NPs exhibited a lower jtotal than all atomically precise NCs, and jtotal for the Au layer was the lowest of all catalysts investigated here (Fig. 3A). We note that the NC catalysts were not activated before electrocatalysis to minimize possible ligand stripping, and the LSV curves taken before and after chronoamperometric CO2RR catalysis (potential range of –0.47 V to –0.77 V vs. RHE) were in close agreement (Fig. S7†). Moreover, the NC-based catalysts display a steady current density over at least 40 min at each applied voltage (–0.47 V, –0.57 V, –0.67 V and –0.77 V vs. RHE; Fig. 3C), and the absorption spectra of NCs recovered from the electrode after catalysis matched those of as-synthesized NCs (Fig. S8†), confirming the stability of the NCs during CO2RR catalysis.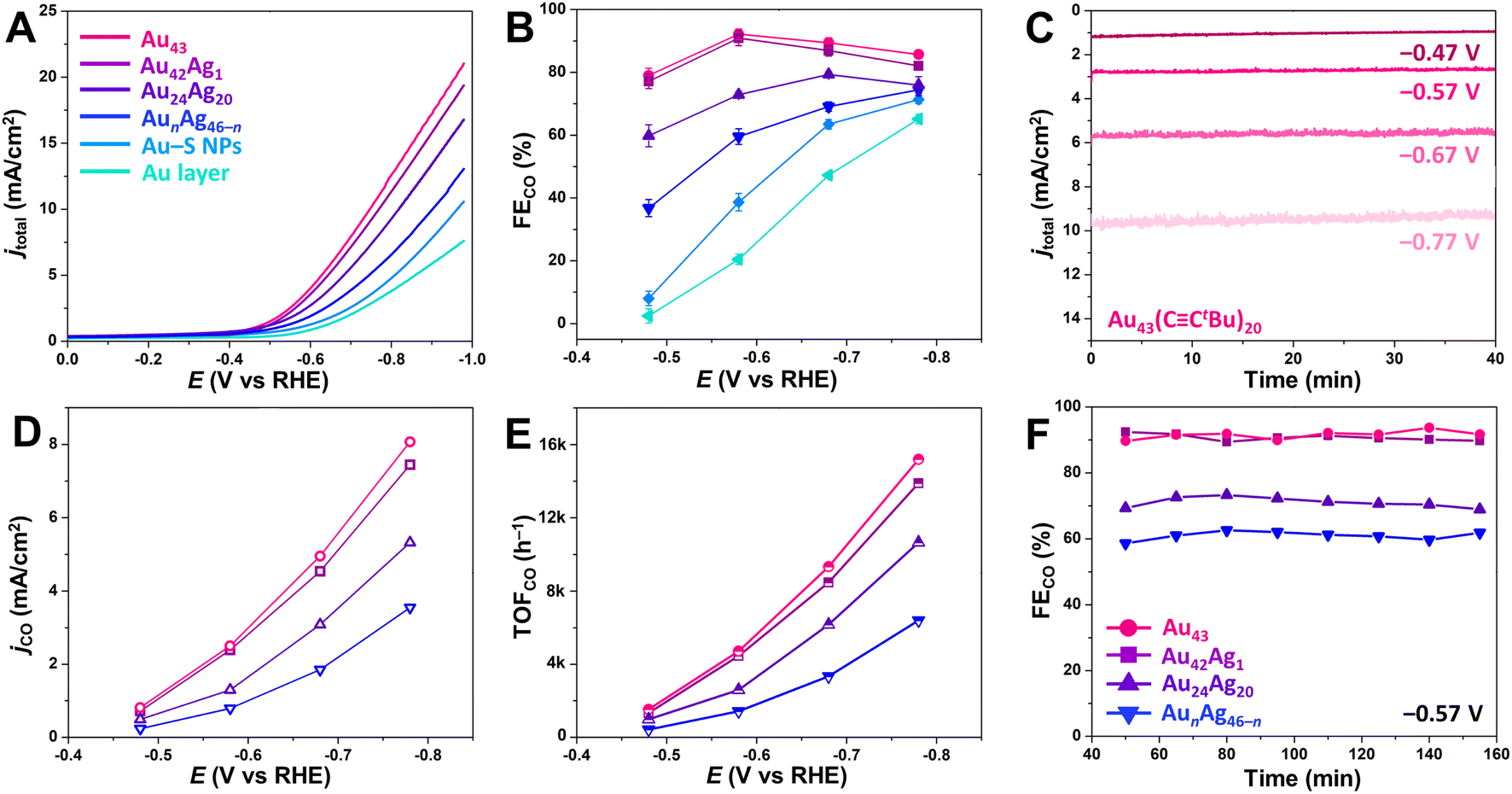 | ||
| Fig. 3 (A) LSV and (B) FECO for CO2RR for the series of NC-based catalysts. (C) Chronoamperometry data for the Au43-based catalyst at different applied potentials. (D) jCO and (E) TOFCO for the NC-based catalysts at different applied potentials during CO2RR. (F) FECO for the NC-based catalysts at –0.57 V vs. RHE during CO2RR over an extended time period. All experiments were conducted in a 0.5 M KHCO3 solution saturated with CO2. In panels A, B, D, E, and F, magenta represents Au43(CCtBu)20, purple represents Au42Ag1(CCtBu)20, indigo represents Au24Ag20(CCPhtBu)24Cl2, blue represents AunAg46−n(CCPh)24Cl4(PPh3)2, sky blue represents Au-SC2H4Ph NPs, and cyan represents the Au layer. | ||
For all atomically precise NC catalysts evaluated here, CO was the major CO2RR product, and H2 was the sole byproduct with no liquid products detected by 1H NMR spectroscopy. The faradaic efficiency for CO production (FECO) was assessed for each catalyst by calculating the percentage of transferred charge that was directed toward CO production (see ESI† for details), and the highest FECO were 92.1 ± 1.7% and 90.9 ± 1.4% for Au43 and Au42Ag1, respectively, at a potential of −0.57 vs. RHE (Fig. 3B). At the same potential, FECO for Au24Ag20 and AunAg46−n were just 72.9 ± 1.0% and 59.5 ± 2.5%, respectively (Fig. 3B). These lower efficiencies can be attributed to more densely packed surface ligands, which likely favors the hydrogen evolution reaction (HER) over CO2RR.25,26,49 The Au–S NPs have an even lower FECO (38.7 ± 0.8%) at the same potential, which is consistent with previously reported studies.27,33 If FECO is related to the density of surface ligands, one might assume that a ligand-free Au layer would have the highest efficiency. However, the FECO for the Au layers is only 20.4 ± 1.6% at –0.57 V vs. RHE, highlighting the important role of the microenvironments created by nanostructured catalysts in driving CO2RR.3,50,51
To further evaluate catalytic performance, CO partial current densities (jCO) were determined and compared across the NC series. Generally, jCO decreases with increasing ligand density on the surface (Fig. 3D): Au43/Ag42Ag1 (20 surface ligands) > Au24Ag20 (26 surface ligands) > AunAg46−n (30 surface ligands). For Au43, jCO is slightly higher than for Au42Ag1, suggesting that substitution of a single surface Au atom for Ag leads to a small decrease in CO2RR performance, particularly at more negative potentials. Furthermore, Au43 shows a high CO turnover frequency (TOFCO) of 4718 h−1 at −0.57 V and 15193 h−1 at −0.77 V vs. RHE (Fig. 3E), which exceeds the values for Au24Ag20 (2597 h−1 at −0.57 V and 10658 h−1 at −0.77 V) and AunAg46−n (1427 h−1 at −0.57 V and 6400 h−1 at −0.77 V). Note that the same mass loading of NCs was used for all experiments. This makes it reasonable to directly compare TOF values since the NCs in this series have similar molecular weights (Table S5†). In addition, differences in catalytic activity cannot be attributed to differences in NC stability as the FECO of all catalysts remained constant for at least 2.5 hours at −0.57 V (Fig. 3F).
While the nature of active sites is regarded as one of the best predictors of catalytic activity,52 it is often challenging to experimentally determine the number of catalytically active sites in a nanostructured material, and theoretical models are required.53,54 Though double-layer capacitance measurements can be used to determine the electrochemically active surface area (ECSA) of catalysts, the ECSA might not reflect the surface area active specifically for CO2RR since CO2RR and HER frequently occur simultaneously. For example, previous studies have shown that Au25(SR)18 and Au38(SR)24 exhibit different CO2RR behavior even though the NCs have almost the same ECSA.33,49 The ligand-to-metal ratio can serve as a proxy for active site density when the sizes of NCs are similar,55 but this ratio is not directly related to the number of active sites owing to the different shapes and surface structures that similarly sized NCs can adopt. Indeed, two isomeric Au38(SR)24 NCs with the same ligand-to-metal ratio have shown significant differences in CO2RR catalysis.34
With these challenges in mind, we sought to establish a simple method for determining the number of metal sites accessible to CO2 in atomically precise NCs that does not rely on computationally intensive density functional theory (DFT) calculations. Briefly, the NC structure determined by crystallography is used to generate a series of several thousand conformers accounting for the different ligand conformations that may arise due to ligand rotation in the absence of crystal packing effects (see ESI† for details). The accessible surface area of each conformation within the conformer series was then calculated for every surface atom using a 1.65 Å spherical probe (the kinetic radius of CO2, Fig. S9†).56 Metal atoms with a positive contact area with CO2 (Fig. S10†) were counted as accessible since these atoms have sufficient space to accommodate a covalent bond with CO2. Accessible metal atoms for the different alkynyl-protected NCs under investigation are highlighted in Fig. 4. Note that the NC conformation with the greatest number of accessible metal atoms (N) was used to represent the surface accessibility of each NC. Using this approach, the number of accessible metal atoms for each NC can be calculated in ∼1 hour. Notably, the number of accessible metal atoms, N, for Au43/Au42Ag1, Au24Ag20, and AunAg46−n are 16, 12, and 5, respectively, which is consistent with a greater density of CO2-accessible surface metal sites driving increased CO2RR activity. The role of CO2-accessible metal sites is further supported by the fact that we observe little variation in TOFCO when it is normalized to the number of accessible metal sites on each NC (TOFCO/N). Specifically, TOFCO/N for Au43, Au42Ag1, Au24Ag20, and AunAg46−n is 294 h−1, 279 h−1, 216 h−1 and 285 h−1, respectively, at –0.57 V vs. RHE (Table 1 and Fig. S11†). This suggests that the number of CO2-accessible metal sites—rather than the degree of Ag doping,57 surface ligand functional groups,58 or the electronic structure59 of the cluster—is the primary driver of catalytic activity, at least for NCs with relatively similar structures and compositions.
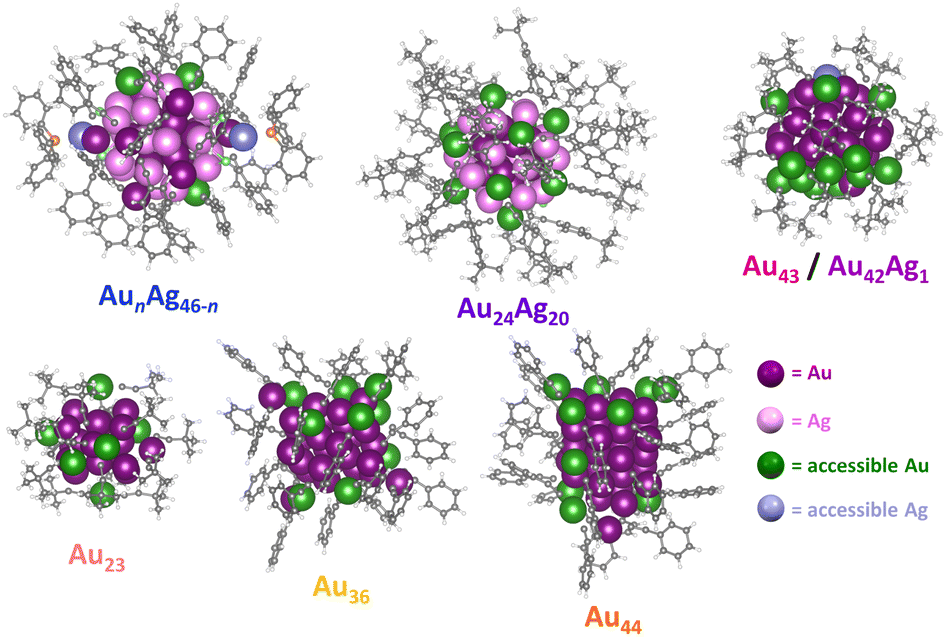 | ||
| Fig. 4 The metal sites accessible to CO2 on the surface of different Au/Ag-alkynyl NCs. | ||
| Catalyst | Number of accessible metals (N) | FECO (%) | j CO (mA cm−2) | TOFCO (h−1) | TOFCO/N (h−1) |
|---|---|---|---|---|---|
| Au43 | 16 | 92.1 ± 1.7 | 2.5 | 4718 | 295 |
| Au42Ag1 | 16 | 90.9 ± 1.4 | 2.4 | 4458 | 279 |
| Au24Ag20 | 12 | 72.9 ± 1.0 | 1.3 | 2597 | 216 |
| AunAg46−n | 5 | 59.5 ± 2.5 | 0.8 | 1427 | 285 |
| Au44 | 12 | 74.5 ± 1.3 | 1.0 | 2045 | 170 |
| Au36 | 9 | 69.7 ± 1.6 | 0.7 | 1325 | 147 |
| Au23 | 6 | 66.7 ± 2.5 | 0.7 | 794 | 132 |
To further investigate the generalizability of accessible metal site number as a predictor of CO2RR activity, we also evaluated a wider range of previously reported alkynyl-protected Au NCs: Au23(CCtBu)15 (Au23),60 Au36(CCPh)24 (Au36), and Au44(CCPh)28 (Au44)61 (Fig. S12†). Within this series, Au44 has the highest number of CO2-accessible metal sites (N = 12) and the highest CO2RR activity, while Au23 has the lowest number of CO2-accessible sites and the lowest CO2RR activity (Fig. 5). The activity of Au44, however, is much lower than that of Au43, which is consistent with the greater number of CO2-accessible metal sites (N = 16) for the latter NC (Table 1 and Fig. S13†). The relationship between the number of accessible metal sites and FECO, jCO is plotted in Fig. S14.† This highlights that even though larger sized Au–alkynyl NCs often have increased CO2RR activities, just like their Au–thiolate counterparts,33 the number of accessible surface metals tends to be more closely related to catalyst performance. Therefore, we conclude that the number of accessible metal sites provides a useful metric for evaluating the likelihood of CO2 binding to a particular atomically precise NC and for predicting trends in catalytic activity. Since Au43 and Au42Ag1 NCs are isostructural, their slight difference in CO2RR activity could be due to replacing a surface Au atom with a more electropositive Ag atom.35 Though Au24Ag20 and Au44 NCs have the same number of metal atoms (44) as well as the number of accessible metals (12), Au44 exhibits a slightly higher FECO and lower jCO than Au24Ag20. Differences in the geometric and electronic structures of the NCs may influence their CO2RR performance, but this is likely a less significant effect than the number of accessible metals. For NCs of the same core structure, more valence electrons in the frontier molecular orbitals elevate the energy of the highest occupied molecular orbital (HOMO), thereby favoring electron transfer from the NC catalyst to the substrate and thus improving the CO2RR activity.59 The performance of Au43 and Au42Ag1 NCs could also be partially attributed to more facile electron transfer during electrocatalysis. However, since the electronic structure of the NCs is determined by the number of metals and ligands, the surface coverage is still of importance. Moreover, since calculating the number of accessible metal sites is straightforward, it can serve as a quick screening tool for identifying the most promising NCs for electrochemical catalysis that is complementary to advanced DFT calculations.
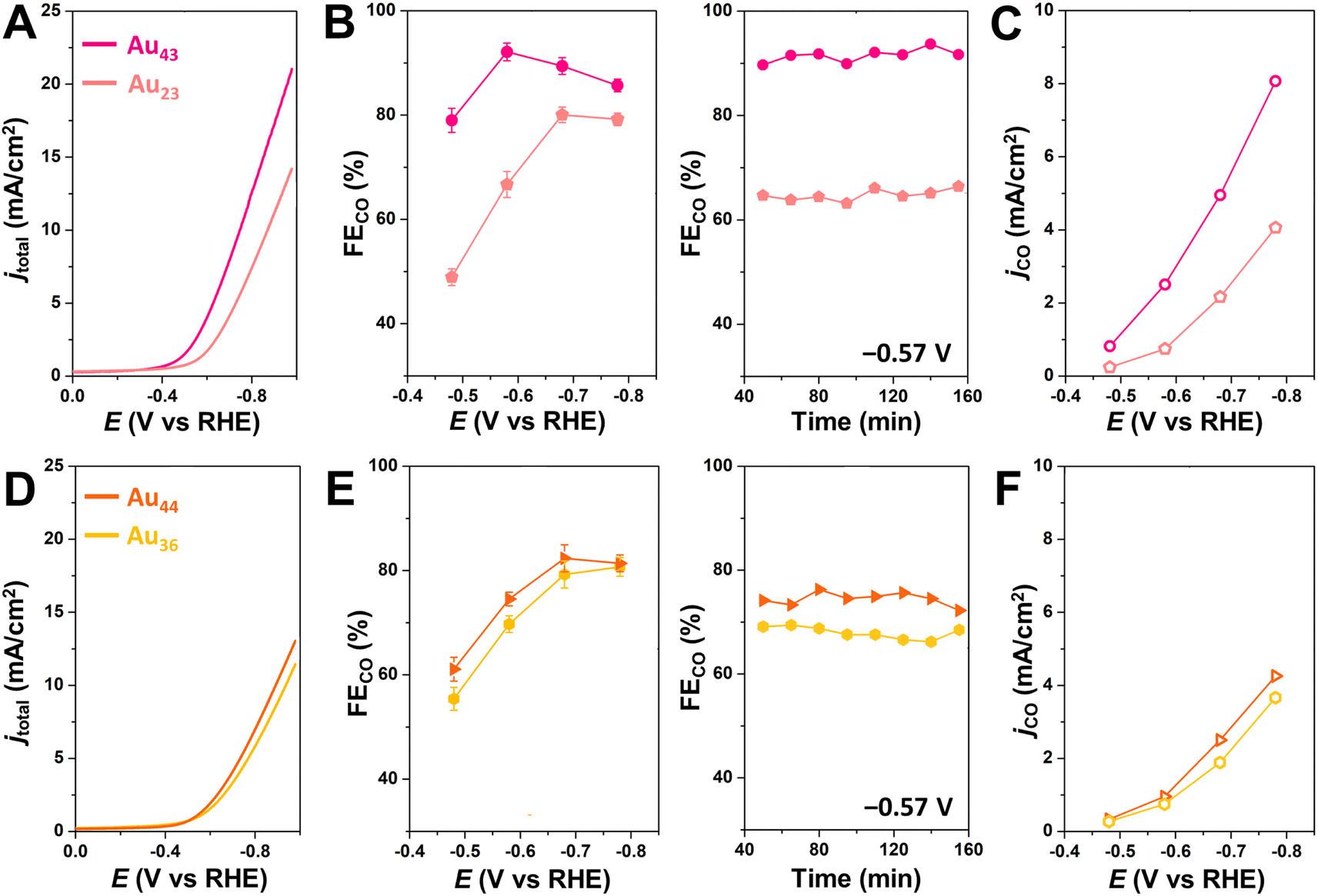 | ||
| Fig. 5 (A/D) LSV, (B/E) FECO at different applied potentials (left), or at –0.57 V vs. RHE for prolonged time (right), and (C/F) jCO at different applied potentials during CO2RR for NC-based catalysts conducted in CO2-saturated 0.5 M KHCO3 solution. Magenta represents Au43(CCtBu)20, pink represents Au23(CCtBu)20, orange represents Au44(CCPh)28, and yellow represents Au36(CCPh)24. | ||
Conclusions
A series of alkynyl-stabilized NCs with similar sizes and core structures but different degrees of surface ligand coverage was used to provide insight into the effect of the number of accessible metal sites on electrochemical CO2RR activity. A simple computational method was developed to calculate the number of metal sites on the NCs that are accessible to CO2. The highest faradaic efficiencies for CO2RR were observed for Au43 and Au42Ag1, which feature the largest number of accessible metal sites. When the TOFCO of the NC-based catalysts was normalized by the number of accessible sites, the differences between NCs were reduced. Collectively, these trends suggest that the number of substrate-accessible metal sites serves as a useful and generalizable predictor for evaluating the potential of atomically precise NCs for CO2RR.Data availability
All the data are shown in the manuscript or the associated ESI.†Author contributions
J. A. M and Y. L. conceived the project. Y. L. synthesized the NCs and grew the single crystals. Y. L. and A. E. T. performed the electrochemical studies and analysed the data. G. J. S performed the computational studies. R. D. M. and S.-L. Z. solved the crystal structures. Y. L and G. J. S wrote the manuscript with contribution from other authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge support from the Major Research Instrumentation (MRI) Program of the National Science Foundation under NSF award no. 2216066 for the X-ray facility. This research was partially supported by the Arnold and Mabel Beckman Foundation through a Beckman Young Investigator grant awarded to J. A. M.References
- L. Liu and A. Corma, Metal Catalysts for Heterogeneous Catalysis: From Single Atoms to Nanoclusters and Nanoparticles, Chem. Rev., 2018, 118, 4981–5079 CrossRef CAS PubMed.
- D. Astruc, Introduction: Nanoparticles in Catalysis, Chem. Rev., 2020, 120, 461–463 CrossRef CAS PubMed.
- A. T. Bell, The Impact of Nanoscience on Heterogeneous Catalysis, Science, 2003, 299, 1688–1691 CrossRef CAS PubMed.
- J. Qiao, Y. Liu, F. Hong and J. Zhang, A Review of Catalysts for the Electroreduction of Carbon Dioxide to Produce Low-Carbon Fuels, Chem. Soc. Rev., 2014, 43, 631–675 RSC.
- W.-H. Wang, Y. Himeda, J. T. Muckerman, G. F. Manbeck and E. Fujita, CO2 Hydrogenation to Formate and Methanol as an Alternative to Photo- and Electrochemical CO2 Reduction, Chem. Rev., 2015, 115, 12936–12973 CrossRef CAS PubMed.
- P. De Luna, C. Hahn, D. Higgins, S. A. Jaffer, T. F. Jaramillo and E. H. Sargent, What Would It Take for Renewably Powered Electrosynthesis to Displace Petrochemical Processes?, Science, 2019, 364, eaav3506 CrossRef CAS PubMed.
- S. Zhao, R. Jin and R. Jin, Opportunities and Challenges in CO2 Reduction by Gold- and Silver-Based Electrocatalysts: From Bulk Metals to Nanoparticles and Atomically Precise Nanoclusters, ACS Energy Lett., 2018, 3, 452–462 CrossRef CAS.
- T. Ishida, T. Murayama, A. Taketoshi and M. Haruta, Importance of Size and Contact Structure of Gold Nanoparticles for the Genesis of Unique Catalytic Processes, Chem. Rev., 2020, 120, 464–525 CrossRef CAS PubMed.
- S. Panigrahi, S. Basu, S. Praharaj, S. Pande, S. Jana, A. Pal, S. K. Ghosh and T. Pal, Synthesis and Size-Selective Catalysis by Supported Gold Nanoparticles: Study on Heterogeneous and Homogeneous Catalytic Process, J. Phys. Chem. C, 2007, 111, 4596–4605 CrossRef CAS.
- X. Zhou, W. Xu, G. Liu, D. Panda and P. Chen, Size-Dependent Catalytic Activity and Dynamics of Gold Nanoparticles at the Single-Molecule Level, J. Am. Chem. Soc., 2010, 132, 138–146 CrossRef CAS PubMed.
- R. Si and M. Flytzani-Stephanopoulos, Shape and Crystal-Plane Effects of Nanoscale Ceria on the Activity of Au–CeO2 Catalysts for the Water–Gas Shift Reaction, Angew. Chem., Int. Ed., 2008, 47, 2884–2887 CrossRef CAS PubMed.
- M. Jin, H. Zhang, Z. Xie and Y. Xia, Palladium Nanocrystals Enclosed by {100} and {111} Facets in Controlled Proportions and Their Catalytic Activities for Formic Acid Oxidation, Energy Environ. Sci., 2012, 5, 6352–6357 RSC.
- L. M. Rossi, J. L. Fiorio, M. A. S. Garcia and C. P. Ferraz, The Role and Fate of Capping Ligands in Colloidally Prepared Metal Nanoparticle Catalysts, Dalton Trans., 2018, 47, 5889–5915 RSC.
- L. Lu, S. Zou and B. Fang, The Critical Impacts of Ligands on Heterogeneous Nanocatalysis: A Review, ACS Catal., 2021, 11, 6020–6058 CrossRef CAS.
- S. Schauermann, N. Nilius, S. Shaikhutdinov and H.-J. Freund, Nanoparticles for Heterogeneous Catalysis: New Mechanistic Insights, Acc. Chem. Res., 2013, 46, 1673–1681 CrossRef CAS PubMed.
- C. Xie, Z. Niu, D. Kim, M. Li and P. Yang, Surface and Interface Control in Nanoparticle Catalysis, Chem. Rev., 2020, 120, 1184–1249 CrossRef CAS PubMed.
- Y. Li and R. Jin, Seeing Ligands on Nanoclusters and in Their Assemblies by X-Ray Crystallography: Atomically Precise Nanochemistry and Beyond, J. Am. Chem. Soc., 2020, 142, 13627–13644 CrossRef CAS PubMed.
- Z. Cao, D. Kim, D. Hong, Y. Yu, J. Xu, S. Lin, X. Wen, E. M. Nichols, K. Jeong, J. A. Reimer, P. Yang and C. J. Chang, A Molecular Surface Functionalization Approach to Tuning Nanoparticle Electrocatalysts for Carbon Dioxide Reduction, J. Am. Chem. Soc., 2016, 138, 8120–8125 CrossRef CAS PubMed.
- Z. Cao, S. B. Zacate, X. Sun, J. Liu, E. M. Hale, W. P. Carson, S. B. Tyndall, J. Xu, X. Liu, X. Liu, C. Song, J. Luo, M.-J. Cheng, X. Wen and W. Liu, Tuning Gold Nanoparticles with Chelating Ligands for Highly Efficient Electrocatalytic CO2 Reduction, Angew. Chem., Int. Ed., 2018, 57, 12675–12679 CrossRef CAS PubMed.
- G. Li and R. Jin, Atomically Precise Gold Nanoclusters as New Model Catalysts, Acc. Chem. Res., 2013, 46, 1749–1758 CrossRef CAS PubMed.
- Y. Du, H. Sheng, D. Astruc and M. Zhu, Atomically Precise Noble Metal Nanoclusters as Efficient Catalysts: A Bridge between Structure and Properties, Chem. Rev., 2020, 120, 526–622 CrossRef CAS PubMed.
- R. Jin, G. Li, S. Sharma, Y. Li and X. Du, Toward Active-Site Tailoring in Heterogeneous Catalysis by Atomically Precise Metal Nanoclusters with Crystallographic Structures, Chem. Rev., 2021, 121, 567–648 CrossRef CAS PubMed.
- Y. Li, T. Higaki, X. Du and R. Jin, Chirality and Surface Bonding Correlation in Atomically Precise Metal Nanoclusters, Adv. Mater., 2020, 1905488 CrossRef CAS PubMed.
- D. R. Kauffman, D. Alfonso, C. Matranga, H. Qian and R. Jin, Experimental and Computational Investigation of Au25 Clusters and CO2: A Unique Interaction and Enhanced Electrocatalytic Activity, J. Am. Chem. Soc., 2012, 134, 10237–10243 CrossRef CAS PubMed.
- Z.-H. Gao, K. Wei, T. Wu, J. Dong, D. Jiang, S. Sun and L.-S. Wang, A Heteroleptic Gold Hydride Nanocluster for Efficient and Selective Electrocatalytic Reduction of CO2 to CO, J. Am. Chem. Soc., 2022, 144, 5258–5262 CrossRef CAS PubMed.
- V. K. Kulkarni, B. N. Khiarak, S. Takano, S. Malola, E. L. Albright, T. I. Levchenko, M. D. Aloisio, C.-T. Dinh, T. Tsukuda, H. Häkkinen and C. M. Crudden, N-Heterocyclic Carbene-Stabilized Hydrido Au24 Nanoclusters: Synthesis, Structure, and Electrocatalytic Reduction of CO2, J. Am. Chem. Soc., 2022, 144, 9000–9006 CrossRef CAS PubMed.
- S.-F. Yuan, R.-L. He, X.-S. Han, J.-Q. Wang, Z.-J. Guan and Q.-M. Wang, Robust Gold Nanocluster Protected with Amidinates for Electrocatalytic CO2 Reduction, Angew. Chem., Int. Ed., 2021, 60, 14345–14349 CrossRef CAS PubMed.
- L. Qin, F. Sun, X. Ma, G. Ma, Y. Tang, L. Wang, Q. Tang, R. Jin and Z. Tang, Homoleptic Alkynyl-Protected Ag15 Nanocluster with Atomic Precision: Structural Analysis and Electrocatalytic Performance toward CO2 Reduction, Angew. Chem., Int. Ed., 2021, 60, 26136–26141 CrossRef CAS PubMed.
- P. Maity, H. Tsunoyama, M. Yamauchi, S. Xie and T. Tsukuda, Organogold Clusters Protected by Phenylacetylene, J. Am. Chem. Soc., 2011, 133, 20123–20125 CrossRef CAS PubMed.
- M. G. Taylor and G. Mpourmpakis, Thermodynamic Stability of Ligand-Protected Metal Nanoclusters, Nat. Commun., 2017, 8, 15988 CrossRef CAS PubMed.
- B. Zhang, J. Chen, Y. Cao, O. J. H. Chai and J. Xie, Ligand Design in Ligand-Protected Gold Nanoclusters, Small, 2021, 17, 2004381 CrossRef CAS PubMed.
- S. Li, X. Du, Z. Liu, Y. Li, Y. Shao and R. Jin, Size Effects of Atomically Precise Gold Nanoclusters in Catalysis, Precis. Chem., 2023, 1, 14–28 CrossRef CAS PubMed.
- H. Seong, V. Efremov, G. Park, H. Kim, J. S. Yoo and D. Lee, Atomically Precise Gold Nanoclusters as Model Catalysts for Identifying Active Sites for Electroreduction of CO2, Angew. Chem., Int. Ed., 2021, 60, 14563–14570 CrossRef CAS PubMed.
- S. Li, A. V. Nagarajan, X. Du, Y. Li, Z. Liu, D. R. Kauffman, G. Mpourmpakis and R. Jin, Dissecting Critical Factors for Electrochemical CO2 Reduction on Atomically Precise Au Nanoclusters, Angew. Chem., Int. Ed., 2022, 61, e202211771 CrossRef CAS PubMed.
- Y. Li, H. K. Kim, R. D. McGillicuddy, S.-L. Zheng, K. J. Anderton, G. J. Stec, J. Lee, D. Cui and J. A. Mason, A Double Open-Shelled Au43 Nanocluster with Increased Catalytic Activity and Stability, J. Am. Chem. Soc., 2023, 145, 9304–9312 CrossRef CAS PubMed.
- J. Xu, L. Xiong, X. Cai, S. Tang, A. Tang, X. Liu, Y. Pei and Y. Zhu, Evolution from Superatomic Au24Ag20 Monomers into Molecular-like Au43Ag38 Dimeric Nanoclusters, Chem. Sci., 2022, 13, 2778–2782 RSC.
- Y. Tang, F. Sun, X. Ma, L. Qin, G. Ma, Q. Tang and Z. Tang, Alkynyl and Halogen Co-Protected (AuAg)44 Nanoclusters: A Comparative Study on Their Optical Absorbance, Structure, and Hydrogen Evolution Performance, Dalton Trans., 2022, 51, 7845–7850 RSC.
- S. Takano, S. Hasegawa, M. Suyama and T. Tsukuda, Hydride Doping of Chemically Modified Gold-Based Superatoms, Acc. Chem. Res., 2018, 51, 3074–3083 CrossRef CAS PubMed.
- S. Takano, S. Ito and T. Tsukuda, Efficient and Selective Conversion of Phosphine-Protected (MAu8)2+ (M = Pd, Pt) Superatoms to Thiolate-Protected (MAu12)6+ or Alkynyl-Protected (MAu12)4+ Superatoms via Hydride Doping, J. Am. Chem. Soc., 2019, 141, 15994–16002 CrossRef CAS PubMed.
- F. Wen, U. Englert, B. S. Gutrath and U. Simon, Crystal Structure, Electrochemical and Optical Properties of [Au9(PPh3)8](NO3)3, Eur. J. Inorg. Chem., 2008, 106–111 CrossRef CAS.
- N. Yan, N. Xia, L. Liao, M. Zhu, F. Jin, R. Jin and Z. Wu, Unraveling the Long-Pursued Au144 Structure by X-Ray Crystallography, Sci. Adv., 2018, 4, eaat7259 CrossRef CAS PubMed.
- Y. Li, R. Juarez-Mosqueda, Y. Song, Y. Zhang, J. Chai, G. Mpourmpakis and R. Jin, Ligand Exchange on Au38(SR)24: Substituent Site Effects of Aromatic Thiols, Nanoscale, 2020, 12, 9423–9429 RSC.
- L. G. AbdulHalim, M. S. Bootharaju, Q. Tang, S. Del Gobbo, R. G. AbdulHalim, M. Eddaoudi, D.-e. Jiang and O. M. Bakr, Ag29(BDT)12(TPP)4: A Tetravalent Nanocluster, J. Am. Chem. Soc., 2015, 137, 11970–11975 CrossRef CAS PubMed.
- Y. Song, Y. Li, M. Zhou, X. Liu, H. Li, H. Wang, Y. Shen, M. Zhu and R. Jin, Ultrabright Au@Cu14 Nanoclusters: 71.3% Phosphorescence Quantum Yield in Non-Degassed Solution at Room Temperature, Sci. Adv., 2021, 7, eabd2091 CrossRef CAS PubMed.
- Y. Li, T.-Y. Luo, M. Zhou, Y. Song, N. L. Rosi and R. Jin, A Correlated Series of Au/Ag Nanoclusters Revealing the Evolutionary Patterns of Asymmetric Ag Doping, J. Am. Chem. Soc., 2018, 140, 14235–14243 CrossRef CAS PubMed.
- Y. Li, M. Zhou, Y. Song, T. Higaki, H. Wang and R. Jin, Double-Helical Assembly of Heterodimeric Nanoclusters into Supercrystals, Nature, 2021, 594, 380–384 CrossRef CAS PubMed.
- M. J. Alhilaly, R.-W. Huang, R. Naphade, B. Alamer, M. N. Hedhili, A.-H. Emwas, P. Maity, J. Yin, A. Shkurenko, O. F. Mohammed, M. Eddaoudi and O. M. Bakr, Assembly of Atomically Precise Silver Nanoclusters into Nanocluster-Based Frameworks, J. Am. Chem. Soc., 2019, 141, 9585–9592 CrossRef CAS PubMed.
- Y. Li, M. J. Cowan, M. Zhou, T.-Y. Luo, Y. Song, H. Wang, N. L. Rosi, G. Mpourmpakis and R. Jin, Atom-by-Atom Evolution of the Same Ligand-Protected Au21, Au22, Au22Cd1, and Au24 Nanocluster Series, J. Am. Chem. Soc., 2020, 142, 20426–20433 CrossRef CAS PubMed.
- Y. Li, S. Li, A. V. Nagarajan, Z. Liu, S. Nevins, Y. Song, G. Mpourmpakis and R. Jin, Hydrogen Evolution Electrocatalyst Design: Turning Inert Gold into Active Catalyst by Atomically Precise Nanochemistry, J. Am. Chem. Soc., 2021, 143, 11102–11108 CrossRef CAS PubMed.
- C. Kim, H. S. Jeon, T. Eom, M. S. Jee, H. Kim, C. M. Friend, B. K. Min and Y. J. Hwang, Achieving Selective and Efficient Electrocatalytic Activity for CO2 Reduction Using Immobilized Silver Nanoparticles, J. Am. Chem. Soc., 2015, 137, 13844–13850 CrossRef CAS PubMed.
- S. Liu, H. Tao, L. Zeng, Q. Liu, Z. Xu, Q. Liu and J.-L. Luo, Shape-Dependent Electrocatalytic Reduction of CO2 to CO on Triangular Silver Nanoplates, J. Am. Chem. Soc., 2017, 139, 2160–2163 CrossRef CAS PubMed.
- C. Vogt and B. M. Weckhuysen, The Concept of Active Site in Heterogeneous Catalysis, Nat. Rev. Chem, 2022, 6, 89–111 CrossRef PubMed.
- J. K. Nørskov, T. Bligaard, B. Hvolbæk, F. Abild-Pedersen, I. Chorkendorff and C. H. Christensen, The Nature of the Active Site in Heterogeneous Metal Catalysis, Chem. Soc. Rev., 2008, 37, 2163–2171 RSC.
- G. Kumar, L. Tibbitts, J. Newell, B. Panthi, A. Mukhopadhyay, R. M. Rioux, C. J. Pursell, M. Janik and B. D. Chandler, Evaluating Differences in the Active-Site Electronics of Supported Au Nanoparticle Catalysts Using Hammett and DFT Studies, Nat. Chem., 2018, 10, 268–274 CrossRef CAS PubMed.
- T. Higaki, Y. Li, S. Zhao, Q. Li, S. Li, X.-S. Du, S. Yang, J. Chai and R. Jin, Atomically Tailored Gold Nanoclusters for Catalytic Application, Angew. Chem., Int. Ed., 2019, 58, 8291–8302 CrossRef CAS PubMed.
- R. T. Yang, Adsorbents: Fundamentals and Applications, John Wiley & Sons, Inc., Hoboken: New Jersey, 2003 Search PubMed.
- G. Deng, J. Kim, M. S. Bootharaju, F. Sun, K. Lee, Q. Tang, Y. J. Hwang and T. Hyeon, Body-Centered-Cubic-Kernelled Ag15Cu6 Nanocluster with Alkynyl Protection: Synthesis, Total Structure, and CO2 Electroreduction, J. Am. Chem. Soc., 2022, 145, 3401–3407 CrossRef PubMed.
- S. Li, A. V. Nagarajan, Y. Li, D. R. Kauffman, G. Mpourmpakis and R. Jin, The Role of Ligands in Atomically Precise Nanocluster-Catalyzed CO2 Electrochemical Reduction, Nanoscale, 2021, 13, 2333–2337 RSC.
- X. Liu, E. Wang, M. Zhou, Y. Wan, Y. Zhang, H. Liu, Y. Zhao, J. Li, Y. Gao and Y. Zhu, Asymmetrically Doping a Platinum Atom into a Au38 Nanocluster for Changing the Electron Configuration and Reactivity in Electrocatalysis, Angew. Chem., Int. Ed., 2022, 61, e202207685 CrossRef CAS PubMed.
- Z.-J. Guan, F. Hu, J.-J. Li, Z.-R. Wen, Y.-M. Lin and Q.-M. Wang, Isomerization in Alkynyl-Protected Gold Nanoclusters, J. Am. Chem. Soc., 2020, 142, 2995–3001 CrossRef CAS PubMed.
- X.-K. Wan, Z.-J. Guan and Q.-M. Wang, Homoleptic Alkynyl-Protected Gold Nanoclusters: Au44(PhCC)28 and Au36(PhCC)24, Angew. Chem., Int. Ed., 2017, 56, 11494–11497 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2260158 and 2260159. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc04085b |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |