DOI:
10.1039/D3SC03996J
(Edge Article)
Chem. Sci., 2023,
14, 10867-10874
Asymmetric Friedel−Crafts reaction of unsaturated carbonyl-tethered heteroarenes via vinylogous activation of Pd0-π-Lewis base catalysis†
Received
1st August 2023
, Accepted 16th September 2023
First published on 19th September 2023
Abstract
The alkyne group can undergo facile transformations under palladium catalysis, such as hydropalladation, Wacker reaction, etc. Here we demonstrate that a chiral Pd0 complex can chemoselectively dihapto-coordinate to the alkyne moiety of 2-indolyl propiolates, and raise the Highest Occupied Molecular Orbital (HOMO)-energy ofthe deactivated heteroarenes via π-Lewis base catalysis. As a result, asymmetric C3-selective Friedel−Crafts addition to activated alkenes occurs, finally affording [3 + 2] or [3 + 4] annulation products with high enantioselectivity and exclusive E-selectivity. Moreover, this π-Lewis base vinylogous HOMO-activation strategy can be extended to remote Friedel−Crafts reaction of diverse five-membered heteroarenes tethered to a 2-enone or 2-acrylate motif with imines or 1-azadienes, and excellent enantiocontrol is generally achieved for the multifunctional adducts, which can be effectively converted to diverse frameworks with higher molecular complexity. In addition, NMR and density functional theory calculation studies are conducted to elucidate the catalytic mechanism.
Introduction
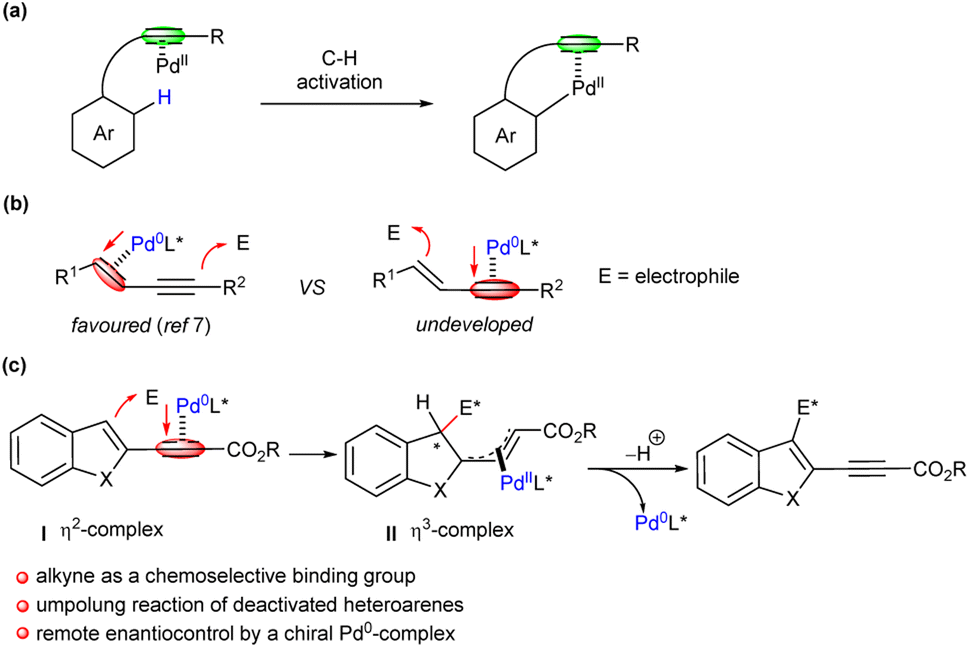
As a fundamental functional group, alkyne is readily available and enables abundant transformations in organic chemistry.1 Possessing π-electrons, alkynes can be activated by complexation with transition metals, and a variety of reactions,2 such as hydrogenation,3 Wacker-type addition,4 carbometallation,5etc., have been facilitated accordingly, furnishing fruitful products in high efficiency. As far as palladium catalysis is concerned, apart from the well-established direct activation and reaction patterns, the alkyne moiety has been introduced in some molecules as a directing group for the C−H activation of aromatic rings under PdII catalysis, affording functionalised aryl substances (Scheme 1a).6
 |
| | Scheme 1 Selected activation modes of alkyne-containing substrates via palladium catalysis. (a) Alkyne as a directing group for C–H activation via PdII catalysis. (b) Chemoselective activation of 1,3-enynes by forming η2-Pd0-alkene complexes. (c) This work: HOMO-activation of alkyne-tethered deactivated heteroarenes via Pd0 catalysis. | |
Recently, our group and others uncovered that Pd0 could chemoselectively form η2-complexes with the alkene moiety of 1,3-enynes.7 As a result, the alkyne group could be HOMO (the Highest Occupied Molecular Orbital)-raised upon π-Lewis base activation of Pd0, and undergo vinylogous nucleophilic attack towards electrophiles, even enantioselectively. Density functional theory (DFT) also supported that regioselective dihapto-coordination to the alkene group by Pd0 was favoured, as outlined in Scheme 1b. Although an array of polyconjugated systems has been successfully utilised via Pd0-π-Lewis base catalysis recently,8 exclusive coordination of Pd0 to the alkene moiety was proposed. It would be highly intriguing that the unsaturated alkyne group could be chemoselectively employed as the binding motif in the presence of other unsaturated systems. It is expected that a five-membered heteroarene tethered to an electron-withdrawing propiolate motif would be deactivated in a Friedel−Crafts (FC) reaction;9 nevertheless, the chemoselective coordination of Pd0 to the alkyne group, rather than the 2,3-double bond of the heteroarene, would be favoured, as illustrated in complex I (Scheme 1c). Therefore, an unusual FC addition reaction at 3-position of the heteroarene might be enhanced upon vinylogous activation of Pd0-π-Lewis base, even enantioselectively through remote control.10 Next, the resultant η3-complex II would undergo aromatisation via a β-H elimination or deprotonation process to give the FC adduct and Pd0 catalyst. Nevertheless, a few challenging issues, as briefly summarised in Scheme 1c, need to be conquered.
Results and discussion
Investigation of reaction conditions
The initial attempt was between 2-indolyl propiolate 1a and α-cyano chalcone 2a. No reaction occurred after heating in toluene at 60 °C for 24 h, indicating the indole moiety was indeed deactivated (Table 1, entry 1).11 Pleasingly, the conversions were smoothly promoted under the catalysis of Pd(PPh3)4, and a [3 + 2] annulation product 3a, via cascade FC reaction and intramolecular Michael addition, was isolated in a moderate yield with exclusive diastereoselectivity and E/Z-selectivity (entry 2). Nevertheless, no product was observed when Pd(OAc)2 was employed, suggesting a Lewis acid catalytic mode might not be involved (entry 3). In addition, no reaction occurred under Pd0 catalysis by using ester 1a′, indicating the alkyne moiety was vital forthe vinylogous-type activation (entry 4). Encouraged by the above results, the asymmetric version was investigated by combining Pd2(dba)3 and various chiral phosphine ligands. Trost’s ligand L1 showed good reactivity as well as enantioselectivity (entry 5), whereas inferior results were obtained with L2 and L3 bearing other aryl groups (entries 6 and 7). The one derived from chiral 1,2-cyclohexanediol (L4) gave a good yield, but poor enantioselectivity was observed, indicating the N-H group of L1 might play an important role as a H-bonding donor (entry 8). After more screenings, interestingly, adding catalytic amounts of tetrabutylammonium bromide (TBAB) significantly improved the yield without apparent effect on the enantioselectivity (entry 9). Further screenings of solvents indicated that using 1,4-dioxane was beneficial for the reactivity (entry 10). In addition, employing N-benzyl indole 1b improved the ee value of corresponding adduct 3b (entry 11), and the yieldalso was significantly improved by adding TBAB (entry 12). A better yield was obtained at a higher concentration (entry 13). Other additives were further tested. Both TBAC and TBAI provided comparable results (entries 14 and 15), but KBr demonstrated to be less effective (entry 16). These results indicated the ammonium cation was important to the reactivity, which was speculated that the ammonium salt might act as a counterion to stablilise intermeidate II after the FC addition to acceptor 2a.12 Moreover, comparable data were gained with 5 mol% of Pd (entry 17), whereas both yield and enantioselectivity were dramatically decreased by further reducing catalyst loadings (entry 18).
Table 1 Screening conditons for the asymmetric FC and Michael addition cascade of 2-indolyl propiolates 1 and α-cyano chalcone 2aa
|

|
| Entry |
1 |
L |
Additive |
Solvent |
Yieldb (%) |
eec (%) |
|
Unless noted otherwise, reactions were carried out with 1 (0.025 mmol), 2a (0.03 mmol), Pd2(dba)3 (5 mol%), L (10 mol%) and additive (20 mol%) in solvent (0.25 mL) at 60 °C under Ar.
Yield of the isolated product.
Determined by HPLC analysis on a chiral stationary phase.
Without catalyst, for 24 h.
With Pd(PPh3)4 (10 mol%).
With Pd(OAc)2 (10 mol%).
With 1b (0.1 mmol) and 2a (0.13 mmol) in 1,4-dioxane (0.5 mL).
With Pd2(dba)3 (2.5 mol%), L1 (5 mol%) at 60 °C for 96 h.
With Pd2(dba)3 (1.25 mol%), L1 (2.5 mol%) at 80 °C for 96 h.
|
| 1d |
1a
|
/ |
/ |
Toluene |
NR |
/ |
| 2e |
1a
|
/ |
/ |
Toluene |
3a, 64 |
/ |
| 3f |
1a
|
/ |
/ |
Toluene |
NR |
/ |
| 4e |
1a′
|
/ |
/ |
Toluene |
NR |
/ |
| 5 |
1a
|
L1
|
/ |
Toluene |
3a, 68 |
89 |
| 6 |
1a
|
L2
|
/ |
Toluene |
3a, 68 |
77 |
| 7 |
1a
|
L3
|
/ |
Toluene |
3a, 46 |
39 |
| 8 |
1a
|
L4
|
/ |
Toluene |
3a, 76 |
41 |
| 9 |
1a
|
L1
|
TBAB |
Toluene |
3a, 86 |
86 |
| 10 |
1a
|
L1
|
TBAB |
1,4-Dioxane |
3a, 89 |
84 |
| 11 |
1b
|
L1
|
/ |
1,4-Dioxane |
3b, 49 |
92 |
| 12 |
1b
|
L1
|
TBAB |
1,4-Dioxane |
3b, 70 |
91 |
| 13g |
1b
|
L1
|
TBAB |
1,4-Dioxane |
3b, 86 |
92 |
| 14g |
1b
|
L1
|
TBAC |
1,4-Dioxane |
3b, 76 |
92 |
| 15g |
1b
|
L1
|
TBAI |
1,4-Dioxane |
3b, 81 |
91 |
| 16g |
1b
|
L1
|
KBr |
1,4-Dioxane |
3b, 52 |
92 |
| 17g,h |
1b
|
L1
|
TBAB |
1,4-Dioxane |
3b, 83 |
91 |
| 18g,i |
1b
|
L1
|
TBAB |
1,4-Dioxane |
3b, 50 |
73 |
Substrate scope exploration
Consequently, the substrate scope and limitations of the asymmetric cascade FC reaction/Michael addition process was explored under the catalysis of Pd/L1 and using TBAB as an additive. As summarised in Scheme 2, a spectrum of α-cyano enones 2 were first evaluated in the reactions with 2-indolyl propiolate 1b. The enones with a variety of β-aryl or -heteroaryl groups were finely compatible, affording corresponding adducts 3b–3k in moderate to good yields with high enantioselectivity, whereas reduced yield and enantiocontrol were obtained for product 3f with an ortho-substituted phenyl group. Comparable results were also attained on a larger scale (product 3b). In addition, enones 2 with diverse α′-aryl and -heteroaryl groups were tolerated as well, providing desired products 3l–3u in high yields and enantioselectivity. In addition, the one with an α′-cyclopropyl-substituent was applicable as well (product 3v). Interestingly, an intramolecular O-Michael reaction instead of C-Michael one took place when some α′-CF3-substituted enones were utilised, generally affording [3 + 4] cycloadducts 4a–4e in moderate yields with excellent enantioselectivity and exclusive E-selectivity. Moreover, the substitution patterns of 2-alkynyl indoles 1 were evaluated. Electron-donating group-substituted indoles showed good reactivity (products 3w–3y), and moderate yields were obtained for chloro-substituted products (3z and 3aa), whereas consistently high enantioselectivity was observed. In addition, excellent stereocontrol was achieved for the indoles having various propiolate or propiolamide moieties (products 3ab–3ae), even for those with complex biologically active scaffolds (products 3af and 3ag).
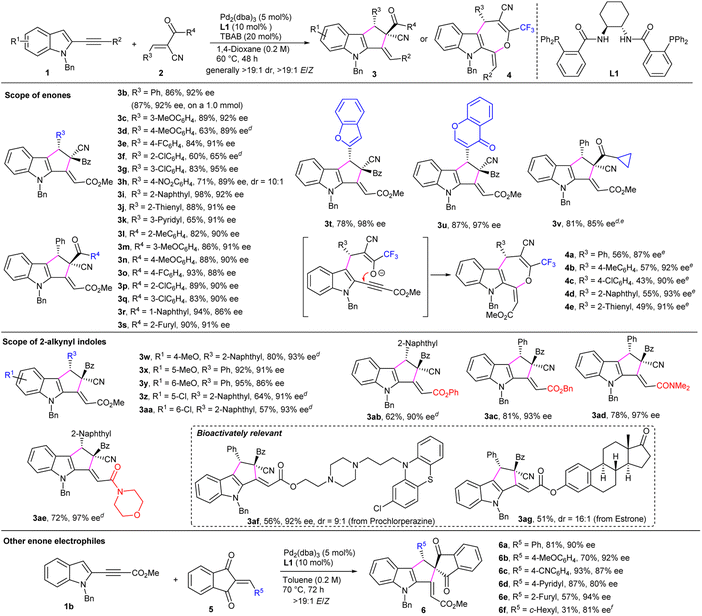 |
| | Scheme 2 Substrate scope of the asymmetric [3+2] or [3+4] annulation reaction of 2-alkynyl indoles and enones.a,b,c a Unless noted otherwise, reactions were carried out with indole 1 (0.10 mmol), enone 2 (0.13 mmol) or 5 (0.12 mmol), Pd2(dba)3 (5 mol%), L1 (10 mol %), and TBAB (20 mol%) in solvent (0.5 mL) under Ar for 48 h. b Yield of the isolated product. c Determined by HPLC analysis on a chiral stationary phase. d With 2 (1.5 equiv) at 80 °C. e Without TBAB. f At 90 °C for 5 d. The absolute configuration of enantiopure 3i, 6c and the structure of rac-4d was determined by X-ray analysis. The other products were assigned by analogy. | |
Apart from α-cyano enones, 1,3-indanedione-derived electrophiles 5 were compatible. As outlined in Scheme 2, enones 5 with an array of β-aryl and -heteroaryl groups were well tolerated in the reactions with indole 1b in toluene under the catalysis of Pd/L1, delivering corresponding products 6a–6e in moderate to good yields with satisfactory enantioselectivity, whereas inferior data were obtained for the one with a c-hexyl group (product 6f).
Furthermore, such a vinylogous HOMO-activation strategy was successfully extended to the indoles with electron-poor 2-alkenyl substitutions. As apparent background [3 + 2] annulation was observed in the assembly of indole 7a (Scheme 3, R1 = H, R2 = Ph) and enone 2a, we turned our attention to less reactive electrophiles, and N-Ts imine 8a was proved to be suitable.13 After screenings, FC adduct 9a was obtained in high yield and excellent enantioselectivity under the catalysis of Pd/bisphosphine L5 and acid A1, which might activate the imine as a Brønsted acid. As summarised in Scheme 3, a few indoles bearing electron-withdrawing or -donating groups were well-tolerated, affording corresponding products 9b–9e in good results. In addition, a variety of aryl and heteroaryl groups could be smoothly introduced into the α′-site of enone moiety of the indole substrates (products 9f–9i), even on a larger scale (for product 9f). Small amounts of Z-isomer were generated for a methyl ketone substrate (product 9j), and apparently reduced yield and stereoselectivity were observed for product 9k with an acrylate motif. Interestingly, an indole with a free acrylic acid moiety still showed good reactivity,8e albeit with moderate enantioselectivity (product 9l). In addition, a spectrum of aryl- and heteroaryl-substituted imine partners were well applied in the reactions with indole 7a, and corresponding products 9m–9t were generally obtained in good yields with excellent enantioselectivity. The imine derived from a chromone aldehyde also performed well (product 9u). Notably, cyclopropanecarbaldehyde- and ethyl glyoxylate-derived imines underwent the FC reaction smoothly, delivering products 9v and 9w, respectively, in good results. Moreover, satisfactory data were obtained for other N-sulfonylimines (products 9x and 9y).
 |
| | Scheme 3 Substrate scope of the asymmetric FC reaction of 2-alkenyl indoles and imines.a,b,c a Unless noted otherwise, reactions were carried out with indole 7 (0.10 mmol), imine 8 (0.20 mmol), Pd2(dba)3 (5 mol%), L5 (10 mol%), 4 Å MS (40.0 mg) and A1 (5 mol%) in xylene (1.0 mL) under Ar for 3−8 d. b Yield of the isolated product. c Determined by HPLC analysis on a chiral stationary phase. d The absolute configuration of enantiopure 9f was determined by X-ray analysis. The other products were assigned by analogy. e At 60 °C. | |
We further explored the challenging remote asymmetric FC reaction with the five-membered heteroarenes functionalised with a 2-enone or 2-acrylate motif. As illustrated in Scheme 4a, the C5-regioselective reaction between diverse pyrroles 10with N-sulfonylimines 8 proceeded effectively in toluene at 50 °C under Pd0 catalysis, and high levels of enantioselectivity were generally obtained by employing a bifunctional phosphine ligand14L6 and salicylic acid A2 as an Brønsted acid additive (products 11a–11g). Moreover, an activated ketimine was compatible, and product 11h was obtained in good yield and enantioselectivity. In addition, excellent yield and enantiocontrol were achieved in the remote FC reaction of furan 12 with 1-azadiene 13 under the catalysis of Pd/bifunctional ligand L7 (product 14, Scheme 4b).
 |
| | Scheme 4 Asymmetric remote FC reaction of functionalised pyrroles and furans.a,b,c (a) Remote FC reaction of pyrrole derivatives. (b) Remote FC reaction of a furan derivative. a Unless noted otherwise, reactions were carried out with 10 (0.12 mmol, 1.2 equiv) or furan 12 (0.2 mmol, 2.0 equiv), 8 (0.1 mmol, 1.0 equiv) or 1-azadiene 13 (0.1 mmol, 1.0 equiv), Pd2(dba)3 (5 mol%), L (10 mol%), 4 Å MS (40.0 mg) and A2 (10 mol%) in toluene (1.0 mL) under Ar. b Yield of the isolated product. c Determined by HPLC analysis on a chiral stationary phase. d The absolute configuration of enantiopure 11g was determined by X-ray analysis. The other products were assigned by analogy. e Using A1 (10 mol%). f The absolute configuration of product 14 was determined by converting to derivative 21. | |
Synthetic transformations and application
The diversely functionalised products enables versatile transformations. As illustrated in Scheme 5, an indole fused fulvene product 15 was obtained in a moderate yield after reduction of cycloadduct 3b with DIBAL-H and the subsequent treatment with dilute HCl.15 After chemoselective reduction of carbonyl of product 9f, an intramolecular N-allylation reaction could be conducted with the resultant allyl alcohol under the catalysis of Pd(PPh3)4 and phosphoric acid (PA), furnishing product 16 in a fair yield with excellent diastereoselectivity.16 In addition, after N-allylation of 9f (some ee losses were observed under basic conditions), anazepino[3,4-b]indole 17 was constructed in a moderate yield via a Metathesis reaction.17 Moreover, piperazine product 18 (ref. 18) and diazepane 19,19 respectively, were obtained efficiently through palladium-catalysed double N-allylations. Besides, an unexpected 20-membered ring system 21 was obtained in a moderate yield with a retained ee value via a Pd-catalysed N-deallylation and N-allylation cascade process of derivative 20.
 |
| | Scheme 5 Synthetic transformations of diverse products. (a) Transformation of [3 + 2] cycloadduct 3b. (b) Transformation of FC adduct 9f. (c) Double allylation of FC adduct 11a. (d) Construction of a 20-membered ring system from adduct 14. | |
Mechanistic studies
To get more insight into the catalytic mechanism, we first conducted frontier molecular orbital (FMO) analysis on the Pd0 complexes of unsaturated indoles. As illustrated in Fig. 1, in comparison with the parent substrate alkyne 1a (−5.69 eV) or alkene 7a (−5.46 eV), the HOMO energy of corresponding η2-Pd0-complex I-1a (−4.71 eV) or I-7a (−4.62 eV) is apparently raised, respectively, supporting the π-Lewis back donation of Pd0 as a Lewis base. Actually, it has been demonstrated that Pd0 could form a stable complex by coordinating to a triple or double bond.20 In our case, the proposed intermediate I-1a or I-7a indeed has been successfully detected by high-resolution mass spectrometry (HRMS) analysis by mixing alkyne 1a or alkene 7a with Pd(PPh3)4, respectively (Fig. 2). Moreover, we carried out NMR studies on the in situ generated complexes. As outlined in Fig. 2a, the 1H NMR analysis showed that 3-H (H1) of alkyne 1a experienced apparent high-field shifts when Pd(PPh3)4 was added (6.06 vs. 6.83 ppm). The possible nucleophilic attack of PPh3 to electron-deficient 1a was not observed by mixing 1a and PPh3. In addition, the 13C NMR analysisexhibited that the signals of the triple bond of 1a disappeared (around 70−80 ppm) after adding Pd(PPh3)4 (Fig. 2b), whereas new peaks were observedat the sp2-carbon region (C2 and C3, around 136 ppm); in contrast, C1 of 1a experienced significant high-field shifts (105.1 vs. 112.4 ppm). Similarly, the signals of H1, H2, H3, C1, C2 and C3 of 7a were all high-field shifted in the presence of Pd(PPh3)4, according to the NMR experiments (Fig. 2c and d). These results well supported that the proposed complexes I-1a and I-7a would be formed, and verified the π-Lewis base activation of Pd0 through strong coordination to the unsaturated group.20
 |
| | Fig. 1 Frontier molecular orbital (FMO) analysis for Pd0-alkyne complex (a) and Pd0-alkene complex (b). The calculations were performed at the B3LYP/6-31G(d)(SDD for Pd) (298.15 K) level of theory. | |
 |
| | Fig. 2 NMR analysis of the unsaturated indoles and corresponding η2-Pd0-complexes. (a) 1H NMR analysis of indole 1a and complex I-1a. (b) 13C NMR analysis of indole 1a and complex I-1a. (c) 1H NMR analysis of indole 7a and complex I-7a. (d) 13C NMR analysis of indole 7a and complex I-7a. | |
We also investigated the origins of enantioselectivity in the reaction of alkyne 1a and enone 2a. Based on the DFT calculation results, the FC addition step in which the first C−C bond was constructed, is the rate- and stereo-determining step.21 As shown in Fig. 3, four related transition states were considered. Transition states (R,S)-TS and (S,S)-TS would lead to product (S,R)-3avia (R,S)-II and (S,S)-II, respectively, and the stereogenic centre at 3-C of indole would disappear through deprotonation/aromatization. On the other hand, (R,R)-TS and (S,R)-TS would lead to enantiomer (R,S)-3avia similar transformations. Notably, H-bonding interaction between the NH group of L1 and the carbonyl group of 1a is observed in (R,S)-TS and (R,R)-TS, while the other two transition states without H-bonding interaction exhibit higher energies [5.2 kcal/mol−1 for (S,S)-TS; 2.9 kcal/mol−1 for (S,R)-TS]. The results indicate that the H-bonding is beneficial for the reaction, which is consistent with the experimental results (Table 1, entry 5 vs. entry 8). Geometric structure analyses show that the forming C−C bond in (R,R)-TS presents a pseudogauche conformation to avoid the steric repulsion between the ester group of 1a and the carbonyl of 2a. As a result, the dihedral angle DC1–C2–C3–H is 45.4 with apparent torsional strain. In contrast, (R,S)-TS possesses smaller torsional strain (referring to the corresponding DC1–C2–C3–H = 57.4), thus leading to lower energy [0 kcal/mol−1 for (R,S)-TSvs. 1.0 kcal/mol−1 for (R,R)-TS]. Therefore, the most favourable transition state (R,S)-TS would afford (S,R)-3a as the major product after annulation, which is consistent with the experimental observation.21
 |
| | Fig. 3 Origins of enantioselectivity for the FC reaction of 1a and 2a. The calculations were performed at the M06/6-311++G(d,p)(SDD for Pd)/SMD//B3LYP/6-31G(d)(SDD for Pd) (298.15 K) level of theory. | |
Conclusion
In summary, we demonstrated that Pd0 could chemoselectively coordinate to the alkyne motif of 2-propiolate-tethered indole substrates, which would raise the HOMO-energy ofthe deactivated heteroarenes to undergo Friedel−Crafts reaction with enone acceptors upon π-Lewis base catalysis, finally furnishing [3 + 2] or [3 + 4] annulation products with exclusive E-selectivity after an intramolecular Michael addition process. Excellent diastereoselectivity and enantioselectivity were generally achieved for substantial substrate assemblies by employing a chiral Trost’s bisphosphine ligand. Moreover, this vinylogous activation strategy via π-Lewis base catalysis was successfully expanded to deactivated indoles tethered to a 2-enone or acrylate motif, and even very remote C5-selective Friedel−Crafts reactions of similarly deactivated pyrroles and furans could be realised with remarkable enantioselectivity by using a chiral bifunctional phosphine ligand, further enriching the structural diversity and skeletal versatility of relevant products. In addition, NMR analysis and DFT calculations were conducted to rationalise the activation and stereocontrol pathways. We believe that this Pd0-π-Lewis base catalysis would find more application in asymmetric synthesis.
Data availability
The data that support the findings of this study are available in the ESI† or on request from the corresponding author.
Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We are grateful for the financial support from the NSFC (92156005, 21931006 and 21921002), the 111 project (B18035) and the Sichuan Science and Technology Program.
Notes and references
- For selected reviews, see:
(a)
F. Diederich, P. J. Stang and R. R. Tykwinski, Acetylene Chemistry: Chemistry, Biology and Material Science, Wiley-VCH, Weinheim, 2005 Search PubMed;
(b)
B. M. Trost and C.-J. Li, Modern Alkyne Chemistry: Catalytic and Atom-Economic Transformations, Wiley, New York, 2014 CrossRef;
(c) J. Lehmann, M. H. Wright and S. A. Sieber, Chem. – Eur. J., 2016, 22, 4666 CrossRef CAS PubMed;
(d) T. T. Talele, J. Med. Chem., 2020, 63, 5625 CrossRef CAS PubMed;
(e) M. Patel, R. K. Saunthwal and A. K. Verma, Acc. Chem. Res., 2017, 50, 240 CrossRef CAS PubMed;
(f) K. Keerthika, S. Nath and K. Geetharani, Catal. Sci. Technol., 2020, 10, 7142 RSC.
- For selected reviews, see:
(a) B. M. Trost and J. T. Masters, Chem. Soc. Rev., 2016, 45, 2212 RSC;
(b) S. Saito and Y. Yamamoto, Chem. Rev., 2000, 100, 2901 CrossRef CAS PubMed;
(c) R. Chinchilla and C. Nájera, Chem. Rev., 2014, 114, 1783 CrossRef CAS PubMed;
(d) V. P. Boyarskiy, D. S. Ryabukhin, N. A. Bokach and A. V. Vasilyev, Chem. Rev., 2016, 116, 5894 CrossRef CAS PubMed;
(e) M. Li, W. Wu and H. Jiang, ChemCatChem, 2020, 12, 5034 CrossRef CAS;
(f) W. Liu and W. Kong, Org. Chem. Front., 2020, 7, 3941 RSC.
-
(a) Á. Molnár, A. Sárkány and M. Varga, J. Mol. Catal. A: Chem., 2001, 173, 185 CrossRef;
(b)
A. M. Kluwer and C. J. Elsevier, Homogeneous Hydrogenation of Alkynes and Dienes, in The Handbook of Homogeneous Hydrogenation, Wiley-VCH, Weinheim, 2007 Search PubMed;
(c)
I. J. Munslow and P. G. Andersson, Alkyne Reductions, in Modern Reduction Methods, Wiley-VCH, Weinheim, 2008 Search PubMed.
- For selected reviews, see:
(a) F. Alonso, I. P. Beletskaya and M. Yus, Chem. Rev., 2004, 104, 3079 CrossRef CAS PubMed;
(b) X. Zeng, Chem. Rev., 2013, 113, 6864 CrossRef CAS PubMed;
(c) N. T. Patil and Y. Yamamoto, Chem. Rev., 2008, 108, 3395 CrossRef CAS PubMed;
(d) L. Chen, K. Chen and S. Zhu, Chem, 2018, 4, 1208 CrossRef CAS.
- For selected reviews and examples, see:
(a) J. Corpas, P. Mauleón, R. G. Arrayás and J. C. Carretero, ACS Catal., 2021, 11, 7513 CrossRef CAS;
(b) B. M. Monks and S. P. Cook, J. Am. Chem. Soc., 2012, 134, 15297 CrossRef CAS PubMed;
(c) X. Zhang, X. Xie and Y. Liu, Chem. Sci., 2016, 7, 5815 RSC;
(d) J. Chen and T. Hayashi, Angew. Chem., Int. Ed., 2020, 59, 18510 CrossRef CAS PubMed.
- For selected reviews and examples, see:
(a) P. Gandeepan and C. H. Cheng, Chem.–Asian J., 2015, 10, 824 CrossRef CAS PubMed;
(b) B. Guo, L. Zheng, L. Zhang and R. Hua, J. Org. Chem., 2015, 80, 8430 CrossRef CAS PubMed;
(c) C. Feng and T.-P. Loh, J. Am. Chem. Soc., 2010, 132, 17710 CrossRef CAS PubMed;
(d) Y. Minami, Y. Shiraishi, K. Yamada and T. Hiyama, J. Am. Chem. Soc., 2012, 134, 6124 CrossRef CAS PubMed;
(e) N. Chernyak and V. Gevorgyan, J. Am. Chem. Soc., 2008, 130, 5636 CrossRef CAS PubMed;
(f) Y. Minami, Y. Noguchi and T. Hiyama, J. Am. Chem. Soc., 2017, 139, 14013 CrossRef CAS PubMed.
-
(a) Q. He, L. Zhu, Z.-H. Yang, B. Zhu, Q. Ouyang, W. Du and Y.-C. Chen, J. Am. Chem. Soc., 2021, 143, 17989 CrossRef CAS PubMed;
(b) Z. Zhao, J.-L. Zheng, W. Du and Y.-C. Chen, Chem Catal., 2022, 2, 3174 CrossRef CAS;
(c) S. Xu, Y. Zhang, B. Li and S.-Y. Liu, J. Am. Chem. Soc., 2016, 138, 14566 CrossRef CAS PubMed;
(d) Y. Zhang, B. Li and S.-Y. Liu, Angew. Chem., Int. Ed., 2020, 59, 15928 CrossRef CAS PubMed;
(e) Z. Wang, J. Wu, W. Lamine, B. Li, J.-M. Sotiropoulos, A. Chrostowska, K. Miqueu and S.-Y. Liu, Angew. Chem., Int. Ed., 2021, 60, 21231 CrossRef CAS PubMed;
(f) Y. Zhang, Z. Wang, W. Lamine, S. Xu, B. Li, A. Chrostowska, K. Miqueu and S.-Y. Liu, J. Org. Chem., 2023, 88, 2415 CrossRef CAS PubMed.
- For selected examples, see:
(a) B.-X. Xiao, B. Jiang, R.-J. Yan, J.-X. Zhu, K. Xie, X.-Y. Gao, Q. Ouyang, W. Du and Y.-C. Chen, J. Am. Chem. Soc., 2021, 143, 4809 CrossRef CAS PubMed;
(b) X.-X. Yang, R.-J. Yan, G.-Y. Ran, C. Chen, J.-F. Yue, X. Yan, Q. Ouyang, W. Du and Y.-C. Chen, Angew. Chem., Int. Ed., 2021, 60, 26762 CrossRef CAS PubMed;
(c) X. Song, J. Zhang, Y.-X. Wu, Q. Ouyang, W. Du and Y.-C. Chen, J. Am. Chem. Soc., 2022, 144, 9564 CrossRef CAS PubMed;
(d) H. Lin, X. Yang, W. Ning, X. Huang, X. Cao, Y. Ge, B. Mao, C. Wang, H. Guo and C. Yuan, Org. Lett., 2022, 24, 9442 CrossRef CAS PubMed;
(e) S.-Z. Tan, P. Chen, L. Zhu, M.-Q. Gan, Q. Ouyang, W. Du and Y.-C. Chen, J. Am. Chem. Soc., 2022, 144, 22689 CrossRef CAS PubMed.
- N. Otero, M. Mandado and R. A. Mosquera, J. Phys. Chem. A, 2007, 111, 5557 CrossRef CAS PubMed.
-
(a) J.-L. Li, C.-Z. Yue, P.-Q. Chen, Y.-C. Xiao and Y.-C. Chen, Angew. Chem., Int. Ed., 2014, 53, 5449 CrossRef CAS PubMed ; for selected recent reviews of asymmetric Friedel–Crafts reactions, see:;
(b) T. Ahmad, S. Khan and N. Ullah, ACS Omega, 2022, 7, 35446 CrossRef CAS PubMed;
(c) D. Gaviña, M. Escolano, J. Torres, G. Alzuet-Piña, M. Sánchez-Roselló and C. del Pozo, Adv. Synth. Catal., 2021, 363, 3439 CrossRef;
(d) M. M. Heravi, V. Zadsirjan, M. Heydari and B. Masoumi, Chem. Rec., 2019, 19, 2236 CrossRef CAS.
- In contrast, a strong background FC reaction was observed when analogous 1-methyl-2-(prop-1-yn-1-yl)-indole was used, see the ESI† for more details.
- For more discussion, see the mechanism study part in the ESI†.
- For more screenings with other electrophiles, see the ESI†.
-
(a) C. Chen, X.-X. Yang, Z. Zhao, B. Han, W. Du and Y.-C. Chen, Chem. Commun., 2022, 58, 5502 RSC;
(b) Y. Zheng, T. Qin and W. Zi, J. Am. Chem. Soc., 2021, 143, 1038 CrossRef CAS PubMed.
- For related intramolecular acyl transfer, see:
(a) S.-Y. Liang, B. Jiang, B.-X. Xiao, Z.-C. Chen, W. Du and Y.-C. Chen, ChemCatChem, 2020, 12, 5374 CrossRef CAS;
(b) C. Parida, R. Maity, S. Chandra Sahoo and S. Chandra Pan, Org. Lett., 2019, 21, 6700 CrossRef CAS PubMed.
- For related frameworks, see:
(a) W. M. Welch, C. A. Harbert and A. Weissman, J. Med. Chem., 1980, 23, 704 CrossRef CAS PubMed;
(b) D. Pagé, H. Yang, W. Brown, C. Walpole, M. Fleurent, M. Fyfe, F. Gaudreault and S. St-Onge, Bioorg. Med. Chem. Lett., 2007, 17, 6183 CrossRef PubMed;
(c) A. Rinderspacher and G. W. Gribble, Molecules, 2020, 25, 261 CrossRef CAS PubMed.
- For related frameworks, see:
(a) C. Schultz, A. Link, M. Leost, D. W. Zaharevitz, R. Gussio, E. A. Sausville, L. Meijer and C. Kunick, J. Med. Chem., 1999, 42, 2909 CrossRef CAS PubMed;
(b) Y. Wan, W. Hur, C. Y. Cho, Y. Liu, F. J. Adrian, O. Lozach, S. Bach, T. Mayer, D. Fabbro, L. Meijer and N. S. Gray, Chem. Biol., 2004, 11, 247 CrossRef CAS PubMed;
(c) H. Tanaka, T. Yasui, M. Uyanik and K. Ishihara, Org. Lett., 2023, 25, 2377 CrossRef CAS PubMed.
- For related frameworks, see:
(a) C.-X. Zhuo, X. Zhang and S.-L. You, ACS Catal., 2016, 6, 5307 CrossRef CAS;
(b) I. Mancini, G. Guella, P. Amade, C. Roussakis and F. Pietra, Tetrahedron Lett., 1997, 38, 6271 CrossRef CAS;
(c) A. Umeyama, S. Ito, E. Yuasa, S. Arihara and T. Yamada, J. Nat. Prod., 1998, 61, 1433 CrossRef CAS PubMed;
(d) R. Rane, N. Sahu, C. Shah and R. Karpoormath, Curr. Top. Med. Chem., 2014, 14, 253 CrossRef CAS PubMed.
- G. V. De Lucca and M. J. Otto, Bioorg. Med. Chem. Lett., 1992, 2, 1639 CrossRef CAS.
- The X-ray structure of the analogous η2-Pd0-complex has been reported, see:
(a) R. Shintani, K. Moriya and T. Hayashi, Org. Lett., 2012, 14, 2902 CrossRef CAS PubMed ; for analogous η2-Pd0-complexes with alkenes, see: ;
(b) M. R. Buchner, B. Bechlars, B. Wahl and K. Ruhland, Organometallics, 2012, 31, 588 CrossRef CAS.
-
(a) For more mechanism elucidation studies, see the ESI†;
(b) The chiral Pd catalyst would not be actively involved in the final diastereoselective Michael addition step..
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, spectroscopic data for new compounds, NMR, HRMS spectra, and HPLC chromatograms, and CIF files of products 3i, rac-4d, 6c, 9f, 11g and 21. CCDC 2251805–2251810. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc03996j |
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence b,
Qin
Ouyang
b,
Qin
Ouyang