
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Highly electron-deficient 3,6-diaza-9-borafluorene scaffolds for the construction of luminescent chelate complexes†
Jan
Adamek
a,
Paulina H.
Marek-Urban
ab,
Krzysztof
Woźniak
 b,
Krzysztof
Durka
*a and
Sergiusz
Luliński
*a
b,
Krzysztof
Durka
*a and
Sergiusz
Luliński
*a
aFaculty of Chemistry, Warsaw University of Technology, Noakowskiego 3, 00-664 Warsaw, Poland. E-mail: krzysztof.durka@pw.edu.pl; sergiusz.lulinski@pw.edu.pl
bDepartment of Chemistry, University of Warsaw, Żwirki i Wigury 101, 02-089 Warsaw, Poland
First published on 5th October 2023
Abstract
The synthesis and characterization of two fluorinated 3,6-diaza-9-hydroxy-9-borafluorene oxonium acids featuring improved hydrolytic stability and the strong electron-deficient character of the diazaborafluorene core is reported. These boracycles served as precursors of fluorescent spiro-type complexes with (O,N)-chelating ligands which revealed specific properties such as delayed emission, white light emission in the solid state and photocatalytic performance in singlet oxygen-mediated oxidation reactions.
Introduction
Boracyclic compounds attract a considerable interest due to their numerous applications in organic synthesis, catalysis and materials chemistry. An important class of these compounds are dibenzo-fused derivatives comprising a central six-membered boracyclic ring with incorporated another heteroatom such as oxa-, aza-, sila-, and thiaborins as well as diboraanthracenes.1 Such compounds are usually more stable than diarylboron derivatives with a non-annulated boron atom. Modifications within a boracycle or adjacent aromatic rings result in varying electron-acceptor properties stemming from the presence of the vacant 2p orbital on the boron atom. Importantly, boracyclic precursors can be easily converted to various chelate complexes featuring the spiro arrangement of a tetracoordinated boron center. In most cases, aromatic chromophore ligands (O,O-, O,N-, and N,N-) were used which enabled fine-tuning of the photophysical properties of respective products.2Recently, the 9-borafluorene scaffold has been extensively used for designing numerous boracycles and offers a useful alternative to its ring-expanded analogues.3 The presence of the five-membered borole ring results in an increase of Lewis acidity which is beneficial for the stability of respective chelate complexes. Further enhancement of the electron-acceptor character of the 9-borafluorene scaffold can be achieved by fluorination of aromatic rings or replacement of one of the benzene rings with the pyridine one.4 The obtained azaborafluorene derivative showed dual-fluorescence behaviour promoted by the formation of a B–N four-coordinate adduct. However, it was prone to hydrolytic cleavage of the boracyclic ring. Herewith, we present a combined strategy involving (i) annulation of a central borole moiety with two pyridine rings and (ii) installation of fluorine substituents as a tool for strong enhancement of electron-acceptor properties (Scheme 1). The designed fluorinated 3,6-diaza-9-borafluorenes feature strong Lewis acidity of the boron atom and thus they were isolated in the form of highly stable water adducts, which were further converted to luminescent (O,N) chelate complexes.
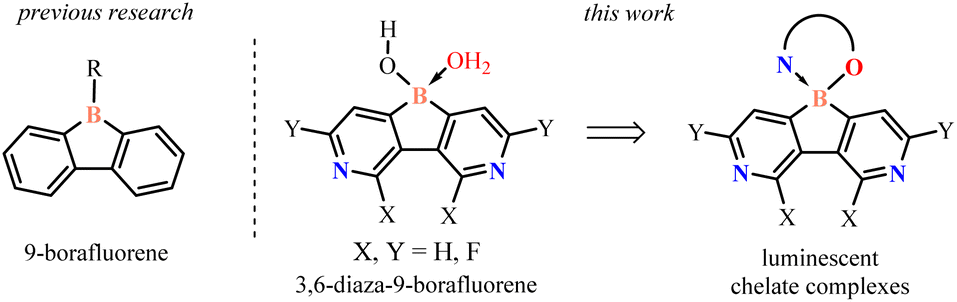 | ||
| Scheme 1 Molecular design of 3,6-diaza-9-borafluorenes and their chelate complexes. | ||
Results and discussion
The synthesis of 3,6-diaza-4,5-difluoro-9-hydroxy-9-borafluorene oxonium complex 2 was accomplished using 2,2′-difluoro-6,6′-diiodo-3,3′-bipyridine 1 as a convenient precursor (Scheme 2).5 Compound 1 was converted to the Grignard reagent via a double I/Mg exchange followed by treatment with B(OSiMe3)3 (1 equiv.)6 and careful hydrolysis with dilute aq. HCl yielding 2. A similar protocol was applied for the synthesis of 5 starting with 2,2′,4,4′-tetrafluoro-6,6′-diiodo-3,3′-bipyridine 4. The products 2 and 5 were isolated as white powders soluble in DMSO and MeOH but insoluble in water, Et2O and acetone. They were characterized by multinuclear 1H, 11B, 13C and 19F NMR spectroscopy. A notable feature of 2 is a large through-space 19F–19F coupling constant of 82 Hz estimated from the simulation of the 13C NMR multiplet of the fluorine-bound carbon atom centered at 157.2 ppm, i.e., the “X” part of the ABX spin system (Fig. S8.3, ESI†). Taking into account the F⋯F distance of 2.579(2) Å, this JFF value is in agreement with the empirical correlation equation proposed by Ernst.7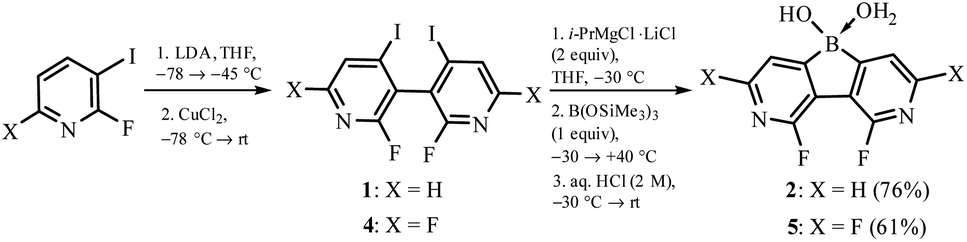 | ||
| Scheme 2 Synthesis of fluorinated 3,6-diaza-9-borafluorenes. | ||
The single-crystal X-ray diffraction analysis of 5 confirmed that the boron atom is tetracoordinate due to the complexation of the water molecule.8 The molecules are assembled into the linear motifs held by very strong O–H⋯O hydrogen bond (HB) interactions (dO⋯O = 2.400(2) Å) formed between coordinated water (HB donor) and the B–OH group (HB acceptor) from a neighbouring molecule (Fig. 1a). In fact, the difference-Fourier density map indicates that the H atom is delocalized between two oxygen atoms (Fig. S2.1, ESI†). This was further confirmed by theoretical calculations (M062X/6-311++G(d,p)) showing that the proton can freely migrate between oxygen atoms (Fig. S3.7, ESI†). The estimated energy of this HB is −84 kJ mol−1 (calculation details are provided in the ESI†) and the amount of electron density at the bond critical point is ρ = 0.73 e·Å−3 which is comparable to the values found in very strong charge-assisted HBs.9 The structure 5 is related to the oxonium acid structures of boronophthalide,8a 3,4,5,6-tetrafluorophenylene-1,2-diboronic acid8c and 1-hydroxy-1H,3H-naphtho[1,8-cd][1,2]oxaborinin-3-one,10 which also feature comparably strong intermolecular HB interactions (dO⋯O = 2.424–2.486 Å, Table S2.3, ESI†) correlating with high Brønsted acidity (pKa = 2–3). Indeed, pKa values for 2 and 5 are 2.4 and 1.4, respectively, as determined by potentiometric titration with 0.1 M aq. NaOH and pH-metric measurements of 0.02 M solutions in H2O/MeOH (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1). In pure water, the pKa values should be lower by ca. 0.3–0.5 units and thus 5 is significantly more acidic than boronophthalide (pKa = 2.0).8a,10 However, it should be noted that 5 is poorly soluble in water which can be rationalized by its strong aggregation as the molecular chains are further interconnected by O–H⋯N interactions (dN⋯O = 2.833(2) Å) producing a very compact and highly symmetric HB network (Fig. 1b). The 11B NMR spectra for 2 and 5 in DMSO-d6 confirmed the presence of a tetracoordinated boron center with chemical shifts of 7.9 and 6.0 ppm, respectively. This indicates that the water adduct observed in the crystal structure of 5 persists in solution which was unambiguously proved by ESI HRMS analysis of both 2 and 5. Finally, the phase purity of 5 was confirmed by PXRD analysis showing perfect overlap between experimental and simulated patterns (Fig S2.6, ESI†). We were unable to grow single crystals of 2, whilst the PXRD pattern of a powder sample roughly resembles that of 5 albeit it shows strongly broadened peaks. Thus, it can be supposed that 2 is isostructural with 5 although the crystallinity of the former is very low.
:1). In pure water, the pKa values should be lower by ca. 0.3–0.5 units and thus 5 is significantly more acidic than boronophthalide (pKa = 2.0).8a,10 However, it should be noted that 5 is poorly soluble in water which can be rationalized by its strong aggregation as the molecular chains are further interconnected by O–H⋯N interactions (dN⋯O = 2.833(2) Å) producing a very compact and highly symmetric HB network (Fig. 1b). The 11B NMR spectra for 2 and 5 in DMSO-d6 confirmed the presence of a tetracoordinated boron center with chemical shifts of 7.9 and 6.0 ppm, respectively. This indicates that the water adduct observed in the crystal structure of 5 persists in solution which was unambiguously proved by ESI HRMS analysis of both 2 and 5. Finally, the phase purity of 5 was confirmed by PXRD analysis showing perfect overlap between experimental and simulated patterns (Fig S2.6, ESI†). We were unable to grow single crystals of 2, whilst the PXRD pattern of a powder sample roughly resembles that of 5 albeit it shows strongly broadened peaks. Thus, it can be supposed that 2 is isostructural with 5 although the crystallinity of the former is very low.
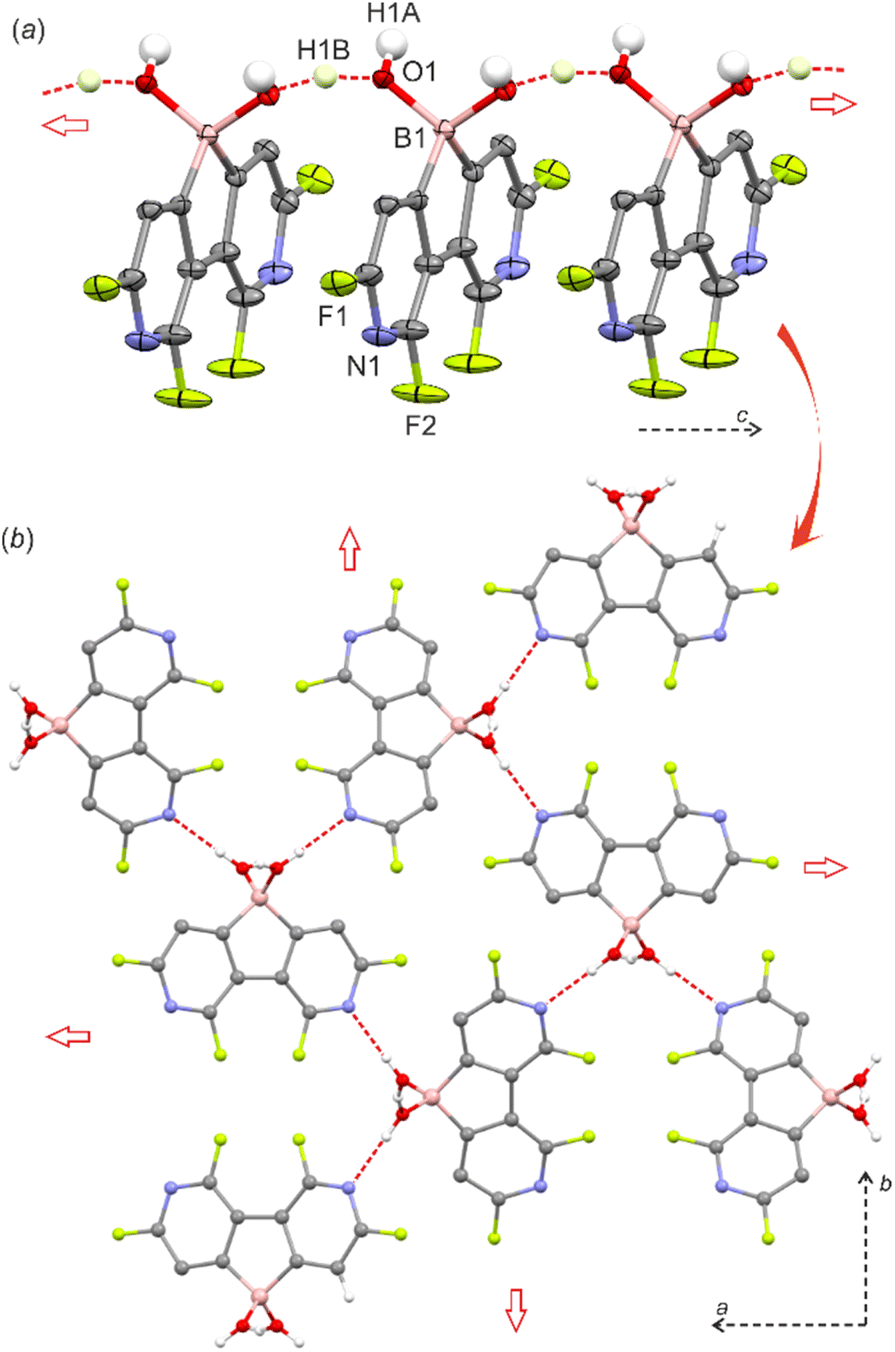 | ||
| Fig. 1 (a) HB linear motif in the crystal structure of 5 (I-42d space group). Thermal ellipsoids were generated at the 50% probability level. Aromatic hydrogen atoms were omitted for clarity. (b) Packing diagram showing the formation of a symmetric tetragonal network based on O–H⋯N HB interactions. | ||
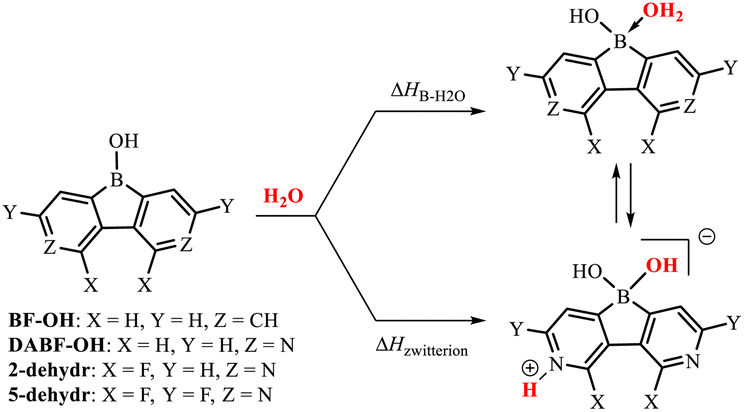
The cyclic voltammetry measurements gave the reduction potentials of −1.71 eV (2) and −0.90 eV (5) vs. the FeCp2/FeCp2+ pair (Fig. S5.1, Table S5.1 and ESI†), which correspond to the LUMO energy levels of −3.39 eV (2) and −4.20 eV (5). This confirms the strong electron-acceptor character of the diazaborafluorene core in 5. It should be noted that the tetracoordinate nature of the boron atom in 2 and 5 must result in strong weakening of the electron-acceptor character compared to related putative systems 2-dehydr and 5-dehydr with the three-coordinate boron atom, i.e., lacking water molecules as donors. The Lewis acidity of 2-dehydr and 5-dehydr was further evaluated by DFT calculations (M062X/6-311++G(d,p) level of theory) of enthalpies of coordination of water molecules to the boron atom (Table 1). For comparison, 9-hydroxyborafluorene (BF-OH) and the parent (non-fluorinated) 9-hydroxy-3,6-diaza-9-borafluorene (DABF-OH) were also studied. Overall, the results point out that the fluorination of the diazaborafluorene system systematically increases boron Lewis acidity. This is also reflected in the shortening of B–OH2 bond distances (Table 1). In general, for 3,6-diaza-9-borafluorenes the oxonium acid species may equilibrate with the zwitterionic tautomer resulting from proton transfer to the pyridine nitrogen atom. According to calculations, the latter form dominates in the case of DABF-OH. In contrast, the oxonium acid tautomer is slightly more stable than the zwitterionic one for 2, however, since the ΔHB–H2O and ΔHzwitterion enthalpies are quite similar, it is expected that both forms equilibrate in solution. The structural lability of 2 could disrupt its crystal packing resulting in a partial amorphization. For 5, the proton transfer to the nitrogen atom is highly unfavourable which is in line with the strongly reduced basicity of pyridine nitrogen flanked by two fluorine substituents (see discussion in ESI, Table S3.2†). Finally, the B–N coordination of the pyridine unit to the boron atom could be also considered11 but DFT calculations indicate that the aggregation through B–OH2⋯N HB interactions is energetically more favoured for all studied diazaborafluorenes (Table S3.4, ESI†). The TGA analyses performed for 2 and 5 showed that both systems apparently lose a water molecule at ca. 150–170 °C (Fig S7.1 and S7.2, ESI†). It can be expected that this would be followed by network reorganization through pyridine–boron coordination. In the case of 2, the resulting material is stable up to 350 °C, while dehydrated 5 decomposes already at 200–300 °C suggesting that its stabilization through N–B coordination is not effective which is consistent with other experimental and theoretical results.
|
|
||||
|---|---|---|---|---|
| BF-OH | DABF-OH | 2-dehydr | 5-dehydr | |
| ΔHB–H2O/kJ mol−1 | −1.0 | −16.4 | −22.8 | −29.2 |
| ΔHzwitterion/kJ mol−1 | — | −47.8 | −17.6 | +13.5 |
| d B–OH2/Å | 1.754 | 1.704 | 1.688 | 1.674 |
UV-Vis spectra of 2 and 5 showed the absorption maxima at λabs = 304 nm in EtOH solution (Fig. 2a). To ensure that the oxonium acid forms persist in solution, the measurements were performed upon the addition of a drop of conc. aq. HCl. According to B3LYP/6-311++G(d,p) calculations, the observed absorption band can be assigned to the π–π* transition (2: λcalcabs = 320 nm, f = 0.109; 5: λcalcabs = 328 nm, f = 0.067) occurring between HOMO and LUMO orbitals (Fig. 2b). Diazaborafluorene 2 exhibits intense sky-blue fluorescence (λem = 450 nm, fluorescence quantum yield QYF = 53%) in EtOH solution. Substitution with two additional fluorine atoms in 5 enhances the fluorescence intensity (QYF = 65%) and leads to the bathochromic shift of the emission band (λem = 472 nm). This is in agreement with TD-DFT calculations performed in the EtOH solvent field (Tables S3.6 and S3.7, ESI†), however the origin of this effect is not fully clear; it may result from the stronger stabilization of the excited state of 5 due to its stronger interaction with the polar solvent. It should be noted that the fluorescence spectra of 2 and 5 are somehow reminiscent of their 9-borafluorene analogue, namely 9-(tert-butoxy)-9-borafluorene.3g However, since the boron center is tetracoordinate, the absorption spectra of diazaborafluorenes lack longer wavelength bands (λabs > 350 nm) of π–B(2p) transitions observed for various 9-borafluorenes.3g Finally, it should be pointed out that fluorescence was almost completely quenched in pure EtOH solutions, i.e., without HCl additive. This indicates that anionic forms of 2 and 5 are not luminescent.
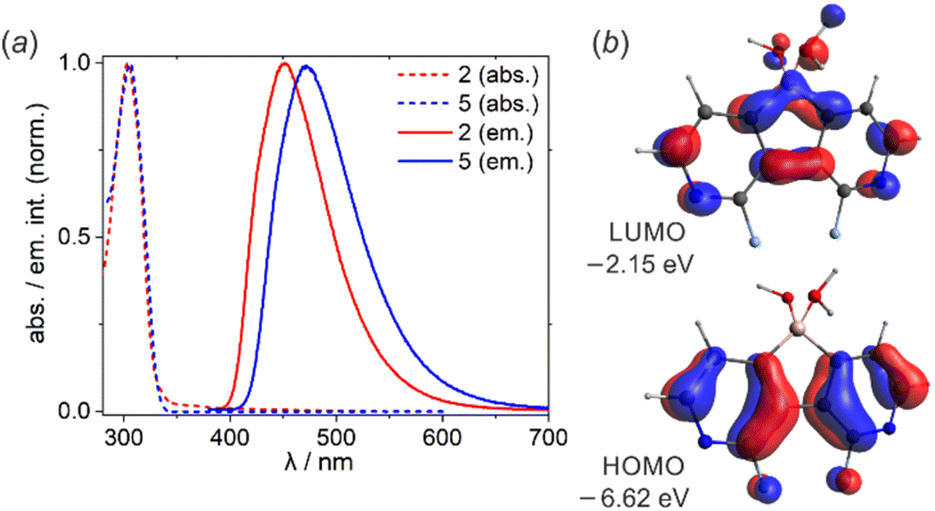 | ||
| Fig. 2 (a) Normalized emission spectra of 2 and 5 in EtOH solution with a drop of conc. aq. HCl. (b) Frontier molecular orbitals in 2 (B3LYP/6-311++G(d,p)). | ||
In the next step, diazaborafluorenes were employed for the preparation of a series of (O,N)-chelate complexes 3a–3g and 6a–6c with selected proligands including 8-hydroxyquinoline, 2-(2-pyridyl)phenol, two salicydeneaniline derivatives and three 2-(hydroxyphenyl)benzoheteroazoles (Het![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, S, NPh) (Scheme 3). All compounds were obtained in reasonable yields (45–78%) as cream-white, pale yellow or intense yellow solids soluble in organic solvents such as CHCl3 and acetone but in most cases insoluble in Et2O and hexane. They are stable in solution as their 1H NMR spectra did not show any visible changes after several weeks. This can be ascribed to the high Lewis acidity of the boron centre which strengthens coordination to the chelating ligands. In fact, 11B NMR chemical shifts are in the range of 4.0–11.0 ppm, i.e., in agreement with the values reported for analogous organoboron complexes.12
O, S, NPh) (Scheme 3). All compounds were obtained in reasonable yields (45–78%) as cream-white, pale yellow or intense yellow solids soluble in organic solvents such as CHCl3 and acetone but in most cases insoluble in Et2O and hexane. They are stable in solution as their 1H NMR spectra did not show any visible changes after several weeks. This can be ascribed to the high Lewis acidity of the boron centre which strengthens coordination to the chelating ligands. In fact, 11B NMR chemical shifts are in the range of 4.0–11.0 ppm, i.e., in agreement with the values reported for analogous organoboron complexes.12
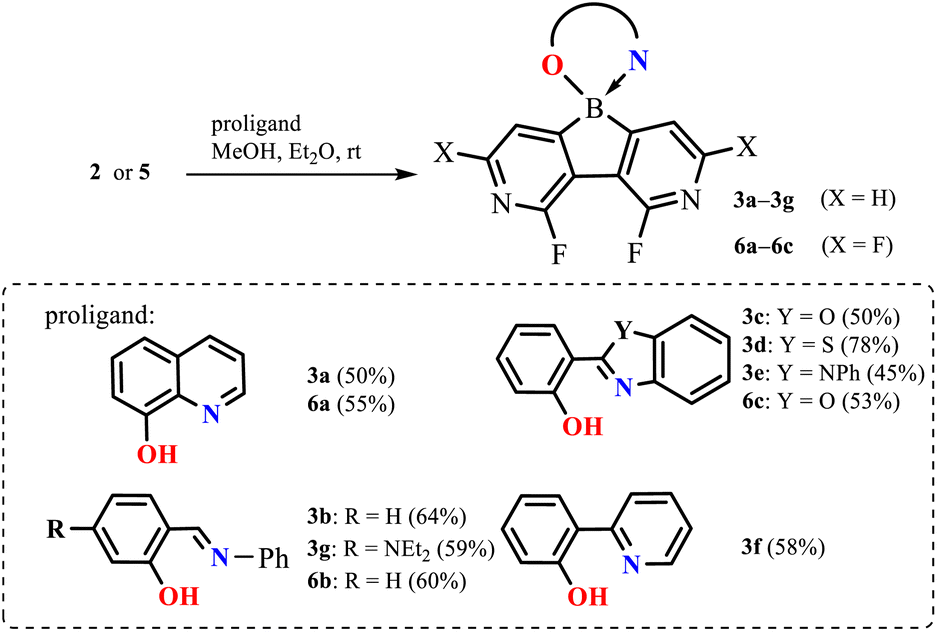 | ||
| Scheme 3 Synthesis of complexes 3a–3g and 6a–6c. | ||
The molecular structures of 3a–3f and 6c were determined by single-crystal X-ray diffraction. Overall, they feature the spiro geometry of boron with an orthogonal arrangement of diazaborafluorene and ligand moieties (Fig. 3a). The B–N, B–O and B–C distances (Table S2.4, ESI†) are within a range typical of organoboron tetracoordinate complexes except for the remarkably short B–N dative bond in 6c (dB–N = 1.569(2) Å). A comprehensive analysis of all structures shows that the molecules remain quite rigid in the diazaborafluorene plane, but regain some additional degree of flexibility of the chelate ligand reflected in the distortion of the B(O,N) heterocyclic ring and ligand in-plane or out-of-plane shifting (Fig. 3b). Such a behaviour was previously observed for crystal structures of related 9-borafluorene chelate complexes.12 Concordantly with these studies, the B(O,N) chelate ring can adopt either flat or half-chair conformations; the latter features boron and/or oxygen atoms distorting out of the ligand plane (Fig. 3c). According to DFT calculations, the conformers have similar electronic energies with low interconversion barriers (below 5 kJ mol−1). Thus, molecules should retain some conformational flexibility in solution.
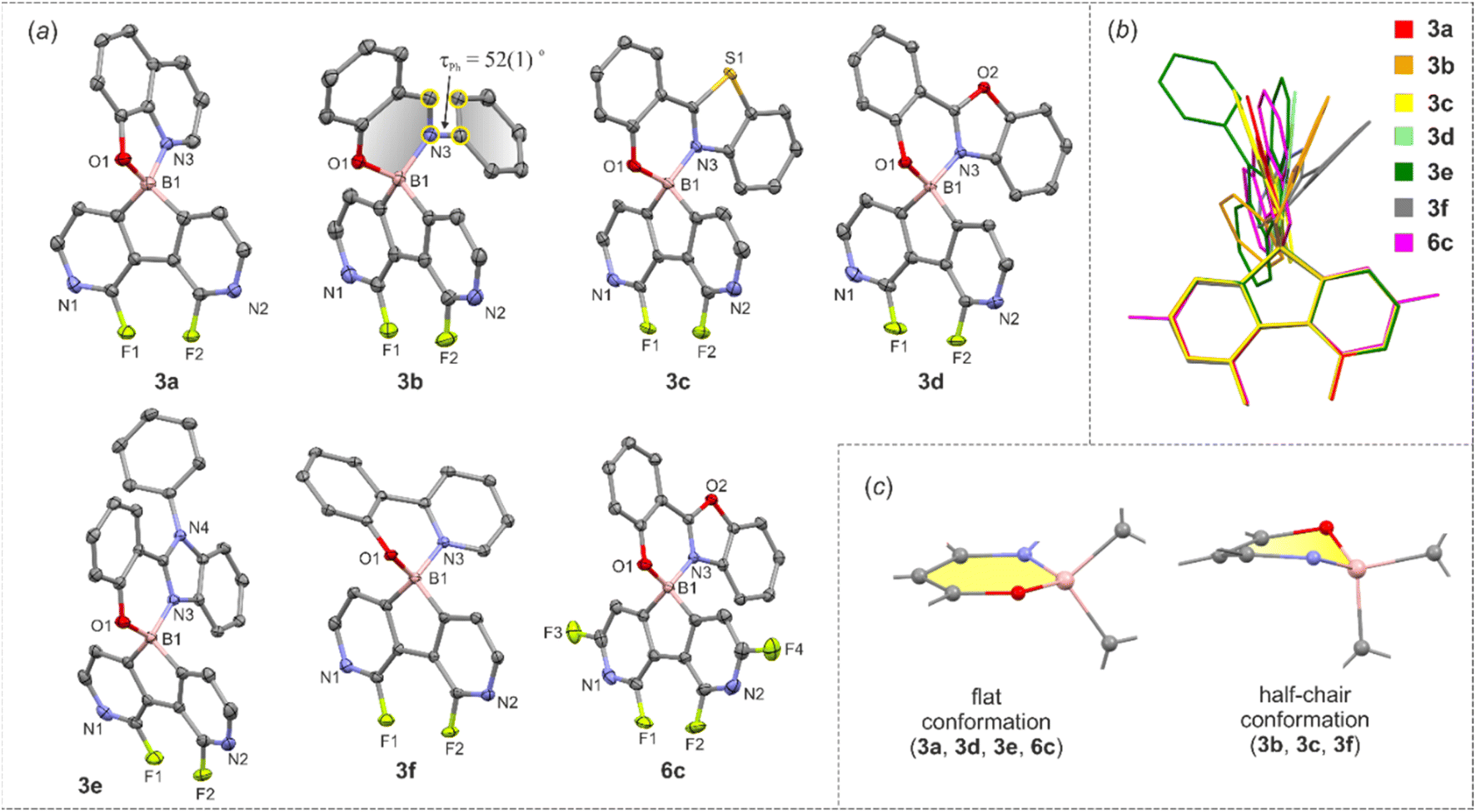 | ||
| Fig. 3 (a) Molecular structures of diazaborafluorene complexes. Thermal ellipsoids were generated at the 50% probability level. Hydrogen atoms were omitted for clarity. (b) Overlay of the molecular structures of 3a–3f, 6c. (c) Two types of B(O,N) chelate ring conformations adopted by the studied complexes. | ||
The supramolecular structures of the studied complexes are dominated by weak HB interactions mostly arranging pyridine nitrogen, chelating oxygen or fluorine atoms as HB acceptors (Fig. 4). The propagation of these contacts results in two types of supramolecular arrangements, i.e., infinite one-dimensional chains (structures 3a, 3c, 3d and 3f) or discrete dimeric motifs (structures 3b, 3e and 6c). The weak HB interactions are usually accompanied by C–H⋯C(π) interactions, although they are not very common as they were observed in the crystal structures of their 9-borafluorene analogues.12 Conversely, diazaborafluorene chelates more likely form π-stacking aggregates (Fig. 5), mainly through mutual interactions between ligands (3c and 3d) or alternating ligand-borafluorene moiety stacking (3f). Notably, J-aggregate motifs, commonly encountered in spiro-organoboron compounds,13 are solely observed for 3e (Fig. S2.4, ESI†).
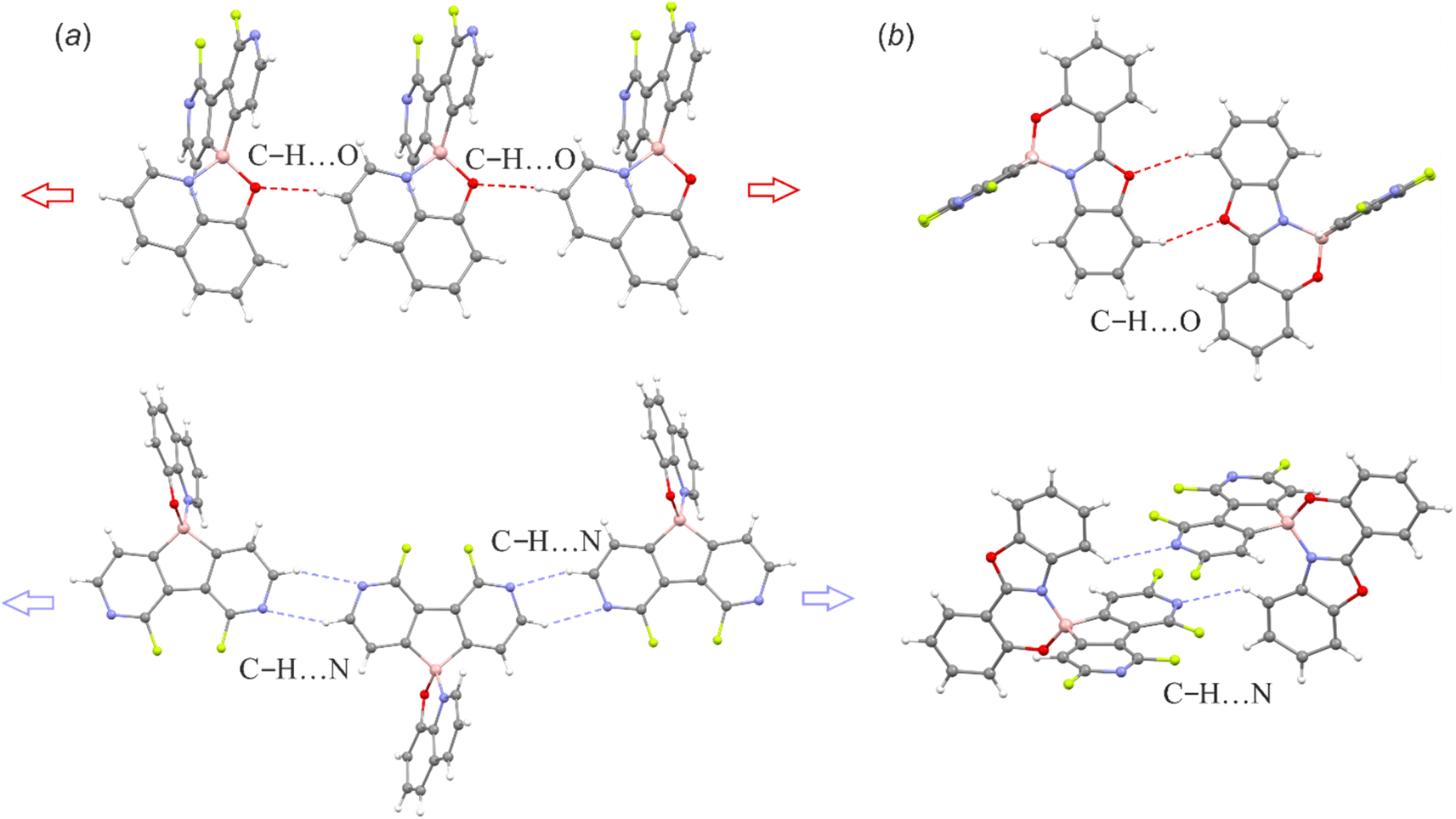 | ||
| Fig. 4 (a) C–H⋯O and C–H⋯N hydrogen-bonded molecular chains in 3a. (b) C–H⋯O and C–H⋯N hydrogen-bonded dimeric motifs in 6c. The supramolecular motifs of remaining structures are presented in the ESI.† | ||
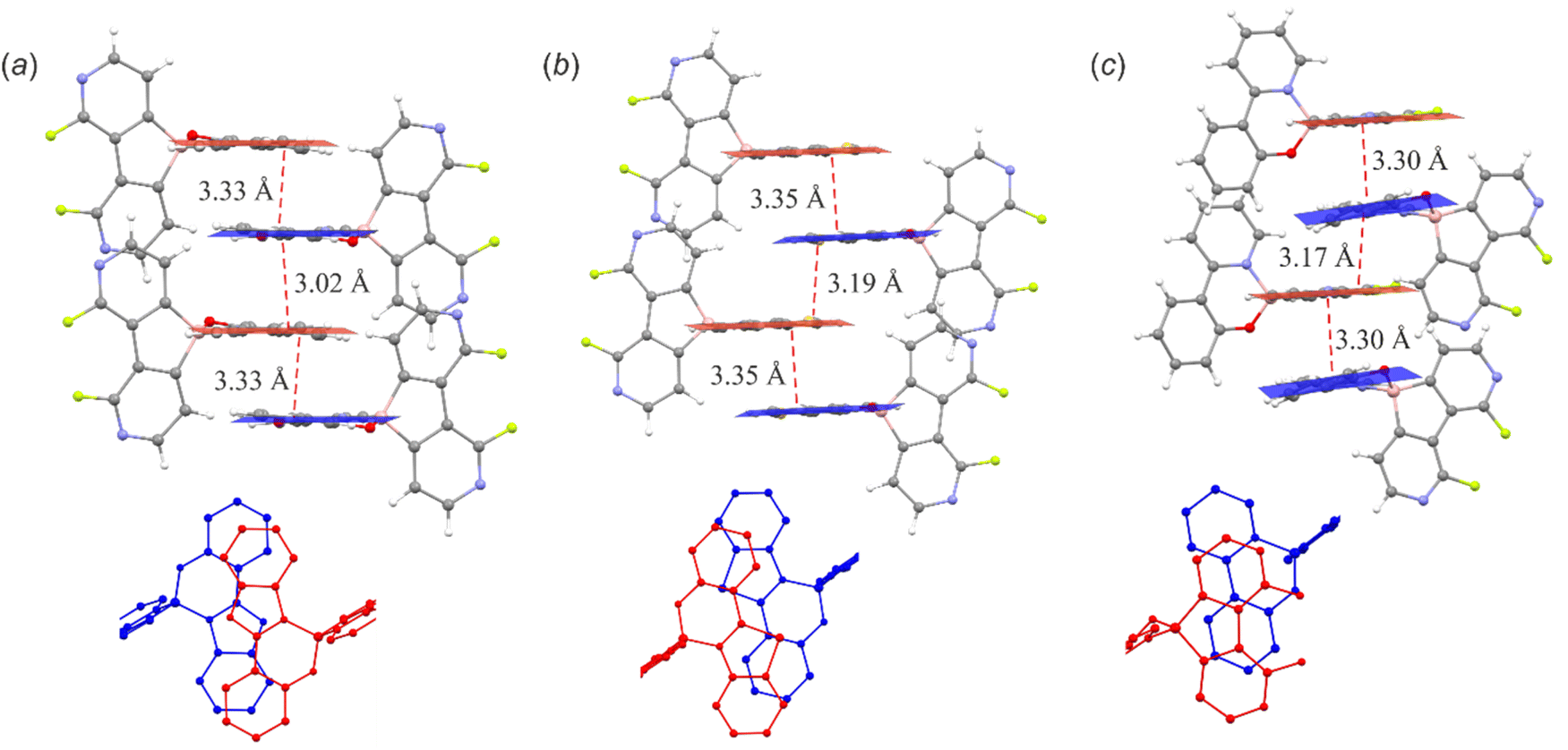 | ||
| Fig. 5 π-Stacking interactions in the crystal structures of (a) 3c (b) 3d and (c) 3f. Distances between stacked planes are additionally provided along with the projection in the direction perpendicular to the planes of the stacking ligands. | ||
The obtained complexes show the longest wavelength absorption bands with maxima in the range of 360–421 nm (CHCl3) with molar extinction values ranging from 2680–16500 M−1 cm−1 (CHCl3) except for 3g showing much higher ε = 109000 M−1 cm−1 (Table 2). Their emission maxima vary in the range of 427–531 nm depending mainly on the ligand type and their luminescence colour can be further tuned by ligand functionalization. For instance, the introduction of the NEt2 group in 3g naturally increases the HOMO energy level, but even more strongly elevates the LUMO (Fig. S3.8, ESI†), leading to an increased band gap and hypsochromic shift of the emission band with respect to 3b (Fig. 6).14 Interestingly, 3g shows the very narrow emission band in the solid state (FWHM = 1700 cm−1). Emission maxima are typically red-shifted in the bulk solid-state (usually up to 20 nm). Exceptionally, complex 3b displays a significant hypsochromic shift of the emission band in the solid state (by 28 nm; 1050 cm−1). This can be connected with the ligand conformational flexibility resulting from the possible rotation of the phenyl group around the single Car–N bond (τPh, Fig. 3) in the less strained solution environment. The TD-DFT calculations for single molecule 3b indicate that the ligand is flattened upon excitation (τPh = 36°), while in the crystal structure it remains twisted around the Car–N bond by τPh = 52(1)° resulting in weakening of π-electron conjugation. In contrast, compound 3e exhibits substantial red-shift of the emission band in the bulk solid-state. The examination of the behaviour of 1 wt% and 5 wt% Zeonex thin films revealed evidence that the emission is systematically shifted as the concentration of the sample is increased. Thus it can be postulated that the observed behaviour is strongly affected by the formation of J-aggregates, which is consistent with the behaviour of other dyes displaying J-aggregate crystal motifs.15 Furthermore, a small shoulder in the emission band of 3e (powder) appears at a wavelength similar to that recorded for respective spectra in solution and Zeonex. This may point to the presence of a fraction of an amorphous or highly disordered phase of 3e in the powder sample.
| CHCl3 solution | Bulk solid-state | Zeonex | ||||
|---|---|---|---|---|---|---|
| λ abs/nm (ε/103 M−1 cm−1) | λ em/nm | QYF/% | λ em/nm | QYF/% | λ em/nm | |
| 3a | 395 (4.26) | 508 | 49 | 510 | 48 | 498 (1%) |
| 3b | 410 (8.38) | 531 | 24 | 503 | 37 | — |
| 3c | 380 (16.5) | 444 | 36 | 463, 539 | 50 | 448 (1%) |
| 3d | 398 (11.4) | 476 | 49 | 493 | 20 | — |
| 3e | 360 (14.6) | 427 | 38 | 494 | 37 | 441 (1%) |
| 458 (5%) | ||||||
| 3f | 364 (6.16) | 466 | 37 | 464 | 33 | — |
| 3g | 421 (109.0) | 476 | 73 | 493 | 50 | — |
| 6a | 396 (2.68) | 505 | 46 | 512 | 66 | 505 (1%) |
| 6b | 407 (6.97) | 525 | 22 | 526 | 16 | — |
| 6c | 377 (9.61) | 439 | 32 | 437, 488 | 37 | 429 (1%) |
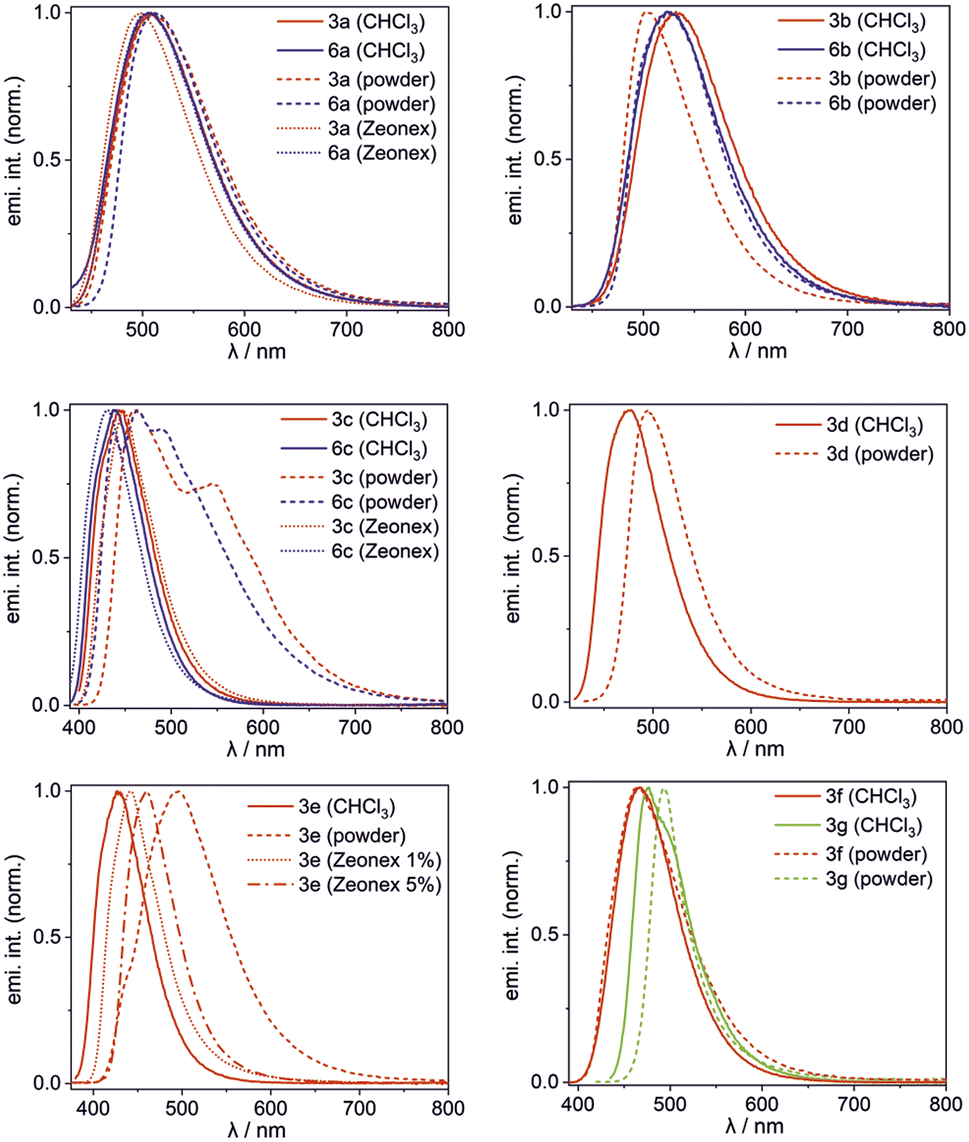 | ||
| Fig. 6 Normalized emission spectra of 3a–3g and 6a–6c in CHCl3 solution, bulk powder and Zeonex thin films. | ||
All complexes are moderate to good emitters with quantum yields in the range of 22–73% (CHCl3) and 16–66% (powder). Notably, in most cases the fluorescence intensities are not affected by solid state aggregation effects. Exceptionally, aggregation-caused quenching was observed for 2-(2-oxyphenyl)benzo[d]thiazole complex 3d (QYFsolution = 49% → QYFpowder = 20%). Although this might be attributed to π-stacking aggregation, the oxazole analogue 3c is characterized by enhanced emission in the solid state (QYFsolution = 36% → QYFpowder = 50%) despite displaying similar π-stacking structural motifs (Fig. 5). The aggregate behaviour of the latter compound (and also its analogue 6c) is also strongly manifested by the appearance of additional intense bathochromically shifted emission bands covering a wide range of the visible spectrum, responsible for net white emission. Even though the TD-DFT calculations may suggest that they result from the emission from the lowest charge transfer state (CT), the emission spectra in Zeonex thin films (1 wt%) are generally retained from the CHCl3 solution confirming the aggregation-caused origin of observed band broadening in the bulk solid-state.
Another interesting luminescent behaviour was observed for quinolate complexes 3a and 6a. The normalized emission spectra in solution, Zeonex thin films and the bulk solid-state perfectly overlap indicating that the emission process is neither dependent on the environment nor on conformational effects. However, we have noted that emission amplifies to some extent upon degassing the CHCl3 solution (Fig. S4.11, ESI†). Furthermore, both systems exhibit biexponential fluorescence decay in CHCl3 with the shorter component attributed to the prompt fluorescence (3a: τPF = 27.7 ns; 6a: τPF = 23.8 ns) and the longer one characteristic for delayed fluorescence (3a: τDF = 10.3 μs; 6a: τDF = 5.7 μs) (Fig. 7).16 The origin of the delayed fluorescence is still not clear, i.e., it may originate either from thermally activated delayed fluorescence (TADF) or triplet–triplet annihilation (TTA). The latter mechanism was recently suggested for related quinolate complexes based on the 9-borafluorene core.17
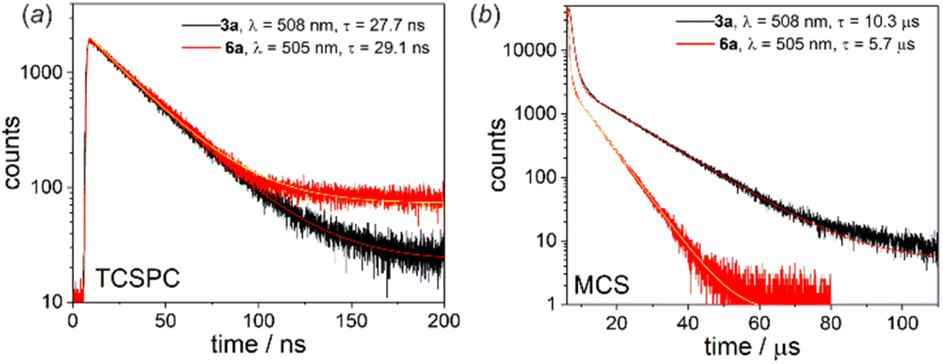 | ||
| Fig. 7 (a) TCSPC and (b) MCS decay traces for 3a and 6a recorded in deoxygenated CHCl3 at RT. | ||
The DFT calculations (B3LYP/6-311++G(d,p)) of 3a–3g and 6a–6c revealed that the HOMO is localized on the diazaborafluorene scaffold whilst the LUMO is spread over the ligand (Fig. 8). Since HOMO−1 is localized on the ligand, the effective π–π* excitation can be described as the HOMO−1 → LUMO transition. This is further confirmed by TD-DFT calculations showing that the observed fluorescence emission is attributed to the second ligand-localized singlet excited state {1LE2(Q)}, while the lowest laying singlet excited state possesses a diazaborafluorene-to-ligand charge transfer character (1CT1) and it is not visible due to its low oscillator strength (Table S3.7, ESI†). In accordance with the above results, the cyclic voltammetry (CV) measurements show that the red-ox processes occur solely on the ligand and are not strongly influenced by the type of organoboron moiety (Fig. S5.1 and Table S5.1, ESI†). It should be noted that reduction and oxidation processes are irreversible, i.e., they are followed by the chemical reactions.
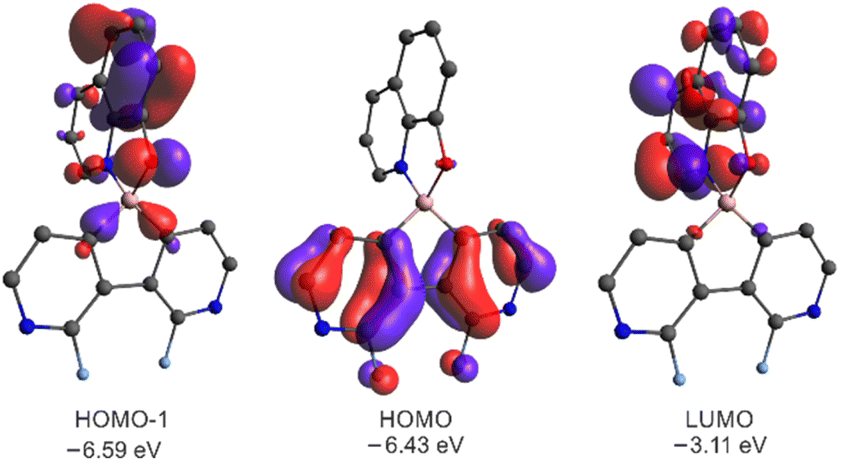 | ||
| Fig. 8 Molecular orbitals for 3a. MOs for remaining systems are presented in Fig. S3.5 and S3.6 in the ESI.† | ||
The calculations of triplet energy levels for 3a and 6a reveal the occurrence of the two lowest triplet excited states with quinoline-localized (3LE1(Q), E = 1.70 eV) and charge transfer (3CT2, E = 2.25 eV) nature, respectively. As initially postulated for boron dipyrromethene (BODIPY) compact donor–acceptor dyads,18 the molecule can transfer to the lowest laying 3LE1(Q) triplet state (E = 1.70 eV) due to direct conversion from the singlet 1CT1 state via the spin–orbit charge transfer intersystem crossing mechanism (SOCT-ISC, Fig. 9). The experimental and theoretical studies confirmed that the SOCT-ISC mechanism operates in a number of BODIPY dyads19 as well as other organoboron complexes based on the borafluorene core2b and it is responsible for the formation of the long-lived triplet state of the molecule.
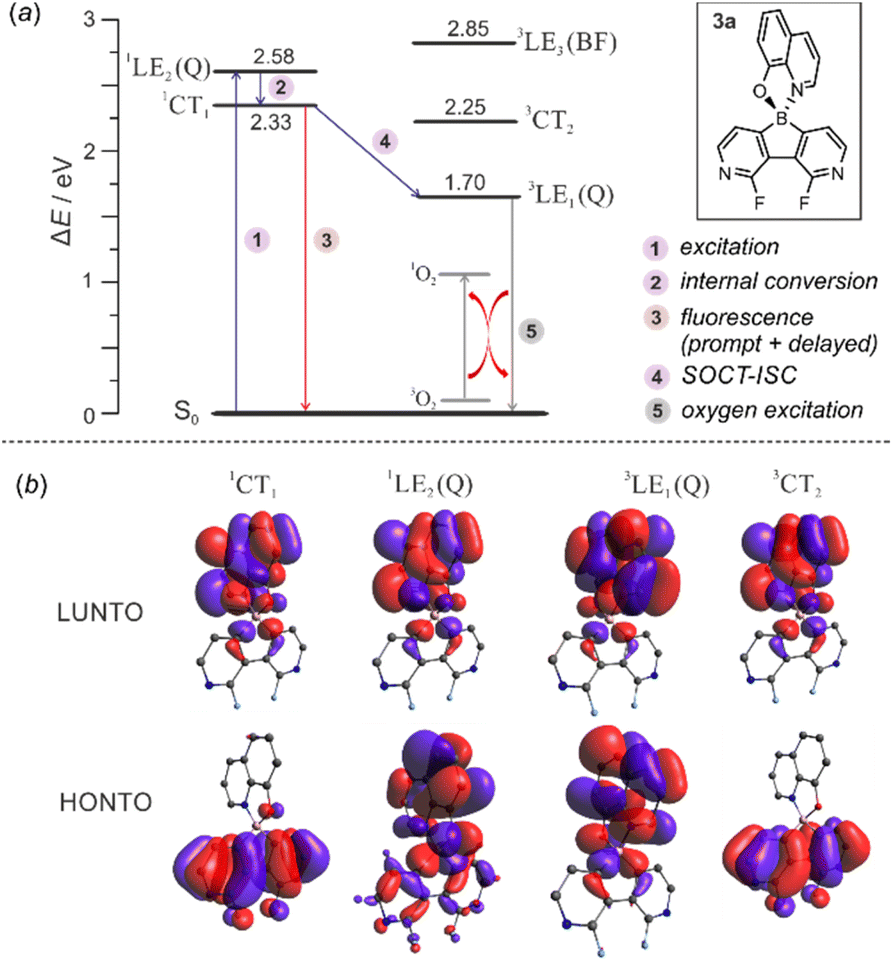 | ||
| Fig. 9 (a) Mechanism underpinning observed photoluminescence and photocatalytic activity in 3a. (b) Visualization of natural transition orbitals in 3a. | ||
The interaction of the photoexcited triplet molecule with naturally abundant triplet oxygen (3O2) leads to the excitation of the latter species to its singlet state (1O2). Since singlet oxygen serves as a powerful oxidant for both small organic molecules and biological macromolecules, it is widely utilized in anticancer photodynamic therapy (PDT),20 organic synthesis,21 and water purification.22 Thus, in the next step we have decided to check the usability of studied diazaborafluorene complexes 3a–3g and 6a–6c as singlet oxygen generators. The photocatalytic activity was quantified by tracking the singlet oxygen-mediated oxidation of 2-furoic acid (FA) – a model reductant. All reactions were performed in CHCl3 using 0.25 mol% photocatalyst loading and the irradiation wavelength was adjusted to respective absorption maxima. The samples were irradiated with a 365 nm (3b, 3c, 3e, 3f, 6b and 6c), 395 nm (3a, 3d and 6a) or 415 nm (3g) LED light source using our home-made reactor (Fig. S6.1, ESI†). All reactions were performed under air at 25 °C and their progress was monitored by 1H NMR spectra analysis of the reaction mixture sampled after a given time. The control experiments showed that the reactions do not proceed in the absence of light or a photocatalyst. We found that quinolate complexes 3a and 6a feature the highest activity with FA conversion reaching 98 and 90%, respectively, after 10 h of irradiation (Fig. 10). Reaction profiles for 3a and 6a show a continuous increase in oxidation product concentration indicating the high stability of photosensitizers under applied conditions. The photostability experiments performed under the same conditions but without FA demonstrate that 6a is characterized by higher stability (half-time decomposition t1/2 = 16 h) with respect to its 3a analogue (t1/2 = 9.2 h). In addition, both complexes are stable in the dark which means that they are not susceptible to chemical degradation (Fig S6.2, ESI†), e.g., hydrolysis resulting from the presence of traces of water in the used solvent.
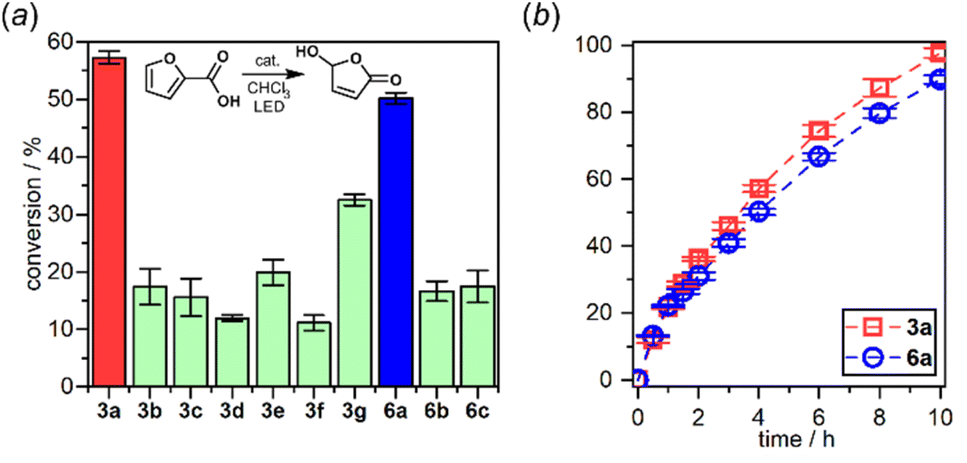 | ||
| Fig. 10 (a) 2-Furoic acid conversion after 4 h of irradiation. (b) Reaction profiles of the best-performing complexes: 3a and 6a. | ||
Conclusions
In summary, two fluorinated diazaborafluorenes 2 and 5 were obtained and characterized as stable water adducts due to the strong Lewis acid properties of the boron atom. DFT calculations confirmed that the oxonium acid form is the most stable, although compound 2 may also equilibrate with its zwitterionic tautomer. Both compounds are characterized by intense blue fluorescence in acidified EtOH solution. In the next step diazaborafluorenes were converted to respective chelate complexes with various (O,N)-ligands. The structural analysis suggests that they are characterized by partial conformational flexibility resulted from B(O,N) chelate ring inversion and ligand in-plane and out-of-plane movements. The molecules interact mainly through C–H⋯O and C–H⋯N hydrogen bonds as well as π-stacking intermolecular interactions, while C–H⋯C(π) contacts are rather avoided. All complexes exhibit moderate-to-good luminescence properties both in solution and the solid state. In most cases the luminescence is red-shifted in the solid state compared to that in solution, but the photoluminescence quantum yields remain at a similar level. In the cases of 3c and 6c, the aggregation leads to the appearance of additional bands covering the wide range of the visible spectrum and resulting in white emission colour. The peculiar nature of electronic excitations and relaxation in quinolate complexes 3a and 6a, manifested by delayed emission and activity in photosensitized 1O2 generation, is the most appealing among other results regarding the optical properties of studied compounds. In fact, such a dual photophysical behaviour was not previously reported for organoboron quinolates. Thus, it seems that the use of proposed boracyclic scaffolds featuring a strong electron-acceptor character can give rise to promising systems for potential diverse applications including organic electronics (electron transport and/or light-emitting materials), photo- and organocatalysis and analytical chemistry (e.g., anion receptors).Data availability
Synthetic procedures, details of crystallographic analyses, characterisation of optical properties and photocatalytic experiments, details of theoretical calculations, NMR and HRMS spectra for all compounds can be found in the ESI.†Author contributions
S. L.: conceptualization of the paper and supervision of the research; S. L., K. D., and P. H. M.-U.: design of the experiments; J. A.: synthesis, performance of photophysical and electrochemical studies; P. H. M.-U.: performance of photocatalytic studies; K. D.: performance of single-crystal X-ray diffraction analyses and theoretical calculations; K. W.: analysis of crystal structures; S. L., and K. D.: analysis of all data; S. L., and K. D.: writing the original draft. All authors have read and agreed to the published version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Science Centre (Poland) within the framework of the OPUS project no. UMO-2020/39/B/ST4/02370 and by Materials_Technologies-3 project granted by Warsaw University of Technology under the programme Excellence Initiative: Research University (ID-UB). Computational facilities were provided by the Wrocław Centre for Networking and Supercomputing (grant no. 285).Notes and references
- (a) A. John, M. Bolte, H.-W. Lerner and M. Wagner, A vicinal electrophilic diborylation reaction furnishes doubly boron-doped polycyclic aromatic hydrocarbons, Angew. Chem., Int. Ed., 2017, 56, 5588–5592 CrossRef CAS; (b) L. G. Mercier, W. E. Piers, R. W. Harrington and W. Clegg, Benzo[b]thiophene-fused boron and silicon ladder acenes, Organometallics, 2013, 32, 6820–6826 CrossRef CAS; (c) S. Ahles, J. Ruhl, M. A. Strauss and H. A. Wegner, Combining bidentate Lewis acid catalysis and photochemistry: formal insertion of o-xylene into an enamine double bond, Org. Lett., 2019, 21, 3927–3930 CrossRef CAS PubMed; (d) S. N. Kessler, M. Neuburger and H. A. Wegner, Domino inverse electron-demand Diels–Alder/cyclopropanation reaction of diazines catalyzed by a bidentate Lewis acid, J. Am. Chem. Soc., 2012, 134, 17885–17888 CrossRef CAS; (e) I. S. Park, K. Matsuo, N. Aizawa and T. Yasuda, High-performance dibenzoheteraborin-based thermally activated delayed fluorescence emitters: molecular architectonics for concurrently achieving narrowband emission and efficient triplet–singlet spin conversion, Adv. Funct. Mater., 2018, 28, 1802031 CrossRef; (f) T. Agou, J. Kobayashi and T. Kawashima, Tuning of the optical properties and Lewis acidity of dibenzopnictogenaborins by modification on bridging main group elements, Inorg. Chem., 2006, 45, 9137–9144 CrossRef CAS PubMed; (g) E. von Grotthuss, A. John, T. Kaese and M. Wagner, Doping polycyclic aromatics with boron for superior performance in materials science and catalysis, Asian J. Org. Chem., 2018, 7, 37–53 CrossRef CAS; (h) K. Durka, I. Głowacki, S. Luliński, B. Łuszczyńska, J. Smętek, P. Szczepanik, J. Serwatowski, U. E. Wawrzyniak, G. Wesela-Bauman, E. Witkowska, G. Wiosna-Sałyga and K. Woźniak, Efficient 8-oxyquinolinato emitters based on a 9,10-dihydro-9,10-diboraanthracene scaffold for applications in optoelectronic devices, J. Mater. Chem. C, 2015, 3, 1354–1364 RSC; (i) A. John, M. Bolte, H.-W. Lerner, G. Meng, S. Wang, T. Peng and M. Wagner, Doubly boron-doped pentacenes as emitters for OLEDs, J. Mater. Chem. C, 2018, 6, 10881–10887 RSC; (j) Y. Ishikawa, K. Suzuki, K. Hayashi, S. Nema and M. Yamashita, Chlorine-substituted 9,10-dihydro-9-aza-10-boraanthracene as a precursor for various boron and nitrogen-containing π-conjugated compounds, Org. Lett., 2019, 21, 1722–1725 CrossRef CAS PubMed; (k) A. Kawachi, H. Morisaka, T. Hirofuji and Y. Yamamoto, Synthesis of silicon-functionalized dibenzosilaborins by intramolecular B–H/C–H dehydrogenative cyclization and their tunable photophysical and chemical properties by silyl groups, Chem.–Eur. J., 2013, 19, 13294–13298 CrossRef CAS PubMed.
- (a) A. C. Murali, P. Nayak and K. Venkatasubbaiah, Recent advances in the synthesis of luminescent tetra-coordinated boron compounds, Dalton Trans., 2022, 51, 5751–5771 RSC; (b) P. H. Marek-Urban, M. Urban, M. Wiklińska, K. Paplińska, K. Woźniak, A. Blacha-Grzechnik and K. Durka, Heavy-atom free spiro organoboron complexes as triplet excited states photosensitizers for singlet oxygen activation, J. Org. Chem., 2021, 86, 12714–12722 CrossRef CAS PubMed; (c) B. M. Bell, T. P. Clark, T. S. De Vries, Y. Lai, D. S. Laitar, T. J. Gallagher, J.-H. Jeon, K. L. Kearns, T. McIntire, S. Mukhopadhyay, H.-Y. Na, T. D. Paine and A. A. Rachford, Boron-based TADF emitters with improved OLED device efficiency roll-off and long lifetime, Dyes Pigm., 2017, 141, 83–92 CrossRef CAS; (d) Y. Tokoro, A. Nagai, K. Tanaka and Y. Chujo, Synthesis of π-conjugated polymers containing aminoquinoline-borafluorene complexes in the main-chain, Macromol. Rapid Commun., 2012, 33, 550–555 CrossRef CAS; (e) D.-G. Chen, R. Ranganathan, J.-A. Lin, C.-Y. Huang, M.-L. Ho, Y. Chi and P.-T. Chou, Ratiometric tuning of luminescence: interplay between the locally excited and interligand charge-transfer states in pyrazolate-based boron compounds, J. Phys. Chem. C, 2019, 123, 4022–4028 CrossRef CAS.
- (a) X. Su, T. A. Bartholome, J. R. Tidwell, A. Pujol, S. Yruegas, J. J. Martinez and C. D. Martin, 9-Borafluorenes: synthesis, properties, and reactivity, Chem. Rev., 2021, 121, 4147–4192 CrossRef CAS PubMed; (b) K. K. Hollister, A. Molino, G. Breiner, J. E. Walley, K. E. Wentz, A. M. Conley, D. A. Dickie, D. J. D. Wilson and R. J. Gilliard Jr, Air-stable thermoluminescent carbodicarbene-borafluorenium ions, J. Am. Chem. Soc., 2022, 144, 590–598 CrossRef CAS PubMed; (c) K. E. Wentz, A. Molino, L. A. Freeman, D. A. Dickie, D. J. D. Wilson and R. J. Gilliard Jr, Approaching dianionic tetraoxadiborecine macrocycles: 10-membered bora-crown ethers incorporating borafluorenate units, Angew. Chem., Int. Ed., 2023, 62, e202215772 CrossRef CAS; (d) S. Fuchs, A. Jayaraman, I. Krummenacher, L. Haley, M. Baštovanović, M. Fest, K. Radacki, H. Helten and H. Braunschweig, Diboramacrocycles: reversible borole dimerisation–dissociation systems, Chem. Sci., 2022, 13, 2932–2938 RSC; (e) H. Budy, S. E. Prey, C. D. Buch, M. Bolte, H.-W. Lerner and M. Wagner, Nucleophilic borylation of fluorobenzenes with reduced arylboranes, Chem. Commun., 2021, 58, 254–257 RSC; (f) M. F. Smith, S. J. Cassidy, I. A. Adams, M. Vasiliu, D. L. Gerlach, D. A. Dixon and P. A. Rupar, Substituent effects on the properties of borafluorenes, Organometallics, 2016, 35, 3182–3191 CrossRef CAS; (g) Z. Zhang, H. Zhang, C. Jiao, K. Ye, H. Zhang, J. Zhang and Y. Wang, 2-(2-Hydroxyphenyl)benzimidazole-based four-coordinate boron-containing materials with highly efficient deep-blue photoluminescence and electroluminescence, Inorg. Chem., 2015, 54, 2652–2659 CrossRef CAS PubMed; (h) S. Yamaguchi, T. Shirasaka, S. Akiyama and K. Tamao, Dibenzoborole-containing π-electron systems: remarkable fluorescence change based on the “on/off” control of the pπ–π* conjugation, J. Am. Chem. Soc., 2002, 124, 8816–8817 CrossRef CAS PubMed; (i) Y. Li, X. Chen, W. Zhang, J. Zhang, L. Xu, Y. Qiao, K. Liu, N. Wang, P. Chen and X. Yin, Substituent modulation for highly bright 9-borafluorene derivatives with carbazole pendant, Org. Lett., 2021, 23, 7236–7241 CrossRef CAS PubMed.
- (a) J. He, F. Rauch, A. Friedrich, J. Krebs, I. Krummenacher, R. Bertermann, J. Nitsch, H. Braunschweig, M. Finze and T. B. Marder, Phenylpyridyl-fused boroles: a unique coordination mode and weak B–N coordination-induced dual fluorescence, Angew. Chem., Int. Ed., 2021, 60, 4833–4840 CrossRef CAS PubMed; (b) J. He, F. Rauch, I. Krummenacher, H. Braunschweig, M. Finze and T. B. Marder, Two derivatives of phenylpyridyl-fused boroles with contrasting electronic properties: decreasing and enhancing the electron accepting ability, Dalton Trans., 2021, 50, 355–361 RSC.
- Y. A. Getmanenko, P. Tongwa, T. V. Timofeeva and S. R. Marder, Base-catalyzed halogen dance reaction and oxidative coupling sequence as a convenient method for the preparation of dihalo-bisheteroarenes, Org. Lett., 2010, 12, 2136–2139 CrossRef CAS.
- H. Matondo, S. Souirti and M. Baboulène, Improved synthesis of azaheteroarylboronic acids using tris(trimethylsilyl)borate under mild conditions, Synth. Commun., 2003, 33, 795–800 CrossRef CAS.
- L. Ernst and K. Ibrom, A new quantitative description of the distance dependence of through-space 19F, 19F spin–spin coupling, Angew. Chem., Int. Ed., 1995, 34, 1881–1882 CrossRef CAS.
- (a) B. J. Graham, I. W. Windsor, B. Gold and R. T. Raines, Boronic acid with high oxidative stability and utility in biological contexts, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2013691118 CrossRef CAS PubMed; (b) A. A. Danopoulos, J. R. Galsworthy, M. L. H. Green, L. H. Doerrer, S. Cafferkey and M. B. Hursthouse, Equilibria in the B(C6F5)3–H2O system: synthesis and crystal structures of H2O·B(C6F5)3 and the anions [HOB(C6F5)3]– and [(F5C6)3B(μ-OH)B(C6F5)3]–, Chem. Commun., 1998, 2529–2560 RSC; (c) K. Durka, S. Luliński, J. Serwatowski and K. Woźniak, Influence of fluorination and boronic group synergy on the acidity and structural behavior of o-phenylenediboronic acids, Organometallics, 2014, 33, 1608–1616 CrossRef CAS.
- S. J. Grabowski, Theoretical studies of strong hydrogen bonds, Annu. Rep. Prog. Chem., Sect. C: Phys. Chem., 2006, 102, 131–165 RSC.
- K. Durka, P. H. Marek-Urban, K. Nowicki, J. Drapała, K. N. Jarzembska, P. Łaski, A. Grzelak, M. Dąbrowski, K. Woźniak and S. Luliński, Expedient synthesis of oxaboracyclic compounds based on naphthalene and biphenyl backbone and phase-dependent luminescence of their chelate complexes, Chem.–Eur. J., 2022, 28, e202104492 CrossRef CAS PubMed.
- (a) D. Salazar-Mendoza, J. Guerrero-Alvarez and H. Höpfl, 3-Pyridineboronic acid → boroxine → pentadecanuclear boron cage → 3D molecular network: a sequence based on two levels of self-complementary self-assembly, Chem. Commun., 2008, 6543–6545 RSC; (b) K. Severin, Boronic acids as building blocks for molecular nanostructures and polymeric materials, Dalton Trans., 2009, 5254–5264 RSC; (c) B. Icli, E. Sheepwash, T. Riis-Johannessen, K. Schenk, Y. Filinchuk, R. Scopelliti and K. Severin, Dative boron–nitrogen bonds in structural supramolecular chemistry: multicomponent assembly of prismatic organic cages, Chem. Sci., 2011, 2, 1719–1721 RSC; (d) E. Sheepwash, V. Krampl, R. Scopelliti, O. Sereda, A. Neels and K. Severin, Molecular networks based on dative boron–nitrogen bonds, Angew. Chem., Int. Ed., 2011, 50, 3034–3037 CrossRef CAS PubMed.
- M. Urban, K. Durka, P. Górka, G. Wiosna-Sałyga, K. Nawara, P. Jankowski and S. Luliński, The effect of locking π-conjugation in organoboron moieties in the structures of luminescent tetracoordinate boron complexes, Dalton Trans., 2019, 48, 8642–8663 RSC.
- (a) K. Yuan, X. Wang, S. K. Mellerup, I. Kozin and S. Wang, Spiro-BODIPYs with a diaryl chelate: impact on aggregation and luminescence, J. Org. Chem., 2017, 82, 13481–13487 CrossRef CAS; (b) P. H. Marek-Urban, K. A. Urbanowicz, K. Wrochna, P. Pander, A. Blacha-Grzechnik, S. T. Hauer, H. R. V. Berens, K. Woźniak, T. J. J. Müller and K. Durka, Bis[1]benzothieno[1,4]thiaborins as a platform for BODIPY singlet oxygen photosensitizers, Chem.–Eur. J., 2023, e202300680 CrossRef CAS PubMed.
- D. Frath, S. Azizi, G. Ulrich, P. Retailleau and R. Ziessel, Facile synthesis of highly fluorescent boranil complexes, Org. Lett., 2011, 13, 3414–3417 CrossRef CAS PubMed.
- (a) J. Mei, N. L. C. Leung, R. T. K. Kwok, J. W. Y. Lam and B. Z. Tang, Aggregation-induced emission: together we shine, united we soar!, Chem. Rev., 2015, 115, 11718–11940 CrossRef CAS; (b) F. Würthner, T. E. Kaiser and C. R. Saha-Möller, J-Aggregates: from serendipitous discovery to supramolecular engineering of functional dye materials, Angew. Chem., Int. Ed., 2011, 50, 3376–3410 CrossRef.
- (a) H. Uoyama, K. Goushi, K. Shizu, H. Nomura and C. Adachi, Highly efficient organic light-emitting diodes from delayed fluorescence, Nature, 2012, 492, 234–238 CrossRef CAS; (b) Y. J. Cho, K. S. Yook and J. Y. Lee, A universal host material for high external quantum efficiency close to 25% and long lifetime in green fluorescent and phosphorescent OLEDs, Adv. Mater., 2014, 26, 4050–4055 CrossRef CAS; (c) B. S. Kim and J. Y. Lee, Engineering of mixed host for high external quantum efficiency above 25% in green thermally activated delayed fluorescence device, Adv. Funct. Mater., 2014, 24, 3970–3977 CrossRef CAS; (d) W. Zeng, H.-Y. Lai, W.-K. Lee, M. Jiao, Y.-J. Shiu, C. Zhong, S. Gong, T. Zhou, G. Xie, M. Sarma, K.-T. Wong, C.-C. Wu and C. Yang, Achieving nearly 30% external quantum efficiency for orange–red organic light emitting diodes by employing thermally activated delayed fluorescence emitters composed of 1,8-naphthalimide-acridine hybrids, Adv. Mater., 2018, 30, 1704961 CrossRef PubMed.
- C. B. Fialho, T. F. C. Cruz, A. I. Rodrigues, M. J. Calhorda, L. F. Vieira Ferreira, P. Pander, F. B. Dias, J. Morgado, A. L. Maçanita and P. T. Gomes, 9-Borafluoren-9-yl and diphenylboron tetracoordinate complexes of F− and Cl− substituted 8-quinolinolato ligands: synthesis, molecular and electronic structures, fluorescence and application in OLED devices, Dalton Trans., 2023, 52, 4933–4953 RSC.
- M. A. Filatov, S. Karuthedath, P. M. Polestshuk, H. Savoie, K. J. Flanagan, C. Sy, E. Sitte, M. Telitchko, F. Laquai, R. W. Boyle and M. O. Senge, Generation of triplet excited states via photoinduced electron transfer in meso-anthra-BODIPY: fluorogenic response toward singlet oxygen in solution and in vitro, J. Am. Chem. Soc., 2017, 139, 6282–6285 CrossRef CAS PubMed.
- (a) Y. Hou, I. Kurganskii, A. Elmali, H. Zhang, Y. Gao, L. Lv, J. Zhao, A. Karatay, L. Luo and M. Fedin, Electronic coupling and spin-orbit charge transfer intersystem crossing (SOCT-ISC) in compact BDP-carbazole dyads with different mutual orientations of the electron donor and acceptor, J. Chem. Phys., 2020, 152, 114701 CrossRef CAS PubMed; (b) Y. Hou, Q. Liu and J. Zhao, An exceptionally long-lived triplet state of red light-absorbing compact phenothiazine-styrylBodipy electron donor/acceptor dyads: a better alternative to the heavy atom-effect?, Chem. Commun., 2020, 56, 1721–1724 RSC; (c) M. Imran, A. M. El-Zohry, C. Matt, M. Taddei, S. Doria, L. Bussotti, P. Foggi, J. Zhao, M. Di Donato, O. F. Mohammed and S. Weber, Intersystem crossing via charge recombination in a perylene–naphthalimide compact electron donor/acceptor dyad, J. Mater. Chem. C, 2020, 8, 8305–8319 RSC; (d) Z. Wang, M. Ivanov, Y. Gao, L. Bussotti, P. Foggi, H. Zhang, N. Russo, B. Dick, J. Zhao, M. Di Donato, G. Mazzone, L. Luo and M. Fedin, Spin–orbit charge-transfer intersystem crossing (ISC) in compact electron donor–acceptor dyads: ISC mechanism and application as novel and potent photodynamic therapy reagents, Chem.–Eur. J., 2020, 26, 1091–1102 CrossRef CAS PubMed; (e) M. A. Filatov, S. Karuthedath, P. M. Polestshuk, S. Callaghan, K. J. Flanagan, M. Telitchko, T. Wiesner, F. Laquai and M. O. Senge, Control of triplet state generation in heavy atom-free BODIPY-anthracene dyads by media polarity and structural factors, Phys. Chem. Chem. Phys., 2018, 20, 8016–8031 RSC; (f) M. A. Filatov, Heavy-atom-free BODIPY photosensitizers with intersystem crossing mediated by intramolecular photoinduced electron transfer, Org. Biomol. Chem., 2020, 18, 10–27 RSC.
- J. P. Celli, B. Q. Spring, I. Rizvi, C. L. Evans, K. S. Samkoe, S. Verma, B. W. Pogue and T. Hasan, Imaging and photodynamic therapy: mechanisms, monitoring, and optimization, Chem. Rev., 2010, 110, 2795–2838 CrossRef CAS PubMed.
- A. A. Ghogare and A. Greer, Using singlet oxygen to synthesize natural products and drugs, Chem. Rev., 2016, 116, 9994–10034 CrossRef CAS PubMed.
- S. Shah, A. Bajaj, A. Shibu, Md. E. Ali and P. P. Neelakandan, Iodo-functionalized salicylideneimine-boron complexes: synthesis and photosensitized degradation of organic water pollutants, Chem.–Eur. J., 2018, 24, 8788–8794 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis, NMR and HRMS data, details of crystal structures, photophysical properties, photocatalytic studies, DFT calculations. CCDC 2243651 (for 3a), 2243652 (for 3b), 2243653 (for 3c), 2243654 (for 3d), 2243655 (for 3e), 2243656 (for 3f), 2243657 (for 6c) and 2293837 (for 5). For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc03876a |
| This journal is © The Royal Society of Chemistry 2023 |