
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePalladium nanoparticles on gallium nitride as a Mott–Schottky catalyst for efficient and durable photoactivation of unactivated alkanes†
Lida
Tan‡
 a,
Xianghua
Kong‡
bc,
Mingxin
Liu‡
ad,
Hui
Su
a,
Hong
Guo
*c and
Chao-Jun
Li
*a
a,
Xianghua
Kong‡
bc,
Mingxin
Liu‡
ad,
Hui
Su
a,
Hong
Guo
*c and
Chao-Jun
Li
*a
aDepartment of Chemistry, FQRNT Centre for Green Chemistry and Catalysis, McGill University, 801 Sherbrooke Street West, Montreal, QC H3A 0B8, Canada. E-mail: cj.li@mcgill.ca
bCollege of Physics and Optoelectronic Engineering, Shenzhen University, 3688 Nanhai Avenue, Nanshan District, Shenzhen 518061, Guangdong, China
cDepartment of Physics, McGill University, Rutherford Building, 3600 University, Montreal, QC H3A 2T8, Canada
dState Key Laboratory of Applied Organic Chemistry, College of Chemistry and Chemical Engineering, Lanzhou University, 222 Tianshui South Road, Chengguan District, Lanzhou 730000, Gansu, China
First published on 28th September 2023
Abstract
The direct functionalization of inert C–H bonds has long been a “holy grail” for the chemistry world. In this report, the direct C(sp3)–N bond formation of unactivated alkanes is reported with a GaN based Mott–Schottky catalyst under photocatalytic reaction conditions. Long term stability and reaction efficiency (up to 92%) were achieved with this photocatalyst. The deposition of a Pd co-catalyst on the surface of GaN significantly enhanced the reaction efficiency. Microscopic investigation suggested a remarkable interaction in the Pd/GaN Schottky junction, giving a significant Pd/GaN depletion layer. In addition, density functional theory (DFT) calculations were performed to show the distinct performance of Pd nanoparticles at the atomic level.
Introduction
The direct conversion of C–H bonds to other functional groups has enabled a rapid route to more complicated molecules from simple structures with advantages of atom economy and reduced carbon emission.1 In particular, C–H amination has received much interest because of the abundance of C–N bonds in natural products, pharmaceuticals and pigments.2,3 Among all types of C–H functionalization, the direct functionalization of unactivated alkyl C(sp3)–H bonds, which are generally inert to chemical transformations, is still facing tremendous challenges due to the high activation energy and low acidity of such bonds.3 Traditionally, a stoichiometric amount of radical initiator is required to overcome this barrier,4 of which the atom economy is particularly disadvantageous. In recent years, a number of photocatalysts for direct C–H functionalization, including mesoporous graphic carbon nitride (mpg-CN),5 tetrabutylammonium decatungstate (TBADT),6–8 and other homogeneous molecular photocatalysts,9–12 have been reported, however, the pursuit of a powerful photocatalytic strategy with high energy efficiency and green practices is still going on.13,14 In 2018, Zuo's group reported a photocatalytic functionalization of light alkanes with cerium salts, which has achieved excellent catalytic efficiency and wide applications.15 While achieving promising results with a light-driven protocol, the cesium/alcohol catalyst used is difficult to be separated and recycled. In 2020, Chudasama's group reported a C(sp3)–H amination of ethereal substrates using atmospheric oxygen as a radical generator.16 Despite its success, the use of hazardous fluorinated alcohols (HFIP) as solvents and operating under high temperature would provide challenges for large-scale application and raise environmental concerns. To pursue the promising potential for light-driven C–H functionalization, the catalyst must be redesigned to better harness light while maintaining enhanced stability for catalyst recycling. Furthermore, it would be highly desirable for the catalyst to be abundant and easily accessible with low cost.Semiconductor photocatalysis is one of the most promising green and renewable energy ideas, as it uses the abundant sunlight or artificial illumination.17 Among them, gallium nitride (GaN), a group III nitride semiconductor, possesses a large energy band gap, great electromigration rate and high stability.18 It is widely employed in optoelectronics as well as next-generation microchips and is touted as a semiconductor of the future. Early in 2014, our group showed the excellent ability of C–H activation over the GaN surface during photocatalytic methane aromatization.19 The reaction proceeds via the H-CH3 polarization induced by the electrostatic force of Ga3+ and N3− on the GaN surface, followed by the heterolytic splitting of H-CH3 into surface-bond CH3− and H+ under irradiation. With the photo-induced hole generated on the surface of the semiconductor, the carbanion will then be oxidized into a methyl radical and initiate the carbon chain elongation. Recently, the design of Mott–Schottky catalysts consisting of a metal–semiconductor heterojunction has made it possible to control the charge transfer between the metal and semiconductor components through the rectifying contact at the interface offering a feasible method for significantly enhancing energy conversion, organic synthesis, photocatalytic reactions, and electrochemical reactions.20–22 So far, the introduction of a Schottky junction in photocatalysts is mainly focused on water splitting,23 CO2 reduction,24 and water treatment.25 However, their potential in the field of C–H activation, especially for the functionalization of unactivated alkanes, has rarely been studied. We envisioned that, with the deposition of noble metal particles to form Schottky junctions,26 the photogenerated charge carriers can be greatly increased, enhancing the performance of catalysts towards overcoming the large energy barrier of alkyl C–H functionalization. For example, Pd deposited on TiO2 has been reported to be an efficient catalyst for photocatalytic C(sp3)–H activation, demonstrating the viability of this approach.27 Herein, we present palladium nanoparticles deposited on GaN as a Mott–Schottky catalyst for photocatalytic C(sp3)–N bond formation of alkanes, proceeding with the outstanding features of high activity, high atom-economy, and high reusability.
Results and discussion
The typical GaN nanoparticles have a Wurtzite crystal structure. The GaN surface is composed of m-planes and c-planes. Transmission Electron Microscopy (TEM) was used to show the detailed structure of the commercially available GaN nanoparticles (Fig. 1a). The high-resolution TEM images and their corresponding diffraction patterns illustrated the different surfaces of GaN (Fig. 1b and c). Note that as for the m-plane, the distance between two adjacent Ga or N lattices is 5.18 Å and the length of the Ga–N bond is 1.95 Å. Compared with the normal sp3 C–H bond (1.09 Å), the dipole generated by the longer Ga–N bond induces an electrostatic stretch, which promotes the polarization of the C–H bond.9 Therefore, the strength of the polarized C–H bond could be weakened and further cleaved under irradiation. | ||
| Fig. 1 (a) Representative TEM image of GaN nanoparticles. (b) High resolution TEM image of the m-plane of GaN nanoparticles with its corresponding electron diffraction patterns. (c) High resolution TEM image of the c-plane of GaN nanoparticles with its corresponding electron diffraction patterns. | ||
Various metal co-catalysts were deposited on the commercially available GaN sample to build Mott–Schottky catalysts via a photodeposition method as follows: various metal precursors (K2PdCl4, K2PtCl4, CuCl2 or AgNO3) were dissolved in 3 mL of distilled water and 1 mL of methanol as a stock solution. 10 mg of the GaN sample was dispersed in 2 mL of the stock solution and stirred under photoirradiation for 7 hours using a xenon lamp (PE300 BUV). Then the suspension was filtered, washed with distilled water and methanol, and dried under vacuum at 100 °C for 12 h. The samples were named as M/GaN, where M represents the deposited metal.
To begin our research, we initially tested between di-tert-butyl azodicarboxylate (DBAD) and 2 equiv. of cyclohexane, with GaN nanoparticles alone as a catalyst for 12 h under argon at room temperature. A quartz tube was used as the reactor with the xenon lamp serving as the light source. The effect of solvent was first examined (Table 1, entries 1–5). The results suggested that the reaction could not happen in solvents with strong UV absorbance such as acetone (Table 1, entry 1) and benzene (Table 1, entry 2). In addition, strongly polar solvents, such as methanol (Table 1, entry 3) or water (Table 1, entry 4), were not compatible with the reaction. To our delight, with the common solvent acetonitrile and the use of commercially available GaN nanoparticles, the yield could reach 5% (Table 1, entry 5). Then, we further investigated the influence of metal deposited (See Fig. S2–S9 in the SI† for more information). Interestingly, the deposition of silver (Table 1, entry 6) and molybdenum (Table 1, entry 7) showed little influence on the activity of the catalyst. Other popular metal decorations such as rhodium (Table 1, entry 8), ruthenium (Table 1, entry 9), copper (Table 1, entry 10), manganese (Table 1, entry 11) and platinum (Table 1, entry 12) would increase the yield of the product 1a. Among all choices of metal decoration, palladium (Table 1, entry 13) was the best choice, affording product 1a in 62% yield. With the positive results using palladium, various amounts of Pd were deposited on GaN (Table 1, entries 13–16) and 2 wt% was found to be the optimal loading. As chlorine was shown to benefit alkyl radical formation,28,29 with the addition of 200 μL dichloromethane (DCM), the yield could reach 78%. Control experiments were also performed, demonstrating the importance of the catalyst (Table 1, entries 18, 19). Moreover, in the absence of photo-irradiation, no product was formed, showing that the reaction is photocatalytic (Table 1, entry 20).
|
|
|||
|---|---|---|---|
| Entry | Solvent (1 mL) | Catalyst | Yield% |
| a Reactions were carried out with 2 mg of the catalyst under Xe lamp irradiation at room temperature and under an inert atmosphere. b 200 μL DCM was added. c Under darkness. | |||
| 1a | Acetone | GaN | Traces |
| 2 | Benzene | GaN | 0 |
| 3 | Methanol | GaN | 0 |
| 4 | Water | GaN | 0 |
| 5 | Acetonitrile | GaN | 5 |
| 6 | Acetonitrile | 2 wt% Ag/GaN | 3 |
| 7 | Acetonitrile | 2 wt% Mo/GaN | 6 |
| 8 | Acetonitrile | 2 wt% Rh/GaN | 39 |
| 9 | Acetonitrile | 2 wt% Ru/GaN | 17 |
| 10 | Acetonitrile | 2 wt% Cu/GaN | 36 |
| 11 | Acetonitrile | 2 wt% Mn/GaN | 26 |
| 12 | Acetonitrile | 2 wt% Pt/GaN | 28 |
| 13 | Acetonitrile | 2 wt% Pd/GaN | 62 |
| 14 | Acetonitrile | 1 wt% Pd/GaN | 49 |
| 15 | Acetonitrile | 3 wt% Pd/GaN | 45 |
| 16 | Acetonitrile | 4 wt% Pd/GaN | 55 |
| 17b | Acetonitrile | 2 wt% Pd/GaN | 78 |
| 18 | Acetonitrile | N/A | 0 |
| 19b | Acetonitrile | N/A | 0 |
| 20b,c | Acetonitrile | 2 wt% Pd/GaN | 0 |
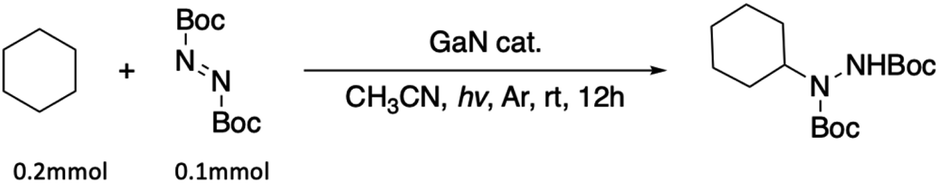
With the optimized conditions in hand, the scope of the reaction was investigated. As shown in Table 2, simple unactivated alkanes (1a–1d) reacted smoothly to form the C–N bond giving moderate to high yields. The widely used dimethyl ether was converted into the corresponding products 1e with moderate yield. For heterocyclic compounds, moderate to high yields were achieved with common chemicals including tetrahydrofuran (1f), tetrahydropyran (1g) and 1,4-dioxane (1h). 2,5-Dihydrofuran (1i) gave the highest yield of 92%. In addition, alkene substrates (1j, 1k) could be tolerated for this reaction. A list of benzyl compounds were also investigated (1l–1o). Dibenzyl methane (1l) could afford product yields of 64%. Toluene (1m) and 1-methylnaphthalene (1n) worked well for the reaction showing that this method could also be applied for the preliminary C(sp3)–H bonds. C–H bonds on bicyclic hydrocarbon indene (1o) gave only 34% yield, which might be due to the rigidity of the C–H bonds. Other nitrogen sources including diisopropyl azodicarboxylate (DIAD) and dibenzyl azodicarboxylate were found to be applicable with this method where 70% (2b) and 53% (2c) of yields were achieved respectively. In addition, the same catalyst could be reused and recycled five times with no significant decrease of the yield showing the long-term stability of the catalyst (Fig. 2).
|
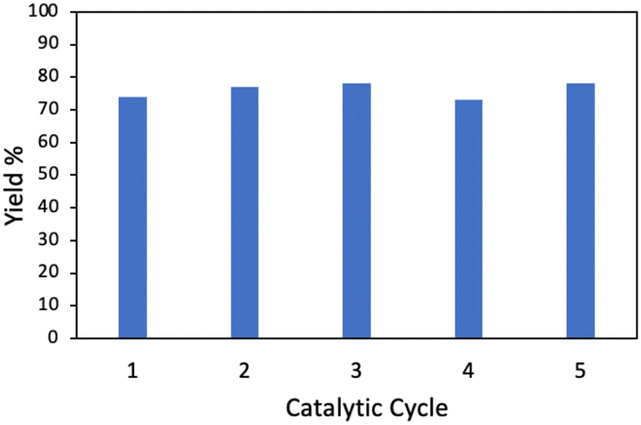 | ||
| Fig. 2 Reusability of 2 wt% Pd/GaN for the photocatalytic animation of cyclohexane. | ||
Microscopic characterization revealed the deposited Pd nanoparticles being embedded on the surface of GaN (Fig. 3a–f, S1†). Fig. 3g demonstrates the high-resolution TEM image for the interface area between GaN and Pd nanoparticles. In the close analysis of inverse fast Fourier transform (IFFT) of the same region (Fig. 3h), some distortions and dislocations could be noticed indicating the defects on the GaN surface adjacent to the deposited Pd nanoparticles. This could be the result of the successful formation of the Pd/GaN Schottky junction. This junction allows electron aggregation from GaN to Pd, which significantly activates the Pd catalyst, and leaves a depletion layer formed on the surrounding GaN.
 | ||
| Fig. 3 (a) Representative TEM image of 2 wt% Pd/GaN. (b) Representative HAADF-STEM images of 2 wt% Pd/GaN. (c) Overall corresponding EDX maps of the HAADF-STEM image b. (d) Corresponding EDX maps of the HAADF-STEM image b showing N atoms. (e) Corresponding EDX maps of the HAADF-STEM image b showing Ga atoms. (f) Corresponding EDX maps of the HAADF-STEM image b showing Pd atoms. (g) High-resolution TEM image of Pd deposited on GaN. (h) Inverse fast Fourier transform (IFFT) of image g. | ||
Photoluminescence spectra (PL) of commercially available GaN and as synthesized 2 wt% Pd/GaN are shown in Fig. 4a. The intensity of the PL emission decreases as Pd nanoparticles are deposited on the surface of GaN. The quenched PL spectra indicate the electron transfer from GaN to Pd nanoparticles, which promotes the charge carrier separation. Additionally, X-ray photoelectron spectroscopy (XPS) analysis was performed to determine the electron transfer in Mott–Schottky catalysts. Pd3d peaks of Pd/C and 2 wt% Pd/GaN are shown in Fig. 4b. 2 wt% Pd/GaN shows strong peaks at 334.4 eV and 339.7 eV. The upshift of Pd3d peaks of 2 wt% Pd/GaN to low binding energy, as compared with Pd3d peaks of Pd/C (337.6 eV and 342.5 eV), verifies the electron transfer from semiconducting GaN to Pd nanoparticles.
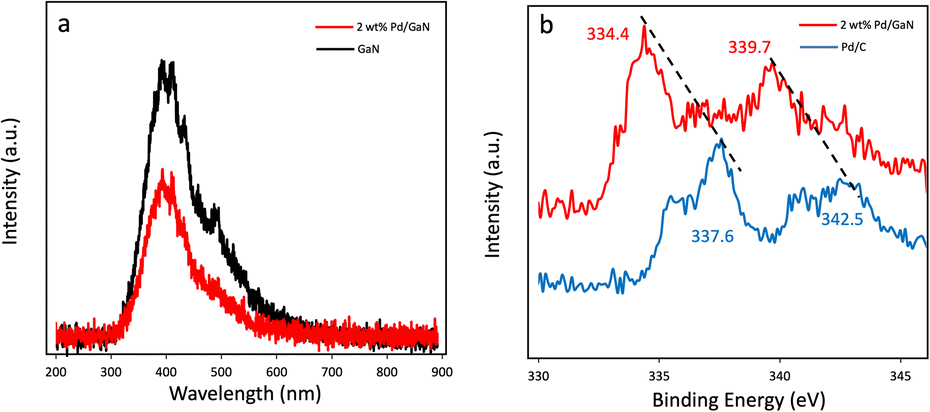 | ||
| Fig. 4 (a) Photoluminescence spectra of GaN and 2 wt% Pd/GaN. (b) Pd3d spectra of Pd/C and 2 wt% Pd/GaN. | ||
To investigate the mechanism of the reported C–N bond formation reaction, an electro paramagnetic resonance (EPR) study was performed. 1 equiv. of 5,5-dimethyl-1-pyrroline N-oxide (DMPO) as a radical trap was added under different reaction conditions. As shown in Fig. 5a, when cyclohexane was not present, the combination of DBAD and DMPO did not give a significant EPR signal under the optimized reaction conditions. In the case of GaN as a catalyst, the EPR signal of the cyclohexyl-DMPO component could be recognized. An obvious EPR signal was identified when DBAD was not present in the reaction mixture under the optimized C–H functionalization conditions with the radical trap, showing the enhancing performance of Pd nanoparticles. The EPR spectrum obtained was consistent with the previous study.30 In addition, in the optimized C–H functionalization of cyclohexane with the additional radical trap, a similar EPR signal could be identified. Providing the result, it is suggested that the reaction proceeds via the formation of an alkyl radical generated by an electrostatic stretch of the alkyl C–H bond by N and Ga atoms on the GaN surface, generating the corresponding proton and carbanion, which were then oxidized by photogenic holes on the semiconductor surface (See Fig. S10 in the ESI† for more information). Then, the alkyl radical was captured by DBAD to afford the desired C–N bond formation (Fig. 5b).
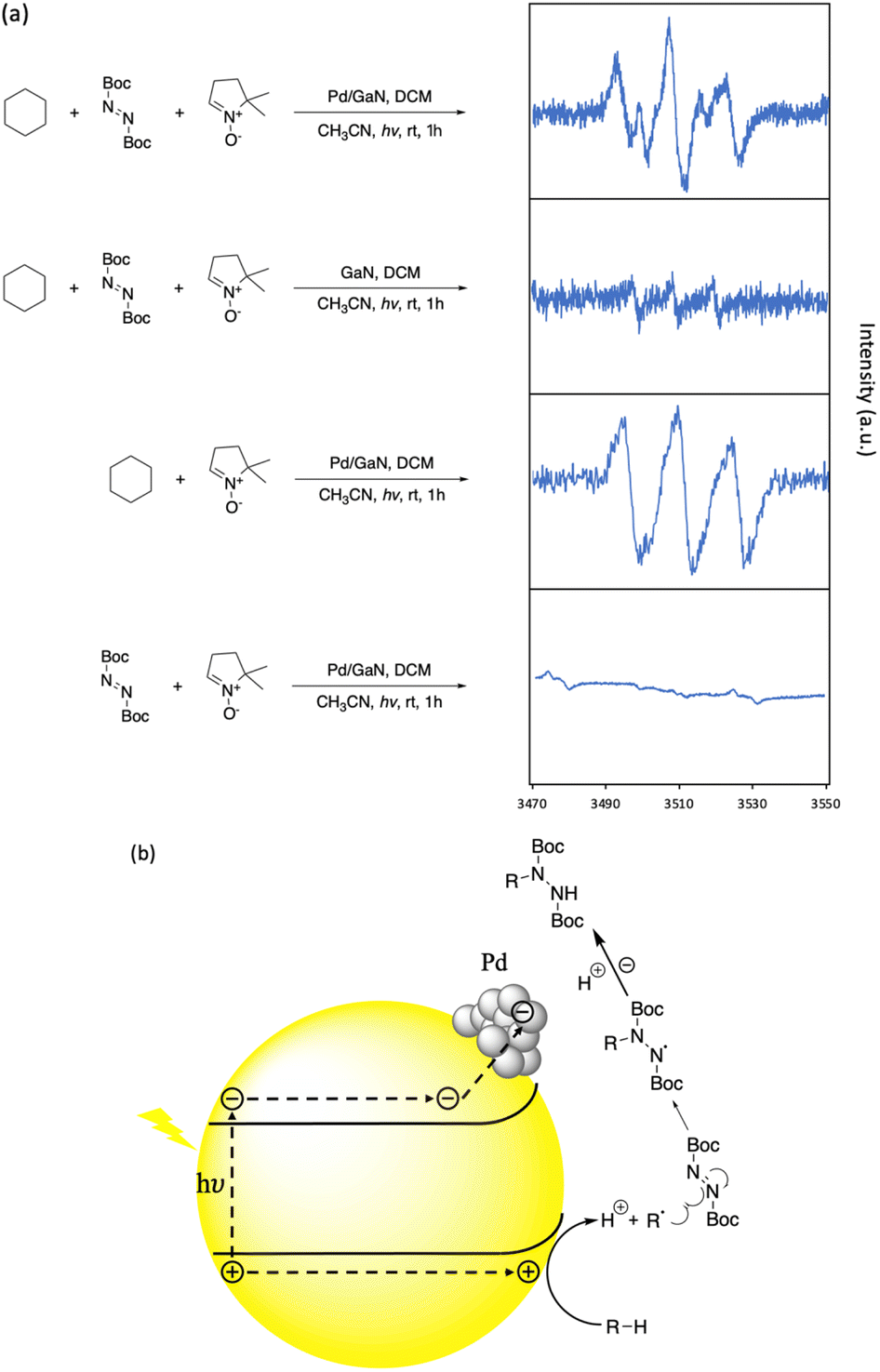 | ||
| Fig. 5 Reaction kinetic studies and proposed mechanism. (a) EPR analysis of hexane C–H functionalization. (b) Proposed reaction mechanism. | ||
Pd is commonly used in photocatalytic C–H activation, but the reason behind this selection requires careful investigation.27,31–33 To further elucidate the distinct performance of the Pd nanoparticles (NPs)/GaN system at the atomic level, density functional theory (DFT) calculations were performed to study the charge transfer between different metal NPs and GaN (Fig. S11†), and the adsorption of the cyclohexane (C6H12) molecule on the surface of the Pd NPs/GaN system (Fig. 6a). Comparably, the adsorption characteristics of C6H12 on bare GaN are plotted in Fig. 6b. Additionally, a general mechanism for the different performances exhibited by different metal NPs deposited on GaN is proposed in Fig. 6c.
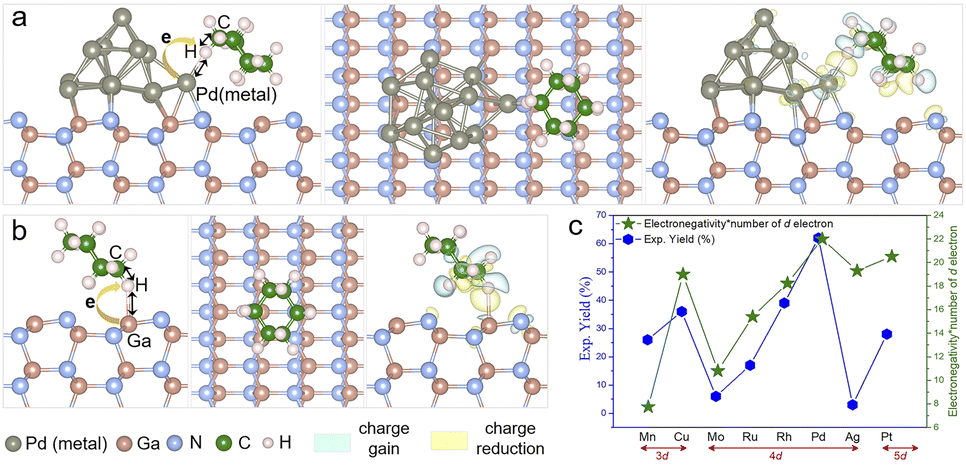 | ||
| Fig. 6 Calculated structural and electronic properties for metal nanoparticles/GaN systems. (a) Side (left panel) and top (middle panel) views of the fully relaxed configuration, and differential charge density (right panel) of C6H12 adsorbed on the Pt NPs/GaN system. (b) Side (left panel) and top (middle panel) views of the fully relaxed configuration, and differential charge density (right panel) of C6H12 adsorbed on the GaN system. (c) Proposed mechanism for the different performances of the metal nanoparticles deposited on GaN. | ||
First, the atomic structures of metal NPs/GaN (10![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 0) systems (Fig. 6a) are established to investigate carrier transport behaviors. After full geometric relaxation, Bader charge analyses are performed as summarized in Fig. S2 and Table S1.† It is found that electrons prefer to migrate from GaN to Pd or Pt nanoparticles, which is in good accordance with the XPS measurements (Fig. 4b).
0) systems (Fig. 6a) are established to investigate carrier transport behaviors. After full geometric relaxation, Bader charge analyses are performed as summarized in Fig. S2 and Table S1.† It is found that electrons prefer to migrate from GaN to Pd or Pt nanoparticles, which is in good accordance with the XPS measurements (Fig. 4b).
This charge transfer, from GaN to NPs, further helps NPs to facilitate the C–N bond formation of unactivated cyclohexane. Because to form the C–N bond, the first and foremost step is the adsorption and deformation of the relatively inert C6H12 on the photocatalyst surface, the electron injection from NPs to cyclohexane is a crucial step to activate the molecule. With more electrons on the NPs due to the charge transfer from GaN, the capability to weaken the C–H bond of alkanes is enhanced. Among the metal NPs investigated in our experiments, Pd and Pt NPs receive electrons from GaN, leading to their better performances in the activation of the cyclohexane molecule.
Subsequently, cyclohexane adsorption on the surface of Pd NPs/GaN, Pt NPs/GaN and bare GaN is studied. The optimized geometry and the differential charge density (DCD) analysis of C6H12/Pd NPs/GaN (100) are presented in Fig. 6a. There is a significant charge accumulation between the Pd atom and H atom (indicated by the light-yellow color) and substantial charge reduction in the middle of the H atom and C atom (marked by the light-blue color). These results are consistent with the following geometric features. Upon adsorption of the cyclohexane molecule on Pd NPs/GaN (Fig. 6a), our calculation showed that the H atom in the molecule forms a Pd–H chemical bond with a value of 2.044 Å, while the C–H bond is weakened by elongation from the original 1.090 Å to 1.130 Å. Besides, it is calculated that the charge on this H atom of the molecule bonded with Pd NPs is 1.041e. Consequently, the weakened C–H bond can be readily broken when the molecule is further excited by photogenerated electrons, setting the stage for subsequent C–N bond formation. Replacing Pd with Pt, namely, the C6H12/Pt NPs/GaN (100) system, the process to weaken the C–H bond is similar. Here, the Pt–H chemical bond length is 2.195 Å while the C–H bond is stretched from 1.090 Å to 1.127 Å; the charge electron on the H atom forming a chemical bond with Pt NPs is 1.002e.
In addition, the geometric and electronic properties are also calculated for the C6H12/GaN (100) system without NPs. As plotted in Fig. 6b, the cyclohexane molecule interacts with the bare GaN surface through the H–Ga covalent bond (2.126 Å), i.e. the C–H bond is elongated from 1.090 Å to 1.118 Å, less than the elongation with NPs. We conclude the rational deposition of metal NPs to be a very effective route for enhancing the chemical reaction of unactivated alkanes as reactants on GaN surfaces. Among the studied metal nanoparticles, Pd performed the best, which is highly consistent with the experimental results.
Notably, it is interesting to investigate the correlation between the electronegativity, charge transfer and chemical reaction of NPs on GaN surfaces, as shown in Fig. S2.† Among eight listed metal NPs, electronegativity of six metal NPs had a positive correlation with the charge transfer, including Pd and Pt. Following the d-band center theory34–36 which determines the catalytic performance, we suggest a quantity, the Product of Electro Negativity and Number of d electron (PENNd) of the metal NPs. As shown in Fig. 6c, PENNd fits the characteristics, such as the variation trend and different d electron classifications, of the catalytic activity curve. This observation gives a qualitative picture by basic material characteristics to catalytic performances of NPs/GaN systems.
Conclusions
In summary, we have reported a GaN based Mott–Schottky catalyst for facile C–H functionalization of unactivated alkanes. The strategy gives long term stability and high reactivity. Density functional theory calculations were conducted to show the uniqueness of Pd nanoparticles. It is shown that the oriented electron migration, facile adsorption of alkanes, and the strong chemical interaction between alkanes and Pd NPs/GaN would weaken the C–H bond and facilitate C–N bond formation. Additionally, the band gap of GaN can be tuned by adding indium (In), thus affording an opportunity to harness virtually the whole solar spectrum and the further investigation of GaN catalysts in photocatalytic reactions is ongoing in our lab.37–40Data availability
Yes, we have deposit the related experimental or computational data in the ESI.†Author contributions
C.-J. L. proposed the research idea and supervised the project. L.-D. T. designed and prepared the catalyst, characterized samples, and performed catalytic measurements. X.-H. K. and H. G. proposed and performed the theoretical calculations as well as result analyses. L.-D. T., X.-H. K., and M.-X. L. wrote the manuscript. H. S. revised the manuscript and participated in critical discussions. All authors interpreted the results and contributed to the development of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful for the Canada Research Chair Foundation, the Natural Science and Engineering Research Council of Canada (Discovery and New Frontiers), the Fond de Rechercher du Quebec – Natural et Technologie, the Canadian Foundations for Innovations, the McGill Sustainability Systems Initiative, the National Natural Science Foundation of China (Grant no. 12104313), Department of Science and Technology of Guangdong Province (No. 2021QN02L820) and Shenzhen Natural Science Fund (the Stable Support Plan Program 20220810161616001) for financial support. The authors also acknowledge HZWTECH, the High-Performance Computing Center of McGill University, Calcul-Quebec, and Compute Canada for their computation facilities.Notes and references
- J. He, M. Wasa, K. S. L. Chan, Q. Shao and J.-Q. Yu, Chem. Rev., 2017, 117, 8754–8786 CrossRef CAS PubMed.
- Y. Park, Y. Kim and S. Chang, Chem. Rev., 2017, 117, 9247–9301 CrossRef CAS PubMed.
- T. Wang and N. Jiao, Acc. Chem. Res., 2014, 47, 1137–1145 CrossRef CAS PubMed.
- R. H. Crabtree and A. Lei, Chem. Rev., 2017, 117, 8481–8482 CrossRef CAS PubMed.
- I. Ghosh, J. Khamrai, A. Savateev, N. Shlapakov, M. Antonietti and B. König, Science, 2019, 365, 360–366 CrossRef CAS PubMed.
- G. Laudadio, Y. Deng, K. van der Wal, D. Ravelli, M. Nuño, M. Fagnoni, D. Guthrie, Y. Sun and T. Noël, Science, 2020, 369, 92–96 CrossRef CAS PubMed.
- Z.-Y. Dai, Z.-S. Nong and P.-S. Wang, ACS Catal., 2020, 10, 4786–4790 CrossRef CAS.
- T. Wan, Z. Wen, G. Laudadio, L. Capaldo, R. Lammers, J. A. Rincón, P. García-Losada, C. Mateos, M. O. Frederick, R. Broersma and T. Noël, ACS Cent. Sci., 2022, 8, 51–56 CrossRef CAS PubMed.
- C.-Y. Huang, J. Li and C.-J. Li, Nat. Commun., 2021, 12, 1–9 CrossRef PubMed.
- J. Lee, S. Jin, D. Kim, S. H. Hong and S. Chang, J. Am. Chem. Soc., 2021, 143, 5191–5200 CrossRef CAS PubMed.
- A. Dewanji, P. E. Krach and M. Rueping, Angew. Chem., Int. Ed., 2019, 58, 3566–3570 CrossRef CAS PubMed.
- Y. Jin, Q. Zhang, L. Wang, X. Wang, C. Meng and C. Duan, Green Chem., 2021, 23, 6984–6989 RSC.
- N. Holmberg-Douglas and D. A. Nicewicz, Chem. Rev., 2022, 122, 1925–2016 CrossRef CAS PubMed.
- B. König, Eur. J. Org Chem., 2017, 2017, 1979–1981 CrossRef.
- A. Hu, J.-J. Guo, H. Pan and Z. Zuo, Science, 2018, 361, 668–672 CrossRef CAS PubMed.
- A. Shamsabadi, A. Maruani, N. Ahmed and V. Chudasama, Org. Biomol. Chem., 2020, 18, 6258–6264 RSC.
- H. Tong, S. Ouyang, Y. Bi, N. Umezawa, M. Oshikiri and J. Ye, Adv. Mater., 2012, 24, 229–251 CrossRef CAS PubMed.
- M. Liu, L. Tan, B. Zhou, L. Li, Z. Mi and C.-J. Li, Chem, 2021, 7, 64–92 CAS.
- L. Li, S. Fan, X. Mu, Z. Mi and C.-J. Li, J. Am. Chem. Soc., 2014, 136, 7793–7796 CrossRef CAS PubMed.
- D. Xu, S.-N. Zhang, J.-S. Chen and X.-H. Li, Chem. Rev., 2023, 123, 1–30 CrossRef CAS PubMed.
- X. Lin, S.-N. Zhang, D. Xu, J.-J. Zhang, Y.-X. Lin, G.-Y. Zhai, H. Su, Z.-H. Xue, X. Liu, M. Antonietti, J.-S. Chen and X.-H. Li, Nat. Commun., 2021, 12, 3882 CrossRef CAS PubMed.
- X. Lin, Z. Zhou, Q.-Y. Li, D. Xu, S.-Y. Xia, B.-L. Leng, G.-Y. Zhai, S.-N. Zhang, L.-H. Sun, G. Zhao, J.-S. Chen and X.-H. Li, Angew. Chem., Int. Ed., 2022, 61, e202207108 CrossRef CAS PubMed.
- Y. Tong, W. Liu, C. Li, X. Liu, J. Liu and X. Zhang, Sustain. Energy Fuels, 2023, 7, 12–30 RSC.
- H.-J. Son, C. Pac and S. O. Kang, Acc. Chem. Res., 2021, 54, 4530–4544 CrossRef CAS PubMed.
- A. Raza, S. Altaf, S. Ali, M. Ikram and G. Li, Sustain. Mater. Technol., 2022, 32, e00406 CAS.
- L. Li, Y. Wang, S. Vanka, X. Mu, Z. Mi and C.-J. Li, Angew. Chem., Int. Ed., 2017, 56, 8701–8705 CrossRef CAS PubMed.
- A. Hainer, N. Marina, S. Rincon, P. Costa, A. E. Lanterna and J. C. Scaiano, J. Am. Chem. Soc., 2019, 141, 4531–4535 CrossRef CAS PubMed.
- S. Rohe, A. O. Morris, T. McCallum and L. Barriault, Angew. Chem., Int. Ed., 2018, 57, 15664–15669 CrossRef CAS PubMed.
- W. Liu, X. Yang, Z.-Z. Zhou and C.-J. Li, Chem, 2017, 2, 688–702 CAS.
- N. Kvasovs, V. Iziumchenko, V. Palchykov and V. Gevorgyan, ACS Catal., 2021, 11, 3749–3754 CrossRef CAS PubMed.
- I. D. Lemir, J. E. Argüello, A. E. Lanterna and J. C. Scaiano, Chem. Commun., 2020, 56, 10239–10242 RSC.
- T. A. Gawargy, P. Costa, A. E. Lanterna and J. C. Scaiano, Org. Biomol. Chem., 2020, 18, 6047–6052 RSC.
- M. Tomás-Gamasa and J. L. Mascareñas, ChemBioChem, 2020, 21, 294–309 CrossRef PubMed.
- B. Hammer and J. K. Nørskov, Surf. Sci., 1995, 343, 211–220 CrossRef CAS.
- B. Hammer and J. K. Nørskov, Adv. Catal., 2000, 45, 71–129 CAS.
- G. Jeff, J. K. Nørskov and M. Mavrikakis, Annu. Rev. Phys. Chem., 2002, 53, 319–348 CrossRef PubMed.
- M. G. Kibria and Z. Mi, J. Mater. Chem. A, 2016, 4, 2801–2820 RSC.
- S. Arafin, X. Liu and Z. Mi, J. Nanophotonics, 2013, 7, 074599 CrossRef CAS.
- P. G. Moses and C. G. Van de Walle, Appl. Phys. Lett., 2010, 96, 021908 CrossRef.
- L. Tan, H. Su, J. Han, M. Liu and C.-J. Li, Sci. Rep., 2022, 12, 18414 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc00688c |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2023 |