
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAnomalous photochromism and mechanochromism of a linear naphthopyran enabled by a polarizing dialkylamine substituent†
Yan
Sun
 ,
Molly E.
McFadden
,
Skylar K.
Osler
,
Ross W.
Barber
and
Maxwell J.
Robb
*
,
Molly E.
McFadden
,
Skylar K.
Osler
,
Ross W.
Barber
and
Maxwell J.
Robb
*
Division of Chemistry and Chemical Engineering, California Institute of Technology, Pasadena, California 91125, USA. E-mail: mrobb@caltech.edu
First published on 4th September 2023
Abstract
In contrast to common angular naphthopyrans that exhibit strong photochromic and mechanochromic behavior, constitutionally isomeric linear naphthopyrans are typically not photochromic, due to the putative instability of the completely dearomatized merocyanine product. The photochemistry of linear naphthopyrans is thus relatively understudied compared to angular naphthopyrans, while the mechanochromism of linear naphthopyrans remains completely unexplored. Here we demonstrate that the incorporation of a polarizing dialkylamine substituent enables photochromic and mechanochromic behavior from polymers containing a novel linear naphthopyran motif. In solution phase experiments, a Lewis acid trap was necessary to observe accumulation of the merocyanine product upon photochemical and ultrasound-induced mechanochemical activation. However, the same linear naphthopyran molecule incorporated as a crosslinker in polydimethylsiloxane elastomers renders the materials photochromic and mechanochromic without the addition of any trapping agent. This study provides insights into the photochromic and mechanochromic reactivity of linear naphthopyrans that have conventionally been considered functionally inert, adding a new class of naphthopyran molecular switches to the repertoire of stimuli-responsive polymers.
Introduction
Naphthopyrans are molecular switches that undergo a 6π electrocyclic ring-opening reaction upon external stimulation to generate highly colored merocyanine dyes.1 The photochemical ring-opening reaction of naphthopyrans upon irradiation with UV light has been extensively studied.2,3 More recently, it was discovered that naphthopyrans respond similarly to mechanical force as the external stimulus,4 enabling new opportunities to use these versatile switches for stress sensing applications in polymers.5 The nascent field of polymer mechanochemistry explores chemical transformations that are promoted selectively by mechanical force in privileged stress-sensitive molecules called mechanophores.6–8 Mechanochromic mechanophores generate colored products upon mechanochemical activation, facilitating the straightforward visualization of stress and/or strain in materials.9 A rapidly growing class of mechanochromic mechanophores includes spiropyran,10–12 spirothiopyran,13 rhodamine,14–16 oxazine,17,18 π-extended anthracene adducts,19,20 triarylmethanes,21 and diarylbibenzofuranone,22,23 among others. Our group has been particularly interested in naphthopyrans due to their synthetic modularity and structural diversity, providing access to mechanochromic mechanophores and materials with a wide range of functional properties.5,24–29The vast majority of studies on the photochromism and mechanochromism of naphthopyran have focused on the so-called angular naphthopyrans (Scheme 1a).1 Angular naphthopyrans have been extensively studied and developed as photoswitches for commercial applications in photochromic lenses.3 The mechanochemical ring-opening reaction of angular 3H-naphtho[1,2-b]pyran in polymeric materials was first reported in 2016.4 In 2021, a closely related angular 2H-naphtho[2,1-b]pyran scaffold was also demonstrated to exhibit mechanochromic reactivity,26,27 complementing the well-known photochromic behavior. However, the mechanochemical reactivity of a structurally isomeric linear 2H-naphtho[2,3-b]pyran scaffold has not been studied (Scheme 1b). These so-called linear naphthopyrans contain a pyran ring that is linearly oriented with respect to the naphthalene nucleus. In contrast to the angular naphthopyrans, linear naphthopyrans do not typically exhibit photochromic behavior at ambient temperature because the ring-opening reaction results in complete dearomatization of the naphthalene core.1 Therefore, linear naphthopyrans have received relatively little attention as photoswitches and their mechanochemical reactivity is unknown.
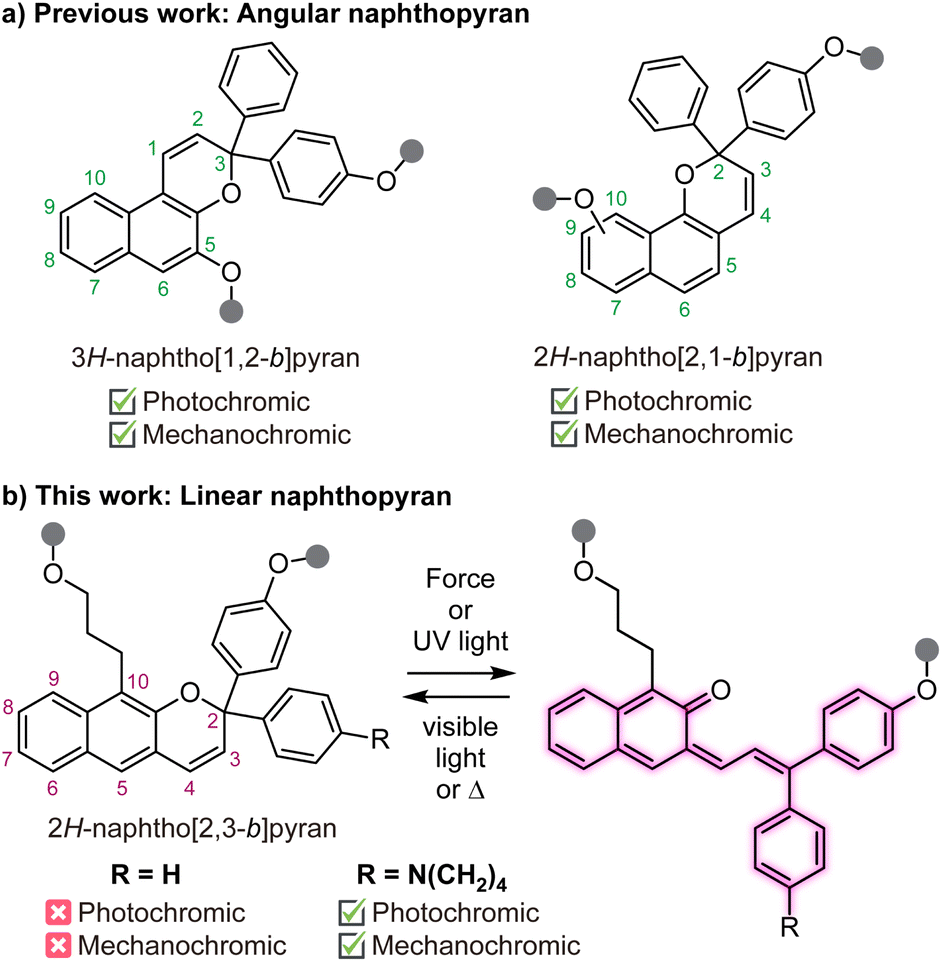 | ||
| Scheme 1 Naphthopyran isomers and their stimuli-responsive behavior. (a) Angular naphthopyrans undergo ring-opening reactions with UV light or mechanical force. (b) Photochromism and mechanochromism of linear 2H-naphthopyran is enabled upon introduction of an electron donating dialkylamine substituent. | ||
We recently demonstrated that mechanical force is capable of promoting the dual ring-opening reaction of naphthodipyran, which diverges from the photochemical reaction where the concurrent ring opening of both pyran units is inaccessible.28 Trapping the putatively unstable dimerocyanine product using a Lewis acid proved critical for observing its formation in solution upon ultrasound-induced mechanochemical activation. Incorporation of an electron rich dialkylamine substituent at the para-position of the pendant aryl groups enables complexation of the merocyanine product with boron trifluoride, which significantly slows or even eliminates thermal reversion.30 Encouraged by these results, we hypothesized that the ring-opening reaction of an appropriately substituted linear naphthopyran may be observable upon trapping with boron trifluoride. Herein, we study the photochemical and mechanochemical reactivity of polymers containing a linear naphthopyran unit. In solution, a polarizing dialkylamine substituent is indeed demonstrated to turn on the photochromic and mechanochromic behavior of linear naphthopyran in the presence of boron trifluoride. Notably, however, silicone elastomers crosslinked with the same dialkylamine-substituted linear naphthopyran motif also exhibit photochromism and mechanochromism in the absence of any trapping agent.
Results and discussion
Density functional theory calculations using the constrained geometries simulate external force (CoGEF) method were first performed to investigate the mechanochemical reactivity of linear naphthopyran.31,32 Models of targeted linear 2H-naphtho[2,3-b]pyrans containing para-H and para-pyrrolidine substituents are both predicted to undergo ring-opening reactions upon mechanical elongation to generate the anticipated merocyanine products (Fig. 1 and S1†). The mechanochemical ring-opening reactions of the linear naphthopyran derivatives with para-H and para-pyrrolidine substitution occur with a maximum force (Fmax) of 4.5 and 4.2 nN, respectively, which are similar to Fmax values predicted by CoGEF for other naphthopyran mechanophores.32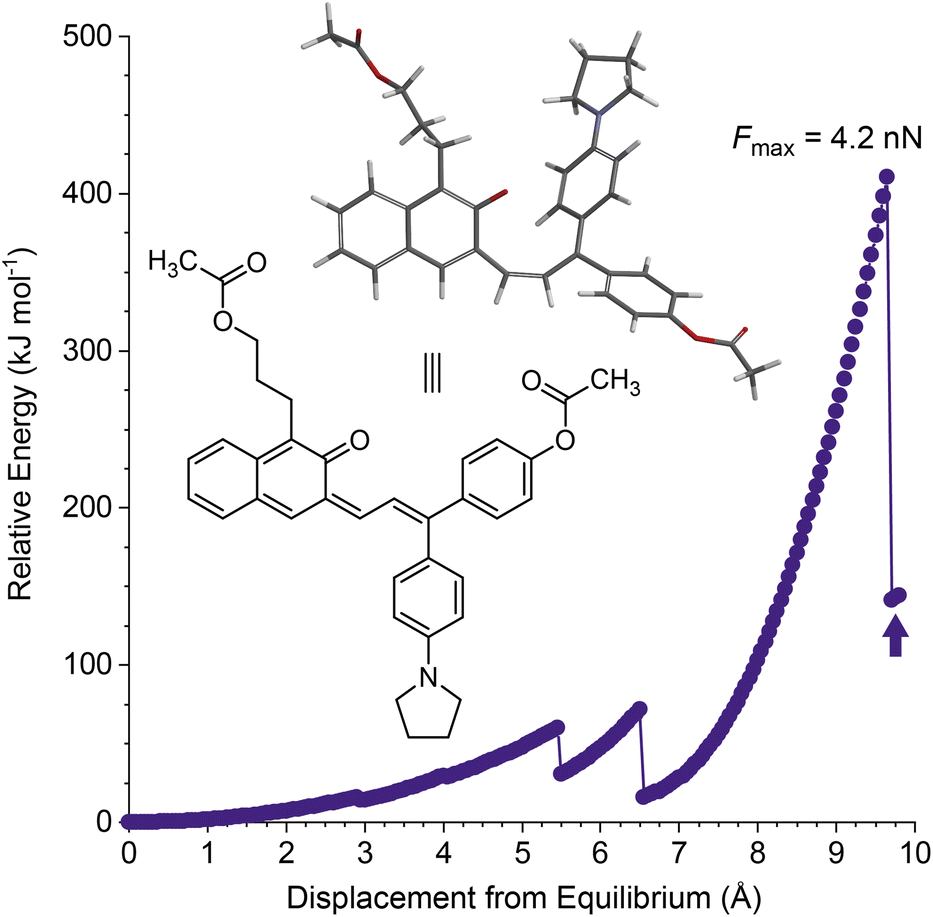 | ||
| Fig. 1 Density functional theory (DFT) calculations using the constrained geometries simulate external force (CoGEF) method performed on a linear 2H-naphtho[2,3-b]pyran model containing a para-pyrrolidine substituent predict a ring-opening reaction upon mechanical elongation. The structure of the predicted merocyanine product is shown, corresponding to the position in the profile denoted by the arrow. The features at ∼5.5 and 6.5 Å displacement correspond to conformational changes. Calculations were performed at the B3LYP/6-31G* level of theory. | ||
Encouraged by these computational results, we next set out to experimentally investigate the photochemical and mechanochemical reactivity of linear naphthopyran. Polymers in dilute solution undergo rapid extension upon ultrasonication with elongational forces maximized near the chain midpoint.33 Therefore, poly(methyl acrylate) polymers containing a chain-centered linear naphthopyran unit (PMA-LNP and PMA-PyLNP) were designed and synthesized, along with polymer PMA-Control containing a pyrrolidine-substituted linear naphthopyran unit at the chain end, which does not experience mechanical force upon ultrasonication (Scheme 2, see the ESI† for details).8 Naphthopyrans are typically synthesized via an acid-catalyzed reaction between a naphthol and a propargyl alcohol.34 However, this protocol is not effective for the synthesis of linear 2H-naphtho[2,3-b]pyrans.35 We therefore employed an alternative Heck coupling strategy for the construction of the linear naphthopyran scaffold.36,37 Synthesis commenced by reacting 1-iodo-2-naphthol with acrylic acid to generate lactone 1, followed by reduction with LAH to afford tethered alcohol 2. Protection of the primary alcohol with tetrahydropyran (THP) furnished naphthol 3, which was engaged in a cross-coupling reaction with allylic alcohols 4 or 5 containing a para-H or para-pyrrolidine substituent, respectively, to afford the corresponding Heck products 6 and 7. Naphthopyran formation and THP removal was then accomplished in a two-step sequence using silica gel in DMF at 135 °C and Amberlyst 15 in methanol at room temperature, respectively. Linear naphthopyran diols 8 and 9 were esterified with bromoisobutyryl bromide to afford bis-initiators 10 and 11, which were employed in the controlled radical polymerization of methyl acrylate using copper wire and Me6TREN in DMSO38 to furnish polymers PMA-LNP (Mn = 132 kDa; Đ = 1.12) and PMA-PyLNP (Mn = 262 kDa; Đ = 1.15) containing a chain-centered linear naphthopyran unit with para-H and para-pyrrolidine substituents, respectively. Polymer PMA-Control (Mn = 238 kDa; Đ = 1.09) containing a pyrrolidine-substituted linear naphthopyran unit at the chain end was synthesized in a similar fashion starting from the naphthopyran with a single α-bromo ester initiating group (see the ESI† for details).
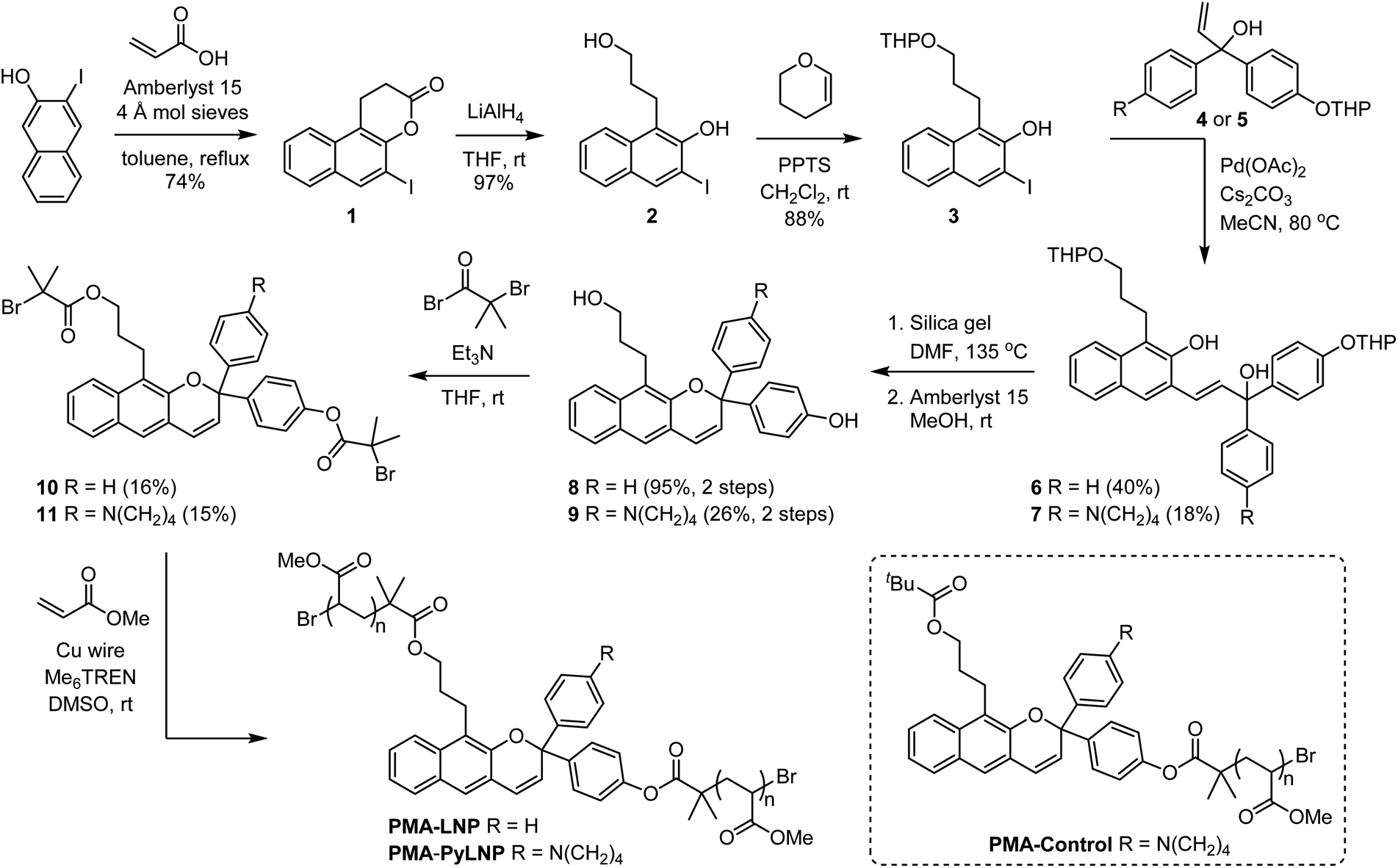 | ||
| Scheme 2 Synthesis of poly(methyl acrylate) polymers incorporating a linear 2H-naphtho[2,3-b]pyran unit. | ||
The photochemical and mechanochemical reactivity of the linear naphthopyrans was evaluated by subjecting dilute solutions of the polymers (2 mg mL−1 in CH3CN with 30 mM BHT) to photoirradiation with 311 nm UV light at −30 °C or continuous ultrasonication at −15 °C, and UV-vis absorption spectra were recorded synchronously using a continuous flow setup (see the ESI† for details).24,26 No photochromic or mechanochromic response was observed for PMA-LNP under these conditions or when similar experiments were performed in the presence of 0.5 mM BF3·Et2O (Fig. S2†). Likewise, no changes in absorption were observed upon photoirradiation or ultrasonication of PMA-PyLNP in the absence of BF3·Et2O (Fig. S3†). However, photoirradiation of PMA-PyLNP in the presence of 0.5 mM BF3·Et2O resulted in a new visible absorption peak at 540 nm (Fig. 2a). An identical absorption spectrum was generated upon ultrasound-induced mechanochemical activation of PMA-PyLNP in the presence of 0.5 mM BF3·Et2O (Fig. 2b). The products were found to be thermally persistent with no thermal reversion of the trapped merocyanine observed upon cessation of ultrasonication (Fig. S4†). Importantly, ultrasonication of PMA-Control under the same conditions in the presence of 0.5 mM BF3·Et2O resulted in negligible changes in absorption, confirming that mechanical force is responsible for the ring-opening reaction of the linear naphthopyran unit in PMA-PyLNP upon ultrasonication in the presence of BF3·Et2O (Fig. 2b and S5†).
 | ||
| Fig. 2 UV-vis absorption spectra of PMA-PyLNP in CH3CN with 0.5 mM BF3·Et2O during (a) photoirradiation at −30 °C with 311 nm UV light, and (b) continuous ultrasonication at −15 °C. Ultrasonication of PMA-Control results in negligible merocyanine formation. Insets show absorbance at 540 nm as a function of photoirradiation or sonication time. | ||
We next explored the potential of the linear naphthopyran mechanophore to serve as a molecular force probe for visualizing stress or strain in solid polymeric materials. Esterification of diol 9 with 4-pentenoic anhydride afforded bis-alkene-functionalized linear naphthopyran Crosslinker-PyLNP, which was covalently incorporated into elastomeric polydimethylsiloxane (PDMS) networks via Pt-catalyzed hydrosilylation (Fig. 3a, see the ESI† for details).12 Interestingly, mechanical activation of the PDMS film via compression using an embossed stamp resulted in purple coloration where force was applied, and a similar chromogenic response was observed upon irradiation with UV light through a simple photomask (Fig. 3b). The purple color generated upon photochemical or mechanochemical activation faded completely after ∼20 min. These results contrast those above for the photochemical and mechanochemical activation of PMA-PyLNP in solution, in which merocyanine accumulation was only observed in the presence of BF3·Et2O. Thermal reversion of naphthopyran-derived merocyanine dyes is significantly slower in bulk polymeric materials,25 but that is unlikely to completely account for the differences between solution and solid state activation. The pyrrolidine substituent presumably favors a zwitterionic form of the merocyanine that regains some degree of aromaticity in comparison to the quinoidal form typical of angular naphthopyran-derived merocyanines. The anionic oxygen atom of the zwitterion may also engage in a stabilizing interaction with the oxophilic silicon atoms in the PDMS network. We note that PDMS materials incorporating a linear naphthopyran crosslinker without a pyrrolidine substituent (i.e., para-H) were prepared in a similar fashion as above, but did not exhibit any photochromic or mechanochromic behavior (Fig. S6).
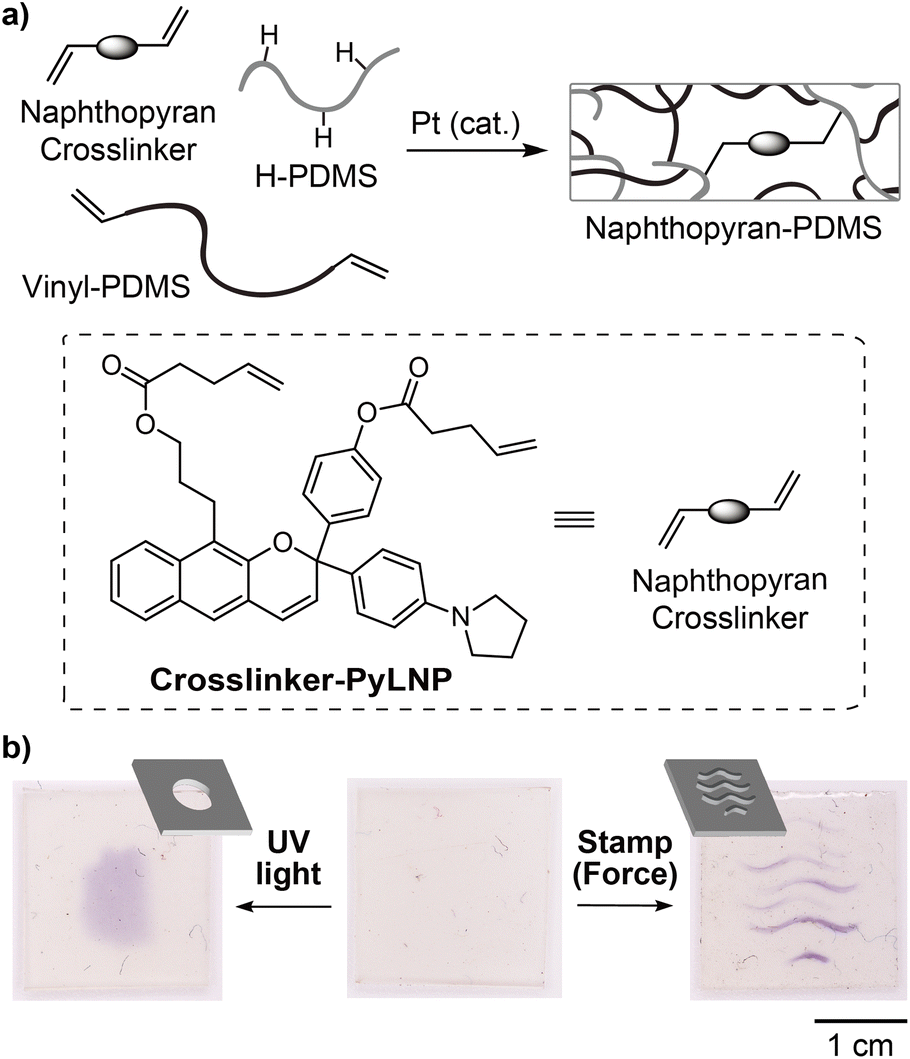 | ||
| Fig. 3 (a) PDMS networks covalently crosslinked with linear 2H-naphthopyran mechanophore Crosslinker-PyLNP (1.5 wt%) prepared via Pt-catalyzed hydrosilylation. (b) Photographs of the material before and after photoirradiation with 365 nm UV light for 120 s through a photomask, and after mechanical force applied via compression (2×) using an embossed stamp. Schematic representations of the photomask and stamp are shown. | ||
Conclusions
Unlike typical angular naphthopyrans that exhibit photochromic and mechanochromic behavior, structurally isomeric linear 2H-naphtho[2,3-b]pyrans are poorly developed because they are commonly considered non-photochromic at ambient temperature, while their mechanochemical activity has not been studied. Here we investigate the photochemical and mechanochemical reactivity of a linear naphthopyran covalently incorporated into polymers and demonstrate that a polarizing dialkylamine substituent enables both photochromic and mechanochromic behavior in solution using UV light and ultrasonication, respectively. Accumulation of the merocyanine dye in solution is attributed to complexation of the ring-opened product with boron trifluoride, which renders it thermally persistent. However, elastomeric polydimethylsiloxane (PDMS) materials incorporating the linear naphthopyran unit as a crosslinker also exhibit photochromic and mechanochromic properties in the absence of any trapping agent. The polarizing dialkylamine substituent may favor the zwitterionic form of the merocyanine dye, regaining some aromaticity and potentially leading to a stabilizing interaction between the anionic oxygen atom and the oxophilic silicon atoms in the PDMS matrix. This study expands knowledge of the reactivity of linear naphthopyrans that have conventionally been overlooked as photoswitches and adds a new class of naphthopyran mechanophores to the catalog for designing stimuli-responsive polymers.Author contributions
M. E. M. conceptualized the project. Y. S., M. E. M., and M. J. R. designed the research. Y. S. performed the experiments. Y. S., M. E. M., S. K. O., R. W. B., and M. J. R. analyzed the data. Y. S. and M. J. R. wrote the manuscript. M. J. R. provided guidance during all stages of the project.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from an NSF CAREER award (CHE-2145791) and the Rose Hills Foundation is gratefully acknowledged. M. E. M. was supported by an NSF Graduate Research Fellowship (DGE-1745301) and a Barbara J. Burger Graduate Fellowship from Caltech. S. K. O. acknowledges support from an Institute Fellowship at Caltech. We thank the Center for Catalysis and Chemical Synthesis of the Beckman Institute at Caltech for access to equipment. M. J. R. gratefully acknowledges the Alfred P. Sloan Foundation for a Sloan Research Fellowship and the Camille and Henry Dreyfus Foundation for a Camille Dreyfus Teacher-Scholar Award.References
- J. D. Hepworth and B. M. Heron, in Functional Dyes, Elsevier Science, 2006, pp. 85–135 Search PubMed.
- B. Van Gemert, in Organic Photochromic and Thermochromic Compounds, Springer, Boston, MA, 2002, pp. 111–140 Search PubMed.
- S. N. Corns, S. M. Partington and A. D. Towns, Color. Technol., 2009, 125, 249–261 CAS.
- M. J. Robb, T. A. Kim, A. J. Halmes, S. R. White, N. R. Sottos and J. S. Moore, J. Am. Chem. Soc., 2016, 138, 12328–12331 CrossRef CAS PubMed.
- M. E. McFadden, R. W. Barber, A. C. Overholts and M. J. Robb, Chem. Sci., 2023 10.1039/D3SC03729K.
- M. K. Beyer and H. Clausen-Schaumann, Chem. Rev., 2005, 105, 2921–2948 CrossRef CAS PubMed.
- M. M. Caruso, D. A. Davis, Q. Shen, S. A. Odom, N. R. Sottos, S. R. White and J. S. Moore, Chem. Rev., 2009, 109, 5755–5798 CrossRef CAS PubMed.
- J. Li, C. Nagamani and J. S. Moore, Acc. Chem. Res., 2015, 48, 2181–2190 CrossRef CAS PubMed.
- R. W. Barber, M. E. McFadden, X. Hu and M. J. Robb, Synlett, 2019, 30, 1725–1732 CrossRef CAS.
- S. L. Potisek, D. A. Davis, N. R. Sottos, S. R. White and J. S. Moore, J. Am. Chem. Soc., 2007, 129, 13808–13809 CrossRef CAS PubMed.
- D. A. Davis, A. Hamilton, J. Yang, L. D. Cremar, D. Van Gough, S. L. Potisek, M. T. Ong, P. V. Braun, T. J. Martínez, S. R. White, J. S. Moore and N. R. Sottos, Nature, 2009, 459, 68–72 CrossRef CAS PubMed.
- G. R. Gossweiler, G. B. Hewage, G. Soriano, Q. Wang, G. W. Welshofer, X. Zhao and S. L. Craig, ACS Macro Lett., 2014, 3, 216–219 CrossRef CAS PubMed.
- H. Zhang, F. Gao, X. Cao, Y. Li, Y. Xu, W. Weng and R. Boulatov, Angew. Chem., Int. Ed., 2016, 55, 3040–3044 CrossRef CAS PubMed.
- Z. Wang, Z. Ma, Y. Wang, Z. Xu, Y. Luo, Y. Wei and X. Jia, Adv. Mater., 2015, 27, 6469–6474 CrossRef CAS PubMed.
- T. Wang, N. Zhang, J. Dai, Z. Li, W. Bai and R. Bai, ACS Appl. Mater. Interfaces, 2017, 9, 11874–11881 CrossRef CAS PubMed.
- M. Wu, Y. Li, W. Yuan, G. De Bo, Y. Cao and Y. Chen, J. Am. Chem. Soc., 2022, 144, 17120–17128 CrossRef CAS PubMed.
- H. Qian, N. S. Purwanto, D. G. Ivanoff, A. J. Halmes, N. R. Sottos and J. S. Moore, Chem, 2021, 7, 1080–1091 CAS.
- Q. Qi, G. Sekhon, R. Chandradat, N. M. Ofodum, T. Shen, J. Scrimgeour, M. Joy, M. Wriedt, M. Jayathirtha, C. C. Darie, D. A. Shipp, X. Liu and X. Lu, J. Am. Chem. Soc., 2021, 143, 17337–17343 CrossRef CAS PubMed.
- R. Göstl and R. P. Sijbesma, Chem. Sci., 2016, 7, 370–375 RSC.
- C. Baumann, M. Stratigaki, S. P. Centeno and R. Göstl, Angew. Chem., Int. Ed., 2021, 60, 13287–13293 CrossRef CAS PubMed.
- J. R. Hemmer, C. Rader, B. D. Wilts, C. Weder and J. A. Berrocal, J. Am. Chem. Soc., 2021, 143, 18859–18863 CrossRef CAS PubMed.
- K. Imato, A. Irie, T. Kosuge, T. Ohishi, M. Nishihara, A. Takahara and H. Otsuka, Angew. Chem., Int. Ed., 2015, 54, 6168–6172 CrossRef CAS PubMed.
- K. Imato, T. Kanehara, T. Ohishi, M. Nishihara, H. Yajima, M. Ito, A. Takahara and H. Otsuka, ACS Macro Lett., 2015, 4, 1307–1311 CrossRef CAS PubMed.
- M. E. McFadden and M. J. Robb, J. Am. Chem. Soc., 2019, 141, 11388–11392 CrossRef CAS PubMed.
- B. A. Versaw, M. E. McFadden, C. C. Husic and M. J. Robb, Chem. Sci., 2020, 11, 4525–4530 RSC.
- S. K. Osler, M. E. McFadden and M. J. Robb, J. Polym. Sci., 2021, 59, 2537–2544 CrossRef CAS.
- M. E. McFadden and M. J. Robb, J. Am. Chem. Soc., 2021, 143, 7925–7929 CrossRef CAS PubMed.
- M. E. McFadden, S. K. Osler, Y. Sun and M. J. Robb, J. Am. Chem. Soc., 2022, 144, 22391–22396 CrossRef CAS PubMed.
- S. K. Osler, M. E. McFadden, T. Zeng and M. J. Robb, Polym. Chem., 2023, 14, 2717–2723 RSC.
- K. Guo and Y. Chen, J. Phys. Org. Chem., 2010, 23, 207–210 CrossRef CAS.
- M. K. Beyer, J. Chem. Phys., 2000, 112, 7307–7312 CrossRef CAS.
- I. M. Klein, C. C. Husic, D. P. Kovács, N. J. Choquette and M. J. Robb, J. Am. Chem. Soc., 2020, 142, 16364–16381 CrossRef CAS PubMed.
- P. A. May and J. S. Moore, Chem. Soc. Rev., 2013, 42, 7497–7506 RSC.
- W. Zhao and E. M. Carreira, Org. Lett., 2003, 5, 4153–4154 CrossRef CAS PubMed.
- C. D. Gabbutt, B. M. Heron, A. C. Instone, D. A. Thomas, S. M. Partington, M. B. Hursthouse and T. Gelbrich, Eur. J. Org Chem., 2003, 1220–1230 CrossRef CAS.
- A. Kicková, J. Donovalová, P. Kasák and M. Putala, New J. Chem., 2010, 34, 1109 RSC.
- J. T. Tee, M. Yaeghoobi, C. F. Chee and N. A. Rahman, Synth. Commun., 2015, 45, 1920–1927 CrossRef CAS.
- N. H. Nguyen, B. M. Rosen, G. Lligadas and V. Percec, Macromolecules, 2009, 42, 2379–2386 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc03790h |
| This journal is © The Royal Society of Chemistry 2023 |