
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhotoswitchable and long-lived seven-membered cyclic singlet diradicals for the bioorthogonal photoclick reaction†
Fuqiang
Hu‡
a,
Cefei
Zhang‡
a,
Zhihao
Liu
a,
Xinyu
Xie
a,
Xiaohu
Zhao
a,
Yanju
Luo
b,
Jielin
Fu
a,
Baolin
Li
a,
Changwei
Hu
 a,
Zhishan
Su
*a and
Zhipeng
Yu
*a
a,
Zhishan
Su
*a and
Zhipeng
Yu
*a
aKey Laboratory of Green Chemistry and Technology of Ministry of Education, College of Chemistry, Sichuan University, 29 Wangjiang Road, Chengdu, 610064, P. R. China. E-mail: suzhishan@scu.edu.cn; zhipengy@scu.edu.cn
bAnalytical & Testing Center, Sichuan University, 29 Wangjiang Road, Chengdu, 610064, P. R. China
First published on 2nd November 2023
Abstract
Annularly 1,3-localized singlet diradicals are energetic and homolytic intermediates, but commonly too short-lived for widespread utilization. Herein, we describe a direct observation of a long-lived and seven-membered singlet diradical, oxepine-3,6-dione-2,7-diyl (OXPID), via spectroscopic experiments and also theoretical evidence from computational studies, which is generated via photo-induced ring-expansion of 2,3-diaryl-1,4-naphthoquinone epoxide (DNQO). The photo-generated OXPID reverts to the thermally stable σ-bonded DNQO with t1/2 in the μs level, thus constituting a novel class of T-type molecular photoswitches with high light-energy conversion efficiency (η = 7.8–33%). Meanwhile, the OXPID is equilibrated to a seven-membered cyclic 1,3-dipole as an electronic tautomer that can be captured by ring-strained dipolarophiles with an ultrafast cycloaddition rate (k2CA up to 109 M−1 s−1). The T-type photoswitchable DNQO is then exploited to be a highly selective and recyclable photoclick reagent, enabling spatiotemporal-resolved bioorthogonal ligation on living cell membranes via a tailored DNQO-Cy3 probe.
Introduction
The exploitation of photochemical transformation1 to establish covalent conjugations in the fashion of click chemistry2–5 has emerged as a cutting-edge research field, named photoclick chemistry.6–9 Photoclick reactions integrate the innate spatiotemporal resolving merits of light rays for non-invasive manipulation over the construction of organic linking frameworks under mild, catalyst-free conditions in a complex environment.10 Therefore, photoclick reactions are elaborate chemical tools for the fabrication or functionalization of materials in 3D space,11–15 tracking of biomolecules in their native context,16–19 and creating hybrid biopolymers in situ.20,21Photoclick reactions employing two inert and exogenous functionalities prior to photoactivated ligation are amenable to single-cell or subcellular bioorthogonal labeling strategies.22–28 During the past decade, unremitting efforts have been made to advance this type of photoclick reaction to meet the strict criteria for utilization in living systems.7,25 The cutting-edge approaches encompass light-triggered copper(I)-catalyzed azide–alkyne cycloaddition;29 photo-uncaged cyclooctyne for cycloaddition with azido-reporters;30 the cycloadditions of nitrile imine (NI) produced by photolysis of tetrazole31 or sydnone32 toward alkene/-yne or cyclic azobenzene; photo-induced hetero-Diels–Alder (HDA) reactions, including photo-oxidative inverse-electron-demand Diels–Alder reactions33 and light-triggered phenanthrenequinone-alkene cycloadditions;34 light-promoted thiol–ene and –yne ligations.35 Since a prolonged exposure of UV light causes photodamage to cells, photoclick reactions triggered by visible or near-IR irradiation with high quantum efficiency are highly advantageous for in vivo applications. The photo-sensitizing approaches can be a good solution for achieving visible or near-IR photoclick reactions.33
The prerequisites for achieving superior spatial resolution and temporal manipulation in photoclick labeling depend on the chemoselectivity, lifetime, and transition pathway of photoexcited intermediates.8,36 Concerning the behaviours of the intermediates, photoclick reactions could be categorized into irreversible and reversible intermediate types,7 among which the reversible type is exemplified by the reversible photo-dehydration of 3-(hydroxymethyl)-2-naphthol (HMNP) in aqueous solution37 and the P-type photoswitchable oxidopyrylium ylide (PY).38 The reversible intermediates can be fully restored to the inert precursor to circumvent undesired conjugation towards native biomolecules and to compensate for molecular diffusion, especially when the target reporter is not in proximity in a crowded microenvironment. Although promising, there are some challenges associated with current approaches, including inefficient suppression of side-reactions toward native thiols and other nucleophiles, an insufficient reaction rate to overcome the accessibility to extremely low-concentrated target reporters and inadequate photo-fatigue resistance of these molecular photoswitches. Therefore, studies on expanding photoswitchable intermediates with robust photo-stability and superb bioorthogonality remain challenging but highly desirable.
The excited triplet states are relatively long-lived and key species during many photochemical conversions8 (e.g., photoisomerization).39–42 They can be indirectly populated from intersystem crossing (ISC) of the excited singlet state that is commonly produced via photoexcited S0 → Sn transitions. However, the excited triplet states might not be ideally orthogonal to native biochemistry because their Dexter energy transfer43,44 to triplet oxygen (3O2), thiols and other redoxes often resulted in reactive oxygen species (ROS),45,46 radicals, and subsequently stress to live cells.
Interestingly, localized singlet diradicals (SDRs) are highly reactive intermediates in σ-bond homolysis promoted by an external stimulus (e.g., light and/or thermal).47–49 And, they are relatively insensitive to triplet species, including 3O2, due to an inefficient ISC at room temperature,50 making them potential bioorthogonal intermediates. The lifetime of SDRs is, however, quite short due to the ultrafast radical–radical coupling back to the σ-bonded state,51 which is adaptive to building T-type photoswitches but difficult to seize.52,53 Over the past decade, breakthroughs have been achieved in the generation of long-lived 1,3-SDRs, which enables direct observation of the chemical equilibrium between the SDR and the σ-bonded species.54,55 Heteroatoms were found to stabilize four-membered annular SDRs and even enable their isolation (Scheme 1a).56–58 Meanwhile ring-strain is also conducive to kinetical stabilization of photo-generated five-membered cyclic SDRs, reported by Abe et al.59 Notably, photo-generated long-lived 4,4-dimethoxy/4-sila-3,5-diphenyl-pyrazolidine-3,5-diyl was discovered to be buffered into a relatively stable cyclo[2,1,0] motif/zwitterion (Scheme 1b), which itself could be trapped by 4-phenyl-1,2,4-triazole-3,5-dione (PTAD) to establish polycyclic adducts.55,60 Despite these breakthroughs, dienophiles/dipolarophiles capable of capturing photoswitchable SDRs to compete against the ultrafast σ-self-coupling are yet to be adequately explored.
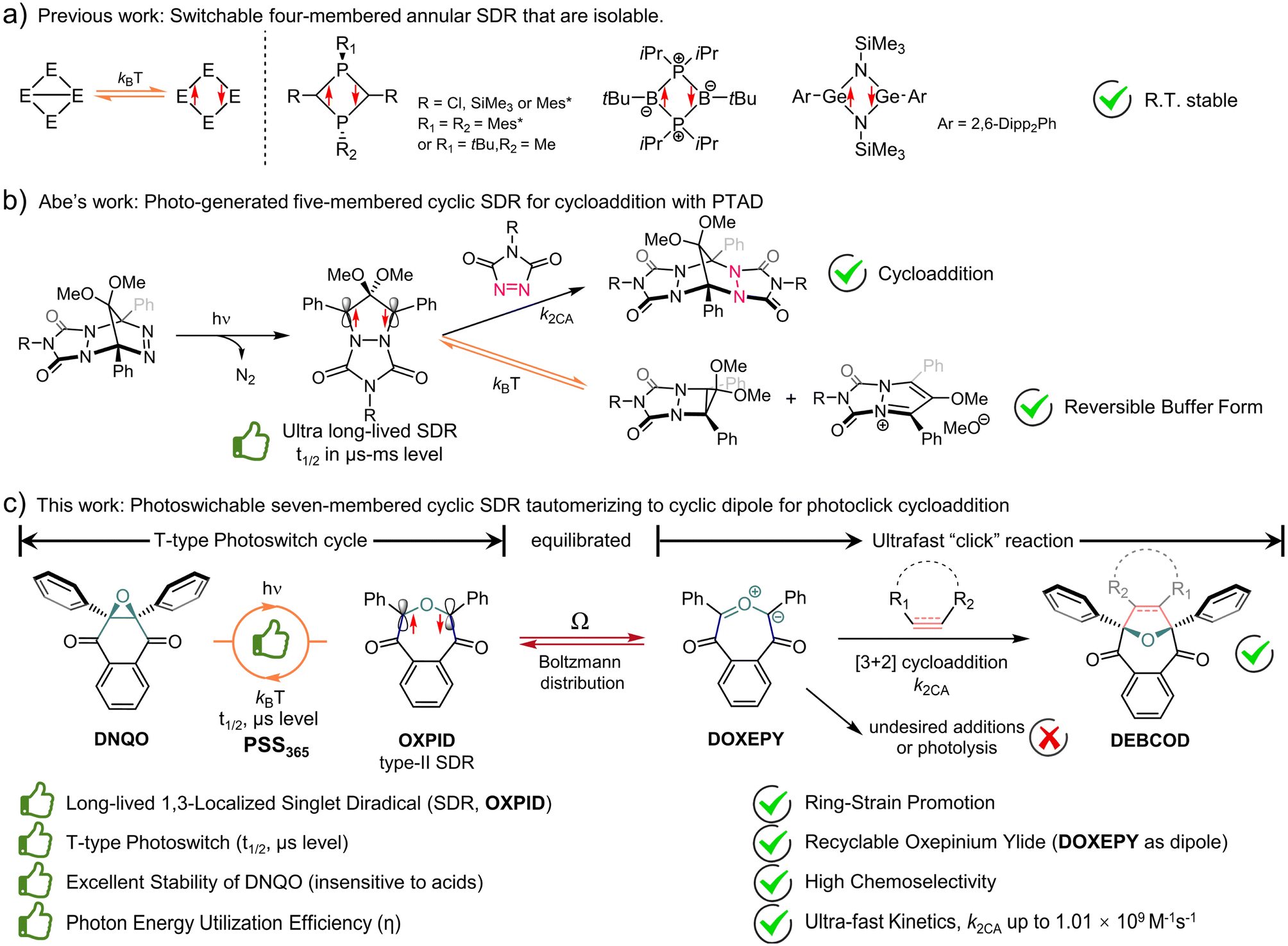 | ||
| Scheme 1 The comparison of the classic (a) four-membered and (b) the switchable five-membered cyclic SDRs versus (c) the seven-membered cyclic SDR which equilibrates to the cyclic 1,3-dipole via electronic tautomerization for subsequent cycloaddition with ring-strained dipolarophiles. The T-type photoswitchable SDRs serve as a novel photoclick reagent. | ||
Herein, a class of ring-expansion/contraction T-type photoswitches between 2,3-diaryl-1,4-naphthoquinone epoxide (DNQO) and oxepine-3,6-dione-2,7-diyl (OXPID, Scheme 1c)61,62 was revealed by nanosecond transient absorption spectroscopic (NTAS) analyses. The photochemical homolysis of the carbon–carbon bond in the epoxy moiety of DNQO and the characters of the 1,3-diaryl seven-membered cyclic SDRs (OXPID) were detailed for the first time via NTAS, electron paramagnetic resonance (EPR), triplet radical capturing, 3O2 quenching and the solvent effect in harmony with computational studies. Engrossingly, we found that the photo-energy harvested SDRs (conversion efficiency: η up to 0.33) is seamlessly tautomerized to 3,6-dioxooxepinium-7-ylide (DOXEPY) as an energetic 1,3-dipole, which can undergo an ultrafast [3 + 2] cycloaddition (k2CA up to 1.01 × 109 M−1 s−1) with endo-bicyclo[6.1.0]nonyn-9-yl-methanol (endo-BCN-OH). The light-driven dynamic cycle of the T-type photoswitching was intercepted by the competitive cycloaddition. Therefore, a novel photoclick reaction was proposed and explored, in which the T-type photoswitchable OXPIDs serve as a recyclable intermediate to minimize unwanted side-reactions (towards, e.g., thiol additions) for bioorthogonal photolabeling with better spatiotemporal resolution.
Results and discussion
Absorbance spectroscopic characterization of DNQO
Back in 1974–1980, photo-triggered cycloadditions between DNQO and alkenes were initially explored by Maruyama, Tezuka, and Osuaka et al.,63 in which the interconversion among DNQO ⇌ DR ⇌ DOXEPY had been hypothesized. In contrast to 2,3-nondiaryl-NQO, 2,3-diaryl-DNQO prefers to photo-react with dipolarophiles and shows resistance to nucleophilic additions and oxygenation,64 which inspire us to conceive bioorthogonality. Therefore, we commenced to establish a library of DNQOs with various heteroaryls or electron donors (eD) and electron acceptors (eA) on the 2,3-diaryl terminals (1b–24b, Fig. 1a and Table S1, ESI†). The UV-Vis spectroscopic characters of the representative DNQOs were summarized with two absorption bands observed (311 ± 20 nm and 340 ± 40 nm) and two extinction coefficients (ε311 and ε365) displayed in Fig. 1b, respectively (also in Table S2, ESI†). TD-DFT calculation and natural transition orbital (NTO) analysis65 indicated that the two absorption bands around 311 nm and 340 nm belong to the S0 → S4 and S0 → S2 transitions of 1b (Fig. 1c, for computational details, Fig. S22, ESI†), respectively, which are π–π* and n–π* type transitions with the oscillator strength, f = 0.211 and 0.009.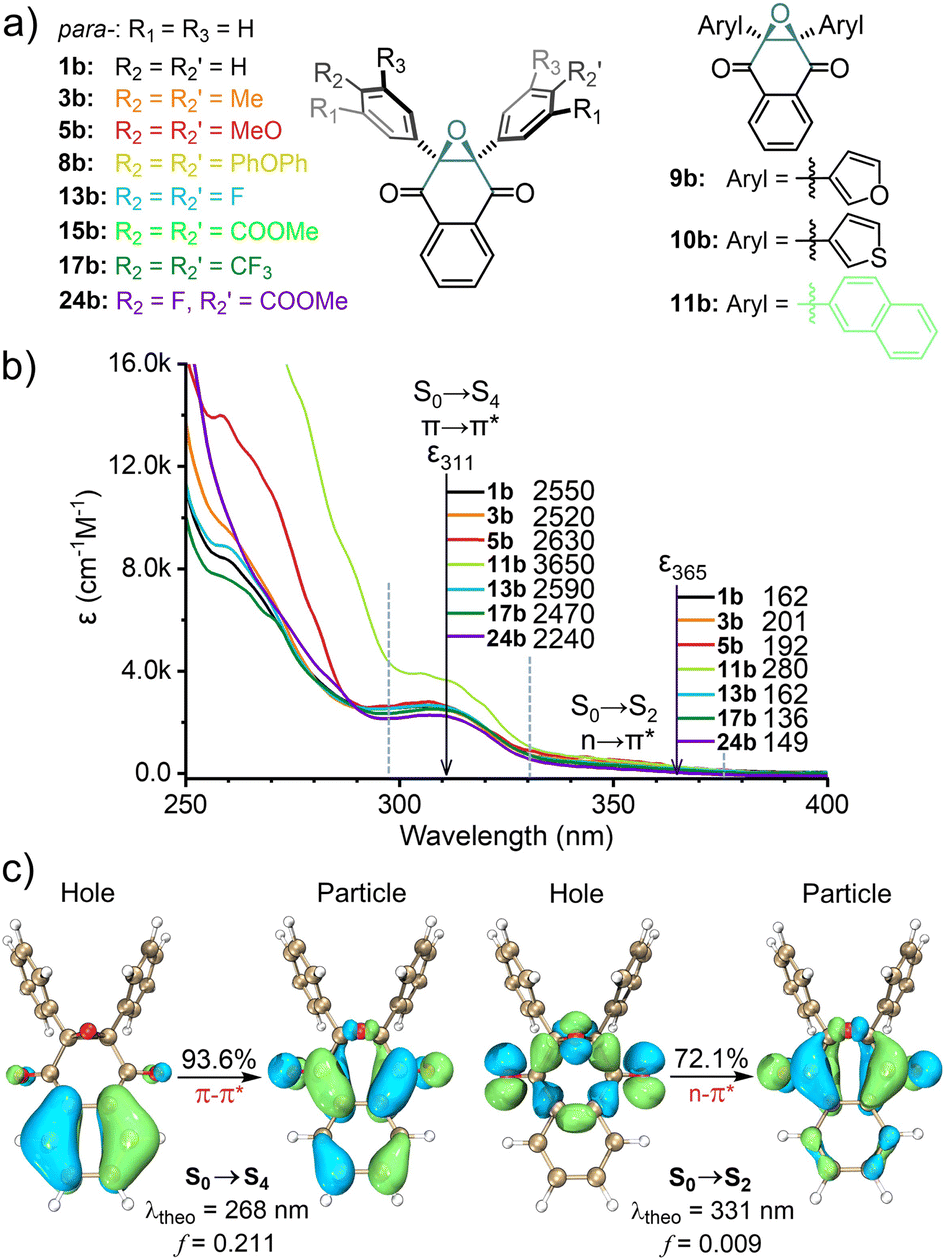 | ||
Fig. 1 (a) The structure of the representative DNQOs. (b) Absorption spectra of 100 μM DNQOs in CH3CN![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2O = 1:1 mixed solvent at 298 K, and the molar absorption coefficients (ε) at the designated wavelengths. (c) Theoretical absorption wavelength (λtheo) and the NTOs of the S0 → S4 and S0 → S2 transitions for 1b, calculated at the TD-M06-2X/6-31G**(SMD, ACN:H2O = 1:1) level of theory. The oscillator strength (f) and eigenvalue of each NTO pair (in percentage) were shown, and the isovalue of NTO plots was 0.04. :H2O = 1:1 mixed solvent at 298 K, and the molar absorption coefficients (ε) at the designated wavelengths. (c) Theoretical absorption wavelength (λtheo) and the NTOs of the S0 → S4 and S0 → S2 transitions for 1b, calculated at the TD-M06-2X/6-31G**(SMD, ACN:H2O = 1:1) level of theory. The oscillator strength (f) and eigenvalue of each NTO pair (in percentage) were shown, and the isovalue of NTO plots was 0.04. | ||
The orbital distributions also suggest that both the π–π* and the n–π* transitions were mainly involved by the oxygen atoms of the epoxide together with the two carbonyl groups, while the 2,3-diaryl terminals have negligible contribution. Therefore, it rationalizes the experimental observation that variation on the diaryl moiety has a trivial effect on the absorption wavelength.
Screening and optimization on the photoclick reactions
To identify a robust reactivity, DNQO (100 μM) was subjected to photo-activation in the presence of typical alkenes/alkynes (500 μM) via irradiation with various light sources (e.g., a 311 nm hand-held mercury lamp and 365 nm or 373 nm LED arrays) for 60 s in a mixed aqueous solvent and comparing with conditions of keeping in the dark for 48 h, respectively (Fig. 2 and Tables S3–S5, ESI†). The alkenes with ring-strain (1c), electron-deficiency (2c–3c), and electron-donors (4c) versus the alkynes with ring-strain (6c and 7c), alkyl substituent (8c), and electron-deficiency (9c) were compared to examine both the electron effect and the ring-strain promotion on the cycloaddition. Based on quantitative HPLC-MS screening (Fig. 2a–c), the results can be summarized as follows: (i) the dipolarophiles without both ring-strain and sufficient electron-deficiency (3c, 4c and 8c) barely reacted with all the DNQOs. Delightedly, the ring-strained dipolarophiles (1c, 6c and 7c) could photo-react efficiently with the majority of the DNQOs, while the highly electron-deficient ones (2c and 9c) were able to afford cycloadducts sluggishly; (ii) in the presence of the ring-strained dipolarophiles, DNQOs possessing electron withdrawing groups (EWGs) or fluorine substituents (e.g., 12b–17b and 19b–20b) tend to show excellent conversions (>80%), compared with very poor conversion for those with multiple electron-donating substituents (e.g., 4b–7b); (iii) without appropriate dipolarophiles, most of the DNQOs appear to be inert under continuous irradiation, especially under a 365 or a 373 nm LED. But severe photolyzes were observed for most of the DNQOs under 311 nm light, which induces mainly the S0 → S4 transition of DNQO. These phenomena reflect the reversible and the short-lived nature of the photogenerated intermediates in the cycloaddition reaction; (iv) there was no background reactions detected in the dark even after incubation for 48 h, implying a high stability of DNQOs; (v) for the ring-strain dipolarophiles, the reaction of 7c towards DNQOs was found to be slower than 1c and 6c, probably due to the steric effects. Therefore, DNQOs 1b, 3b, 5b, 13b, 17b, and 24b with representative substituents were preferred candidates for in-depth evaluation of the performance of the photoclick reaction toward the ring-strained BCN-OH. 365 nm LED was chosen as an optimal light source, considering the high phototoxicity, the photolysis of DNQOs under 311 nm light as well as the poor photo-conversion under 373 nm light (Table S2, ESI†). After a synthetical scale preparation (0.48 mmol scale and 71% yield) and crystallization, the structure of the endo-cycloadduct (1DEBCOD, Fig. 2d) from 1b and BCN-OH was identified via single crystal XRD (Table S7, ESI†).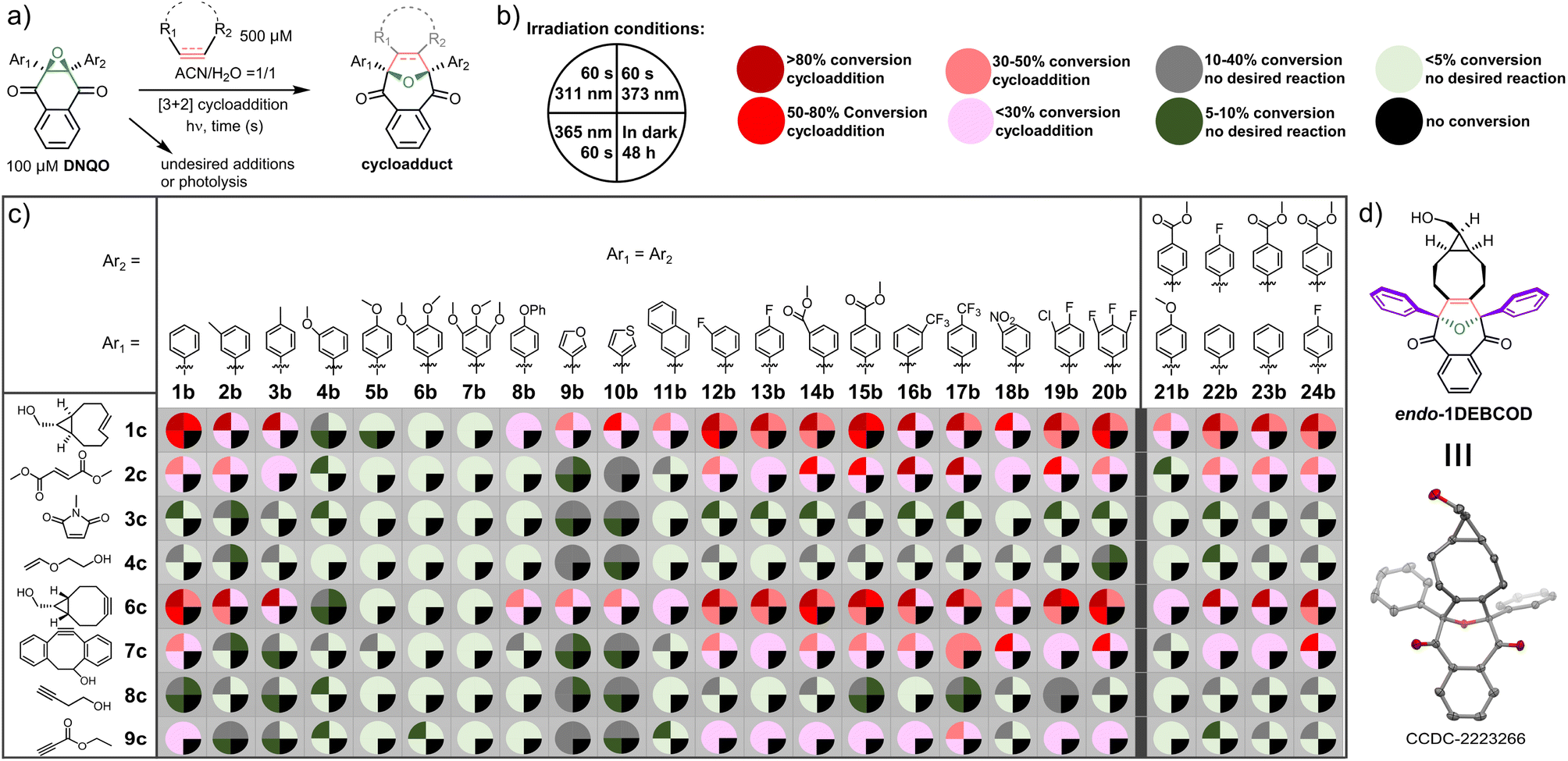 | ||
| Fig. 2 Screening on DNQO/dipolarophile combinations for the photo-initiated or thermal cycloaddition reactions. Experiments were carried out at 298 K in ACN:H2O = 1:1, irradiated with the indicated light sources. [DNQO] = 100 μM and [dipolarophile] = 500 μM. (a) Reaction schemes. (b) The four irradiation conditions for screening, and the colour codes for the characteristics of each photo-induced [3 + 2] cycloaddition analysed via HPLC-MS. (c) Colour-coded screening results. (d) X-Ray structure of the endo-cycloadduct between 1b and 6c. | ||
Proofs for the presence of SDRs and the substituent effects
Given the relatively short-lived intermediates (SDRs, Fig. 3a) from the photochemical conversion of DNQO 1b, we adopted NTAS measurement (λex. = 355 nm, 1 ns pulse duration, 7 μJ, and 1k Hz) to unravel the absorption spectrum and the decaying of the intermediates in CH3CN:H2O = 1:1. Intriguingly, a strong absorption band was observed in the visible region (λmax = 550 nm, Fig. 3b) with an exponential decay to the original state (t1/2 = 18 μs, Fig. 3c) under an air atmosphere. This transient species was then discovered to be a seven-membered cyclic singlet diradical, 1SDR, based on the following experimental evidence:55,59 (i) this species absorbing at 550 nm was EPR-silent in a methanol matrix at 140 K (Fig. S6, ESI†); (ii) the lifetime of this transient species was scarcely altered in the presence of saturated 3O2 or by typical triplet quenchers (Fig. S4, ESI†); (iii) spin trapping of this transient intermediates (5.0 mM 1b in ACN/H2O at 298 K) by radical scavengers,66e.g., 5,5-dimethyl-1-pyrroline N-oxide (DMPO), under continuous 365 nm irradiation resulted in failures in identifying the corresponding spin adducts based on HPLC-MS analysis (Fig. S7, ESI†).
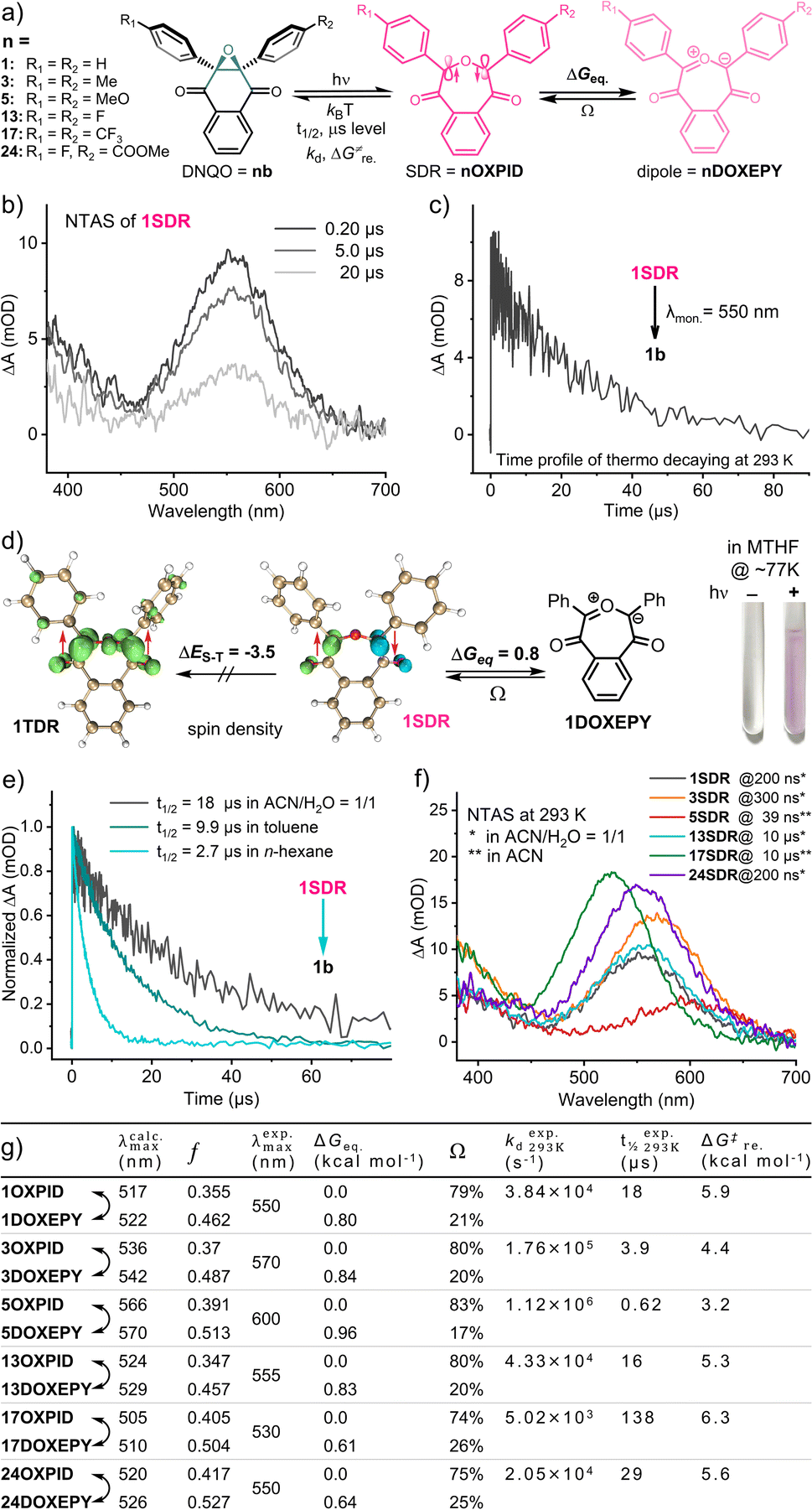 | ||
| Fig. 3 (a) The scheme for the T-type photoswitching of DNQOs and the substitution effect on the tautomerization of SDRs ⇌ DOXEPYs in CH3CN:H2O = 1:1 at 293 K. (b) Transient absorption spectrum of 1SDR at various delay times, generated from the flash photolysis of 1b (λex. = 355 nm). (c) Time-dependent decaying profile of 1SDR (λmon. = 550 nm). (d) Visualization of the spin density analysis (isovalue = 0.01), the relative energies for 1TDR ⇌ 1SDR ⇌ 1DOXEPY, and the photograph of in situ formed 1SDR in an NMR tube. (e) Transient decaying traces of 1SDR in various solvents and corresponding half-life. (f) Transient absorption spectra of the selected SDRs at 293 K. (g) The tabular parameters for the spectral characters of the transient species and their thermodynamic properties. | ||
As theoretical proofs, the optimized geometry of 1SDR was obtained by the broken-symmetry wavefunction method (for computational details, see the ESI†).67,68 Spin density analysis (by using Multiwfn 3.8dev69 and visualization by using VMD 1.9.370) support the existence of single electrons with opposite spin on the two bond-broken carbon atoms (Fig. 3d). It is also calculated that the Gibbs free energy of 1SDR is lower than that of the triplet diradical 1TDR, exhibiting a prominent singlet-triplet energy gap, ΔES–T = −3.5 kcal mol−1, and S2 = 0.32 at the M06-2X/6-31G** level of theory (by using Shermo 2.3,71Fig. 3d and Tables S9 and S14, ESI†). The predicted λmax for the absorption of 1SDR in the visible region (λcalc.max = 517 nm and f = 0.355) at the TD-UM06-2X/6-31G** level of theory72 also matches well with the experimental value, λexp.max = 550 nm (Fig. 3g, Table S11, ESI†). According to the most stable electronic configuration in the ground-singlet-state, SDRs can be classified into two types. Type-I has the symmetric orbital ψS as the highest occupied molecular orbital (HOMO) and the antisymmetric orbital ψA as the lowest unoccupied molecular orbital (LUMO).59 In contrast, type-II adopts ψA as the HOMO and ψS as the LUMO. CASSCF calculations at the CAS(2,2)/6-31G**(SMD, ACN:H2O = 1:1) level of theory indicated that 1SDR is a type-II SDR with the HOMO of antisymmetric ψA (Fig. 4a). The coefficients (c) of electronic configurations I and III were 0.92 and −0.38, respectively. Therefore, the numbers of occupied electrons on the ψA and ψS orbitals were 1.69 (= 2 × 0.922) and 0.29 [= 2 × (−0.38)2], respectively (Fig. 4a). And the diradical character y of 1SDR was determined to be 0.29.
 | ||
| Fig. 4 (a) The molecular orbitals of the two active spaces (ψA and ψS) of 1SDR are shown (isovalue = 0.05) with the values of the occupation numbers of the two electrons displayed in parentheses. The three electronic configurations (I–III) with the corresponding coefficients “c” are also given. (b) Gibbs free energy profile for the TS and intermediates in the photoswitching and the cycloaddition reaction pathways. The red pathway is the radical σ-coupling of 1SDR, and the blue pathway is the cycloaddition between BCN-OH and 1DOXEPY. (c) Free energy barrier of the σ-coupling of SDRs (ΔG‡TS3, left) vs. the 1,3-dipolar cycloaddition between BCN-OH and DOXEPYs (ΔG‡TS2, right) with various substituents. | ||
Taking the seven-membered cyclic dipole (1DOXEPY, Fig. 3a) into account, we subsequently reassessed the absorption spectrum and decaying of the transient 1SDR in various solvents because the predicted absorbance maximum of 1DOXEPY (λcalc.max = 522 nm, f = 0.462, Fig. 3g) is also in good agreement with the experimental value. As a result of the solvent effect,66 the lifetime of 1SDR was notably prolonged in more polar solvent (Fig. 3e) implying that the radical–radical σ-coupling energy barrier (ΔG‡re.) increased, but the λexp.max was almost unchanged (Fig. S5a, ESI†). The higher ΔG‡re. supports the presence of 1DOXEPY, which is calculated to be in equilibrium via electronic tautomerism with 1SDR (Fig. 3d) and stabilized by polar solvents (Fig. S5b, ESI†).59 This absorbance peak (λexp.max = 550 nm, Fig. 3b) was assigned to the characteristic π–π* (HOMO–LUMO) electronic transition of 1SDR and its electronic tautomer 1DOXEPY after comparing with their TD-DFT results. The major electronic transitions were located at 517 nm (f = 0.355) and 522 nm (f = 0.462, Table S11, ESI†), respectively. Based on their free energy difference (ΔGeq. = 0.80 kcal mol−1, Fig. 3d), the Boltzmann distribution between 1SDR and 1DOXEPY (Ω, Fig. 3g and Table S11, ESI†) at 298 K was obtained, indicating a component of 79% 1SDRversus 21% 1DOXEPY. And, this distribution is constant during the thermal decay of 1SDR → 1b (kd = 3.8 × 104 s−1) because the electronic tautomerism is an ultrafast equilibrium with no transition states73 and only the reconstitution in the electronic structure. At about 77 K, we were able to photograph the pink-coloured 1SDR and its thermo-relaxation process in a MTHF matrix, clarifying the T-type photoswitching nature (Fig. 3d).
In view of the apparent substitution effect on the cycloaddition yield (Fig. 2), we further investigated the properties of the selected SDRs (Fig. 3f). With strong electron-donors (eDs), the absorption maximum of 5SDR was red-shifted to 600 nm in contrast to 17SDR with strong electron-acceptors (eAs) showing a hypochromic shift to 530 nm. And these experimental spectra were consistent with the predicted spectra obtained via TD-DFT calculation (λexp.maxvs. λcalc.max, Fig. 3g). Accordingly, the thermal ring-contraction rate (kd) of 3SDR and 5SDR with eDs accelerated to 1.73 × 105 s−1 (4.6-fold) and 1.12 × 106 s−1 (29-fold), respectively, while that of 17SDR with eAs decelerated to 5.02 × 103 s−1 (0.13-fold, Fig. 3g and S3, ESI†) by using 1SDR as a benchmark. As the transition states (TS) searching explicated (Table S11, ESI†), the thermodynamic barrier (ΔG‡re.) for the relaxation of SDR → DNQO descended from 6.3 to 3.2 kcal mol−1 along with a substituent order from eAs (17SDR > 1SDR > 13SDR) to eDs (3SDR > 5SDR), which was also in conformity to the experimental texp.1/2 (Fig. 3g). By comparing the ΔGeq. with the ΔG‡re. in Fig. 3g, we speculate that the thermal decaying observed in the NTAS was presumably rate-determined by the radical–radical σ-coupling process of SDR → DNQO rather than the faster tautomerization between SDR ⇌ DOXEPY. Since there is only small variation in the ΔGeq. of the equilibrium of SDR⇌DOXEPY among various substitutions, the Boltzmann distribution was always dominated by the SDRs (a content of 74–83%, Fig. 3g).
DFT calculation on the competitive mechanism of the photoswitch cycles versus the cycloaddition
To decipher the mechanism of the photoclick process mediated via the photoswitchable 1SDR, further studies on the photochemical pathways were performed via (TD-)DFT (Fig. 4b). Once 1b was excited to the first singlet state (1b-S1) by 365 nm light (both S0 → S1 and S0 → S2 transitions), 1SDR could be obtained as a converged structure via a homolytic cleavage of the C–C bond in the epoxy via1b-TS1-S1 with a free energy barrier (ΔG‡TS1) of 4.4 kcal mol−1 (Fig. 4b). Without BCN-OH, 1SDR would primarily undergo a thermodynamic radical–radical coupling, returning to 1bvia transition state 1b-TS3 (ΔG‡TS3 = ΔGre.‡ = 5.9 kcal mol−1) to complete an ultrafast photoswitch cycle. In the presence of BCN-OH, a competitive pathway leading to a 1,3-dipolar cycloaddition between BCN-OH and 1,3-dipole (1DOXEPY, as the electronic tautomer of 1SDR) was involved, which affords the final cycloadduct 1DEBCODvia transition state 1b-TS2. Notably, the ΔG‡TS2 of the cycloaddition step was substantially lower than that of the radical coupling process (3.8 vs. 5.9 kcal mol−1), suggesting that BCN-OH is capable of capturing the transient 1DOXEPY if [BCN-OH] is given at 0.5 mM (theoretical kd/k2CA = 58). Considering the bioorthogonality of these reactive intermediates, we have further searched for the nucleophilic addition pathways of the 1,3-dipole (1DOXEPY) with respect to thiols, e.g., glutathione (GSH, Fig. S23 and Table S9, ESI†). As a result, the energy barrier in the nucleophilic addition by GSH was incredibly higher than that of the ring-strain promoted cycloaddition with BCN-OH (17.6 vs. 3.8 kcal mol−1) and therefore implies a superior chemoselectivity toward the cycloaddition.To understand the electronic effects on the efficiency of the cycloaddition, five exemplary DNQOs (3b, 5b, 13b, 17b and 24b, Fig. 4c) were subjected to mechanism interpretation at the same level of theory. The computational results illustrated that the introduction of various substituents (either eD or eA) displayed comparable energy barriers during the ring-strain promoted cycloaddition step (ΔG‡TS2, 3.5–4.0 kcal mol−1, Table S12, ESI†). However, the energy barrier associated with the radical–radical coupling processes (ΔG‡TS3) became differentiated. The eA groups (such as trifluoromethyl in 17SDR) dramatically elevated the ΔG‡TS3 = 6.3 kcal mol−1, prolonging the half-life of the SDR and facilitating the cycloaddition with BCN-OH (ΔG‡TS2 = 3.8 kcal mol−1, Fig. 4c). In contrast, the eD group, e.g., fluorine atoms in 13SDR, methyl groups in 3SDR and methoxyl groups in 5SDR, lowered the ΔG‡TS3 in the radical-coupling by 0.6, 1.5 and 2.7 kcal mol−1, respectively, shortening the half-life of the SDRs. Especially for 5SDR with the shortest half-life (t1/2 = 620 ns, Fig. 3g), the ΔG‡TS3 of 3.2 kcal mol−1 was even slightly lower than that in the cycloaddition step (ΔG‡TS2 = 3.5 kcal mol−1), leaving few chances for trapping the SDR via the cycloaddition (theoretical kd/k2CA = 3320 when [BCN-OH] = 0.5 mM). The theoretical calculation was consistent with the experimental observations that DNQOs with eD groups showed very poor photo-conversion (4b–7b, Fig. 2c). Upon further calculation, there was no significant alteration found in both the orbital characteristics and the electron occupied numbers by the various substituents in the five SDRs (Fig. S24, S25 and Table S13, ESI†). The relatively long-lived SDRs55 were likely stabilized by both the carbonyl and the diaryl moieties, thus enabling the cycloaddition with BCN-OH to establish an ultrafast photoclick reaction.
Key parameters for the photoswitching and the photoclick reaction
As a proof-of-concept for the photoclick strategy utilizing T-type photoswitchable SDRs, the quantum yield (Φmax) and the light-to-chemical-energy conversion efficiency (η) for the photochemical transformation from DNQO to SDRs as well as the cycloaddition rate (k2CA) are essential parameters. Since the apparent quantum yield (Φ) for a photoswitchable intermediate is dependent on the concentration of [BCN-OH] under given irradiation conditions, we were able to derive both the Φmax and the kd/k2CA (based on experimental studies) by varying [BCN-OH], according to reported methods (Fig. S8 and S9, ESI†).74 Herein, the kd is directly obtained by monitoring the decay of SDRs from NTAS measurements. Then, the k2CA is calculated based on the competition reactions between the thermal decay and the cycloaddition of DOXEPY. As an energetic SDR molecule, the η is the ratio of the potential energy difference (ΔGSDR-DNQO) between DNQO and the “metastable” SDR harvested during the photochemical conversion with a quantum yield (Φmax) versus the incident photon energy (Elight = hνNA, Table S15, ESI†).75 These parameters for the photoclick reaction between the five DNQOs and BCN-OH are summarized in Table 1, except 5b due to a very poor reactivity. Firstly, we found that the [3 + 2] cycloaddition rates between DOXEPYs and the ring-strained BCN-OH are ultrafast, ranging from 4.86 × 107 M−1 s−1 for unsymmetric 24b to 1.01 × 109 M−1 s−1 for 13b with p-F-phenyl. The cycloaddition rates at this level imply promising kinetics to ligate to extremely diluted BCN reporters on biomolecules. Regardless of the substitutions on the diaryl, all the SDRs hold a high and consistent ΔGSDR-DNQO energy, about 32.1–33.8 kcal mol−1. Therefore, a higher η depends mainly on a better Φmax. Symmetric p-FPh (13b), p-CF3Ph (17b) on 1,3-positions of DNQO and unsymmetric p-FPh combined with p-COOMePh (24b) terminals were favourable to a high Φmax (= 0.56, 0.58 and 0.76) in the photo-ring-opening step, and accordingly the value of η can be gained as 24%, 25% and 33%, respectively. Thus, the light irradiation not only manages the process of the photoclick reaction, but its energy is also efficiently converted to the chemical energy of the long-lived SDR and DOXEPY during the T-type photoswitching, powering an ultrafast cycloaddition with BCN-OH.| DNQO | Φ max | k d/k2CA (mM) | k 2CA (M−1 s−1) | t 1/2 (μs) | ΔGSDR-DNQO (kcal mol−1) | η (%) |
|---|---|---|---|---|---|---|
|
a 20 μM DNQOs upon 365 nm LED irradiation for a specific period of time (3–28 s, Fig. S8, ESI) in CH3CN:H2O = 1:1 at 298 K in the presence of [BCN-OH] = 0.4 mM, 0.6 mM, 1.0 mM or 1.6 mM, respectively.
b The half-life of DOXEPY in the pseudo-first-order cycloaddition reaction, when [BCN-OH] = 500 μM. N.A. = not available.
|
||||||
| 1b | 0.25 | 0.114 | 3.38 × 108 | 4.1 | 33.2 | 10.6 |
| 3b | 0.19 | 0.278 | 6.32 × 108 | 2.2 | 32.1 | 7.8 |
| 5b | N.A. | N.A. | N.A. | N.A. | 31.9 | N.A. |
| 13b | 0.56 | 0.043 | 1.01 × 109 | 1.4 | 33.2 | 23.7 |
| 17b | 0.58 | 0.031 | 1.62 × 108 | 8.6 | 33.8 | 25.0 |
| 24b | 0.76 | 0.422 | 4.86 × 107 | 29 | 33.2 | 33.1 |
Evaluation of the chemoselectivity and the stability of DNQOs in a biological medium
In a complex living context, photolysis or quenching of the photo-generated intermediates (type-I, Fig. 5a) by the abundant chemicals before coming into contact with desired reporters are a major reason for undesired side-reactions in photoclick chemistry. The reversion of the reactive 1SDR ⇌ 1DOXEPY back to 1bvia the superfast σ-coupling provides an alternative pathway to circumvent these undesired processes, and the SDR is naturally insensitive to 3O2. To evaluate the biocompatibility (Fig. 5a), we rescreened the DNQO-BCN photoclick reactions by moving to a biological mimicking medium containing both 10.0 mM GSH and nucleophilic natural amino acids (1.0 mM each, Fig. S10, ESI†). Without BCN-OH, the concentration of DNQOs (1b, 13b, 15b and 24b) was almost unchanged after incubation in the dark for 24 h or under 365 nm irradiation for 60 s, verifying a superior photostability of the DNQOs against a variety of nucleophiles as well as an excellent recovery through photoswitching cycles. Afterward, temporally controlled cycloadditions were then realized in the presence of BCN-OH by using incremental irradiation with the cycloadduct yield increased along the accumulation of the irradiation time (Fig. 5b and S11, ESI†). Although the formation of by-products became notable after prolonged irradiation (0–14.3% for 30–180 s, the orange columnar segments, Fig. 5b), they were attributed to the photolysis of the desired cycloadducts (type-II, Fig. 5a) rather than the type-I photolysis or nucleophilic quenching of the SDR or DOXEPY (Fig. S12, S13, ESI†). To further monitor type-I photolysis, BCN-OH has to be omitted from the mixture. However, by-products produced by type-I photolysis after 180 s exposure in simply CH3CN/PBS = 1/1 mixed solvent remain negligible. Furthermore, a high concentration of GSH (10–50 mM) as well as mammalian cell culture medium were included to reassess the photostability of 1SDR (Fig. 5c and S14–S18, ESI†). Gratifyingly, the remaining 1b could be kept up to 80% in the presence of 50.0 mM GSH (almost saturated solution) or even in the Dulbecco's modified Eagle's medium (DMEM with 10% fetal bovine serum + 1% antibiotics) after 3.0 min irradiation of a 365 nm LED. It is worth noting that DNQO 1b did not deteriorate after storage at 298 K for more than 1 year or under strong acid treatment for 5 h (CF3COOH:CH2Cl2 = 1:1) or even after heating to 100 °C for 10 h in DMF, making it a robust precursor for photoclick ligation in living systems.
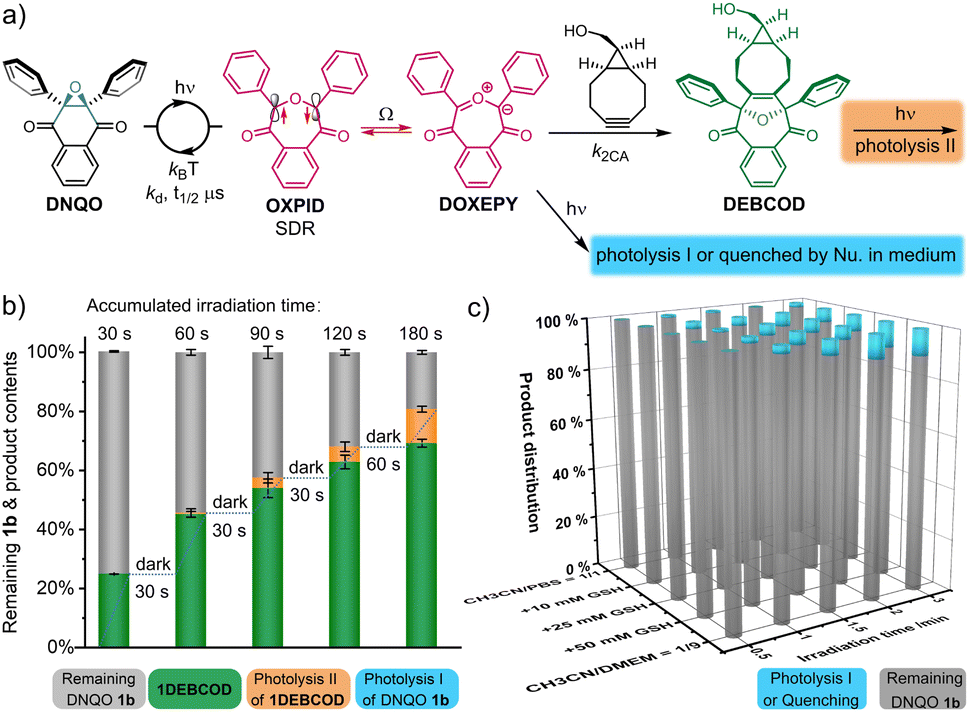 | ||
| Fig. 5 Studies on the chemoselectivity of the photoclick reaction between 1b with BCN-OH upon 365 nm LED irradiation at 298 K under biocompatible conditions. (a) The schematic illustration. (b) Percentage distribution of each component after the photoclick reactions between 1b (40 μM) + BCN-OH (500 μM) with GSH (5.0 mM) in CH3CN/PBS = 1/1. (c) The remaining 1b and photolysis products after the irradiation in the absence of BCN-OH with various concentrations of GSH in CH3CN/PBS = 1/1 or without GSH in CH3CN/DMEM = 1/9 with 10% FBS + 1% antibiotics. DMEM = Dulbecco's modified Eagle's medium. | ||
Spatiotemporally resolved fluorescence labeling on the membrane of living cells via the photoclick reaction
To explore the bioorthogonal decoration on the cell surface via the T-type photoswitchable SDR (Fig. 6a), we then tailored a water-soluble fluorophore conjugate (DNQO-Cy3, Fig. 6b) in which a sulfo-Cy3 was linked to the core of 1bvia oligo ethylene glycol to avoid cell permeability. To incorporate the BCN reporter onto the membrane of a living A549 cancer cell via immuno-recognition toward the epidermal growth factor receptor (EGFR+), a chemically modified chimeric monoclonal antibody, cetuximab-FITC-BCN (20 μg mL−1 = 0.13 μM Cex-FITC-BCN, Fig. S19, ESI†) was attached via incubation for 60 min. After exposure to a 365 nm LED for 90 s with the treatment of 20 μM DNQO-Cy3 in air (Fig. 6c), the concomitant appearance of both Cy3 and FITC fluorescence was observed on the cell surface, colocalizing precisely (Fig. 6c, upper row), suggesting a temporal controllability of this photoclick reaction. Without the exposure, there was no Cy3 signal detected on the cell surface but only the FITC signal was observed due to binding of Cex-FITC-BCN (Fig. 6c, lower row). Without the treatment with Cex-FITC-BCN, both Cy3 and FITC signals were negligible even after identical exposure (for other stringent control conditions, Fig. S20, ESI†). | ||
| Fig. 6 Spatiotemporally resolved fluorescence labeling on the membrane of living mammalian cells. (a) Schematic illustration for the photoclick labeling procedure on the cell membrane via 365 nm light (90 s) to induce the photoswitching of DNQO-Cy3 → DOXEPY-Cy3 (total 20 μM); (b) chemical structure of the tailored DNQO-Cy3 probe; (c) confocal microscopic imaging to identify the colocalization of both Cy3 and FITC signals on cell surface, Hoechst-33342 staining for nucleus of the living cells, BF = bright field, and scale bar = 10 μm; (d) the merged fluorescence imaging of the cells after exposure to the patterned 365 nm LED stimulation (purple colour) projected through the DMD (via Polygon 400, Mightex, 25% 2.0 W LED light power) for 30 s, with the white arrows pointing to the labelled membrane segments and the yellow arrows to the membrane segments outside the stimulation pattern. | ||
Encouraged by the ultrafast reaction rates and the high chemoselectivity of the DNQO-BCN photoclick reaction, we attempted spatial-resolved fluorescence labeling of cetuximab-BCN on single or even partial cell membranes. To achieve lithographic patterning with sub-micron resolution, we programmed a 30 s continuous 365 nm illumination sequence via a digital microarray device (DMD) embedded in an epifluorescence microscope through a 100× objective lens. Meanwhile a lithographic pattern of a grid matrix (58.7 × 32.1 μm, 6 × 6 purple lines, Fig. 6d) with two rectangle columns at the terminal (light blue regions) filled was drawn as the input shape. As a result, the DNQO-Cy3 probe in PBS (20 μM and pH = 7.4) was photo-switched on to generate SDR-Cy3 within the stimulation region, and subsequently conjugated toward the bound cetuximab-BCN (incubated at 120 μg mL−1 in DMEM for 1 h) in proximity (Fig. 6d, for details, refers to Fig. S21, ESI†). Unreacted SDR-Cy3 was mostly recovered to DNQO-Cy3 instantaneously with minimal molecule diffusion because of its μs level half-life. Consequently, most of the cell membrane segments overlaying within the grid-patterned region were covalently marked with the Cy3 fluorophore, resulting in curve-shaped signals (white arrows). But the unexposed portion of the membranes (yellow arrows) scarcely showed any signal, indicating a subcellular spatial-resolving power of the photoclick reaction. However, off-site labeling could also be observed (Fig. S21, ESI†), which might be ascribed to scattering of the light-ray in the specimen or dynamic change of the membrane morphology during the extensive washing procedure or unexpected off-target labeling.
Conclusions
Inspired by the transient SDR intermediate produced from the photoactivation of DNQO, we present a novel T-type photoswitch based on the homolytic ring-expansion in association with the counteracting thermal σ-coupling of the SDR in dynamic cycles. The unique seven-membered cyclic OXPID (SDR) as well as its long-lived character revealed by spectroscopic and computational characterization constitute an efficient photo-energy transducer, which tautomerize into an energetic 1,3-dipole, DOXEPY. Therefore, the energy harvesting nature of the T-type molecular photoswitch makes the OXPID a highly useful species in a limited repertoire of SDRs that can drive an ultrafast cycloaddition with endo-BCN-OH. In this way, we established a type of bioorthogonal photoclick reaction for spatiotemporal labeling on living cells, which is underpinned by the unique features of the SDR, including insensitivity to triplet species and regeneration without capturing by BCN-OH. The facile two-step synthesis of DNQO facilitates an easy access for versatile utilities of the photoswitchable SDR in photoclick chemistry. Currently, the requirement of 365 nm UV light to induce the photoswitching is the limiting factor. Therefore, research on visible-light-triggered bioorthogonal reactions with temporal and spatial control by using DNQO is now underway in our lab.Data availability
Additional figures as described in the main text, all experimental procedures, synthetic procedures, copies of spectral data for the compounds, data processing, and computational details are available in the ESI.†Author contributions
F. H. and Z. Y. initiated the idea, designed the experiments, and wrote and revised the manuscript. F. H. synthesized the main compounds and conducted all of the experiments. C. Z., Z. S., and C. H. carried out the theoretical calculations. Z. L., X. X., X. Z., J. F., and B. L. helped with compound synthesis and in vitro cell experiments. Y. L. assisted with the NTAS experimental studies. All authors contributed to the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Dr Hanjiao Chen (Analytical & Testing Center, SCU) for help with the EPR tests. We appreciate the Xiaoming Feng laboratory (SCU) for access to equipment, including NMR collection and single crystal XRD analysis by Dr Yuqiao Zhou. We also express our gratitude to Yanhong Liu (the Comprehensive Training Platform of Specialized Laboratory, College of Chemistry) for her assistance with the confocal image collection. Financial support was provided by the National Natural Science Foundation of China (22001181 and 22077090) and the Fundamental Research Funds for the Central Universities (20826041D4117) and the Institutional Research Fund from Sichuan University (2020SCUNL105).Notes and references
- N. J. Turro, V. Ramamurthy and J. C. Scaiano, Modern Molecular Photochemistry of Organic Molecules, University Science Books, Mill Valley, 2010 Search PubMed.
- C. W. Tornøe, C. Christensen and M. Meldal, J. Org. Chem., 2002, 67, 3057–3064 CrossRef PubMed.
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 CrossRef CAS PubMed.
- G. J. Brighty, R. C. Botham, S. Li, L. Nelson, D. E. Mortenson, G. Li, C. Morisseau, H. Wang, B. D. Hammock, K. B. Sharpless and J. W. Kelly, Nat. Chem., 2020, 12, 906–913 CrossRef CAS PubMed.
- N. J. Agard, J. A. Prescher and C. R. Bertozzi, J. Am. Chem. Soc., 2004, 126, 15046–15047 CrossRef CAS PubMed.
- M. A. Tasdelen and Y. Yagci, Angew. Chem., Int. Ed., 2013, 52, 5930–5938 CrossRef CAS PubMed.
- G. S. Kumar and Q. Lin, Chem. Rev., 2021, 121, 6991–7031 CrossRef CAS PubMed.
- B. D. Fairbanks, L. J. Macdougall, S. Mavila, J. Sinha, B. E. Kirkpatrick, K. S. Anseth and C. N. Bowman, Chem. Rev., 2021, 121, 6915–6990 CrossRef CAS PubMed.
- P. Klán, T. Šolomek, C. G. Bochet, A. Blanc, R. Givens, M. Rubina, V. Popik, A. Kostikov and J. Wirz, Chem. Rev., 2013, 113, 119–191 CrossRef PubMed.
- C. Brieke, F. Rohrbach, A. Gottschalk, G. Mayer and A. Heckel, Angew. Chem., Int. Ed., 2012, 51, 8446–8476 CrossRef CAS PubMed.
- S. Arumugam, S. V. Orski, J. Locklin and V. V. Popik, J. Am. Chem. Soc., 2012, 134, 179–182 CrossRef CAS PubMed.
- C. Wang, H. Zhang, T. Zhang, X. Zou, H. Wang, J. E. Rosenberger, R. Vannam, W. S. Trout, J. B. Grimm, L. D. Lavis, C. Thorpe, X. Jia, Z. Li and J. M. Fox, J. Am. Chem. Soc., 2021, 143, 10793–10803 CrossRef CAS PubMed.
- S. Arumugam and V. V. Popik, J. Am. Chem. Soc., 2012, 134, 8408–8411 CrossRef CAS PubMed.
- E. Blasco, M. Wegener and C. Barner-Kowollik, Adv. Mater., 2017, 29, 1604005 CrossRef PubMed.
- V. X. Truong, J. Bachmann, A. Unterreiner, J. P. Blinco and C. Barner-Kowollik, Angew. Chem., Int. Ed., 2022, 61, e202113076 CrossRef CAS PubMed.
- A. Jemas, Y. Xie, J. E. Pigga, J. L. Caplan, C. W. am Ende and J. M. Fox, J. Am. Chem. Soc., 2022, 144, 1647–1662 CrossRef CAS PubMed.
- G. S. Kumar, S. Racioppi, E. Zurek and Q. Lin, J. Am. Chem. Soc., 2022, 144, 57–62 CrossRef CAS PubMed.
- Y. Fu, H. Helbert, N. A. Simeth, S. Crespi, G. B. Spoelstra, J. M. van Dijl, M. van Oosten, L. R. Nazario, D. van der Born, G. Luurtsema, W. Szymanski, P. H. Elsinga and B. L. Feringa, J. Am. Chem. Soc., 2021, 143, 10041–10047 CrossRef CAS PubMed.
- A. Guo, D. Wei, H. Nie, H. Hu, C. Peng, S. Li, K. Yan, B. Zhou, L. Feng, C. Fang, M. Tan, R. Huang and X. Chen, Nat. Commun., 2022, 11, 5472 CrossRef PubMed.
- D. Fong, A. Lang, K. Li and A. Adronov, Macromolecules, 2020, 53, 1760–1766 CrossRef CAS.
- V. X. Truong, ChemPhotoChem, 2020, 4, 564–570 CrossRef CAS.
- H. C. Hang, C. Yu, D. L. Kato and C. R. Bertozzi, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 14846 CrossRef CAS PubMed.
- J. Li, H. Kong, C. Zhu and Y. Zhang, Chem. Sci., 2020, 11, 3390–3396 RSC.
- D. M. Patterson, L. A. Nazarova and J. A. Prescher, ACS Chem. Biol., 2014, 9, 592–605 CrossRef CAS PubMed.
- E. M. Sletten and C. R. Bertozzi, Angew. Chem., Int. Ed., 2009, 48, 6974–6998 CrossRef CAS PubMed.
- N. K. Devaraj, ACS Cent. Sci., 2018, 4, 952–959 CrossRef CAS PubMed.
- L. Liu, D. Zhang, M. Johnson and N. K. Devaraj, Nat. Chem., 2022, 14, 1078–1085 CrossRef CAS PubMed.
- W. Song, Y. Wang, J. Qu and Q. Lin, J. Am. Chem. Soc., 2008, 130, 9654–9655 CrossRef CAS PubMed.
- B. J. Adzima, Y. Tao, C. J. Kloxin, C. A. DeForest, K. S. Anseth and C. N. Bowman, Nat. Chem., 2011, 3, 256–259 CrossRef CAS PubMed.
- (a) A. A. Poloukhtine, N. E. Mbua, M. A. Wolfert, G. J. Boons and V. V. Popik, J. Am. Chem. Soc., 2009, 131, 15769–15776 CrossRef CAS PubMed; (b) S. V. Mayer, A. Murnauer, M. K. von Wrisberg, M. L. Jokisch and K. Lang, Angew. Chem., Int. Ed., 2019, 58, 15876–15882 CrossRef CAS PubMed.
- (a) A. Herner and Q. Lin, Top. Curr. Chem., 2016, 374, 1 CrossRef CAS PubMed; (b) S. Arndt and H. A. Wagenknecht, Angew. Chem., Int. Ed., 2014, 53, 14580–14582 CrossRef CAS PubMed; (c) P. An, T. M. Lewandowski, T. G. Erbay, P. Liu and Q. Lin, J. Am. Chem. Soc., 2018, 140, 4860–4868 CrossRef CAS PubMed.
- (a) L. Zhang, X. Zhang, Z. Yao, S. Jiang, J. Deng, B. Li and Z. Yu, J. Am. Chem. Soc., 2018, 140, 7390–7394 CrossRef CAS PubMed; (b) J. Gao, Q. Xiong, X. Wu, J. Deng, X. Zhang, X. Zhao, P. Deng and Z. Yu, Commun. Chem., 2020, 3, 29 CrossRef CAS PubMed.
- (a) H. Zhang, W. S. Trout, S. Liu, G. A. Andrade, D. A. Hudson, S. L. Scinto, K. T. Dicker, Y. Li, N. Lazouski, J. Rosenthal, C. Thorpe, X. Jia and J. M. Fox, J. Am. Chem. Soc., 2016, 138, 5978–5983 CrossRef CAS PubMed; (b) V. X. Truong, K. M. Tsang, F. Ercole and J. S. Forsythe, Chem. Mater., 2017, 29, 3678–3685 CrossRef CAS.
- (a) J. Li, H. Kong, L. Huang, B. Cheng, K. Qin, M. Zheng, Z. Yan and Y. Zhang, J. Am. Chem. Soc., 2018, 140, 14542–14546 CrossRef CAS PubMed; (b) Y. Fu, N. A. Simeth, R. Toyoda, R. Brilmayer, W. Szymanski and B. L. Feringa, Angew. Chem., Int. Ed., 2023, 62, e202218203 CrossRef CAS PubMed.
- (a) A. R. Davis, J. A. Maegerlein and K. R. Carter, J. Am. Chem. Soc., 2011, 133, 20546–20551 CrossRef CAS PubMed; (b) X. Meng, J. Hu, Z. Chao, Y. Liu, H. Ju and Q. Cheng, ACS Appl. Mater. Interfaces, 2018, 10, 1324–1333 CrossRef CAS PubMed.
- N. R. B. Boase, Macromol. Rapid Commun., 2020, 41, 2000305 CrossRef CAS.
- (a) S. Arumugam and V. V. Popik, J. Am. Chem. Soc., 2009, 131, 11892–11899 CrossRef CAS PubMed; (b) S. Arumugam and V. V. Popik, J. Am. Chem. Soc., 2011, 133, 15730–15736 CrossRef CAS PubMed.
- X. Xie, F. Hu, Y. Zhou, Z. Liu, X. Shen, J. Fu, X. Zhao and Z. Yu, Angew. Chem., Int. Ed., 2023, 62, e202300034 CrossRef CAS PubMed.
- J. Boelke and S. Hecht, Adv. Opt. Mater., 2019, 7, 1900404 CrossRef.
- I. M. Welleman, M. W. H. Hoorens, B. L. Feringa, H. H. Boersma and W. Szymański, Chem. Sci., 2020, 11, 11672–11691 RSC.
- P. Bharmoria, S. Ghasemi, F. Edhborg, R. Losantos, Z. Wang, A. Mårtensson, M. Morikawa, N. Kimizuka, Ü. İşci, F. Dumoulin, B. Albinsson and K. Moth-Poulsen, Chem. Sci., 2022, 13, 11904–11911 RSC.
- S. Crespi, N. A. Simeth and B. König, Nat. Rev. Chem, 2019, 3, 133–146 CrossRef CAS.
- J. B. Geri, J. V. Oakley, T. Reyes-Robles, T. Wang, S. J. McCarver, C. H. White, F. P. Rodriguez-Rivera, D. L. Parker Jr, E. C. Hett, O. O. Fadeyi, R. C. Oslund and D. W. C. MacMillan, Science, 2020, 367, 1091–1097 CrossRef CAS PubMed.
- S. S. Skourtis, C. Liu, P. Antoniou, A. M. Virshup and D. N. Beratan, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 8115–8120 CrossRef CAS.
- (a) C. C. Winterbourn, Nat. Chem. Biol., 2008, 4, 278–286 CrossRef CAS PubMed; (b) H. Sies and D. P. Jones, Nat. Rev. Mol. Cell Biol., 2020, 21, 363–383 CrossRef CAS.
- T. C. Pham, V. Nguyen, Y. Choi, S. Lee and J. Yoon, Chem. Rev., 2021, 121, 13454–13619 CrossRef CAS PubMed.
- T. S. Chambers and G. B. Kistiakowsky, J. Am. Chem. Soc., 1934, 56, 399–405 CrossRef CAS.
- (a) J. A. Berson, L. D. Pedersen and B. K. Carpenter, J. Am. Chem. Soc., 1976, 98, 122–143 CrossRef CAS; (b) J. A. Berson, Acc. Chem. Res., 1997, 30, 238–244 CrossRef CAS.
- L. Salem and C. Rowland, Angew. Chem., Int. Ed., 1972, 11, 92–111 CrossRef CAS.
- W. Adam, H. Platsch and J. Wirz, J. Am. Chem. Soc., 1989, 111, 6896–6898 CrossRef CAS.
- (a) S. J. Getty, D. A. Hrovat and W. T. Borden, J. Am. Chem. Soc., 1994, 116, 1521–1527 CrossRef CAS; (b) W. Adam, G. Fragale, D. Klapstein, W. M. Nau and J. Wirz, J. Am. Chem. Soc., 1995, 117, 12578–12592 CrossRef CAS.
- S. Crespi, N. A. Simeth, M. D. Donato, S. Doria, C. N. Stindt, M. F. Hilbers, F. L. Kiss, R. Toyoda, S. Wesseling, W. J. Buma, B. L. Feringa and W. Szymański, Angew. Chem., Int. Ed., 2021, 60, 25290–25295 CrossRef CAS PubMed.
- M. Kathan and S. Hecht, Chem. Soc. Rev., 2017, 46, 5536–5550 RSC.
- (a) T. Nakagaki, T. Sakai, T. Mizuta, Y. Fujiwara and M. Abe, Chem.–Eur. J., 2013, 19, 10395–10404 CrossRef CAS PubMed; (b) M. Abe, S. Tada, T. Mizuno and K. Yamasaki, J. Phys. Chem. B, 2016, 120, 7217–7226 CrossRef CAS PubMed.
- M. Abe, E. Kubo, K. Nozaki, T. Matsuo and T. Hayashi, Angew. Chem., Int. Ed., 2006, 45, 7828–7831 CrossRef CAS PubMed.
- (a) E. Niecke, A. Fuchs, F. Baumeister, M. Nieger and W. W. Schoeller, Angew. Chem., Int. Ed., 1995, 34, 555–557 CrossRef CAS; (b) H. Sugiyama, S. Ito and M. Yoshifuji, Angew. Chem., Int. Ed., 2003, 42, 3802–3804 CrossRef CAS PubMed; (c) M. Sebastian, M. Nieger, D. Szieberth, L. Nyulászi and E. Niecke, Angew. Chem., Int. Ed., 2004, 43, 637–641 CrossRef CAS.
- D. Scheschkewitz, H. Amii, H. Gornitzka, W. W. Schoeller, D. Bourissou and G. Bertrand, Science, 2002, 295, 1880–1881 CrossRef CAS PubMed.
- C. Cui, M. Brynda, M. M. Olmstead and P. P. Power, J. Am. Chem. Soc., 2004, 126, 6510–6511 CrossRef CAS PubMed.
- (a) M. Abe, C. Ishihara and M. Nojima, J. Org. Chem., 2003, 68, 1618–1621 CrossRef CAS PubMed; (b) S. Yoshidomi, M. Mishima, S. Seyama, M. Abe, Y. Fujiwara and T. Ishibashi, Angew. Chem., Int. Ed., 2017, 56, 2984–2988 CrossRef CAS; (c) S. Yoshidomi and M. Abe, J. Am. Chem. Soc., 2019, 141, 3920–3933 CrossRef CAS PubMed.
- T. Nakamura, A. Takegami and M. Abe, J. Org. Chem., 2010, 75, 1956–1960 CrossRef CAS PubMed.
- S. Arakawa, J. Org. Chem., 1977, 42, 3800–3811 CrossRef CAS.
- K. Maruyama and A. Osuka, J. Org. Chem., 1980, 45, 1898–1901 CrossRef CAS.
- (a) H. Kato, K. Yamaguchi and H. Tezuka, Chem. Lett., 1974, 3, 1089–1090 CrossRef; (b) A. Osuka, H. Suzuki and K. Maruyama, Chem. Lett., 1981, 10, 201–204 CrossRef.
- K. Maruyama, A. Osuka and H. Suzuki, J. Chem. Soc. Chem. Commun., 1980, 723–724 RSC.
- R. L. Martin, J. Chem. Phys., 2003, 118, 4775 CrossRef CAS.
- M. Abe, W. Adam, T. Heidenfelder, W. M. Nau and X. Zhang, J. Am. Chem. Soc., 2000, 122, 2019–2026 CrossRef CAS.
- L. Noodleman, J. Chem. Phys., 1981, 74, 5737 CrossRef CAS.
- K. Yamaguchi, F. Jensen, A. Dorigo and K. N. Houk, Chem. Phys. Lett., 1988, 149, 537–542 CrossRef CAS.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33–38 CrossRef CAS.
- T. Lu and Q. Chen, Comput. Theor. Chem., 2021, 1200, 113249 CrossRef CAS.
- (a) Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed; (b) M. M. Francl, W. J. Pietro, W. J. Hehre, J. S. Binkley, M. S. Gordon, D. J. DeFrees and J. A. Pople, J. Chem. Phys., 1982, 77, 3654–3665 CrossRef CAS.
- M. B. Smith, March's Advanced Organic Chemistry: Reactions, Mechanisms, and Structure, Wiley, Hoboken, NJ, 8th edn, 2020, pp. 96–97 Search PubMed.
- K. Maruyama and T. Ogawa, J. Org. Chem., 1983, 48, 4968–4976 CrossRef CAS.
- W. Moormann, T. Tellkamp, E. Stadler, F. Röhricht, C. Näther, R. Puttreddy, K. Rissanen, G. Gescheidt and R. Herges, Angew. Chem., Int. Ed., 2020, 59, 15081–15086 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Details on experimental procedures, spectral properties, HPLC traces, computational data, and characterization of all new compounds. CCDC 2246955 and 2223266. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc03675h |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |