
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTargeting mitochondrial degradation by chimeric autophagy-tethering compounds†
Zhenqi
Liu
ab,
Geng
Qin
ab,
Jie
Yang
ab,
Wenjie
Wang
ab,
Wenting
Zhang
ab,
Boxun
Lu
 *c,
Jinsong
Ren
ab and
Xiaogang
Qu
*ab
*c,
Jinsong
Ren
ab and
Xiaogang
Qu
*ab
aLaboratory of Chemical Biology, State Key Laboratory of Rare Earth Resource Utilization, Changchun Institute of Applied Chemistry, Chinese Academy of Sciences, Changchun, Jilin 130022, P. R. China. E-mail: xqu@ciac.ac.cn
bSchool of Applied Chemistry and Engineering, University of Science and Technology of China, Hefei, Anhui 230026, P. R. China
cNeurology Department at Huashan Hospital, State Key Laboratory of Medical Neurobiology, Ministry of Education Frontiers Center for Brain Science, School of Life Sciences, Fudan University, Shanghai, China. E-mail: luboxun@fudan.edu.cn
First published on 26th September 2023
Abstract
The ability to regulate mitophagy in a living system with small molecules remains a great challenge. We hypothesize that adding fragments specific to the key autophagosome protein LC3 to mitochondria will mimic receptor-mediated mitophagy, thus engaging the autophagy-lysosome pathway to induce mitochondrial degradation. Herein, we develop a general biochemical approach to modulate mitophagy, dubbed mito-ATTECs, which employ chimera molecules composed of LC3-binding moieties linked to mitochondria-targeting ligands. Mito-ATTECs trigger mitophagy via targeting mitochondria to autophagosomes through direct interaction between mito-ATTECs and LC3 on mitochondrial membranes. Subsequently, autophagosomes containing mitochondria rapidly fuse with lysosomes to facilitate the degradation of mitochondria. Therefore, mito-ATTECs circumvent the detrimental effects related to disruption of mitochondrial membrane integrity by inducers routinely used to manipulate mitophagy, and provide a versatile biochemical approach to investigate the physiological roles of mitophagy. Furthermore, we found that sustained mitophagy lead to mitochondrial depletion and autophagic cell death in several malignant cell lines (lethal mitophagy). Among them, apoptosis-resistant malignant melanoma cell lines are particularly sensitive to lethal mitophagy. The therapeutic efficacy of mito-ATTECs has been further evaluated by using subcutaneous and pulmonary metastatic melanoma models. Together, the mitochondrial depletion achieved by mito-ATTECs may demonstrate the general concept of inducing cancer cell lethality through excessive mitochondrial clearance, establishing a promising therapeutic paradigm for apoptosis-resistant tumors.
Introduction
Mitophagy, the degradation of mitochondria via a specialized form of macroautophagy (hereafter referred to as autophagy), is a major mechanism for mitochondrial quality control.1 During mitophagy, dysfunctional or redundant mitochondria are recognized and engulfed by autophagosomes, followed by mitophagosomal fusion with lysosomes and autophagic degradation.2 Depending on the physiological context, mitophagy is classified as basal, programmed or stress-induced. Basal mitophagy has been demonstrated to be required for routine mitochondrial maintenance.3,4 Programmed mitophagy is activated for the adjustment of mitochondrion numbers for changing metabolic requirements under developmental conditions.5–7 Stress signals affect mitochondrial homeostasis and may induce acute mitochondrial degradation, by stress-induced mitophagy.8 Besides, mitophagy is also associated with various pathological conditions, such as neurodegeneration, metabolic disorders, aging, and cancer.9–12 Thus, discovering mitophagy inducers may lead to therapeutic strategies targeting mitophagy-associated physiological and pathological conditions.13We sought a simple and versatile approach to modulate mitophagy. This design was inspired by the growing body of literature focused on identifying selective mitophagy receptors that are important for mitochondrial degradation. For example, the mitochondrial proteins NIX, BNIP3 and FUNDC1 are mitophagy receptors that sustain mitochondrial homeostasis following diverse stimuli.1 NIX (NIP3-like protein X) contains both a transmembrane (TM) domain that is anchored to the outer mitochondrial membrane (OMM) and a conserved LC3-interacting region (LIR) that acts to promote the formation of autophagosomes surrounding mitochondria.14 Similar to NIX, OMM-localized BNIP3 (ref. 15) (BCL2 interacting protein 3) and FUNDC1 (ref. 16) (FUN14 domain containing protein 1) interact directly with LC3-II-positive autophagosomes through their LIR motifs, inducing mitochondrial degradation. Broadly, these mitophagy receptors participate in the formation of bridges between mitochondria and LC3-II-positive autophagosomes to trigger mitophagy.17 These give us confidence that a chimera molecule, which can bind to both mitochondria and autophagy protein LC3, could target mitochondria directly to autophagosomes for degradation (Scheme 1).18–22
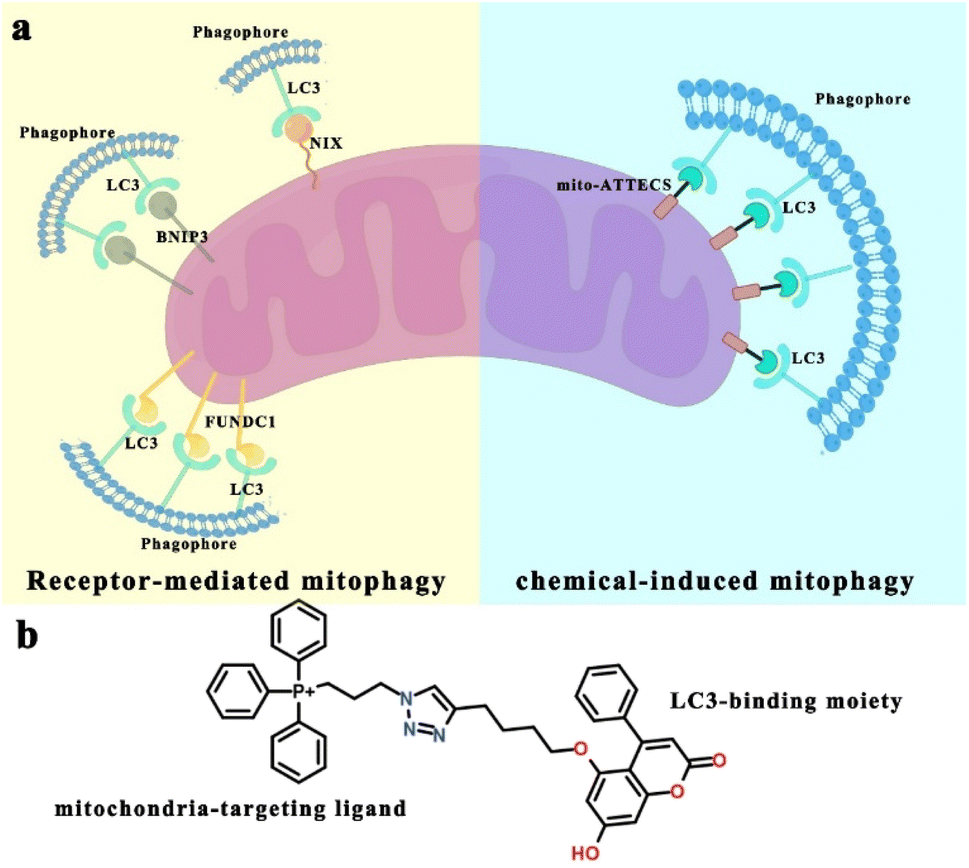 | ||
| Scheme 1 (a) Mechanism of mito-ATTEC-triggered mitophagy. Receptor-mediated mitophagy (left): OMM-anchored mitophagy receptors (such as BNIP3, NIX and FUNDC1) interact directly with LC3 to mediate mitochondrial elimination. Inspired by this process, we developed a general chemical tool to modulate mitophagy, mito-ATTECs, which employ chimera molecules composed of LC3-binding moieties linked to mitochondria-targeting ligands. Mito-ATTECs targeted mitochondria and interact directly with LC3 to trigger mitophagy (right). (b) Chemical structure of a mito-ATTEC. | ||
The concept of autophagosome-tethering compound (ATTEC) illustrates a novel therapeutic modality for directly hijacking the autophagy pathway to degrade pathogenic biomolecules or organelles.23–25 ATTEC molecules interact with both the protein of interest (POI) and key autophagosome protein LC3, bringing the POI to the autophagosomes directly for subsequent degradation. Inspired by this strategy, in the present work, we design and synthesize a novel mitophagy inducer, termed mito-ATTECs, which are engineered by linking a mitochondria binder with a LC3-binding molecule. Mito-ATTECs would target mitochondria and form a complex with LC3 to promote autophagosome formation surrounding mitochondria. Subsequently, autophagosomes containing mitochondria rapidly fuse with lysosomes to facilitate the degradation of mitochondria. Therefore, mito-ATTECs overcome the detrimental effects associated with the depolarization of the mitochondrial membrane potential (MMP) by inducers routinely used to trigger mitophagy. Our results identify the function of mito-ATTECs in recruiting autophagosomes to mitochondria, demonstrating a general strategy for chemical-induced mitophagy. Furthermore, the excessive mitophagy activation (lethal mitophagy) leads to complete mitochondrial degradation and autophagy-dependent cell death in tumor cells.18,26,27 Specifically, our results indicate that melanoma is substantially sensitive to mito-ATTECs. The therapeutic efficacy of mito-ATTECs has been further evaluated by using subcutaneous and pulmonary metastatic melanoma models. Our results indicate that mito-ATTEC treatments lead to mitophagy hyperactivation and irreversible cell death in melanoma murine models with no adverse side effects. Thus, targeting lethal mitophagy by mito-ATTECs may offer an attractive therapeutic approach to circumvent therapeutic resistance and enhance the effects of anticancer therapies.27
Results
Compound design and synthesis
A derivative of 5,7-dihydroxy-4-phenylcoumarin (alk-DP) was chosen as the LC3-targeting moiety according to previous literature.23,24 For the mitochondria-targeting moiety, we chose a derivative of triphenylphosphonium (azi-TPP), which is widely used as a specific warhead for mitochondrial targeting.28 Then, alk-DP and azi-TPP were conjugated by the azide–alkyne cycloaddition (CuAAC) reaction. So, the synthesized bifunctional mito-ATTECs could interact with both mitochondria and LC3-II-positive autophagosomes simultaneously, leading to autophagic degradation of mitochondria (Scheme 1).First, we characterized the interactions between mito-ATTECs and mitochondria using the fluorescence colocalization experiment. MitoTracker Red CMXRos (MTR) dye was used to label mitochondria in living cells. We observed that mito-ATTECs localized to mitochondria (Fig. S1a†) without affecting the mitochondrial function (Fig. S1b and c†). Similar results were obtained from in vitro MMP experiments29 (Fig. S1d†). Taken together, targeting mito-ATTECs to mitochondria did not disrupt mitochondrial integrity.19
Selective degradation of mitochondria by mito-ATTECs
Since mito-ATTECs bind to mitochondria in cells, we next explored whether mito-ATTECs might enhance the degradation of mitochondria as expected. Mitochondrial content was first determined by CLSM using MTR dye.30 We observed no change in MTR signals in cells treated with two separated warheads (alk-DP and azi-TPP, 30 μM), indicating that the separated warheads were incapable of reducing cellular mitochondrial content (Fig. 1a). In contrast, we observed a dose-dependent decrease in MTR fluorescence levels on treatment with mito-ATTECs for 24 h, reaching a near complete mitochondrial depletion at a concentration of 30 μM (Fig. 1b). Similarly, prolonged exposure durations at a fixed concentration of mito-ATTECs (30 μM) resulted in a time-dependent decrease in cellular mitochondrial content (Fig. 1c and d). Collectively, the conjugation between the mitochondrion-targeting warhead and LC3-binding moiety in the bifunctional molecule was indispensable for the depletion of mitochondria.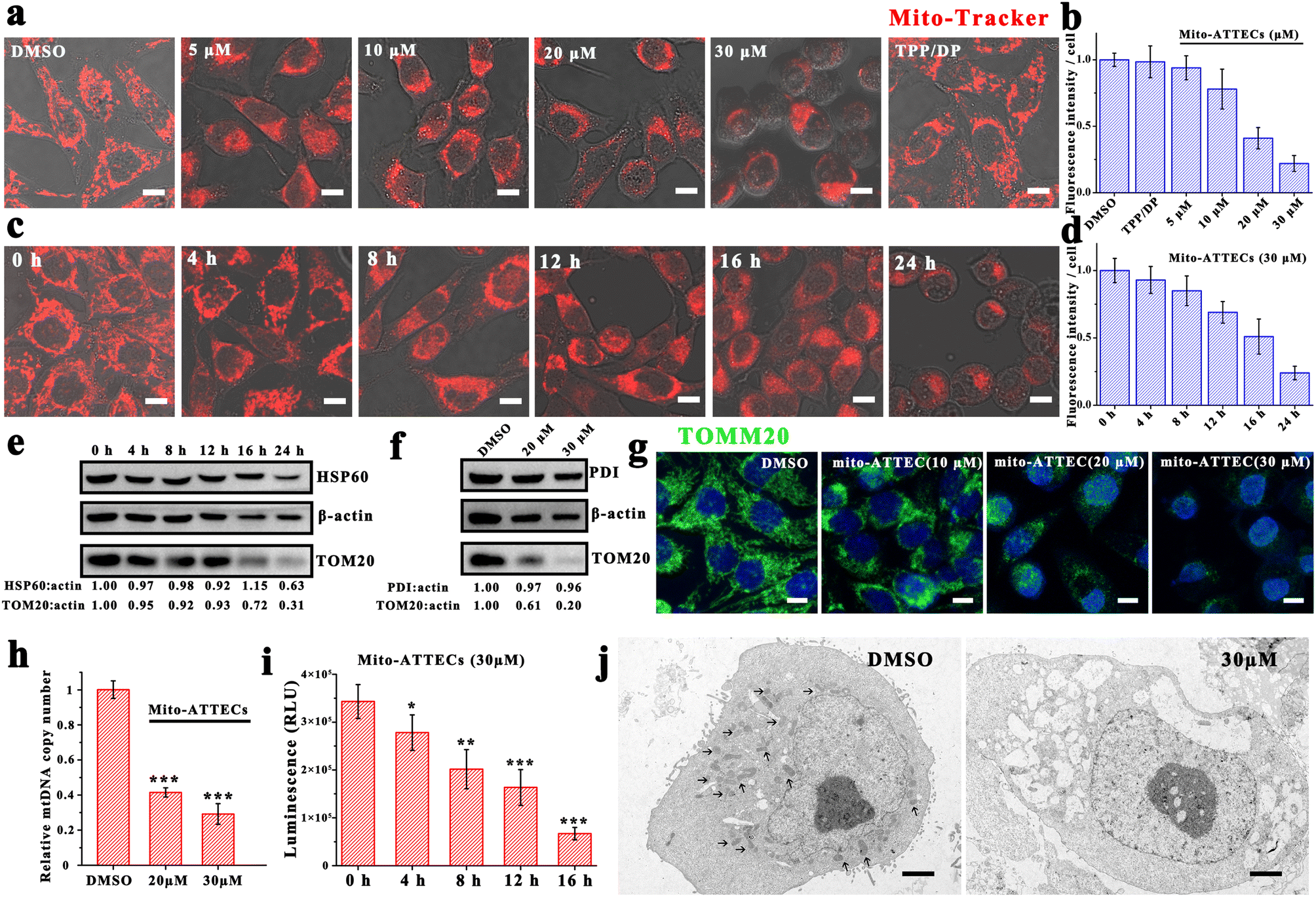 | ||
| Fig. 1 Mito-ATTEC treatment leads to mitochondrial depletion in MCF-7 cells. (a and b) MTR signals were reduced in a dosage-dependent manner (24 h treatment). Scale bars, 10 μm. (c and d) MTR signals were reduced in a time-dependent manner (mito-ATTECs: 30 μM). Scale bars, 10 μm. (e) Western blotting measuring HSP60 and Tom20 in mito-ATTEC (30 μM)-treated MCF-7 cells. (f) Western blotting measuring PDI and Tom20 in mito-ATTEC (24 h)-treated MCF-7 cells. (g) Representative images of immunofluorescence staining of TOM20 in mito-ATTEC-treated cells (0, 10, 20 or 30 μM, 16 h). Scale bars, 10 μm. (h) Copy number of mitochondrial DNA (mtDNA) in DMSO or mito-ATTEC-treated MCF-7 cells (24 h) was measured by PCR amplification. (i) ATP levels were reduced in a time-dependent manner (mito-ATTECs: 30 μM). (j) Representative TEM images of DMSO or mito-ATTEC (30 μM, 24 h) treated MCF-7 cells, arrows indicate mitochondria. Scale bars, 2 μm. Each experiment contained at least three independent biological replicates. | ||
These findings were corroborated by lower mitochondrial protein levels (OMM protein TOM20 as well as the mitochondrial matrix 60 kDa heat shock protein (HSP60)) after mito-ATTEC treatment (Fig. 1e). Immunofluorescence staining analysis showed similar results (Fig. 1g). However, no significant change was found in the level of endoplasmic reticulum luminal protein disulfide isomerase (PDI) in mito-ATTEC-treated cells (Fig. 1f), indicating that mitochondria were selectively affected after mito-ATTEC treatment.26 Moreover, the mtDNA copy number decreased gradually with increasing concentrations of mito-ATTEC molecules (Fig. 1h). We also observed lower ATP contents after mito-ATTEC treatment compared with that in DMSO-treated cells (Fig. 1i). Furthermore, the acute loss of mitochondria was directly observed (Fig. 1j) by transmission electron microscopy (TEM). Quantitation of TEM images showed that 55% of MCF-7 cells lacked mitochondria completely after mito-ATTEC treatment. In conclusion, these results suggested that mito-ATTEC treatment could reduce pools of functional mitochondria; a similar process is observed during erythrocyte maturation.5
Mito-ATTECs target mitochondria to autophagosomes for degradation
Mito-ATTECs were predicted to interact with both mitochondria and LC3, thus bridging the targeted mitochondria with autophagosomes for subsequent degradation. To verify this, we evaluated the accumulation of autophagosome protein LC3 in the mitochondrial fraction.24 Upon 6 h mito-ATTEC treatment, LC3 puncta were mainly colocalized with OMM protein Tom20 compared to puncta in the untreated control (Fig. 2a), suggesting that mito-ATTEC treatment facilitated the engulfment of mitochondria by autophagosomes. In contrast, LC3B puncta were not colocalized with Tom20 in response to alk-DP/azi-TPP (30 μM) treatment, indicating that the induced interaction between mitochondria and LC3 has important roles in targeting LC3-II-positive autophagosomes to mitochondria.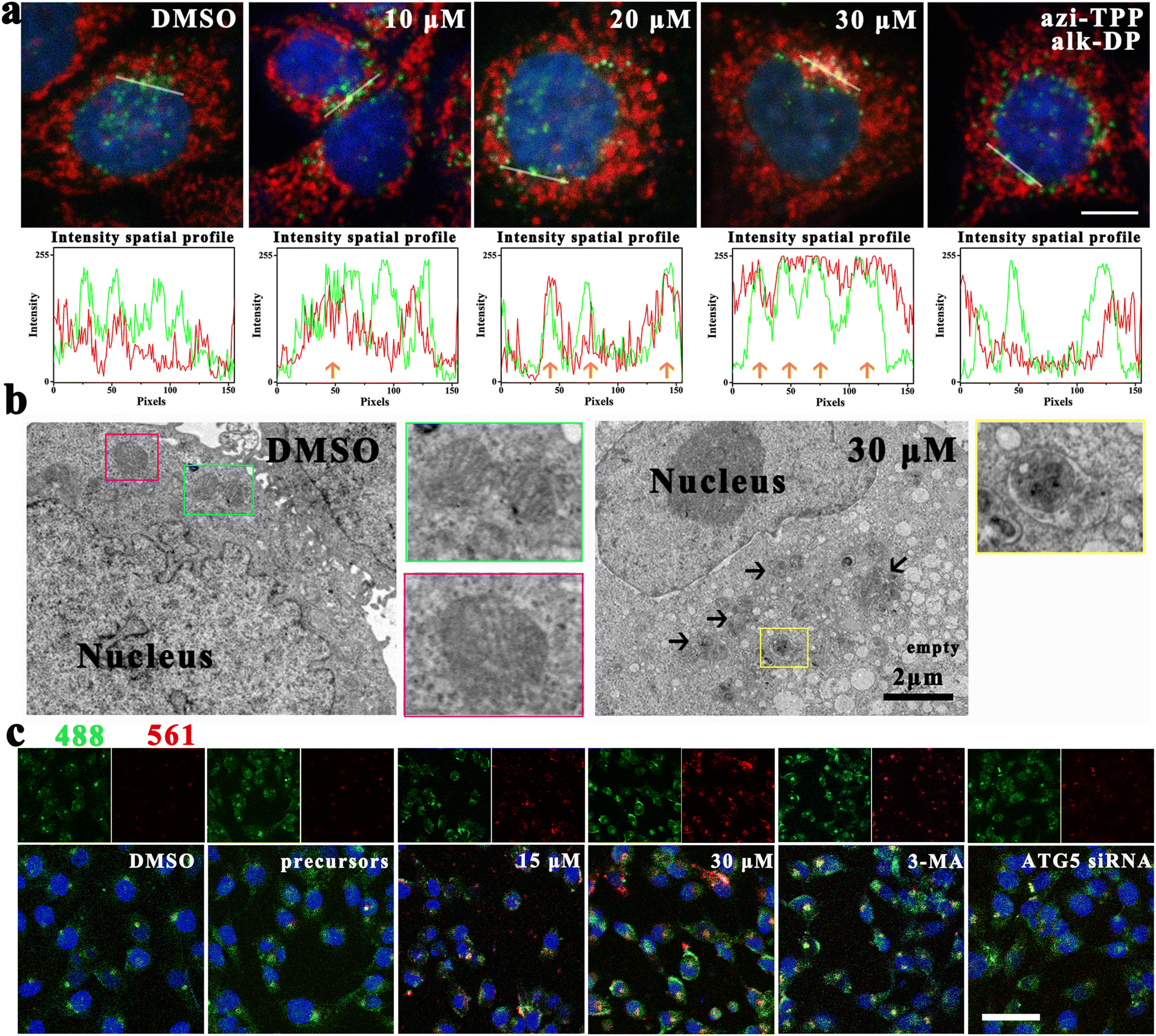 | ||
| Fig. 2 Mito-ATTECs trigger autophagic degradation of mitochondria. (a) Mito-ATTEC treatment (6 h) led to a significant increase in colocalizations of mitochondria (TOM20, red) and autophagosomes (LC3B puncta, green). Scale bars, 10 μm. (b) TEM analysis was performed in DMSO and mito-ATTEC (30 μM, 24 h)-treated MCF-7 cells. The blue and violet boxes indicated the normal mitochondria in DMSO-treated cells. The yellow box showed the autophagosomes engulfing mitochondria. Black arrows indicated autolysosomes (ASS). Empty vacuoles were late-stage autolysosomes in which cellular content was degraded. Scale bars, 2 μm. (c) Assessment of mitophagy in mito-Keima-transfected MCF-7 cells. Our laser sources for CLSM were 488 and 561 nm rather than 458 and 561 nm, which resulted in the overlap of the ‘‘red’’ signal in the ‘‘green’’ images. Scale bars, 50 μm. Data are representative of three independent biological replicates. | ||
Next, potential co-localization of mitochondria and autolysosomes was visualized by CLSM using MCF-7 cells stained with MTR and LysoTracker Green (LTG).18,31 Compared to the untreated control, the colocalization of mitochondria and lysosome obviously increased after mito-ATTEC treatment, indicating the induction of mitophagy (Fig. S2a†). Whereas knockdown of ATG5, which is essential for autophagosome maturation, obviously decreased the co-localization of mitochondria with lysosomes. Moreover, accumulation of the sequestered mitochondrion could be detected within the autolysosomes in the mito-ATTEC-treated cells under a TEM (Fig. 2b). In conclusion, the designed mito-ATTECs induced the mitochondria–LC3 interaction and tethered mitochondria to autolysosomes for subsequent degradation.
We further ascertained the activation of mitophagy using the mito-Keima protein, a mitochondria-targeted reporter that allows for assessment of mitophagy (Fig. 2c).32 Keima is a lysosomal protease-resistant fluorescent protein and also exhibits pH-sensitive excitation. At a neutral pH in the cytosol, the shorter-wavelength excitation predominates (458 nm). At an acidic pH following delivery of mitochondria to acidic lysosomes, mt-Keima undergoes a gradual shift to longer-wavelength excitation (561 nm).32 Compared to DMSO-treated cells, a robust increase in red fluorescence was observed in cells treated with mito-ATTECs (12 h), suggesting the induction of mitophagy. 3-Methyladenine (3-MA, 2 mM) co-treated MCF-7 cells showed a modest reduction in red fluorescence compared with only mito-ATTEC-treated cells, consistent with decreased mitophagic flux in these cells. Similarly, ATG5 knockdown resulted in a significant decrease in the red signal in MCF-7 cells, reflecting autophagy's dominant role in mito-ATTEC-induced mitochondrial degradation.
The role of autophagy in elimination of mitochondria was further supported by the result that there is a partial retention of normal mitochondria (Fig. S2b and c†), when the same treatments were conducted in the presence of 3-MA (2 mM).21 Similarly, the knockdown of ATG5 (ref. 24) also abolished mito-ATTEC-mediated mitochondrial depletion (Fig. S2d†). In contrast, co-incubation with rapamycin (100 nM) resulted in a more pronounced decrease in mitochondrial content (Fig. S2e and f†).24 These results further ascertained that mito-ATTECs eliminated mitochondria via autophagy, which could be enhanced with autophagy activators.
While mito-ATTECs enhanced engulfment of mitochondria by autophagosomes, we inferred that they probably did not influence global autophagy, since the LC3-targeting warhead of the synthesized chimera molecules did not influence global autophagy.23,24 As shown in Fig. S2g,† compared to DMSO-treated cells, we observed no significant change in the numbers of lysosomes in mito-ATTEC-treated cells. Moreover, the levels of autophagosomal marker LC3-II were not affected by mito-ATTEC treatment (Fig. S2h†). Collectively, mito-ATTECs could activate mitophagy without influencing the global autophagy activity.
Mitophagy induced by mito-ATTEC blocked cancer migration and growth
Given that mitochondrial function is strongly associated with tumor invasiveness and metastasis,33 we investigated the potential effect of mito-ATTECs on cancer invasion via a Boyden chamber invasion assay. As compared with DMSO-treated cells, mito-ATTECs effectively suppressed the migration of MCF-7 cells at a relatively low concentration, whereas treatment with separated warheads (30 μM) were insufficient to suppress cancer invasion (Fig. 3a and b).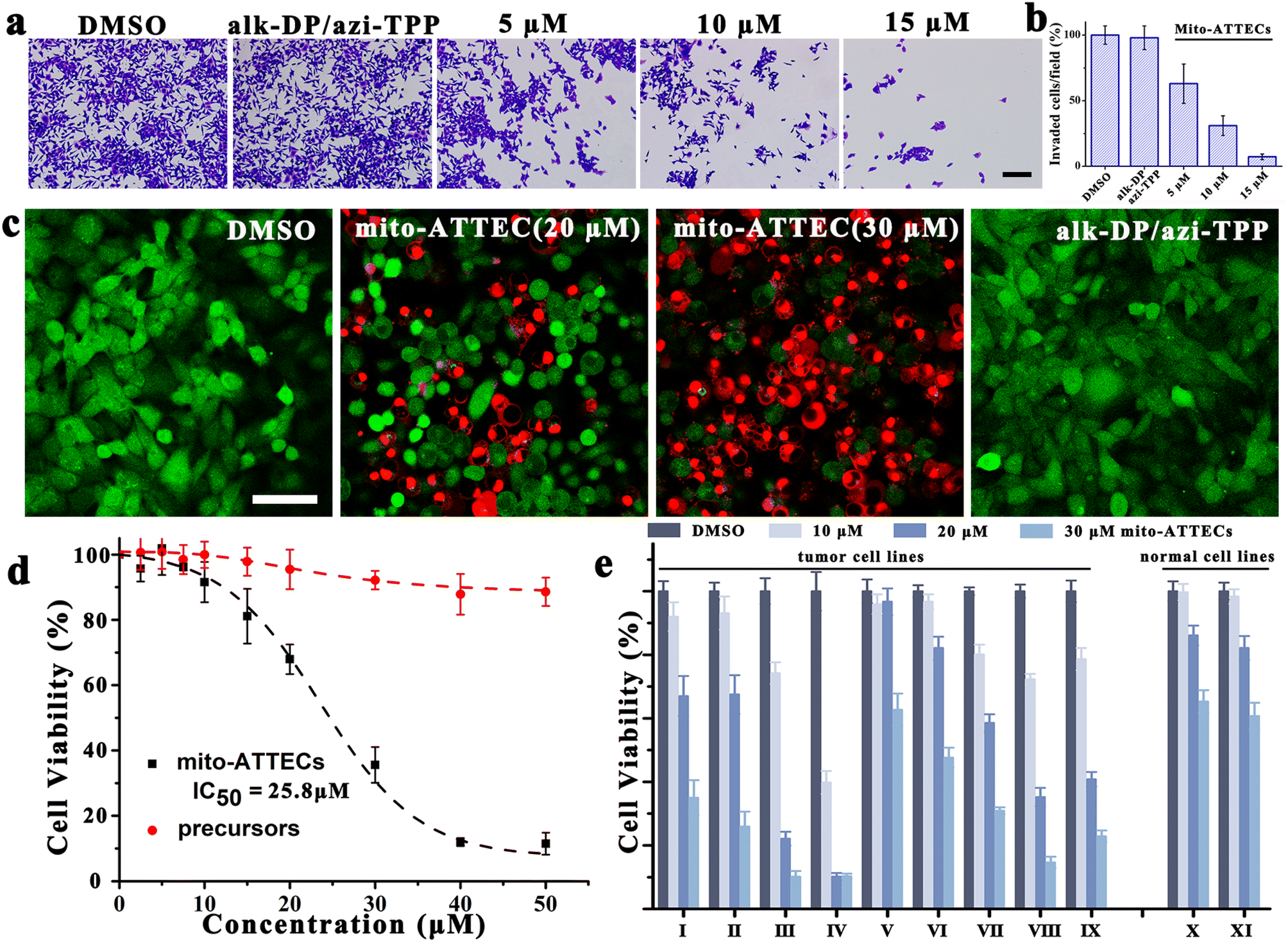 | ||
| Fig. 3 Mito-ATTEC treatment inhibited invasion and growth of tumor cells. (a and b) Effect of mito-ATTECs on the invasion of MCF-7 cells. Representative images from the invasion assay are shown in (a) and quantitative analysis of the data is shown in (b). Scale bars, 200 μm. (c) Live/dead cell analysis after different treatments (24 h). Scale bars, 50 μm. (d) Cell viability of MCF-7 cells treated with mito-ATTECs or precursors (alk-DP and azi-TPP). (e) Antiproliferative activity of mito-ATTECs in tumor and normal cells. The cell viability was measured by MTT assay. MCF-7 cell line (I), MDA-MB-231 cell line (II), A375 cell line (III), B16 cell line (IV), Panc-1 cell line (V), HeLa cell line (VI), HepG2 cell line (VII), U87-MG cell line (VIII), K562 cell line (IX), HEK 293T cell line (X), and L929 cell line (XI). The results shown are representative of at least three independent repeats. | ||
Next, we evaluated the effects of hyperactivating mitophagy on the viability of malignant cells.18,27 As shown in Fig. 3c, live/dead staining clearly demonstrated the anti-proliferative and cytotoxic effects of mito-ATTECs on tumor cells. Fig. 3d demonstrates the dose-dependent decrease in growth and viability of mito-ATTEC-treated MCF-7 cells. The IC50 values for inhibiting cell proliferation were obviously lower for mito-ATTECs than precursors. Thus, linking alk-DP to the azi-TPP via click chemistry is essential to increase its anti-tumor efficiency. Furthermore, mito-ATTECs also exhibited potent cytotoxic activities against different types of tumors (Fig. 3e). Specifically, melanoma cells were more sensitive to mito-ATTECs than other tumors or normal cells.
Mito-ATTECs kill cancer cells through an autophagy-dependent mechanism
Next, we characterized the mechanism of cell death induced by mito-ATTECs. With regard to apoptosis, we observed that Z-VAD-FMK (40 μM), an inhibitor of caspases and apoptosis, failed to block mito-ATTEC-triggered cell death (Fig. 4a). In addition, the necrosis inhibitor necrostatin-1 (10 μM) also failed to protect MCF-7 cells from mito-ATTEC-induced cell death, indicating that apoptotic or necroptotic death machinery was not involved here.34,35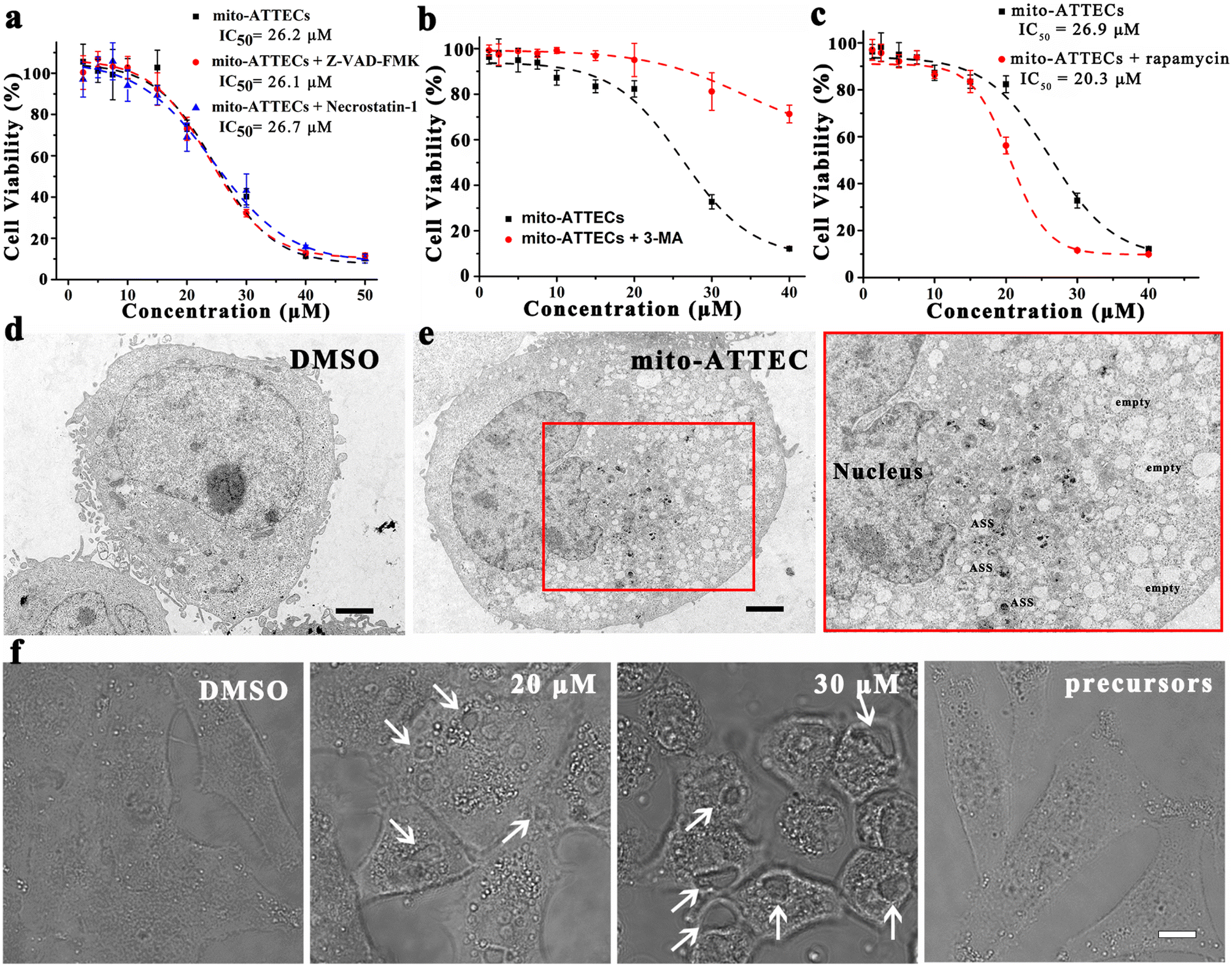 | ||
| Fig. 4 Mito-ATTEC induced autophagic cell death resulting in reduced cell viability. (a) Pretreatment with apoptosis (Z-VAD-FMK) or necrosis inhibitors (necrostatin-1) had no impact on mito-ATTEC-induced cell death. (b) Co-incubation with an autophagy inhibitor (3-MA, 2 mM) significantly reduced mito-ATTEC induced cell death. (c) Autophagy activation by rapamycin (100 nM) sensitized MCF-7 cells to mito-ATTEC-induced cell death. (d and e) Morphological analysis of mito-ATTEC-induced autophagic cell death. DMSO-treated cells showed a normal ultrastructure. (d) Mito-ATTEC (30 μM, 24 h)-treated cells showed reduced numbers of cellular mitochondria, increased number of autolysosomes (ASS), and increased vacuolization. (e) Panels shown on the right are magnifications of boxed areas shown on the left. (f) Mito-ATTEC (24 h) treatment induced autophagic cell death in MCF-7 cells. Arrows indicated autophagic vacuoles in bright field images of mito-ATTEC-treated cells. Scale bars, 10 μm. Data obtained from three representative experiments. | ||
Having excluded apoptosis and necrosis as determinants of the mito-ATTEC-triggered cell death, we sought to evaluate the role of autophagy in cell death. As shown in Fig. S3,† mito-ATTECs reduced the growth and viability of MCF-7 cells. In contrast, ATG5 knockdown protected MCF-7 cells from mito-ATTEC-induced cell death. Alk-DP and azi-TPP did not obviously decrease the growth and viability of MCF-7 or ATG5-KD MCF-7 cells. These results indicated that mito-ATTECs kill malignant cells through an autophagy-dependent mechanism.18,27
To substantiate this conclusion, MCF-7 cells were co-treated with autophagy inhibitors or activators. As shown in Fig. 4b, 3-MA (2 mM) co-treatment obviously abrogated tumor suppression in response to mito-ATTEC treatment, suggesting that activation of autophagy is functionally important for cell death induced by mito-ATTECs.18 In contrast, coincubation with rapamycin (100 nM) in MCF-7 cells sensitized cells to mito-ATTEC-induced cell death (Fig. 4c). These results indicated that mito-ATTECs kill cancer cells via autophagy, and may have cooperative effects with autophagy activators.24
We further ascertained the dominant role of autophagy in mito-ATTEC-triggered cell death using morphological analysis. Ultrastructural examination by TEM of mito-ATTEC (30 μM, 24 h)-treated cells exhibited unique characteristics of autophagy-dependent cell death.36 Namely, mito-ATTEC-treated MCF-7 cells showed increased vacuolization and a decreased number of mitochondria, yet retained normal nuclear morphology (Fig. 4d).35,37 Moreover, increased vacuolization was also observed in CLSM images and was unique to mito-ATTEC-triggered autophagy-dependent cell death (Fig. 4e). Collectively, these findings indicated that mito-ATTEC-induced excessive mitophagy led to autophagic cell death.27
Broad activity of mito-ATTECs against tumor cell lines
Recent studies have indicated that functional mitochondria are indispensable for the growth of malignant cells in several tumor types.38 To determine how widely applicable the lethal mitophagy might be, we further assessed the killing efficacy of mito-ATTECs on several tumor cell lines. Mito-ATTECs reduced viable cell numbers in all of these 9 tumor cell lines after 24 h treatment (Fig. 3e). The mechanisms of mito-ATTEC-induced killing of malignant cells identified above were highly relevant to other cancers. As depicted in Fig. S4a–d,† mito-ATTEC treatment obviously suppressed the growth of MDA-MB-231 cells by complete mitochondrial depletion. Of note, it was found that low concentration of mito-ATTECs exhibited powerful bioactivity and cytotoxicity on human malignant melanoma cell line A375 (Fig. S4e–h†). The IC50 value of mito-ATTECs against A375 cells was about 11.9 μM. Similarly, mito-ATTECs were also active against mouse melanoma cell lines (Fig. S4i–k†), inducing autophagic death of B16-F10 cells with an IC50 value of 9.1 μM. The elevated protein expression of LC3B in melanoma may sensitize melanoma to lethal mitophagy (Fig. S5†). In summary, the mito-ATTEC-induced mitochondrial depletion and cell death were confirmed in several cell lines using different methods, indicating that induction of lethal mitophagy may be a general therapeutic approach.Maximum tolerated dose of mito-ATTECs
First, a 21 day toxicology study of mito-ATTECs was conducted on C57BL/6J mice. As shown in Fig. S6a,† the administration of mito-ATTECs showed negligible impact on the growth of mice. Moreover, no adverse effects were observed in mice by hematology analysis or blood biochemical assay (Fig. S6b†). Therefore, mito-ATTECs, even at a high dose (34 mg kg−1), was well tolerated and did not show any detectable toxicity.In vivo evaluation on the anti-melanoma activity
To further evaluate the therapeutic potential of mito-ATTECs in melanoma, two mouse models of melanoma were generated to evaluate the efficacy of mito-ATTECs. B16F10 melanoma cells were injected subcutaneously into male C57BL/6J mice that were treated 7 days later by injection of saline, precursors (alk-DP and azi-TPP) or mito-ATTECs (17 mg kg−1) over a period of 14 days. Dramatic tumor growth was observed in saline- or precursor-treated groups, whereas mito-ATTEC treatment obviously reduced the growth of subcutaneous transplanted B16F10 tumors (Fig. 5a–c). Excessive mitophagy was also confirmed in the tumor tissues dissected from mito-ATTEC-treated mice (Fig. 5d), demonstrating that the mechanism confirmed in vitro also occurred in the B16F10-melanoma-bearing mice model. Moreover, mice exhibited normal behavior and no apparent toxicity was observed during mito-ATTEC treatment (Fig. 5b). Hematoxylin and eosin (H&E) staining images also confirmed that no organ damage occurred after mito-ATTEC treatment (Fig. S6c†).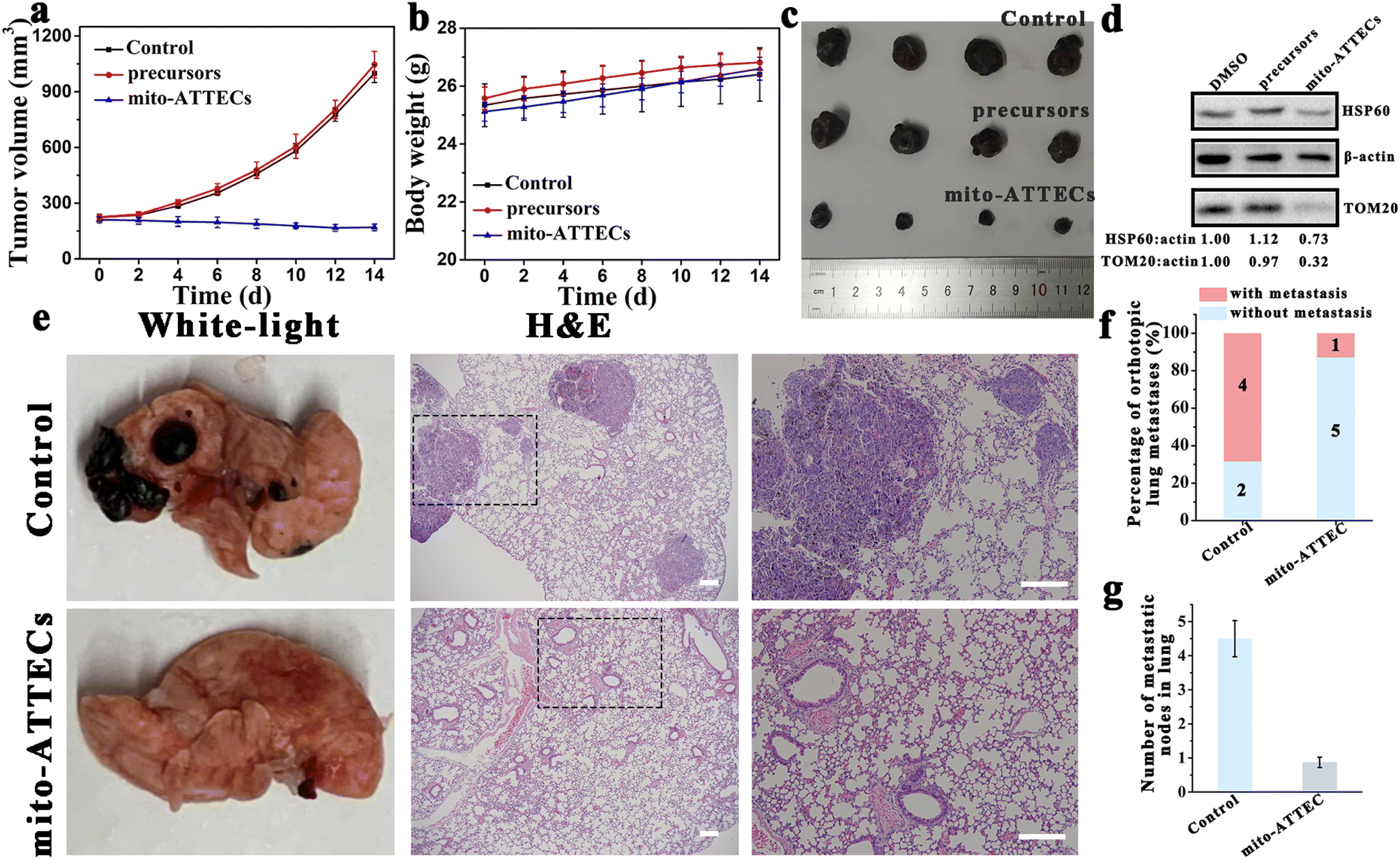 | ||
| Fig. 5 Mito-ATTEC treatment (17 mg kg−1) suppressed the growth of tumors in B16F10-melanoma-bearing mice. (a) Average tumor volume of the subcutaneous melanoma-bearing mice under different conditions. (b) The body weight changes in subcutaneous melanoma-bearing mice treated under different conditions. (c) Representative images of the dissected tumors from subcutaneous melanoma-bearing mice after different treatments (14 days). (d) Mito-ATTECs induced lethal mitophagy in tumor tissues from subcutaneous melanoma-bearing mice. Images are representative of two replicates. (e) Mito-ATTEC treatment suppressed the lung metastasis of the B16F10 cells. Representative images of H&E-stained lung tissue slices after different treatments. Scale bars, 200 μm. (f) Percentage of lung metastases (%) in DMSO or mito-ATTEC-treated mice. (g) Number of metastatic nodes in the lungs of DMSO or mito-ATTEC-treated mice. | ||
Next, we evaluated the effects of mito-ATTECs in a C57BL/6J mouse model of lung metastasis. As shown in Fig. 5e–g and S6d,† when B16F10 melanoma cells were injected into the lateral tail vein of C57BL/6J mice, where cells would transfer to the lung to form tumors, treatment with mito-ATTECs effectively reduced the numbers of melanoma cells in the lung.27 These results clearly suggested the potent anti-melanoma activity of mito-ATTECs in vivo.
Discussion
Bioavailable mitophagy inducers are scarce. Pharmacological screenings for mitophagy chemical inducers are ongoing, and several natural and synthetic compounds have been shown to manipulate mitophagy in mitophagy-defective models.1 Unfortunately, high-throughput screening for drug discovery is time-consuming and also has a high failure rate.11 Moreover, the effectiveness of these inducers may only be applicable to the specified cell line or a certain species (e.g., mouse).12 Thus, a simple and universal strategy for mitophagy modulation is needed. Lessons from targeted protein degradation and a fresh understanding of mitophagy have the potential to facilitate the development of novel mitophagy chemical inducers. Inspired by the receptor-mediated mitophagy, we developed a general chemical tool to modulate mitophagy, mito-ATTECs, which employs chimera molecules composed of LC3-recruiting molecules linked to mitochondria-binding moieties. Mitochondria-targeted mito-ATTEC interacts with LC3 to promote autophagosome formation surrounding mitochondria. Subsequently, autophagosomes containing mitochondria rapidly fuse with lysosomes to facilitate the degradation of mitochondria. Our results indicate that chimera molecules bind to both mitochondria and autophagosome protein LC3, and target mitochondria directly to autophagosomes for degradation. Thus, mito-ATTECs overcome the toxicity related to dissipation of the MMP by agents routinely used to activate mitophagy, and provide a versatile chemical tool to manipulate mitophagy. Besides, LC3/ATG8 family proteins are evolutionarily conserved, existing in all eukaryotic cells. Therefore, mito-ATTECs would be functional in multiple species, including fungi, plants and animals. More importantly, by varying the mitochondrion binding moieties, it is possible to activate mitophagy in a tissue-specific manner. For example, monoamine oxidase (MAO), mainly expressed in the brain, is an OMM-anchored, well-characterized senescence marker.39 We thus hypothesized that MAO inhibitor-based mito-ATTECs would localize at the OMM and interact with LC3 directly to induce mitophagy in the brain, specifically. Collectively, the described methodology may become a universal chemical tool for mitophagy modulation.19Activated mitophagy oftentimes has a complex impact on the cell fate, promoting either cell survival or death.1 Several chemical inducers have been verified to facilitate the elimination of dysfunctional mitochondria through mitophagy activation, such as urolithin A (UA),9,10 kaempferol,11 T-271,13 and AUTAC.12 Supplementation of these inducers results in cytoprotective and anti-ageing effects in several mitophagy-defective models. On the other hand, sustained mitophagy induced by pharmacological or genetic methods may eventually lead to cell death.1 For example, the depletion of AAA-ATPase ATAD3 hyperactivated mitophagy in mouse hematopoietic cells. Affected mice displayed decreased bone-marrow cellularity, erythroid anemia, and reduced survival.40 Moreover, C18-ceramide, a bioactive sphingolipid, has been shown to localize to the mitochondria, induce excessive mitophagy, and induce autophagy-dependent tumor cell death.18,26 However, the effects of chemical inducers on mitophagy hyperactivation and the roles of mitophagy hyperactivation in cell death remain unclear. To this end, we design and synthesize the first lethal mitophagy inducer, which to our knowledge, can hyperactivate mitophagy by bridging mitochondria and LC3-II-positive autophagosomes. We have demonstrated that sustained mitophagy leads to mitochondrial depletion and autophagy-dependent cell death. Our results pave the way towards a deeper understanding of lethal mitophagy in living systems.
Mitochondria lie at the nexus of cancer genesis and progression, such as metabolic reprogramming, invasive capability, and apoptosis.38 As such, mitochondria-damage-induced apoptosis is emerging as a vital mechanism by which chemotherapeutic drugs induce cell death.41 However, numerous studies have demonstrated that the anti-tumor effects of chemotherapy are attenuated due to increased mitophagy, which is closely related to chemoresistance and tumor recurrence.42,43 Consequently, it is of great importance to explore alternative approaches for anti-tumor therapy other than destroy mitochondrial function. In the present work, lethal mitophagy induced by mito-ATTECs may provide an alternative modality, fundamentally distinct from current strategies, to completely eliminate the cellular mitochondrial content and induce autophagic cell death. We have verified that mito-ATTEC-induced dose-dependent cell death can be prevented by genetic (ATG5 knockdown) and pharmacological (3-MA) interference with autophagy, but not apoptosis or necroptosis.27 Moreover, detailed ultrastructural analysis also reveals unique death characteristics such as increased vacuolization and a decreased number of mitochondria.37 Our results have demonstrated the predominant role of autophagy in mito-ATTEC-mediated mitochondrial depletion and autophagic cell death. Taken together, this work provides a compelling rationale for proficiently harnessing mitophagy in anti-cancer therapy.
Our present work indicated that melanoma is substantially sensitive to mito-ATTECs. There are at least two potential explanations for such a result. First, mito-ATTECs bind to both mitochondria and autophagosome protein LC3, initiating mitophagy. Thus, the expression level of the autophagosome protein LC3 is closely related to mito-ATTEC activity.24 Several tumor samples and normal control samples were analyzed to assess LC3 expression levels. As depicted in Fig. S5,† LC3 expression was significantly higher in melanoma compared to in other tumor types and normal control samples. The increased protein expression of LC3 in melanoma may enhance susceptibility to lethal mitophagy. Second, the bioactivity and cytotoxicity of mito-ATTECs is deeply influenced by autophagy. Autophagy inhibition (3-MA co-incubation or ATG5 knockdown) impeded both the degradation of mitochondria and the suppression of tumor growth following treatment with mito-ATTECs. Conversely, autophagy activation (rapamycin co-incubation) sensitized cells to mito-ATTEC-induced mitochondrial depletion and subsequent cell death. A high autophagic index is a significant mechanism of apoptosis resistance in melanomas.44,45 Patients with a high autophagic index showed reduced responsiveness to chemotherapy and experienced shorter survival rates when compared to those with a low autophagic index.46 The high autophagic index may sensitize melanoma cells to mito-ATTEC-induced mitochondrial depletion and autophagic cell death. Collectively, our current study represents proof of principle that anti-melanoma effects can be obtained through lethal mitophagy activation, and as such warrant further investigation.
Conclusion
In summary, we develop a general biochemical approach to modulate mitophagy, mito-ATTECs, which employs changeable chimera molecules composed of LC3-targeting moieties linked to mitochondria-binding compounds. Mito-ATTECs target mitochondria and directly interact with LC3 to promote autophagosome formation surrounding mitochondria. Subsequently, autophagosomes containing mitochondria rapidly fuse with lysosomes to facilitate the degradation of mitochondria. The mito-ATTEC-triggered mitochondrial degradation has been validated in several cell lines using multiple biochemical assays. Our results verify the function of mito-ATTECs for recruiting autophagolysosomes to mitochondria, demonstrating a general strategy for chemical-induced mitophagy. More intriguingly, the sustained mitophagy leads to complete mitochondrial depletion and autophagy-dependent cell death in tumor cells (lethal mitophagy). Apoptosis-resistant malignant melanoma cell lines are even more sensitive to lethal mitophagy, and the therapeutic efficacy of mito-ATTECs has been further evaluated in subcutaneous and pulmonary metastatic melanoma models. Our results indicate that mito-ATTEC treatments lead to mitophagy hyperactivation and irreversible cell death in melanoma murine models with no adverse side effects. Therefore, targeting lethal mitophagy by mito-ATTECs provides a promising therapeutic paradigm for apoptosis-resistant tumors.Ethical statement
The C57BL/6 mice were purchased from Jilin University and acclimatized for 1 week before the experiments. All animal experiments and maintenance protocols complied with the Institutional Animal Care and Use Committee of Jilin University.Data availability
All data are available upon request from the authors.Author contributions
X. Q., J. R. and B. L. conceived the project. Z. L. and G. Q. performed the experimental study and wrote the manuscript. J. Y., W. W. and W. Z. assisted with with the experiments and data analysis. All authors approved the final version.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support was provided by the National Key R&D Program of China (2019YFA0709202 and 2021YFF1200700), and National Natural Science Foundation of China (91856205, 21820102009, and 22237006).References
- K. Palikaras, E. Lionaki and N. Tavernarakis, Nat. Cell Biol., 2018, 20, 1013–1022 CrossRef CAS PubMed.
- M. Onishi, K. Yamano, M. Sato, N. Matsuda and K. Okamoto, EMBO J., 2021, 40, e104705 CrossRef CAS PubMed.
- T. G. McWilliams, A. R. Prescott, L. Montava-Garriga, G. Ball, F. Singh, E. Barini, M. M. K. Muqit, S. P. Brooks and I. G. Ganley, Cell Metab., 2018, 27, 439–449 CrossRef CAS PubMed.
- F. Le Guerroue, F. Eck, J. Jung, T. Starzetz, M. Mittelbronn, M. Kaulich and C. Behrends, Mol. Cell, 2017, 68, 786–796 CrossRef CAS.
- H. Sandoval, P. Thiagarajan, S. K. Dasgupta, A. Schumacher, J. T. Prchal, M. Chen and J. Wang, Nature, 2008, 454, 232–235 CrossRef CAS PubMed.
- M. Sato and K. Sato, Science, 2011, 334, 1141–1144 CrossRef CAS PubMed.
- S. Al Rawi, S. Louvet-Vallee, A. Djeddi, M. Sachse, E. Culetto, C. Hajjar, L. Boyd, R. Legouis and V. Galy, Science, 2011, 334, 1144–1147 CrossRef CAS PubMed.
- M. Lazarou, D. A. Sliter, L. A. Kane, S. A. Sarraf, C. Wang, J. L. Burman, D. P. Sideris, A. I. Fogel and R. J. Youle, Nature, 2015, 524, 309–314 CrossRef CAS PubMed.
- D. Ryu, L. Mouchiroud, P. A. Andreux, E. Katsyuba, N. Moullan, A. A. Nicolet-Dit-Felix, E. G. Williams, P. Jha, G. Lo Sasso, D. Huzard, P. Aebischer, C. Sandi, C. Rinsch and J. Auwerx, Nat. Med., 2016, 22, 879–888 CrossRef CAS PubMed.
- E. F. Fang, Y. Hou, K. Palikaras, B. A. Adriaanse, J. S. Kerr, B. Yang, S. Lautrup, M. M. Hasan-Olive, D. Caponio, X. Dan, P. Rocktaschel, D. L. Croteau, M. Akbari, N. H. Greig, T. Fladby, H. Nilsen, M. Z. Cader, M. P. Mattson, N. Tavernarakis and V. A. Bohr, Nat. Neurosci., 2019, 22, 401–412 CrossRef CAS PubMed.
- C. Xie, X. X. Zhuang, Z. Niu, R. Ai, S. Lautrup, S. Zheng, Y. Jiang, R. Han, T. S. Gupta, S. Cao, M. J. Lagartos-Donate, C. Z. Cai, L. M. Xie, D. Caponio, W. W. Wang, T. Schmauck-Medina, J. Zhang, H. L. Wang, G. Lou, X. Xiao, W. Zheng, K. Palikaras, G. Yang, K. A. Caldwell, G. A. Caldwell, H. M. Shen, H. Nilsen, J. H. Lu and E. F. Fang, Nat. Biomed. Eng., 2022, 6, 76–93 CrossRef CAS PubMed.
- D. Takahashi, J. Moriyama, T. Nakamura, E. Miki, E. Takahashi, A. Sato, T. Akaike, K. Itto-Nakama and H. Arimoto, Mol. Cell, 2019, 76, 797–810 CrossRef CAS PubMed.
- H. Katayama, H. Hama, K. Nagasawa, H. Kurokawa, M. Sugiyama, R. Ando, M. Funata, N. Yoshida, M. Homma, T. Nishimura, M. Takahashi, Y. Ishida, H. Hioki, Y. Tsujihata and A. Miyawaki, Cell, 2020, 181, 1176–1187 CrossRef CAS PubMed.
- I. Novak, V. Kirkin, D. G. McEwan, J. Zhang, P. Wild, A. Rozenknop, V. Rogov, F. Lohr, D. Popovic, A. Occhipinti, A. S. Reichert, J. Terzic, V. Dotsch, P. A. Ney and I. Dikic, EMBO Rep., 2010, 11, 45–51 CrossRef CAS PubMed.
- J. Zhang and P. A. Ney, Cell Death Differ., 2009, 16, 939–946 CrossRef CAS PubMed.
- L. Liu, D. Feng, G. Chen, M. Chen, Q. Zheng, P. Song, Q. Ma, C. Zhu, R. Wang, W. Qi, L. Huang, P. Xue, B. Li, X. Wang, H. Jin, J. Wang, F. Yang, P. Liu, Y. Zhu, S. Sui and Q. Chen, Nat. Cell Biol., 2012, 14, 177–185 CrossRef CAS PubMed.
- L. Liu, K. Sakakibara, Q. Chen and K. Okamoto, Cell Res., 2014, 24, 787–795 CrossRef CAS.
- R. D. Sentelle, C. E. Senkal, W. Jiang, S. Ponnusamy, S. Gencer, S. P. Selvam, V. K. Ramshesh, Y. K. Peterson, J. J. Lemasters, Z. M. Szulc, J. Bielawski and B. Ogretmen, Nat. Chem. Biol., 2012, 8, 831–838 CrossRef CAS.
- X. Y. Fan, L. Guo, L. N. Chen, S. Yin, J. Wen, S. Li, J. Y. Ma, T. Jing, M. X. Jiang, X. H. Sun, M. Chen, F. Wang, Z. B. Wang, C. F. Zhang, X. H. Wang, Z. J. Ge, C. Hu, L. Zeng, W. Shen, Q. Y. Sun, X. H. Ou and S. M. Luo, Nat. Biomed. Eng., 2022, 6, 339–350 CrossRef CAS.
- B. Ding, L. Zhang, Z. Li, Y. Zhong, Q. Tang, Y. Qin and M. Chen, Cell Host Microbe, 2017, 21, 538–547 CrossRef CAS.
- X. Qin, R. Wang, H. Xu, L. Tu, H. Chen, H. Li, N. Liu, J. Wang, S. Li, F. Yin, N. Xu and Z. Li, Autophagy, 2022, 18, 2178–2197 CrossRef CAS PubMed.
- M. Liu, Z. Liu, G. Qin, J. Ren and X. Qu, Nano Lett., 2023, 23, 4965–4973 CrossRef CAS PubMed.
- Z. Li, C. Wang, Z. Wang, C. Zhu, J. Li, T. Sha, L. Ma, C. Gao, Y. Yang, Y. Sun, J. Wang, X. Sun, C. Lu, M. Difiglia, Y. Mei, C. Ding, S. Luo, Y. Dang, Y. Ding, Y. Fei and B. Lu, Nature, 2019, 575, 203–209 CrossRef CAS PubMed.
- Y. Fu, N. Chen, Z. Wang, S. Luo, Y. Ding and B. Lu, Cell Res., 2021, 31, 965–979 CrossRef CAS.
- Y. Ding, D. Xing, Y. Fei and B. Lu, Chem. Soc. Rev., 2022, 51, 8832–8876 RSC.
- M. Dany, S. Gencer, R. Nganga, R. J. Thomas, N. Oleinik, K. D. Baron, Z. M. Szulc, P. Ruvolo, S. Kornblau, M. Andreeff and B. Ogretmen, Blood, 2016, 128, 1944–1958 CrossRef CAS PubMed.
- W. J. Wang, Y. Wang, H. Z. Chen, Y. Z. Xing, F. W. Li, Q. Zhang, B. Zhou, H. K. Zhang, J. Zhang, X. L. Bian, L. Li, Y. Liu, B. X. Zhao, Y. Chen, R. Wu, A. Z. Li, L. M. Yao, P. Chen, Y. Zhang, X. Y. Tian, F. Beermann, M. Wu, J. Han, P. Q. Huang, T. Lin and Q. Wu, Nat. Chem. Biol., 2014, 10, 133–140 CrossRef CAS.
- J. Zielonka, J. Joseph, A. Sikora, M. Hardy, O. Ouari, J. Vasquez-Vivar, G. Cheng, M. Lopez and B. Kalyanaraman, Chem. Rev., 2017, 117, 10043–10120 CrossRef CAS PubMed.
- J. M. Suski, M. Lebiedzinska, M. Bonora, P. Pinton, J. Duszynski and M. R. Wieckowski, Methods Mol. Biol., 2012, 810, 183–205 CrossRef CAS PubMed.
- M. Mauro-Lizcano, L. Esteban-Martinez, E. Seco, A. Serrano-Puebla, L. Garcia-Ledo, C. Figueiredo-Pereira, H. L. Vieira and P. Boya, Autophagy, 2015, 11, 833–843 CrossRef CAS PubMed.
- N. J. Dolman, K. M. Chambers, B. Mandavilli, R. H. Batchelor and M. S. Janes, Autophagy, 2013, 9, 1653–1662 CrossRef CAS PubMed.
- N. Sun, D. Malide, J. Liu, I. I. Rovira, C. A. Combs and T. Finkel, Nat. Protoc., 2017, 12, 1576–1587 CrossRef CAS PubMed.
- T. Saha, C. Dash, R. Jayabalan, S. Khiste, A. Kulkarni, K. Kurmi, J. Mondal, P. K. Majumder, A. Bardia, H. L. Jang and S. Sengupta, Nat. Nanotechnol., 2021, 17, 98–106 CrossRef PubMed.
- G. Cheng, Q. Zhang, J. Pan, Y. Lee, O. Ouari, M. Hardy, M. Zielonka, C. R. Myers, J. Zielonka, K. Weh, A. C. Chang, G. Chen, L. Kresty, B. Kalyanaraman and M. You, Nat. Commun., 2019, 10, 2205 CrossRef PubMed.
- K. Shchors, A. Massaras and D. Hanahan, Cancer Cell, 2015, 28, 456–471 CrossRef CAS PubMed.
- D. Denton and S. Kumar, Cell Death Differ., 2019, 26, 605–616 CrossRef CAS PubMed.
- S. K. Dasari, S. Bialik, S. Levin-Zaidman, V. Levin-Salomon, A. H. Merrill Jr, A. H. Futerman and A. Kimchi, Cell Death Differ., 2017, 24, 1288–1302 CrossRef CAS PubMed.
- S. E. Weinberg and N. S. Chandel, Nat. Chem. Biol., 2015, 11, 9–15 CrossRef CAS PubMed.
- Y. Santin, J. Resta, A. Parini and J. Mialet-Perez, Ageing Res. Rev., 2021, 66, 101256 CrossRef CAS PubMed.
- G. Jin, C. Xu, X. Zhang, J. Long, A. H. Rezaeian, C. Liu, M. E. Furth, S. Kridel, B. Pasche, X. W. Bian and H. K. Lin, Nat. Immunol., 2018, 19, 29–40 CrossRef CAS PubMed.
- X. Guo, N. Yang, W. Ji, H. Zhang, X. Dong, Z. Zhou, L. Li, H. M. Shen, S. Q. Yao and W. Huang, Adv. Mater., 2021, 33, e2007778 CrossRef PubMed.
- J. Y. Guo, B. Xia and E. White, Cell, 2013, 155, 1216–1219 CrossRef CAS PubMed.
- J. M. M. Levy, C. G. Towers and A. Thorburn, Nat. Rev. Cancer, 2017, 17, 528–542 CrossRef CAS PubMed.
- V. Gray-Schopfer, C. Wellbrock and R. Marais, Nature, 2007, 445, 851–857 CrossRef CAS PubMed.
- M. Corazzari, F. Rapino, F. Ciccosanti, P. Giglio, M. Antonioli, B. Conti, G. M. Fimia, P. E. Lovat and M. Piacentini, Cell Death Differ., 2015, 22, 946–958 CrossRef CAS PubMed.
- X. H. Ma, S. Piao, D. Wang, Q. W. McAfee, K. L. Nathanson, J. J. Lum, L. Z. Li and R. K. Amaravadi, Clin. Cancer Res., 2011, 17, 3478–3489 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc03600f |
| This journal is © The Royal Society of Chemistry 2023 |