DOI:
10.1039/D3SC03932C
(Edge Article)
Chem. Sci., 2023,
14, 11749-11760
Ru(II)/Os(II)-based carbonic anhydrase inhibitors as photodynamic therapy photosensitizers for the treatment of hypoxic tumours†
Received
29th July 2023
, Accepted 21st September 2023
First published on 22nd September 2023
Abstract
Photodynamic therapy (PDT) is a medical technique for the treatment of cancer. It is based on the use of non-toxic molecules, called photosensitizers (PSs), that become toxic when irradiated with light and produce reactive oxygen specious (ROS) such as singlet oxygen (1O2). This light-induced toxicity is rather selective since the physician only targets a specific area of the body, leading to minimal side effects. Yet, a strategy to improve further the selectivity of this medical technique is to confine the delivery of the PS to cancer cells only instead of spreading it randomly throughout the body prior to light irradiation. To address this problem, we present here novel sulfonamide-based monopodal and dipodal ruthenium and osmium polypyridyl complexes capable of targeting carbonic anhydrases (CAs) that are a major target in cancer therapy. CAs are overexpressed in the membrane or cytoplasm of various cancer cells. We therefore anticipated that the accumulation of our complexes in or outside the cell prior to irradiation would improve the selectivity of the PDT treatment. We show that our complexes have a high affinity for CAs, accumulate in cancer cells overexpressing CA cells and importantly kill cancer cells under both normoxic and hypoxic conditions upon irradiation at 540 nm. More importantly, Os(II) compounds still exhibit some phototoxicity under 740 nm irradiation under normoxic conditions. To our knowledge, this is the first description of ruthenium/osmium-based PDT PSs that are CA inhibitors for the selective treatment of cancers.
Introduction
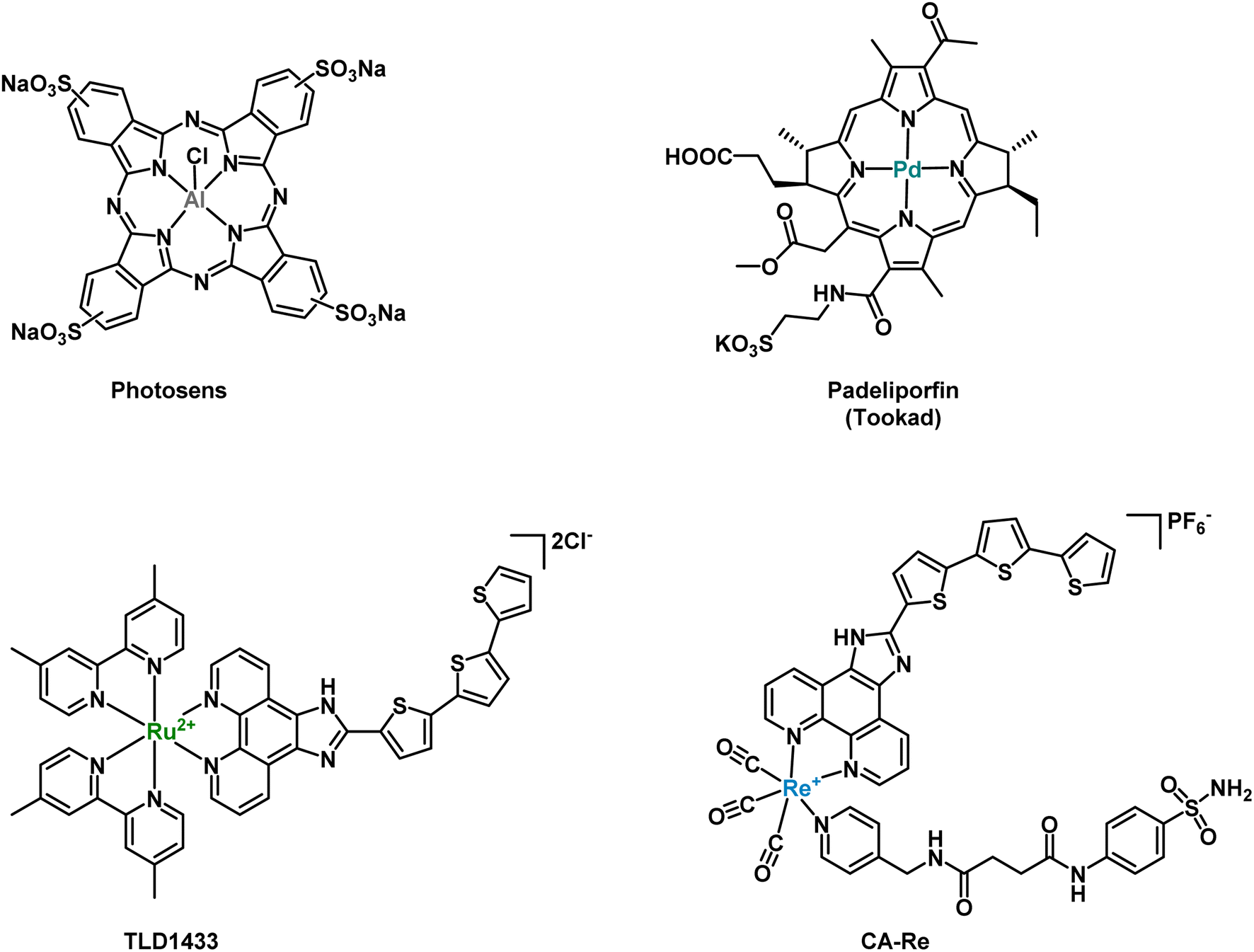
Photodynamic therapy (PDT) has recently emerged as a promising medical technique to treat certain forms of cancer due to its high spatiotemporal precision, leading to minimal side effects.1,2 A PDT treatment mainly relies on a photosensitizer (PS) to produce cytotoxic reactive oxygen species (ROS) upon light irradiation. The ROS produced usually include superoxide anion radicals (O2˙−), hydroxyl radicals (OH˙), or hydrogen peroxide (H2O2) in type I PDT and singlet oxygen (1O2) in type II PDT.3–6 Apart from classical porphyrins or other organic-based PSs, transition metal-based PDT PSs, such as Ru(II) or Os(II) polypyridyl complexes, among others, have been extensively studied in the last few years due to their high stability, low photobleaching rate and high 1O2 production.7–16 For example, the metal-based PSs Photosens (Al-phthalocyanines) and Tookad (padeliporfin) have successively been approved for clinical use and the Ru(II)-based PS TLD1433 has entered phase II clinical trials against bladder cancer (see Fig. 1).17 We note that, compared to Ru(II) polypyridyl complexes, Os(II) polypyridyl complexes typically exhibit absorption maxima in the near-infrared region, which is closer to the most favorable photodynamic therapeutic window if deep-seated or large tumors are targeted.7,18–21
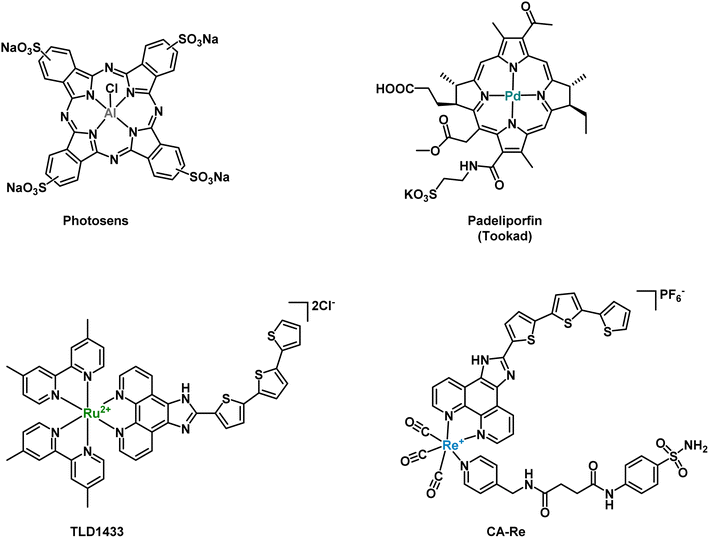 |
| | Fig. 1 Metal-based PDT PSs, which have been approved or are currently in clinical trials, and rhenium-CA inhibitor based PDT PS developed by Mao et al.48 | |
However, the common Ru(II) or Os(II) polypyridine complexes reported so far in the literature are not inherently selective for cancer cells.22 To overcome these limitations, metal complexes have been combined with some cancer cell-selective biomolecules (e.g., peptides, aptamers, and antibodies).22–30 It is also noteworthy that the tumor microenvironment (TME) is under hypoxic conditions due to the uncontrolled growth of tumor cells that consume a massive quantity of O2, reducing the efficacy of PDT treatments.31,32 Therefore, it is of great interest to discover PSs that can equally or more effectively work against the hypoxic TME to improve the therapeutic potency of PDT.
Carbonic anhydrases (CAs) are a group of metallo-enzymes, which catalyze, in a reversible manner, carbon dioxide and water to bicarbonate and a proton.33 This simple chemical reaction is widely distributed in various living organisms and has important implications for physiological activities, such as ion exchange, CO2 exchange and pH balance. There are 16 known isoforms of CAs in human, which play different roles in tissue distribution, cell localization and kinetics.34 As a representative of the cytosolic form, CAII mainly exists in red blood cells and plays a crucial role in maintaining physiological blood pH and metabolism, and is considered as a drug target for various diseases.35–37 CAIX is a transmembrane protein directly overexpressed in the presence of hypoxia or transcription factors (HIF-1). Therefore, it has been recognized as an important marker of hypoxia.38,39 In addition, CAs can regulate the pH of the plasma membrane by promoting ion transport, thereby preventing the accumulation of intracellular acidity leading to toxicity. CAIX inhibitors have shown promise as potential candidates for targeted hypoxia solid antitumor therapy.40,41 Two main classes of CAIX inhibitors have been investigated: sulfonamides and coumarin compounds. Some of them are currently in clinical trials (SLC-0111 and DTP-348).42–48 For example, Alberto and coworkers reported on rhenium- and technetium-based human carbonic anhydrase IX inhibitors for therapy and imaging (theranostic).40 Recently, Mao and co-workers designed a carbonic anhydrase IX (CAIX)-anchored rhenium(I) PS (CA-Re, Fig. 1), which was localized on cell membranes and demonstrated nanomolar phototoxicity under normoxic and hypoxic (2.5 times decrease vs. normoxia) conditions at 425 nm and 525 nm (ca. 10 times decrease vs. 425 nm).49
In addition, Ward and coworkers have extensively explored the addition of metal-based cofactors in human carbonic anhydrase (hCAII) for catalysis.35,50–54 Therefore, inspired by their pioneering work on the utilization of surface-displayed hCA to accumulate sulfonamide-bearing cofactors,55 we set out to merge CA-targeting, normoxic anti-tumoral activity and a higher absorption wavelength by using metal complexes. In this spirit, we designed and evaluated four CA-inhibitors anchored Ru(II)/Os(II) PSs. These PSs, based on sulfonamide ligands, demonstrated phototoxicity at specific wavelengths on MDA-MB-231 and A549 cells, both under normoxic and hypoxic conditions. Notably, by transitioning from Ru to Os while maintaining the same ligand framework, we achieved a significant red-shift in the absorption spectrum, enabling near-infrared PDT (NIR-PDT) applications. The resulting Os(II) compounds maintained their cytotoxicity at a wavelength of 740 nm.
Results and discussion
Synthesis and characterization
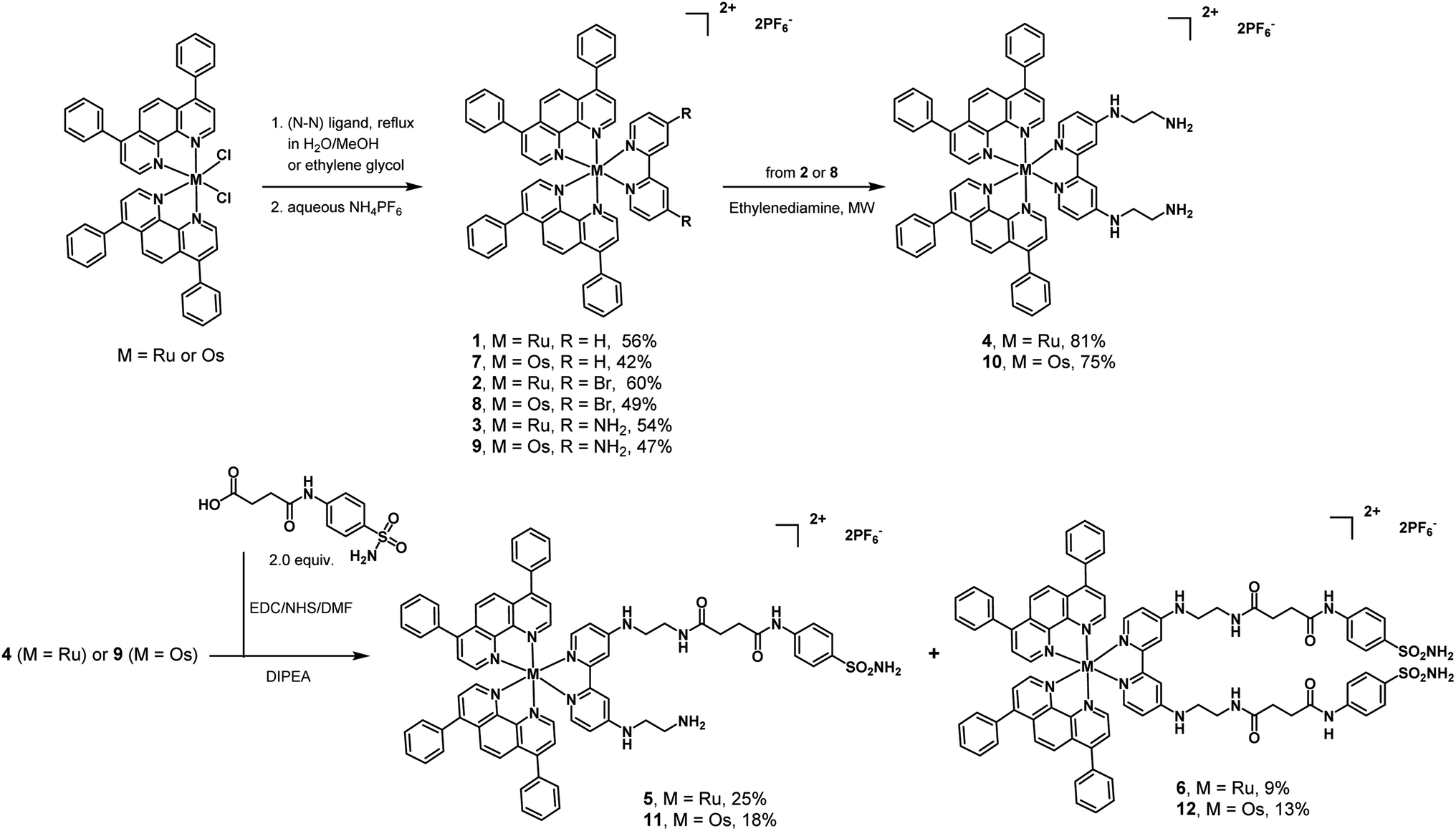
Ruthenium complexes 1–3 (Scheme 1) were synthesized according to previous reports.16,56–58 Slight modifications were undertaken. Ru(Bphen)2Cl2 (Bphen = 4,7-diphenyl-1,10-phenanthroline) was refluxed with various substituted bipyridine ligands (R1/R2 = H, Br, NH2) in a mixture of CH3OH/H2O (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v) and purified using silica gel chromatography. The preparation of complex 4 involved the reaction of complex 2 and ethylenediamine under microwave heating. Complex 4 was then coupled with N4-succinoylsulfanilamide, which possesses the targeting sulfonamide group found in CAIX inhibitors. The carboxylic acid was activated as an N-hydroxysuccinimide and reacted with 4. As a result, from the same reaction, two complexes were obtained and isolated: the Ru(II)-monosulfonamide complex 5 and the Ru(II)-disulfonamide complex 6. The same successful strategies were employed to synthesize the Os(II) polypyridine complexes 7–10, Os(II)-monosulfonamide complex 11 and Os(II)-disulfonamide complex 12. All complexes were characterized by 1H and 13C NMR and HR-MS (Fig. S1–S36†) and their purity was confirmed by HPLC (Fig. S37 and S38†). It is worth noting that all octahedral complexes studied are isolated as a racemic mixture of the Λ and Δ enantiomers. In the case of 5 and 11 that bear a non-symmetrical bi-pyridine ligand, this leads to the formation of a mixture of diastereoisomers. To ensure better solubility of complexes in biological fluids, the cell-based experiments of the complexes were performed with the chloride counter-anion, using an Amberlite IRA-410 to exchange the hexafluorophosphate anion, and the replacements were confirmed by 19F NMR (Fig. S39–S42†).
:1, v/v) and purified using silica gel chromatography. The preparation of complex 4 involved the reaction of complex 2 and ethylenediamine under microwave heating. Complex 4 was then coupled with N4-succinoylsulfanilamide, which possesses the targeting sulfonamide group found in CAIX inhibitors. The carboxylic acid was activated as an N-hydroxysuccinimide and reacted with 4. As a result, from the same reaction, two complexes were obtained and isolated: the Ru(II)-monosulfonamide complex 5 and the Ru(II)-disulfonamide complex 6. The same successful strategies were employed to synthesize the Os(II) polypyridine complexes 7–10, Os(II)-monosulfonamide complex 11 and Os(II)-disulfonamide complex 12. All complexes were characterized by 1H and 13C NMR and HR-MS (Fig. S1–S36†) and their purity was confirmed by HPLC (Fig. S37 and S38†). It is worth noting that all octahedral complexes studied are isolated as a racemic mixture of the Λ and Δ enantiomers. In the case of 5 and 11 that bear a non-symmetrical bi-pyridine ligand, this leads to the formation of a mixture of diastereoisomers. To ensure better solubility of complexes in biological fluids, the cell-based experiments of the complexes were performed with the chloride counter-anion, using an Amberlite IRA-410 to exchange the hexafluorophosphate anion, and the replacements were confirmed by 19F NMR (Fig. S39–S42†).
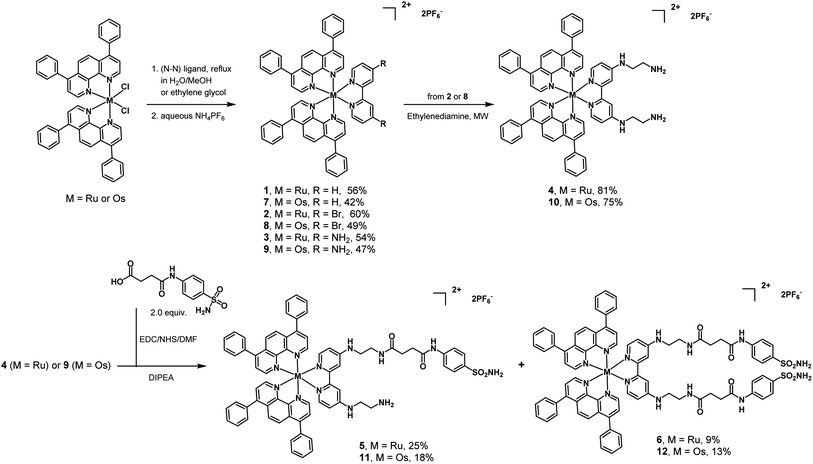 |
| | Scheme 1 Synthetic route and molecular structures of the complexes studied in this work. All complexes were isolated as PF6 salts. Biological experiments of the complexes were performed with the chloride counter-anion. | |
Spectroscopic properties and singlet oxygen production
To better understand the effect of various bipyridine substituents and metal ions on the photophysical properties of the complexes, their UV-Vis absorption spectra and emission spectra were recorded in acetonitrile (MeCN). As displayed in Table 1, all complexes exhibit typical ligand-centered (LC) transition absorption around 280 nm, while metal-to-ligand charge transfer (1MLCT) absorption was observed in the visible region (400–650 nm). Similar spectra were obtained in H2O (Fig. S43†). The presence of different substituent groups on the pyridine ligand affects the absorption bands of the complexes. Replacing hydrogen with bromine causes a red shift in the visible region, and changing the R group to an amino group further enhances this shift. Although the substitution of bromine and amino groups also shifts the emission band to longer wavelengths, it leads to a noticeable decrease in luminescence intensity. The sulfonamide-based osmium complexes 11 and 12 did not present any luminescence, while the ruthenium complexes 5 and 6 presented very low luminescence with a quantum yield of 2%. In addition, a broad absorption band (600–800 nm) was observed for the Os(II) polypyridine complexes owing to the spin-forbidden absorption of the 3MLCT state.18,59
Table 1 Spectroscopic properties and 1O2 quantum yields in MeCNa
|
|
Spectroscopic properties |
1O2 quantum yield |
|
λ
abs/nm (ε/M−1 cm−1 × 10−3) |
λ
em/nm |
Φ
em/% |
τ/ns |
Φ
Δ/% |
|
λ
abs absorption maximum, λem emission maximum, Φem luminescence quantum yield, τ fluorescence lifetime, ΦΔ1O2 quantum yield at 450 nm, and n.d. not detectable.
|
|
5
|
280(66.4), 452(17.9), 495(16.3) |
674 |
2 |
122 |
77 |
|
6
|
280(66.4), 452(16.0), 505(14.8) |
675 |
2 |
117 |
77 |
|
11
|
280(48.7), 463(11.9), 531(12.0) |
n.d. |
0 |
n.d. |
11 |
|
12
|
278(51.4), 463(11.9), 528 (11.9) |
n.d. |
0 |
n.d. |
10 |
| Ru(bpy)3 |
— |
608 |
8 |
159 |
77 |
The ability of the sulfonamide complexes to produce 1O2 upon irradiation at 450 nm in MeCN was evaluated by direct observation of 1O2 phosphorescence at about 1270 nm using [Ru(bpy)3]Cl2 as a reference. As shown in Table 1, the number of sulfonamide moieties had almost no effect on the production of 1O2, in contrast to the metal center that played an important role in the ability to produce 1O2. Overall, for the Ru(II) compounds, the highest 1O2 quantum yield was 77% (5/6), compared to ca. 10% for the Os(II) compounds (11/12). This observation can be explained by the 3MLCT lifetimes of Os(II) compounds that are shorter than for the corresponding Ru(II) compounds, which is consistent with the literature.60
Hydrophobicity and dispersion
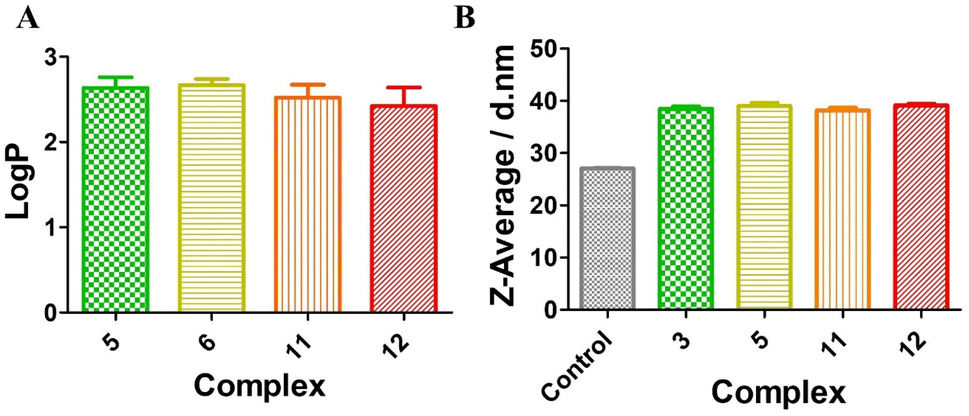
Lipophilicity plays an important role in the cellular uptake, which can be evaluated with the organic/water partition coefficient (expressed as logP).61,62 PBS buffer was used here instead of water to reduce the effect of pH on the logP value. As shown in Fig. 2A, for the Ru(II)-sulfonamide complexes 5 and 6 or the Os(II)-sulfonamide complexes 11 and 12, their logP values were greater than 1, indicating that all complexes are lipophilic. However, as shown in Fig. S44 and S45,† both Ru(II)-sulfonamide complexes 5 and 6 and Os(II)-sulfonamide complexes 11 and 12 have a good solubility in water. Compared with the complexes having PF6− as the counteranion, the corresponding complexes with Cl− as the counteranion generally have better solubility in water. However, we note that when these compounds were dispersed in the cell environment PBS supplemented with 10% FBS, the hydrodynamic size of complexes increased from 27.2 nm (control: 10% FBS in PBS without compounds) to 37 nm, indicating that they formed nanoparticles after binding with serum albumin proteins (Fig. 2B and S46†), as previously shown with other Ru(II) polypyridine complexes.63 The fluorescence quantum yields of the complexes were significantly higher in the aggregated state (10% FBS in PBS, 1% DMSO) than in the solution state (PBS, 1% DMSO) (see Fig. S57†). In order to gain a deeper understanding of the photophysical properties upon irradiation, a series of photobleaching experiments were conducted using 10% FBS in PBS as the solvent. The results detailed in Fig. S55† revealed that all complexes demonstrated a relatively stable behavior when exposed to 540 nm irradiation for up to 40 min, which corresponds to the duration of the irradiation for photo-toxicity experiments at this wavelength.
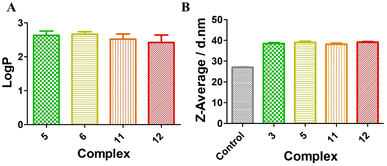 |
| | Fig. 2 (A) Octanol/PBS partition coefficients of the Ru(II)-sulfonamide complexes 5 and 6 and Os(II)-sulfonamide complexes 11 and 12. All complexes were performed as PF6− salts; (B) dynamic light scattering data: Z-average of particle size distribution by the intensity of complexes 5 and 6 and 11 and 12 (10 μM) in 10% FBS in PBS, and the control group is 10% FBS in PBS. | |
Competitive binding affinity for the inhibition of hCA II
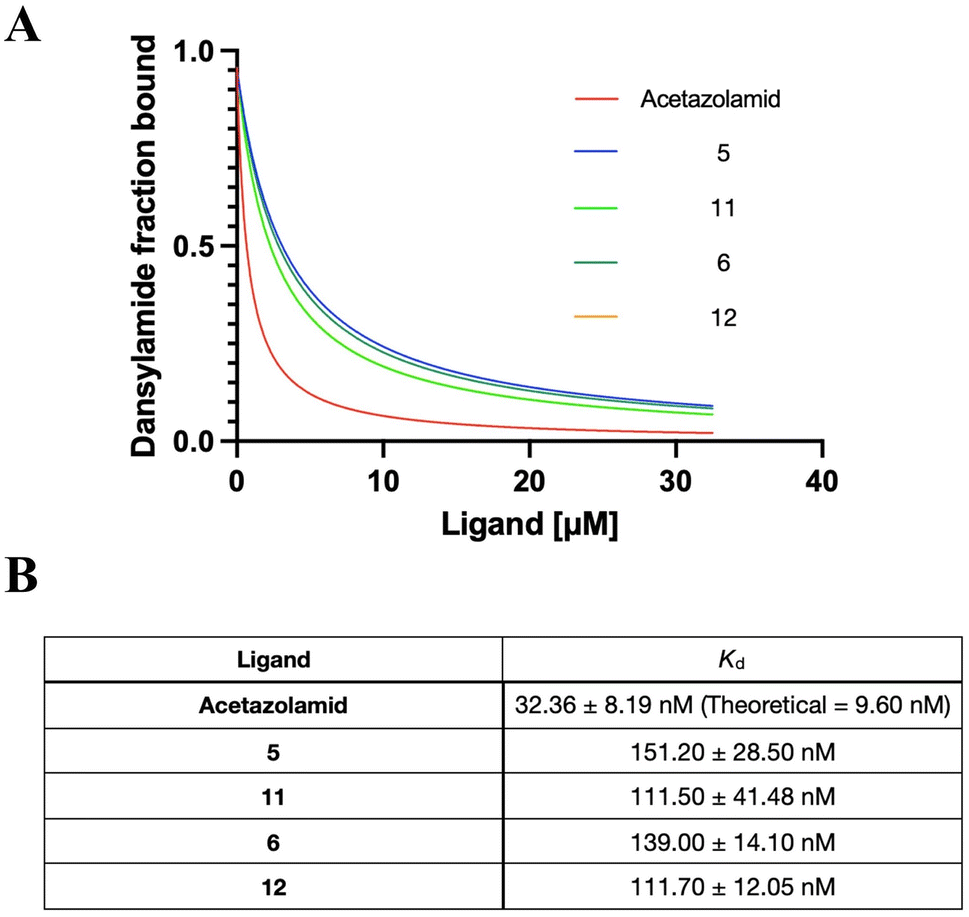
Sulfonamide derivatives have been reported as effective CA inhibitors, with dissociation constants ranging from micromolar to nanomolar concentrations.64–66 Since hCAII is widely used in mechanistic and high-resolution structure studies and easier to obtain from commercial sources than other forms of CAs, hCAII was chosen for the binding affinity studies.67,68 Acetazolamide was used as a control since it is a clinically approved CA inhibitor.69 Competitive binding affinity for the inhibition of hCAII was determined using the fluorophore Dansylamide (DNSA), which increases fluorescence emission intensity through Förster resonance energy transfer (FRET) with tryptophan residues near the active site when it binds to CAII. However, the presence of sulfonamide complexes would cause the dissociation of DNSA, which leads to a decrease in fluorescence. The monitoring of DNSA dissociation can therefore characterize the dissociation of a compound from hCA II, providing insight into its behavior and therapeutic potential.52 As shown in Fig. 3 and S47,† both the ruthenium-based (5 and 6) and osmium-based complexes (11 and 12) bind to hCAII effectively with dissociation constants (Kd) of 151.2 nM, 139.0 nM, 111.5 nM and 111.7 nM, respectively. This demonstrated the excellent hCAII-binding ability of our metal sulfonamide complex. Of note, the Os sulfonamide complex exhibits slightly stronger inhibition compared to the Ru sulfonamide complex and the bis-sulfonamide substitution does not appear to affect the binding. We speculate that this may be due to steric hindrance, which would prevent binding to a second CA once one binding is operative as the two sulfonamides lay in relatively close vicinity.
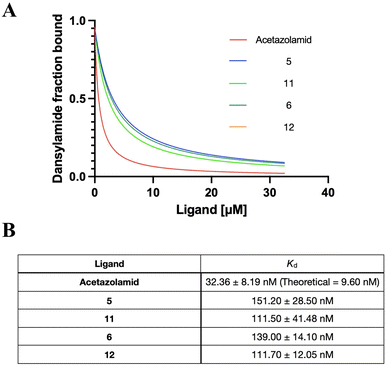 |
| | Fig. 3 (A) Competitive binding affinity for the inhibition of hCAII. The equilibrium dissociation constants for ligands were determined by competitive binding with DNSA. Fixed concentration 20 μM DNSA and 0.25 μM hCA II Wt (180 μL) were titrated against complexes from 0–32.5 μM (5–15 μL). All the titration experiments were performed in triplicate. (B) Summary of the dissociations constants. | |
The overexpression of CA in different cell lines
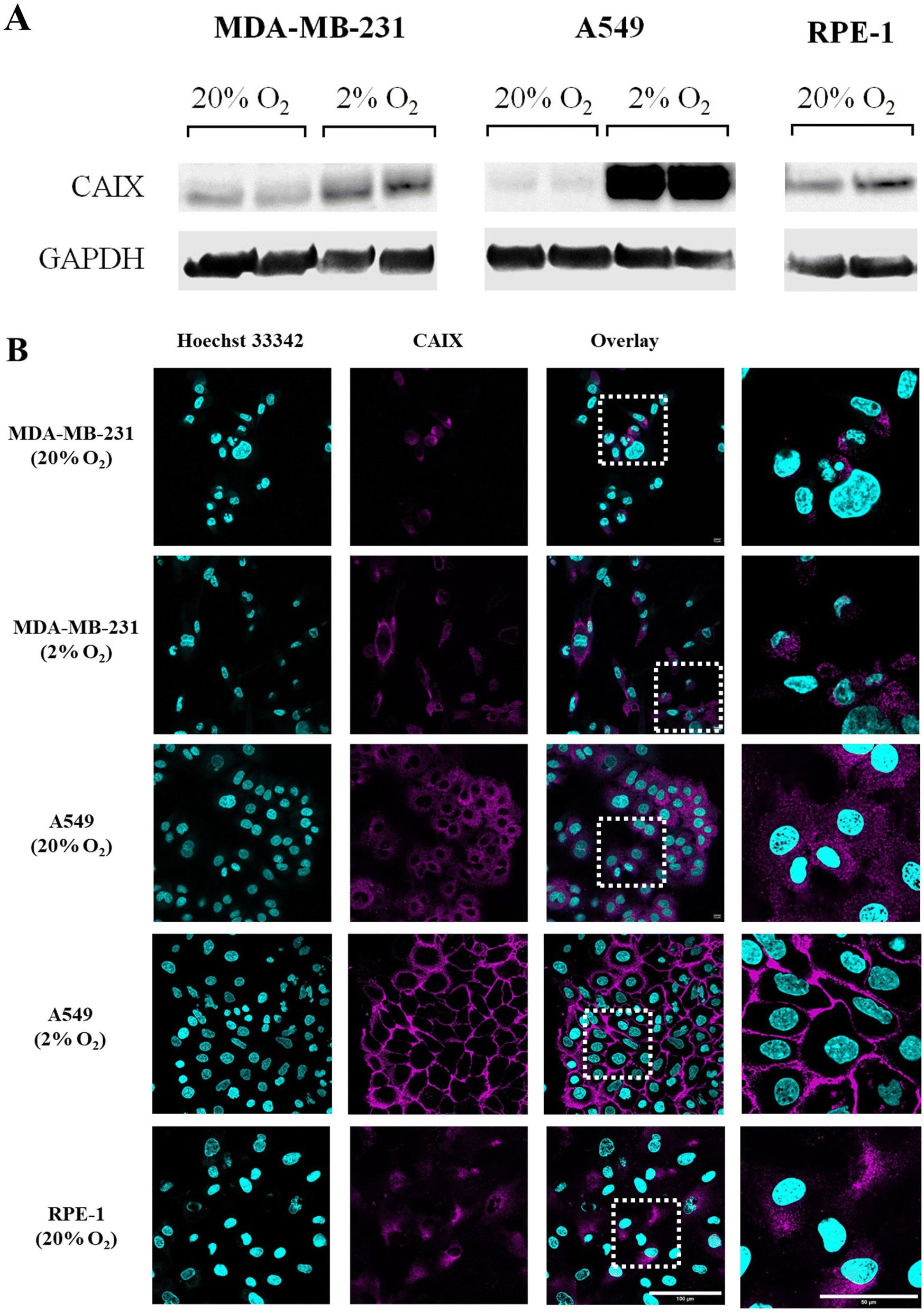
The endogenous expression of CAIX in different kinds of cell lines was studied by western blotting. GAPDH (glyceraldehyde-3-phosphate dehydrogenase) was used as an internal reference to assess whether similar amounts of proteins were loaded in each lane. As shown in Fig. 4A and S56,† the basal (20% O2) expression of CAIX was similar in tumor cells (MDA-MB-231 and A549) and non-tumor cells (RPE-1). Moreover, according to previous reports,48 it was increased by hypoxia (48 h at 2% O2) in both tumor cell lines. Of note, CAIX upregulation by hypoxia was visibly much stronger in A549 cells than in MD-MB-231. These results were confirmed by CAIX immunostaining and confocal imaging, which also allowed assessing the subcellular localization of the protein (see Fig. 4B). In RPE-1 cells, normoxic or hypoxic MDA-MB-231 cells and normoxic A549 cells, we observed a faint intracellular signal. Considering that CAIX is a transmembrane protein, this signal likely corresponds to an intracellular pool of the protein localized in the endoplasmic reticulum and Golgi apparatus. By contrast, hypoxic A549 cells displayed a strong signal localized at the plasma membrane (Fig. 4B and S53† for the staining specificity control). Therefore, A549 cells were selected as a positive cell line towards CAIX, while MDA-MB-231 cells and RPE-1 cells were used as negative cell lines.
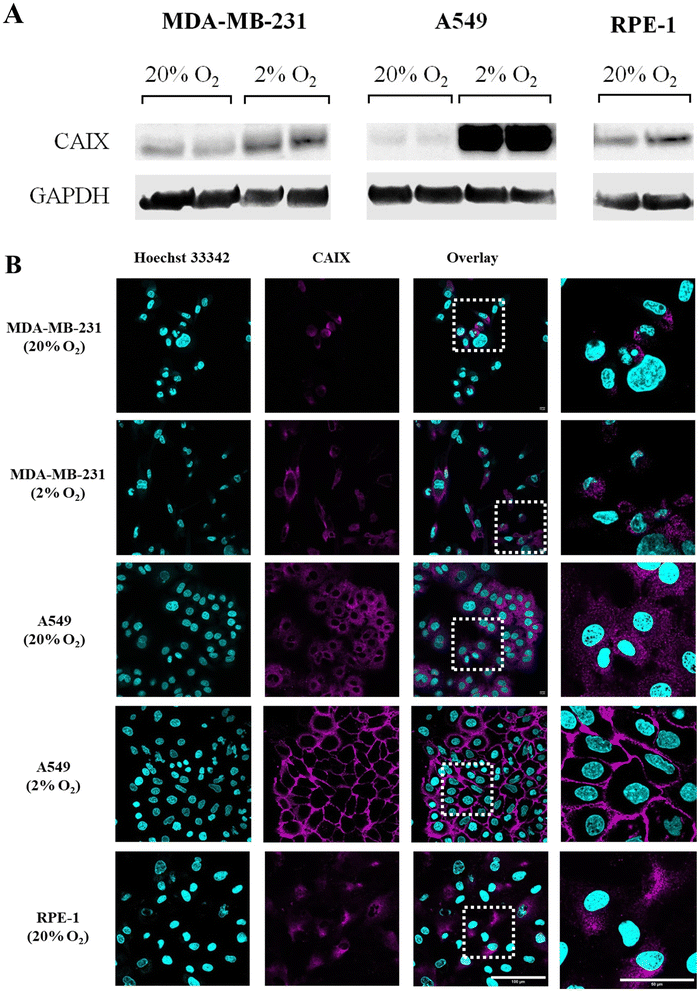 |
| | Fig. 4 Endogenous expression of CAIX in cancerous (MDA-MB-231, A549) and non-cancerous (RPE-1) cell lines. (A) Western blot analysis. GAPDH is used as a loading control. (B) Confocal microscopy images of A549, MDA-MB-231 and RPE1 cells immunostained for CAIX (magenta) and labelled with the Hoechst 33342 nuclear stain (cyan). In each lane, the right panel shows a high magnification image of the boxed area. | |
(Photo-)toxicity
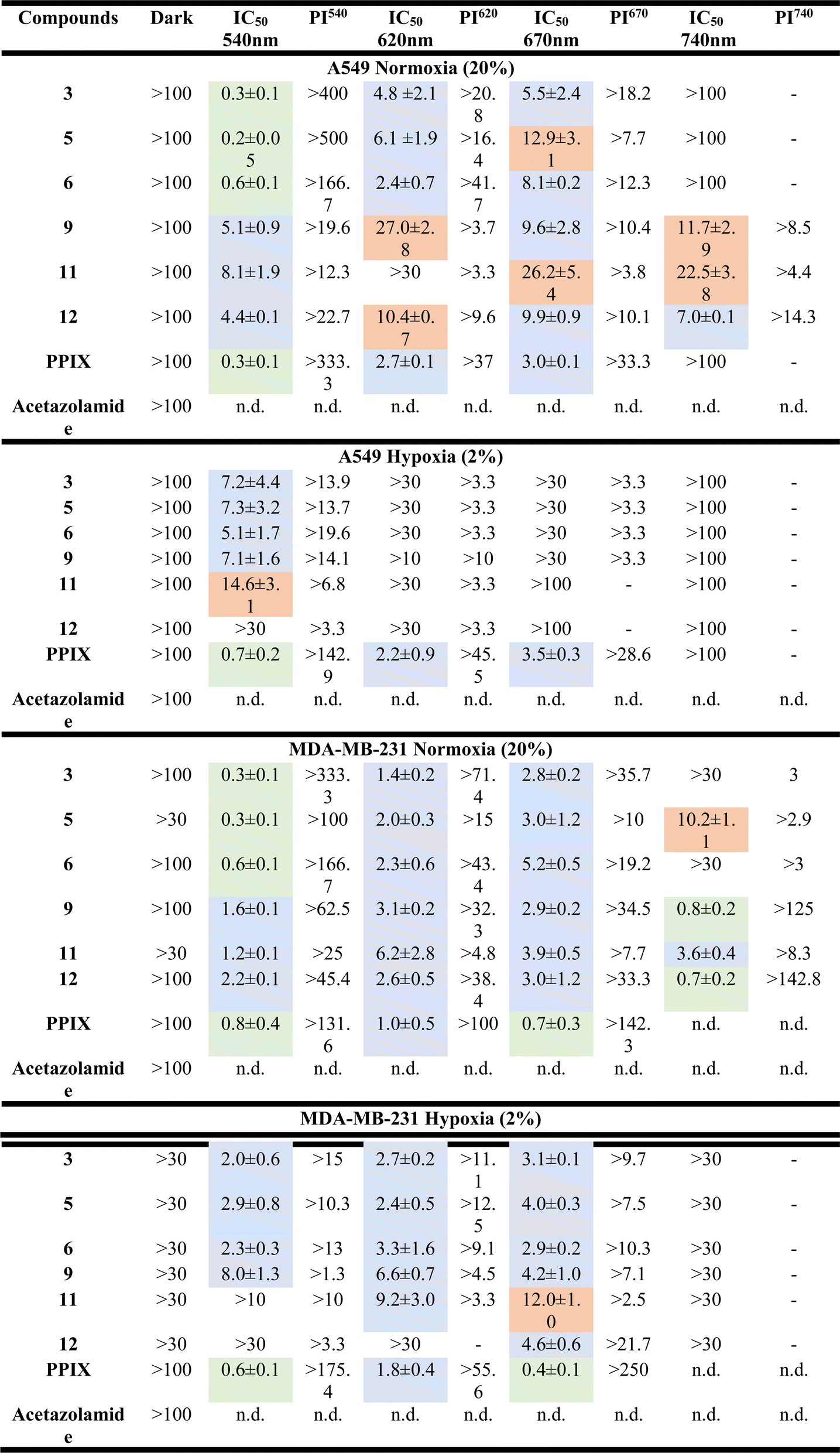
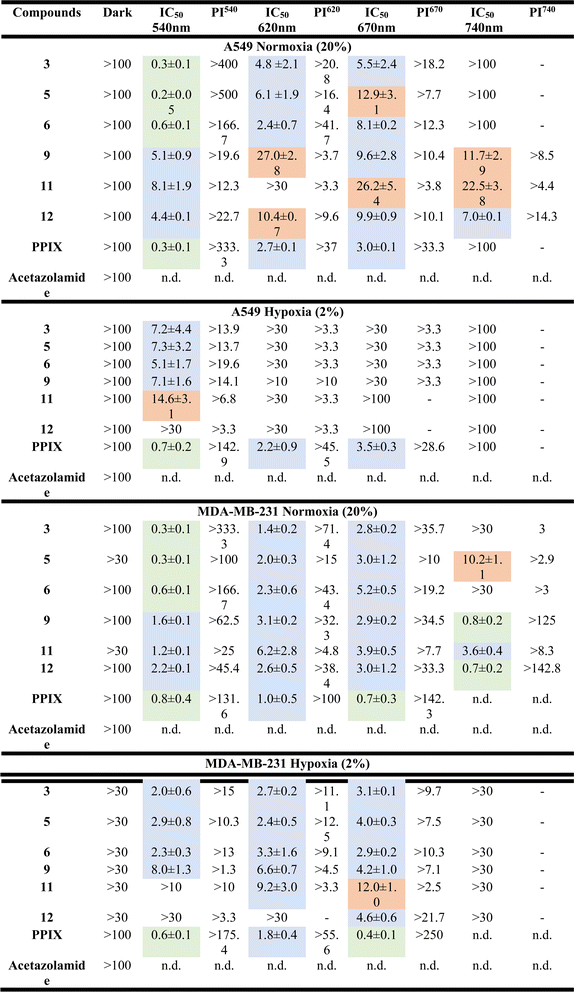
The cytotoxic activities of the metal-based sulfonamide compounds (5, 6, 11 and 12) towards cancerous MDA-MB-231 and A549 cell lines and the non-cancerous RPE1 cell line were investigated using a fluorometric cell viability assay (Resazurin).70 Complexes 3 and 9 were used as control complexes, and since they do not bear a sulfonamide group, along with the clinically approved PDT PS, protoporphyrin IX (PPIX) and acetazolamide, an approved CA inhibitor, were used for comparison purposes.71,72 The phototoxicity of these compounds was assessed using standard procedures with irradiation at 540 nm for 40 min (light dose 9.0 J cm−2), 620 nm for 60 min (light dose 6.7 J cm−2), 670 nm for 60 min (light dose 13.5 J cm−2) and 740 nm for 60 min (light dose 12.6 J cm−2), respectively. To determine the dark cytotoxicity and evaluate the phototoxicity index (PI), the cells were treated identically and incubated in the dark. As shown in Table 2 and S48–S51,† all compounds did not show any cytotoxicity in the dark under both normoxic and hypoxic conditions (IC50 values >100 μM). Upon light irradiation at 540 nm, all metal-based sulfonamide compounds (5, 6, 11 and 12) and sulfonamide-free compounds (3 and 9, R = NH2) exhibited a high cytotoxicity level within the low micromolar range under normoxic and even under hypoxic conditions. Due to the nature of PSs, which induce toxicity by reacting with oxygen, IC50 values under hypoxic conditions were significantly higher than those under normoxic conditions. While Ru(II)-based compounds (3, 5, and 6) could induce phototoxicity upon irradiation up to 670 nm wavelength only, Os(II)-based compound (9, 11, and 12) still proved phototoxic upon irradiation at 740 nm. Similar results were obtained for MDA-MB-231 cells and there was no dark toxicity towards non-cancerous RPE-1 cells (see Table 3 and S52†). In view of these results, complexes 6 and 12 were selected as lead compounds for further study.
Table 2 (Photo-)toxicity (IC50, μM) of the tested compounds against cancerous A549 cells and MDA-MB-231 cells for 4 h in the absence and presence of 540 nm (9.0 J cm−2, 40 min), 620 nm (6.7 J cm−2, 60 min), 670 nm (13.5 J cm−2, 60 min) or 740 nm (12.6 J cm−2, 60 min) irradiationa,b
|
PPIX stands for protoporphyrin; PI stands for the phototoxicity index at the mentioned wavelengths in comparison with dark toxicity.
Note: the value below 1 μM in green, between 1 and 10 μM in blue and between 10 and 29 μM in orange, and the rest in white.
|
|
Table 3 Cytotoxicity (IC50, μM) of the tested compounds against non-cancerous RPE-1 cells in the dark as a control
| RPE-1 normoxia (20%) |
| Compounds |
3
|
5
|
6
|
9
|
11
|
12
|
PPIX |
Acetazolamide |
| Dark |
>100 |
>100 |
>100 |
>30 |
>100 |
>100 |
>100 |
>100 |
Cellular uptake and localization studies
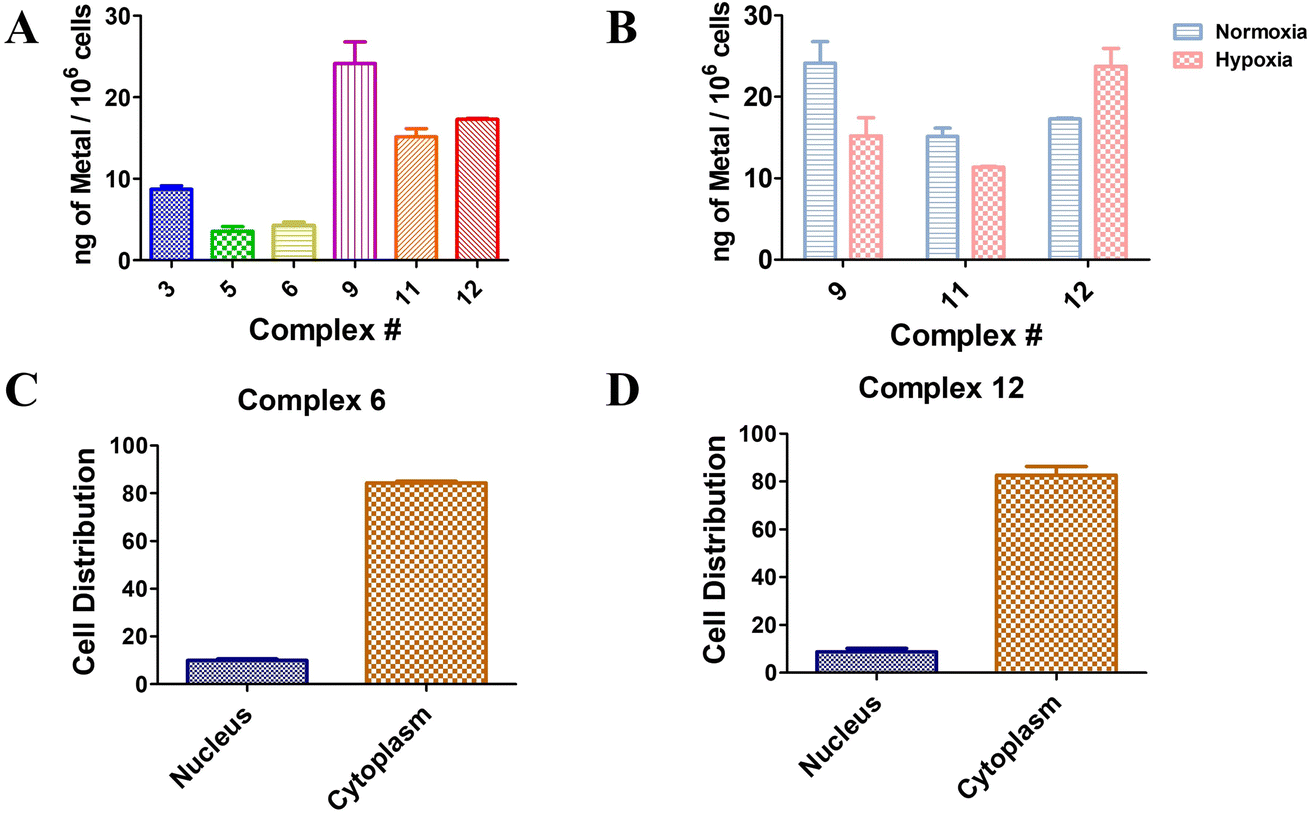
The cellular uptake capacity of a target small molecule is an essential aspect to obtain the expected PDT effect. Herein, 5 μM of Ru(II) complexes 3, 5, and 6 and Os(II) complexes 9, 11, and 12 were co-incubated with A549 cells for 4 h, respectively, and the cellular uptake of the complexes in cells was evaluated by inductively coupled plasma mass spectrometry (ICP-MS) based on the concentration of Ru(II) or Os(II). As shown in Fig. 5A, Ru(II) sulfonamide complexes 5 and 6 were taken up in a comparable manner. In contrast, complex 3, which does not bear a sulfonamide moiety, appeared to accumulate 2 times more than Ru(II) sulfonamide-containing complexes 5 and 6. Notably, Os(II) complexes were 2.5 times better taken up by the cells compared to Ru(II) complexes with the same ligands. The Os(II) complexes exhibited comparable results, with sulfonamide complexes 11 and 12 demonstrating similar uptake properties. Again, complex 9, which lacks the sulfonamide moiety, exhibited 1.5 times higher accumulation compared to the other compounds. Although the sulfonamide compounds displayed a lower uptake rate compared to the control complexes 3 and 9, their phototoxicity levels were found to be similar. Moreover, it was observed that the disubstituted sulfonamide exhibited a slightly higher uptake than the monosubstituted sulfonamide compounds, suggesting a positive association with enhanced phototoxicity. Os(II) complexes 9, 11 and 12 were chosen for further investigation under hypoxic conditions. As depicted in Fig. 5B, sulfonamide-free complex 9 exhibited notably reduced cellular uptake under hypoxic conditions compared to normoxic conditions. The mono-sulfonamide complex 11 has a lower cellular uptake compared to complex 9 under normoxic conditions, but the decrease of uptake under hypoxic conditions is somewhat mitigated. In contrast, the cellular uptake of the di-sulfonamide complex 12 in a hypoxic environment was found to be 1.3 times higher than under normoxic conditions. This implied that complexes bearing sulfonamide groups have an increased cellular uptake capacity in hypoxic environments. This correlates, although it is too early to establish a clear causality, with the overexpression of CA under hypoxic conditions. However, it should be noted that the relationship between the structure and toxicity has not been fully established at this stage. Further research is required to better understand the precise connection between the structure, cell penetration and (photo)toxic effects.
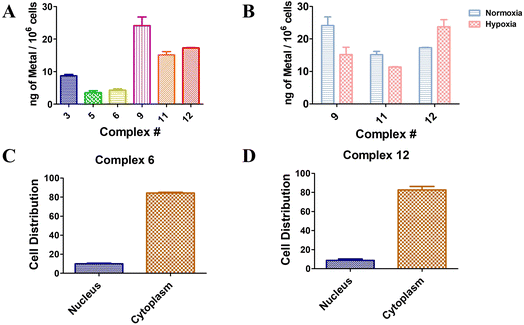 |
| | Fig. 5 (A) Cellular uptake of Ru(II) complexes 3, 5, and 6 and Os(II) complexes 9, 11, and 12 (5 μM and 4 h) in A549 cells determined by ICP-MS; (B) cellular uptake of Os(II) complexes 9, 11, and 12 (5 μM and 4 h) in A549 cells determined by ICP-MS under normoxic conditions and hypoxic conditions; (C) cellular fraction of complex 6 in different parts of A549 cells; (D) cellular fraction of complex 12 in different parts of A549 cells. All the experiments were performed in triplicate. | |
To investigate the subcellular localization of disubstituted sulfonamide complexes 6 and 12, which were co-incubated with cells for 24 h, the nuclei and cytoplasm were isolated, and the contents were analyzed using ICP-MS. The results, depicted in Fig. 5C and D, revealed a comparable pattern of cellular localization for both complexes. Remarkably, over 80% of the compounds were found to be localized within the cytoplasm.
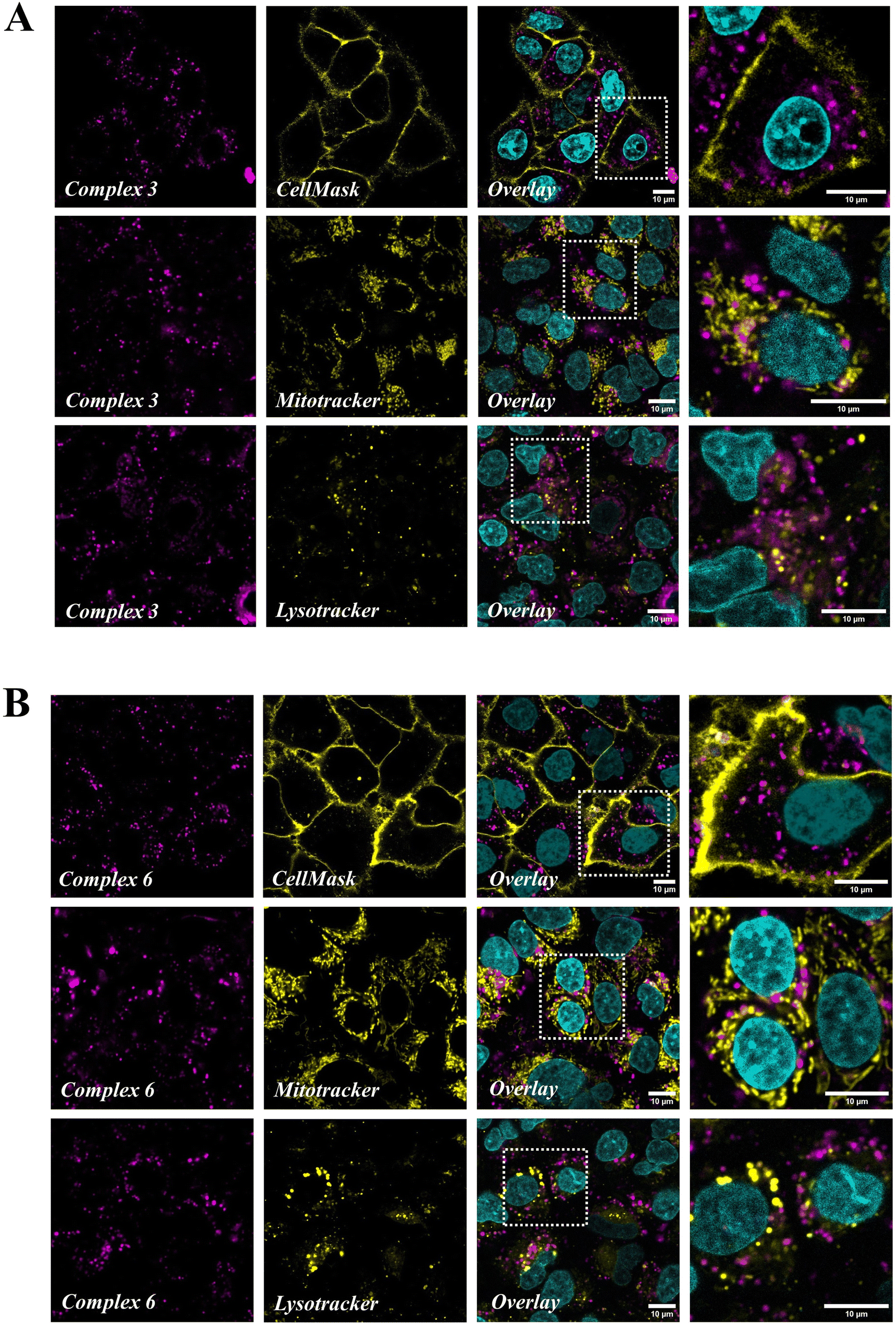
Confocal microscopy was used to get further insight into the subcellular localization of the internalized complexes, using the intrinsic luminescence of Ru(II) compounds. Green CellMask, MitoTracker Green (MTG) and LysoTracker Green (LTG) were used to specifically label the plasma membrane, mitochondria and lysosomes respectively, while Hoechst 33342 allowed nuclear staining. There was no crosstalk between acquisition channels as assessed by single staining experiments (Fig. S54†). Both complexes 3 (Fig. 6A) and 6 (Fig. 6B) were visualized as an intracellular dot pattern except in the nucleus. In high magnification images (see the enlargement of boxed areas in Fig. 6), complexes (magenta) and MTG or LTG (yellow) staining patterns were clearly distinct, indicating that the complexes do not significantly accumulate in mitochondria or lysosomes. This was confirmed by colocalization analysis using the coloc2 plugin in Image J, which yielded Pearson colocalization coefficients (PCC) with a MTG and LTG of respectively −0.33 and −0.11 with complex 3, and −0.24 and −0.08 with complex 6. Similarly, no colocalization of complexes 3 and 6 was found with Hoechst (PCC −0.08 and −0.06 respectively) and CellMask (PCC −0.25 and −0.27), indicating that they do not accumulate in the nucleus or plasma membrane. Altogether confocal imaging shows that complexes 3 and 6 are internalized in A549 cells where they accumulate as free cytoplasmic aggregates, possibly in the endoplasmic reticulum, the Golgi apparatus or other vesicles (excluding mitochondria and lysosomes).
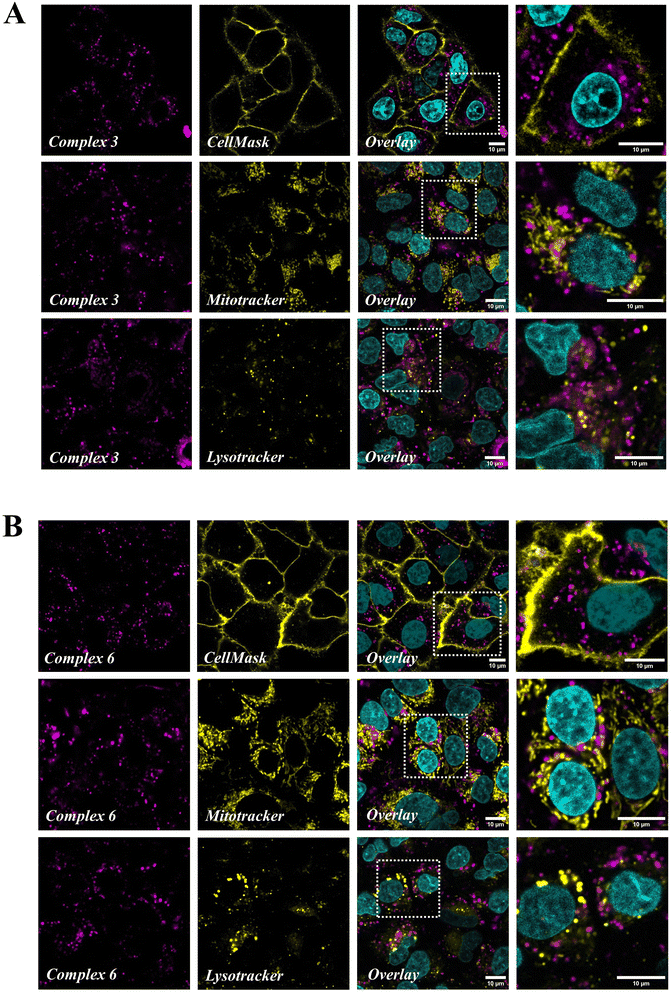 |
| | Fig. 6 Confocal microscopy images of A549 cells incubated with complex 3 (A) (5 μM and 4 h) or complex 6 (B) (10 μM and 4 h) and labelled with the plasma membrane stain Green CellMask (100 nM and 10 min), mitochondria stain MTG (100 nM and 10 min) or lysosome stain LTG (100 nM and 40 min), and nuclear stain Hoechst 33342. Excitation/emission wavelengths are 405/420–450 nm (Hoechst), 488/670–800 nm (complexes 3 and 6), and 488/500–550 nm (green CellMask, Mitotracker Green and Lysotracker Green). In each lane, the right panel shows a high magnification image of the boxed area. The scale bar is 10 μm. | |
Sulfonamide inhibition
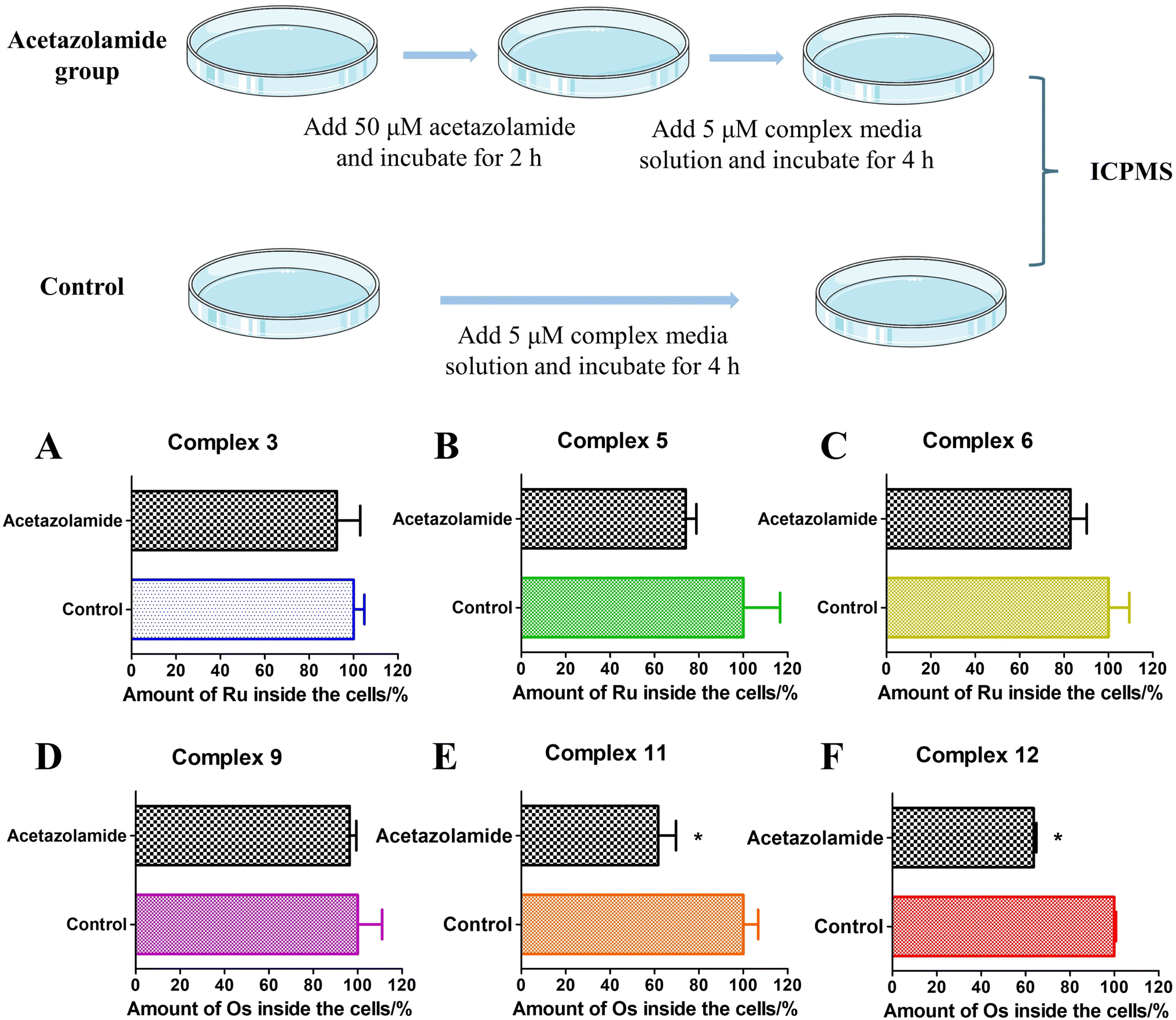
Based on the above experiments, it was demonstrated that sulfonamide-containing compounds have the ability to bind to CAII. The cellular uptake of these complexes in A549 cells under various conditions (in the presence or absence of acetazolamide) was determined using ICP-MS analysis (Fig. 7). For complexes 3 and 9, the impact of acetazolamide pre-incubation was negligible. However, for Ru(II) sulfonamide complexes (5 and 6) or Os(II) sulfonamide complexes (11 and 12), the cellular uptake was notably reduced when A549 cells were pre-incubated with acetazolamide. This indicates that acetazolamide effectively affects cellular uptake capacity. To gain a better understanding and evaluate the statistical significance of this observation, a two-way ANOVA test followed by a Bonferroni post-test was used to calculate the effect of acetazolamide on the cellular uptake of different compounds. Among them, complexes 11 and 12 showed a significant decrease of cellular uptake by acetazolamide pre-treatment, which was demonstrated by the Bonferroni post-test.73 The addition of acetazolamide inhibitors may lead to intracellular CA binding, which thus limits the available binding sites for the metallic complexes, thereby curbing their cellular uptake.
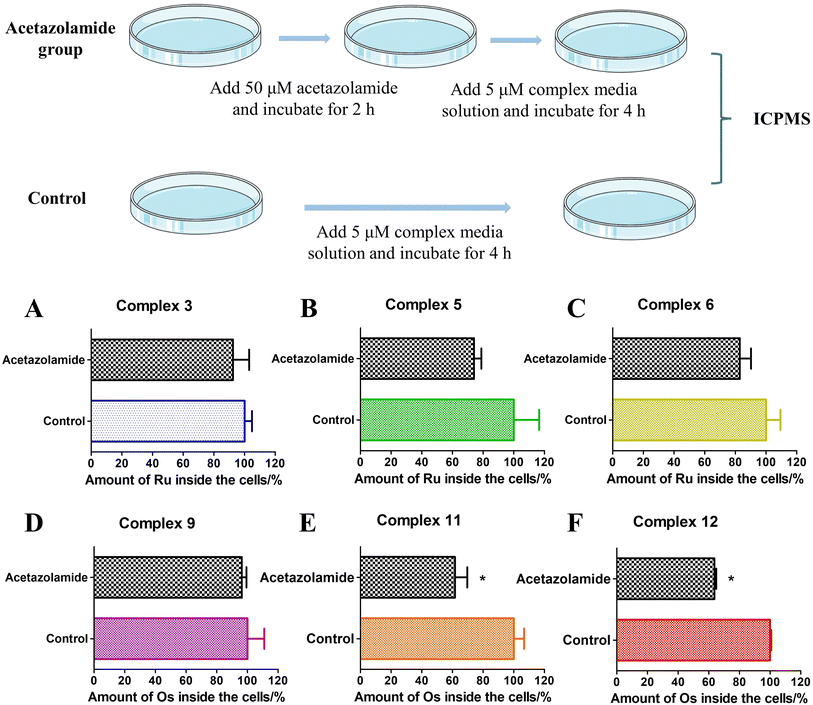 |
| | Fig. 7 Cellular uptake inhibition mechanism study in the A549 cell line of different compounds (5 μM, 1% DMSO, v%, and 4 h) in the presence/absence of the acetazolamide inhibitor (50 μM and 2 h). (A) Complex 3; (B) complex 5; (C) complex 6; (D) complex 9; (E) complex 11; complex 12 (F). “*” represent a p value <0.05 for the Bonferroni post-test. | |
Conclusions
In this work, we mainly designed and synthesized four complexes bearing sulfonamide moieties and explored their photophysical and photochemical properties. The sulfonamide-containing Ru(II) complexes (5 and 6) displayed a 1O2 production quantum yield of 77%, while the absorption spectrum of the sulfonamide-containing Os(II) complexes (11 and 12) exhibited a notable red shift. Furthermore, these complexes have good binding ability to CAII. More importantly, A549 cells were considered as CAIX positive cells and MDA-MB-231 cells as negative cells through western blotting and immunofluorescence experiments. The biological evaluation was performed on these compounds, which exhibited micromolar phototoxicity against the A549 cells under both normoxic and hypoxic conditions, particularly at 540 nm. Notably, as the wavelength increased, the cellular phototoxicity decreased. However, compared with Ru(II) compounds, Os(II) compounds still showed significant phototoxicity upon irradiation at 740 nm under normoxic conditions. These findings demonstrate the potential of sulfonamide-containing Ru(II)/Os(II)-based PSs as an effective drug for PDT. Their ability to induce phototoxicity in the visible and near-infrared regions, as well as under hypoxic conditions, holds promise for targeted cancer therapy. Further refinement and exploration of these compounds may contribute to the development of new therapies that exploit unique features of the tumor microenvironment, such as hypoxia and enzyme overexpression, and red-shifted absorption, to enhance therapeutic efficacy.
Data availability
Data available upon request.
Author contributions
K. C., T. R. and G. G. conceived the research. Y. W. synthesized and characterized the complexes. P. A. and C. F. investigated the photophysical and photochemical properties of the compounds. Y. W., P. M. and B. S. performed confocal microscopy studies. Y. W., P. M. and K. P. performed the cell biology experiments. P. B. performed the ICP-MS measurements. Y. W. wrote the first draft of the manuscript, which was then corrected by all authors. All authors have approved the final version of the manuscript.
Conflicts of interest
The authors declare no conflict of interest.
Acknowledgements
This work was financially supported by an ERC Consolidator Grant PhotoMedMet to G. G. (GA 681679), by the ITMO Cancer AVIESAN (Alliance Nationale pour les Sciences de la Vie et de la Sante/National Alliance for Life Sciences & Health) within the framework of the Cancer Plan (G. G.) and has received support under the program Investissements d’Avenir launched by the French Government and implemented by the ANR with the reference ANR-10-IDEX-0001-02 PSL (G. G.). Y. W. thanks the China Scholarship Council for financial support. We thank Prof. Thomas Ward from the University of Basel for providing us with hCAII.
References
- L. Benov, Med. Princ. Pract., 2015, 24, 14–28 CrossRef PubMed.
- M. Lan, S. Zhao, W. Liu, C. Lee, W. Zhang and P. Wang, Adv. Healthcare Mater., 2019, 8, 1900132 CrossRef PubMed.
- P. Agostinis, K. Berg, K. A. Cengel, T. H. Foster, A. W. Girotti, S. O. Gollnick, S. M. Hahn, M. R. Hamblin, A. Juzeniene, D. Kessel, M. Korbelik, J. Moan, P. Mroz, D. Nowis, J. Piette, B. C. Wilson and J. Golab, Ca-Cancer J. Clin., 2011, 61, 250–281 CrossRef PubMed.
- S. S. Lucky, K. C. Soo and Y. Zhang, Chem. Rev., 2015, 115, 1990–2042 CrossRef CAS PubMed.
- F. Heinemann, J. Karges and G. Gasser, Acc. Chem. Res., 2017, 50, 2727–2736 CrossRef CAS PubMed.
- Y.-Y. Wang, Y.-C. Liu, H. Sun and D.-S. Guo, Coord. Chem. Rev., 2019, 395, 46–62 CrossRef CAS.
- R. Lincoln, L. Kohler, S. Monro, H. Yin, M. Stephenson, R. Zong, A. Chouai, C. Dorsey, R. Hennigar, R. P. Thummel and S. A. McFarland, J. Am. Chem. Soc., 2013, 135, 17161–17175 CrossRef CAS PubMed.
- S. M. Meier-Menches, C. Gerner, W. Berger, C. G. Hartinger and B. K. Keppler, Chem. Soc. Rev., 2018, 47, 909–928 RSC.
- P. Zhang and H. Huang, Dalton Trans., 2018, 47, 14841–14854 RSC.
- J. Shum, P. K.-K. Leung and K. K.-W. Lo, Inorg. Chem., 2019, 58, 2231–2247 CrossRef CAS PubMed.
- P. S. Felder, S. Keller and G. Gasser, Adv. Ther., 2020, 3, 1900139 CrossRef.
- L. M. Lifshits, J. A. Roque III, P. Konda, S. Monro, H. D. Cole, D. von Dohlen, S. Kim, G. Deep, R. P. Thummel, C. G. Cameron, S. Gujar and S. A. McFarland, Chem. Sci., 2020, 11, 11740–11762 RSC.
- J. Li and T. Chen, Coord. Chem. Rev., 2020, 418, 213355 CrossRef CAS.
- A. Gandosio, K. Purkait and G. Gasser, Chimia, 2021, 75, 845 CrossRef PubMed.
- J. Karges, Angew. Chem., Int. Ed., 2022, 61, e202112236 CrossRef CAS PubMed.
- A. Mani, T. Feng, A. Gandioso, R. Vinck, A. Notaro, L. Gourdon, P. Burckel, B. Saubaméa, O. Blacque, K. Cariou, J. Belgaied, H. Chao and G. Gasser, Angew. Chem., 2023, 135, e202218347 CrossRef.
- S. A. McFarland, A. Mandel, R. Dumoulin-White and G. Gasser, Curr. Opin. Chem. Biol., 2020, 56, 23–27 CrossRef CAS PubMed.
- Y. Sun, L. E. Joyce, N. M. Dickson and C. Turro, Chem. Commun., 2010, 46, 6759 RSC.
- P. Zhang, Y. Wang, K. Qiu, Z. Zhao, R. Hu, C. He, Q. Zhang and H. Chao, Chem. Commun., 2017, 53, 12341–12344 RSC.
- S. Lazic, P. Kaspler, G. Shi, S. Monro, T. Sainuddin, S. Forward, K. Kasimova, R. Hennigar, A. Mandel, S. McFarland and L. Lilge, Photochem. Photobiol., 2017, 93, 1248–1258 CrossRef CAS PubMed.
- J. A. Roque, P. C. Barrett, H. D. Cole, L. M. Lifshits, G. Shi, S. Monro, D. von Dohlen, S. Kim, N. Russo, G. Deep, C. G. Cameron, M. E. Alberto and S. A. McFarland, Chem. Sci., 2020, 11, 9784–9806 RSC.
- M. Martínez-Alonso and G. Gasser, Coord. Chem. Rev., 2021, 434, 213736 CrossRef.
- S. M. Meier, M. Novak, W. Kandioller, M. A. Jakupec, V. B. Arion, N. Metzler-Nolte, B. K. Keppler and C. G. Hartinger, Chem.–Eur. J., 2013, 19, 9297–9307 CrossRef CAS PubMed.
- I. Neundorf, Curr. Med. Chem., 2017, 24, 1853–1861 CrossRef CAS PubMed.
- V. del Solar and M. Contel, J. Inorg. Biochem., 2019, 199, 110780 CrossRef CAS PubMed.
- J. Karges, M. Jakubaszek, C. Mari, K. Zarschler, B. Goud, H. Stephan and G. Gasser, ChemBioChem, 2020, 21, 531–542 CrossRef CAS PubMed.
- L. K. McKenzie, M. Flamme, P. S. Felder, J. Karges, F. Bonhomme, A. Gandioso, C. Malosse, G. Gasser and M. Hollenstein, RSC Chem. Biol., 2022, 3, 85–95 RSC.
- M. J. S. A. Silva, R. Vinck, Y. Wang, B. Saubaméa, M. Tharaud, E. Dominguez-Jurado, J. Karges, P. M. P. Gois and G. Gasser, ChemBioChem, 2023, 24, e202200647 CrossRef CAS PubMed.
- E. Bortolamiol, F. Visentin and T. Scattolin, Appl. Sci., 2023, 13, 5561 CrossRef CAS.
- N. E. S. Tay, K. A. Ryu, J. L. Weber, A. K. Olow, D. C. Cabanero, D. R. Reichman, R. C. Oslund, O. O. Fadeyi and T. Rovis, Nat. Chem., 2023, 15(1), 101–109 CrossRef CAS PubMed.
- Y. Jiang, J. Li, Z. Zeng, C. Xie, Y. Lyu and K. Pu, Angew. Chem., Int. Ed., 2019, 58, 8161–8165 CrossRef CAS PubMed.
- M.-Z. Jin and W.-L. Jin, Signal Transduction Targeted Ther., 2020, 5, 166 CrossRef PubMed.
- A. Angeli, F. Carta and C. T. Supuran, Catalysts, 2020, 10, 1008 CrossRef CAS.
- C. T. Supuran, Nat. Rev. Drug Discovery, 2008, 7, 168–181 CrossRef CAS PubMed.
- F. W. Monnard, E. S. Nogueira, T. Heinisch, T. Schirmer and T. R. Ward, Chem. Sci., 2013, 4, 3269 RSC.
- V. M. Krishnamurthy, G. K. Kaufman, A. R. Urbach, I. Gitlin, K. L. Gudiksen, D. B. Weibel and G. M. Whitesides, Chem Rev., 2008, 108(3), 946–1051 CrossRef CAS PubMed.
- S. Kumar, S. Rulhania, S. Jaswal and V. Monga, Eur. J. Med. Chem., 2021, 209, 112923 CrossRef CAS PubMed.
- S. Kaluz, M. Kaluzova and E. J. Stanbridge, Cancer Res., 2004, 64, 978 Search PubMed.
- P. C. McDonald, J.-Y. Winum, C. T. Supuran and S. Dedhar, Oncotarget, 2012, 3, 84–97 CrossRef PubMed.
- D. Can, B. Spingler, P. Schmutz, F. Mendes, P. Raposinho, C. Fernandes, F. Carta, A. Innocenti, I. Santos, C. T. Supuran and R. Alberto, Angew. Chem., Int. Ed. Engl., 2012, 51(14), 3354–3357 CrossRef CAS PubMed.
- S. Angerani and N. Winssinger, J. Am. Chem. Soc., 2020, 142(28), 12333–12340 CrossRef CAS PubMed.
- C. T. Supuran, F. Briganti, S. Tilli, W. R. Chegwidden and A. Scozzafava, Bioorg. Med. Chem., 2001, 9, 703–714 CrossRef CAS PubMed.
- A. Cecchi, A. Hulikova, J. Pastorek, S. Pastoreková, A. Scozzafava, J.-Y. Winum, J.-L. Montero and C. T. Supuran, J. Med. Chem., 2005, 48, 4834–4841 CrossRef CAS PubMed.
- J.-Y. Winum, Expert Opin. Ther. Pat., 2007, 17, 1393–1396 CrossRef CAS.
- L. Dubois, N. G. Lieuwes, A. Maresca, A. Thiry, C. T. Supuran, A. Scozzafava, B. G. Wouters and P. Lambin, Radiother. Oncol., 2009, 92, 423–428 CrossRef CAS PubMed.
- T. Rogez-Florent, S. Meignan, C. Foulon, P. Six, A. Gros, C. Bal-Mahieu, C. T. Supuran, A. Scozzafava, R. Frédérick, B. Masereel, P. Depreux, A. Lansiaux, J.-F. Goossens, S. Gluszok and L. Goossens, Bioorg. Med. Chem., 2013, 21, 1451–1464 CrossRef CAS PubMed.
- S. Carradori, Expert Opin. Ther. Pat., 2013, 23, 751–756 CrossRef CAS PubMed.
- C. T. Supuran and J.-Y. Winum, Future Med. Chem., 2015, 7, 1407–1414 CrossRef CAS PubMed.
- X. Su, W. Wang, Q. Cao, H. Zhang, B. Liu, Y. Ling, X. Zhou and Z. Mao, Angew. Chem., Int. Ed., 2022, 61, e202115800 CrossRef CAS PubMed.
- A. Stein, D. Chen, N. V. Igareta, Y. Cotelle, J. G. Rebelein and T. R. Ward, ACS Cent. Sci., 2021, 7(11), 1874–1884 CrossRef CAS PubMed.
- J. G. Rebelein, Y. Cotelle, B. Garabedian and T. R. Ward, ACS Catal., 2019, 9(5), 4173–4178 CrossRef CAS PubMed.
- J. Zhao, A. Kajetanowicz and T. R. Ward, Org. Biomol. Chem., 2015, 13(20), 5652–5655 RSC.
- F. W. Monnard, T. Heinisch, E. S. Nogueira, T. Schirmer and T. R. Ward, Chem. Commun., 2011, 47(29), 8238–8240 RSC.
- M. Schmid, E. S. Nogueira, F. W. Monnard, M. Meuwly and T. R. Ward, Chem. Sci., 2012, 3(3), 690–700 RSC.
- A. Baiyoumy, J. Vallapurackal, F. Schwizer, T. Heinisch, T. Kardashliev, M. Held, S. Panke and T. R. Ward, ACS Catal., 2021, 11(17), 10705–10712 CrossRef CAS PubMed.
- S. Mehanna, N. Mansour, H. Audi, K. Bodman-Smith, M. A. Mroueh, R. I. Taleb, C. F. Daher and R. S. Khnayzer, RSC Adv., 2019, 9, 17254–17265 RSC.
- A. Notaro, M. Jakubaszek, N. Rotthowe, F. Maschietto, R. Vinck, P. S. Felder, B. Goud, M. Tharaud, I. Ciofini, F. Bedioui, R. F. Winter and G. Gasser, J. Am. Chem. Soc., 2020, 142, 6066–6084 CrossRef CAS PubMed.
- R. Vinck, J. Karges, M. Tharaud, K. Cariou and G. Gasser, Dalton Trans., 2021, 50, 14629–14639 RSC.
- R. E. Holmlin, J. A. Yao and J. K. Barton, Inorg. Chem., 1999, 38, 174–189 CrossRef CAS.
- A. A. Abdel-Shafi, D. R. Worrall and A. Y. Ershov, Dalton Trans., 2004, 30 RSC.
- C. A. Puckett and J. K. Barton, J. Am. Chem. Soc., 2007, 129, 46–47 CrossRef CAS PubMed.
- C. Tan, S. Lai, S. Wu, S. Hu, L. Zhou, Y. Chen, M. Wang, Y. Zhu, W. Lian, W. Peng, L. Ji and A. Xu, J. Med. Chem., 2010, 53, 7613–7624 CrossRef CAS PubMed.
- R. Vinck, A. Gandioso, P. Burckel, B. Saubaméa, K. Cariou and G. Gasser, Inorg. Chem., 2022, 61, 13576–13585 CrossRef CAS PubMed.
- T. Mann and D. Keilin, Nature, 1940, 146, 164–165 CrossRef CAS.
- S. K. Nair, J. F. Krebs, D. W. Christianson and C. A. Fierke, Biochemistry, 1995, 34, 3981–3989 CrossRef CAS PubMed.
- P. Koutnik, E. G. Shcherbakova, S. Gozem, M. G. Caglayan, T. Minami and P. Anzenbacher, Chem, 2017, 2, 271–282 CAS.
- S. Lindskog, Pharmacol. Ther., 1997, 74, 1–20 CrossRef CAS PubMed.
- B. C. Tripp, K. Smith and J. G. Ferry, J. Biol. Chem., 2001, 276, 48615–48618 CrossRef CAS PubMed.
- R. B. Mokhtari, S. Kumar, S. S. Islam, M. Yazdanpanah, K. Adeli, E. Cutz and H. Yeger, BMC Cancer, 2013, 13, 378 CrossRef PubMed.
- S. Ansar Ahmed, R. M. Gogal and J. E. Walsh, J. Immunol. Methods, 1994, 170, 211–224 CrossRef CAS PubMed.
- W. G. Reiss and K. S. Oles, Ann. Pharmacother., 1996, 30, 514–519 CrossRef CAS PubMed.
- L. Yan, J. Miller, M. Yuan, J. F. Liu, T. M. Busch, A. Tsourkas and Z. Cheng, Biomacromolecules, 2017, 18, 1836–1844 CrossRef CAS PubMed.
- C. Albers, Nat. Commun., 2019, 10, 1921 CrossRef PubMed.
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence d,
Kevin
Cariou
d,
Kevin
Cariou