
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIsoacridone dyes with parallel reactivity from both singlet and triplet excited states for biphotonic catalysis and upconversion†
Björn
Pfund‡
 ,
Valeriia
Hutskalova‡
,
Christof
Sparr
* and
Oliver S.
Wenger
*
,
Valeriia
Hutskalova‡
,
Christof
Sparr
* and
Oliver S.
Wenger
*
Department of Chemistry, University of Basel, St. Johanns-Ring 19, 4056 Basel, Switzerland. E-mail: oliver.wenger@unibas.ch; christof.sparr@unibas.ch
First published on 2nd October 2023
Abstract
Metal-based photosensitizers commonly undergo quantitative intersystem crossing into photoactive triplet excited states. In contrast, organic photosensitizers often feature weak spin–orbit coupling and low intersystem crossing efficiencies, leading to photoactive singlet excited states. By modifying the well-known acridinium dyes, we obtained a new family of organic photocatalysts, the isoacridones, in which both singlet- and triplet-excited states are simultaneously photoactive. These new isoacridone dyes are synthetically readily accessible and show intersystem crossing efficiencies of up to 52%, forming microsecond-lived triplet excited states (T1), storing approximately 1.9 eV of energy. Their photoactive singlet excited states (S1) populated in parallel have only nanosecond lifetimes, but store ∼0.4 eV more energy and act as strong oxidants. Consequently, the new isoacridone dyes are well suited for applications requiring parallel triplet–triplet energy transfer and photoinduced electron transfer elementary steps, which have become increasingly important in modern photocatalysis. In proof-of-principle experiments, the isoacridone dyes were employed for Birch-type arene reductions and C–C couplings via sensitization-initiated electron transfer, substituting the commonly used iridium or ruthenium based photocatalysts. Further, in combination with a pyrene-based annihilator, sensitized triplet–triplet annihilation upconversion was achieved in an all-organic system, where the upconversion quantum yield correlated with the intersystem crossing quantum yield of the photosensitizer. This work seems relevant in the greater contexts of developing new applications that utilize biphotonic photophysical and photochemical behavior within metal-free systems.
Introduction
Triplet-excited states have fundamentally different photochemical reactivities and photophysical behavior than singlet-excited states but are often inaccessible by direct excitation of organic substrates.1–3 Triplet-sensitization via energy transfer has therefore become relevant in many areas of photochemistry, such as photodynamic therapy,4 photocatalysis,5–7 and triplet–triplet annihilation upconversion (sTTA-UC).7–11 In recent years, triplet sensitization has furthermore become important for photochemical reactions requiring particularly high energy input in the form of multiple photons,12–16 to overcome the inherent limitations of traditional monophotonic excitation processes.17,18 The development of photosensitizers (PS) enabling efficient intersystem crossing (ISC), often focuses on transition metal complexes containing precious elements such as iridium or ruthenium,19–23 raising cost and sustainability issues.24,25 Against this background, there is increased interest in developing photosensitizers made from abundant first-row transition metal complexes,26–30 and purely organic (metal-free) alternatives.31–35 Organic PS could be more amenable to larger scale applications,36–38 and numerous organic PS such as xanthene-type dyes (eosin Y, fluorescein),39,40 cyanoarenes,31,41 anthraquinones,42,43 flavins,44–46 BODIPY,47–50 and others are already known.51–55 The performance of organic photosensitizers can be limited by several factors including modest excited-state redox potentials,56 visible light absorption,51–55 or (photo)stability,57 but inefficient ISC to access photoactive triplet-excited states appears to be among the primary limitations.58 BODIPY compounds have found application in sensing, biological labeling,59 potential cancer treatment,4 and for upconversion,60 but clear molecular design concepts to enhance ISC have remained scarce for organic photosensitizers. In the case of BODIPY dyes, efficient ISC has been achieved by introducing heavy atoms such as iodine,61 twisted π-conjugated structures,62 or by radical-enhanced ISC.50 However, the high intersystem crossing efficiencies of these modified BODIPY dyes lead to comparatively low excited state redox potentials, limiting their application potential for photoredox catalysis.47,57Acridinium dyes are highly tunable and widely used organic photosensitizers, with excited-state oxidation potentials often exceeding 2.3 V vs. SCE,63,64 making them powerful photo-oxidants. However, the electron-deficient nature of the heteroaromatic acridinium core results in susceptibility to nucleophilic attack at their C-9 position, often leading to photocatalyst decomposition, which can be prevented by the introduction of sterically demanding substituents such as phenyl or mesityl (Fig. 1a, compound 1).65 In the past, the stability of the acridinium photosensitizers was further improved by the attachment of bulky substituents at positions 2 and 7, or at positions 3 and 6, and further by replacing the N-methyl substituent with an N-phenyl unit (Fig. 1a, compound 2).66–69 Some of the more photostable acridinium photosensitizers were later successfully applied for biphotonic processes such as the ConPET mechanism,12,70 in which the photoactive singlet excited state of the acridinium dye was reductively quenched by an excess of sacrificial donor. The resulting one-electron reduced form was then promoted to a short-lived but highly reactive excited state by absorption of a second photon, to accomplish reductive substrate activations requiring reduction potentials as negative as −2.9 V vs. SCE.71 Such biphotonic excitation mechanisms are attractive, because they can give rise to much broader chemical reactivities and substrate scopes, due to the combined energy input from two photons.13,14 However, other biphotonic mechanisms such as sensitized triplet–triplet annihilation upconversion (sTTA-UC),23,72–74 sensitization-initiated electron transfer (SeniET),16 or Birch-type arene reductions,15 all rely on triplet–triplet energy transfer and as such necessitate photosensitizers with photoactive triplet excited states, which are not efficiently accessible with common acridinium dyes. Recently, it was shown that electron-donating dimethylamino groups on the acridinium core enhance the intersystem crossing efficiency, yet the quantum yield for this process was less than 7%.75 However, more efficient intersystem crossing is required for effective triplet-based photoreactions, but the rational design of enhanced intersystem crossing efficiencies remains challenging.50,58,62,76–79
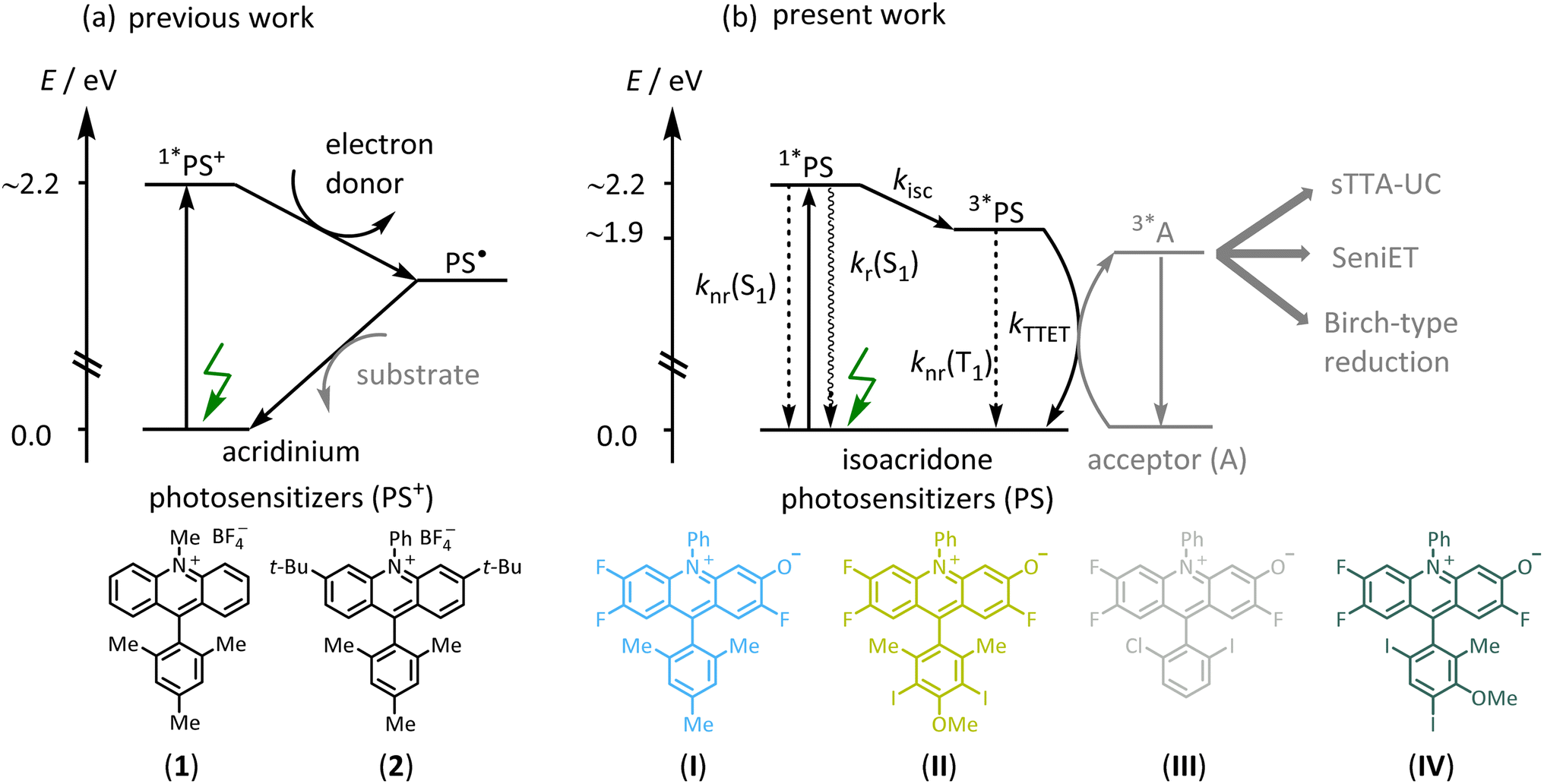 | ||
| Fig. 1 (a) Acridinium photosensitizers reacting from singlet-excited states for traditional (mono-photonic) photoredox catalysis. (b) New isoacridone dyes with enhanced intersystem crossing quantum yields for biphotonic catalysis requiring parallel singlet and triplet photoreactions, and for sensitized triplet–triplet annihilation upconversion (sTTA-UC). | ||
In this work, we designed a range of isoacridone-based photosensitizers that were readily accessible by utilizing an expedient three-step route consisting of acridane synthesis via aryne-imine-aryne coupling, its subsequent oxidation, followed by nucleophilic aromatic substitution of one fluorine atom (Fig. 1b, see ESI† for more details).80–82 Targeting high intersystem crossing quantum yields (ISC-QYs), we introduced iodine atoms at different positions of the aryl-substituent at C-9 of the acridinium core, resulting in four different isoacridone dyes (shown in Fig. 1b) that were explored in comparative fashion (PS(I)–PS(IV)). We anticipated that the position of the iodine atoms strongly affects the ISC-QY.61,83 Iodine at meta- and para-positions were expected to have a relatively minor influence on intersystem crossing,49,60,84 whereas the ortho-position was predicted to lead to a substantial increase of the ISC-QY due to the close proximity and its strong electronic coupling to the isoacridone core. Thus, we anticipated that both PS(I) and PS(II) show low ISC-QY, whereas PS(III) and PS(IV) should feature much enhanced ISC-QY. The predicted correlation of this rational design approach was indeed largely fulfilled, resulting in a new family of triplet photosensitizers with ISC-QYs between 0.26 to 0.52. These new photocatalysts were then successfully applied in multiphoton catalysis, including Birch-type arene photoreductions and a sensitization-initiated electron transfer process, where both a single electron transfer, mainly from the lowest singlet-excited state (S1), and a triplet energy transfer from the lowest triplet-excited state (T1), is required. Furthermore, the new isoacridone dyes were successfully applied in sTTA-UC using a pyrene-based annihilator, providing the proof-of-concept that isoacridones could act as surrogates for precious-metal based triplet-sensitizers in light conversion.
Results and discussion
Photophysical characterization
Calibrated UV-vis absorption, steady-state emission spectra (recorded at room temperature and at 77 K), time-resolved emission measurements, and fluorescence quantum yield measurements were performed with all four new PS. The key results are shown in Fig. 2 and summarized in Table 1. The reference compound without any iodine atoms (PS(I)) shows a broad absorption band with a local maximum (λabs) at 485 nm and a molar extinction coefficient (ε) of 11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500 M−1 cm−1. The iodo-substituted photosensitizers show minor red-shifts while retaining similar molar extinction coefficients at the respective absorption band maxima (PS(II), λabs = 491 nm, ε = 10400 M−1 cm−1; PS(III), λabs = 492 nm, ε = 11200 M−1 cm−1; PS(IV), λabs = 492 nm, ε = 9800 M−1 cm−1). The steady-state emission spectra display an analogous red shift with fluorescence band maxima from 556 nm for PS(I) to 570 nm for PS(IV). The S1 energies were estimated by determining the intersection of room-temperature emission and absorption spectra, resulting in essentially identical S1 energies (ES1) in all four compounds (Table 1). The fluorescence lifetime (τS1) decreases along the series from PS(I) to PS(IV) (Table 1), due to non-radiative deactivation by ISC (Fig. 1b), as discussed below. The fluorescence quantum yields (ΦFL) decrease from 0.227 for PS(I) to 0.169 (PS(II)), 0.060 (PS(III)), and 0.062 for PS(IV) (Table 1), in line with enhanced ISC to the lowest triplet excited state.
500 M−1 cm−1. The iodo-substituted photosensitizers show minor red-shifts while retaining similar molar extinction coefficients at the respective absorption band maxima (PS(II), λabs = 491 nm, ε = 10400 M−1 cm−1; PS(III), λabs = 492 nm, ε = 11200 M−1 cm−1; PS(IV), λabs = 492 nm, ε = 9800 M−1 cm−1). The steady-state emission spectra display an analogous red shift with fluorescence band maxima from 556 nm for PS(I) to 570 nm for PS(IV). The S1 energies were estimated by determining the intersection of room-temperature emission and absorption spectra, resulting in essentially identical S1 energies (ES1) in all four compounds (Table 1). The fluorescence lifetime (τS1) decreases along the series from PS(I) to PS(IV) (Table 1), due to non-radiative deactivation by ISC (Fig. 1b), as discussed below. The fluorescence quantum yields (ΦFL) decrease from 0.227 for PS(I) to 0.169 (PS(II)), 0.060 (PS(III)), and 0.062 for PS(IV) (Table 1), in line with enhanced ISC to the lowest triplet excited state.
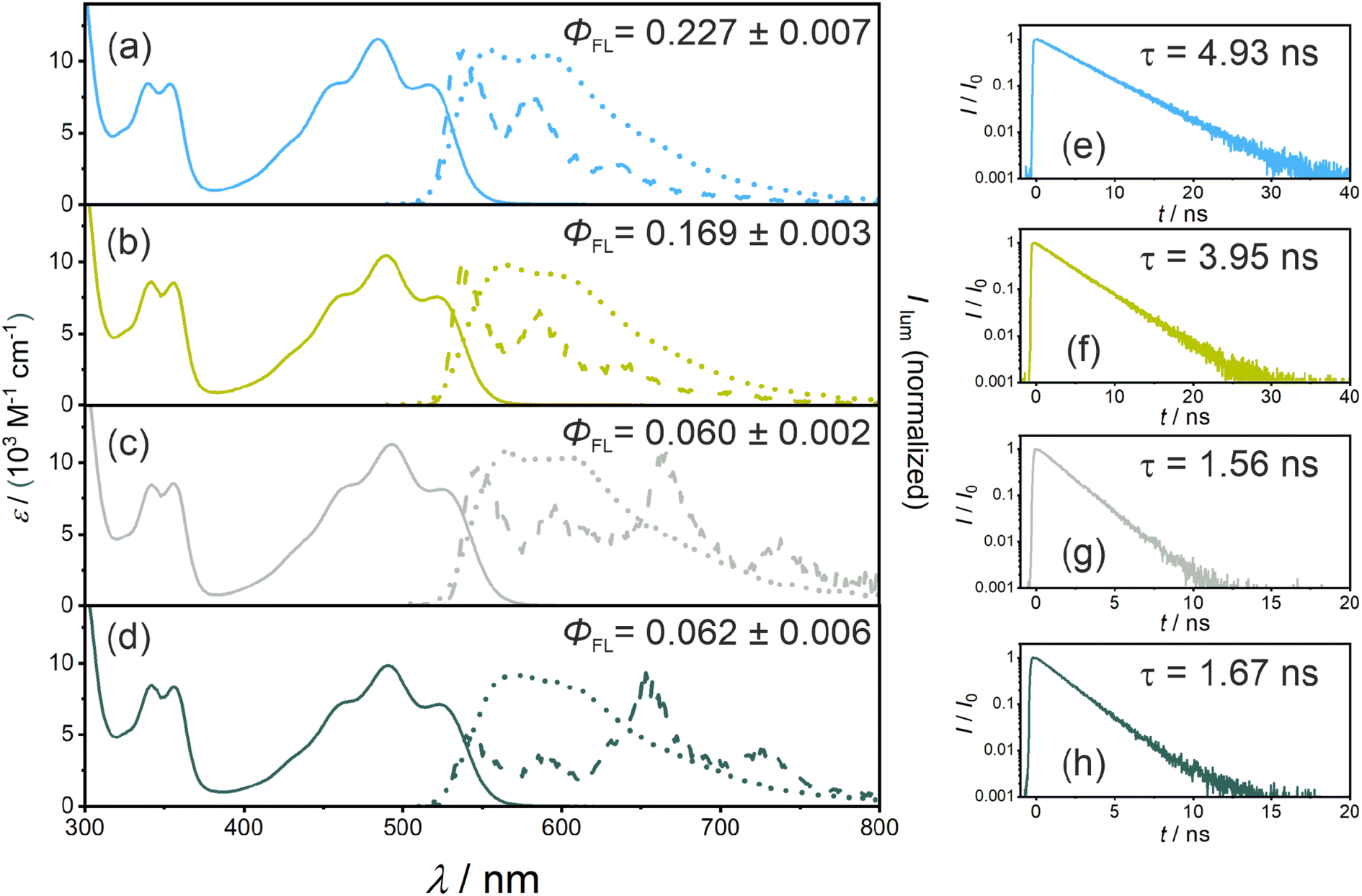 | ||
| Fig. 2 Photophysical properties of the fluorescent singlet excited states of PS(I) (a and e), PS(II) (b and f), PS(III) (c and g), and PS(IV) (d and h). (a–d) calibrated absorption (solid lines) and normalized fluorescence spectra (dotted lines) in deaerated THF at 20 °C (upon excitation at 470 nm) containing 1 × 10−5 M of the respective PSs. The phosphorescence spectra (dashed lines) were obtained in frozen 2-methyltetrahydrofuran at 77 K upon 470 nm excitation. The fluorescence quantum yields of the respective photosensitizers (ΦFL) were measured using an integration sphere. (e–h) Upon 472 nm excitation, the singlet excited state lifetimes were measured at the emission band maxima of the corresponding PSs in deaerated THF at 20 °C (c = 1 × 10−5 M) using the TCSPC technique. | ||
| PS | E S1 /eV | τ S1 /ns | Φ FL | Φ ISC | E T1 /eV | τ T1 /μs | k ISC /107 s−1 | k r(S1)h/107 s−1 | k nr(S1)i/108 s−1 | k nr(T1)j/104 s−1 |
|---|---|---|---|---|---|---|---|---|---|---|
| a Singlet excited state energies (ES1) were determined by the intersections between absorption and fluorescence spectra. b Singlet excited state lifetimes (τS1) were obtained by the TCSPC technique using deaerated THF solutions at 20 °C, containing 1 × 10−5 M of the corresponding photosensitizers. c The fluorescence quantum yields (ΦFL) in THF at 20 °C were measured using an integration sphere. d Intersystem crossing quantum yields (ΦISC) were determined by monitoring the formation of triplet-excited anthracene after excitation of the photosensitizers in the presence of 10 mM anthracene, as described in the text. e Triplet excited state energies (ET1) were determined by the onset of the highest-energy phosphorescence band measured at 77 K (where the phosphorescence intensity is 10% compared to the maximum). f Triplet excited state lifetimes (τT1) were detected using laser flash photolysis by exciting with a 450 nm pulsed laser. g Rate constants for ISC (kISC) were calculated using kISC = 1/τS1 × ΦISC. h Rate constants of the radiative singlet-excited state decay (kr(S1)) were calculated using kr(S1) = 1/τS1 × ΦFL. i Rate constants of the non-radiative singlet-excited state decay (knr(S1)) were calculated using τS1 = 1/ktot(S1) = 1/(kr(S1) + knr(S1) + kISC). j Rate constants of the non-radiative triplet-excited state decay (knr(T1)) were calculated using τT1 = 1/ktot(T1) = 1/(kr,(T1) + knr(T1)). | ||||||||||
| (I) | 2.33 | 4.93 | 0.23 | 0.26 | n/d | 43.7 | 5.3 | 4.7 | 1.0 | 2.3 |
| (II) | 2.30 | 3.95 | 0.17 | 0.30 | n/d | 42.9 | 7.6 | 4.3 | 1.3 | 2.3 |
| (III) | 2.29 | 1.56 | 0.06 | 0.43 | 1.88 | 25.7 | 28 | 3.9 | 3.3 | 3.9 |
| (IV) | 2.30 | 1.67 | 0.06 | 0.52 | 1.88 | 33.2 | 31 | 3.6 | 2.5 | 3.0 |
Luminescence spectra recorded from frozen solvent matrices at 77 K (Fig. 2a–d, dashed lines) are similar to room temperature fluorescence spectra in the case of PS(I) and PS(II), whereas for PS(III) and PS(IV) additional emission bands at 660 nm and 730 nm indicate a triplet-based phosphorescence. This observation is in line with the expected increase of the ISC-QY when iodo-substituents are attached in ortho-position relative to the isoacridone core. Based on the onset of the highest-energy phosphorescence band at 660 nm (corresponding to the point, at which the phosphorescence intensity is 10% compared to the maximum), a triplet excited state energy (ET1) of ∼1.88 eV was estimated for both PS(III) and PS(IV). It seems plausible to assume that the ET1 of PS(I) and PS(II) are similar to those of PS(III) and PS(IV).
Nanosecond transient UV-vis absorption spectroscopy was applied to probe the triplet excited states of the photosensitizers (Fig. 3a–d). All four photosensitizers show ground state bleaches (GSB) at ∼490 nm and at ∼355 nm (where the UV-vis (ground-state) absorption spectra have the strongest bands), as well as excited state absorption (ESA) signals at ∼410 nm and ∼560 nm. Both GSB and ESA show essentially identical lifetimes of 43.7 μs for PS(I), 42.9 μs for PS(II), 25.7 μs for PS(III), and 33.2 μs for PS(IV) (Fig. 3e–h), indicating the presence of a non-emissive (dark) T1 state at room temperature in all four photosensitizers. When comparing the changes in optical density at 415 nm, an increase from ∼15 mΔOD for PS(I) and PS(II) to ∼35 mΔOD for PS(III) and PS(IV) is observed, in line with the expected improved ISC-QY of PS(III) and PS(IV) compared to PS(I) and PS(II). Additionally, the T1 lifetimes (τT1) of PS(I) and PS(II) (43.7 μs and 42.9 μs, respectively) are significantly longer compared to those of PS(III) (25.7 μs) and PS(IV) (33.2 μs) (Table 1), compatible with the expected enhancement of ISC. The increased spin–orbit coupling increases not only the singlet-triplet conversion but also the reverse process when the T1 state relaxes back to the singlet ground state (S0).
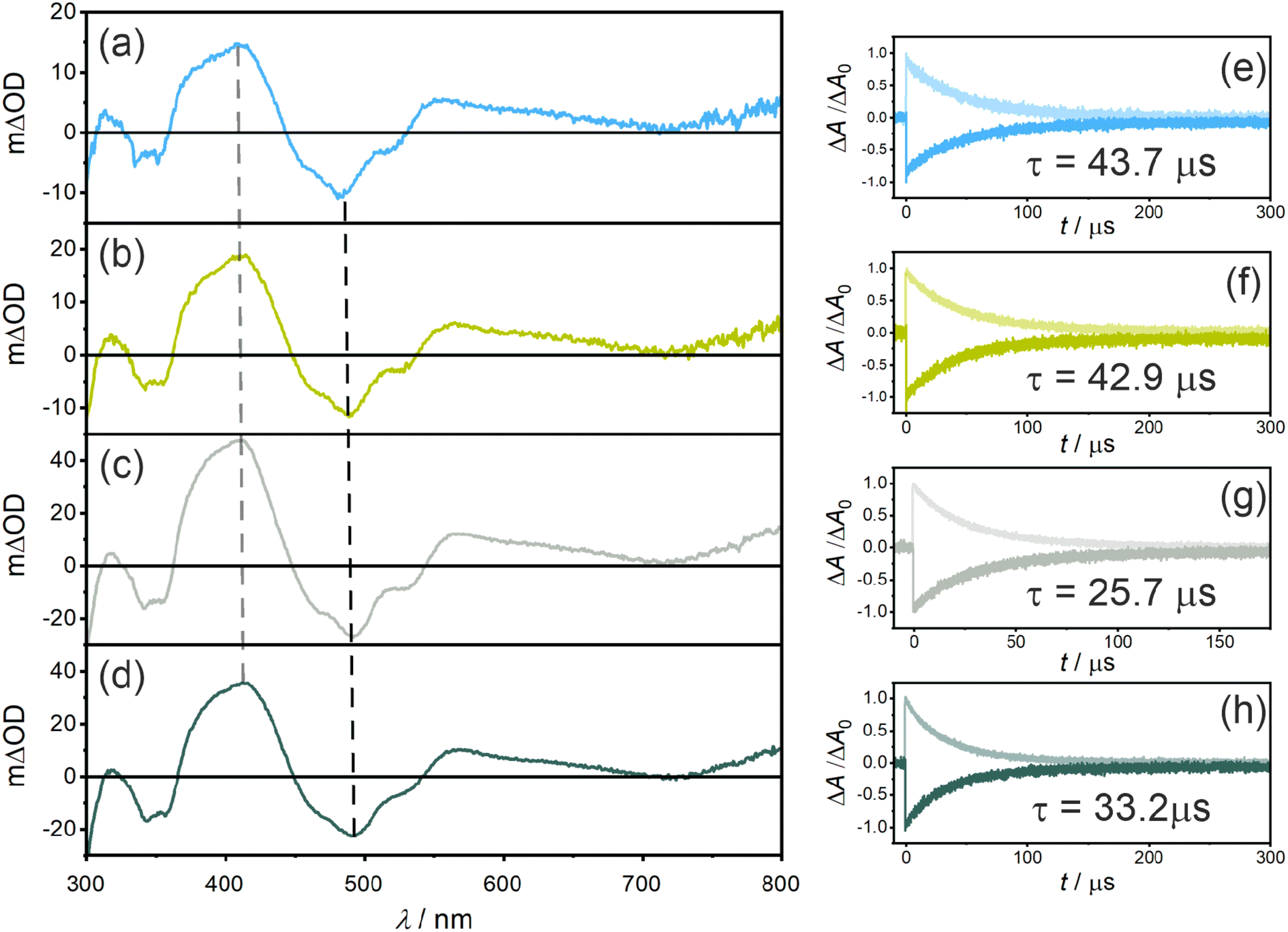 | ||
| Fig. 3 Triplet excited-state properties of the four photosensitizers PS(I) (a and e), PS(II) (b and f), PS(III) (c and g), and PS(IV) (d and h) in deaerated THF at 20 °C containing 2 × 10−5 M of the corresponding dyes. (a–d) Transient UV-vis absorption spectra were recorded with a time delay of 100 ns and a time integration over 200 ns after 450 nm pulsed excitation (15 mJ energy per pulse). The vertical dashed lines in panels (a–d) indicate the detection wavelengths of the ESA and GSB kinetics shown in panels (e–f). | ||
Relative actinometry experiments were performed to determine the absolute ISC-QYs. Selective excitation of the individual photosensitizers in the presence of excess anthracene leads to the formation of triplet-excited anthracene after ISC and bimolecular triplet–triplet energy transfer (TTET). Given the driving-force of ca. 0.1 eV for TTET and the large excess of anthracene (10 mM), this process is assumed to be essentially quantitative. Consequently, by determining the concentration of triplet-excited anthracene by transient UV-vis absorption spectroscopy (based on the measured molar extinction coefficient of triplet anthracene at 423 nm, 82000 M−1 cm−1 in THF, see ESI†), the molar quantity of triplet excited photosensitizers was estimated. Comparing the formed molar quantity of triplet excited isoacridone dyes with an aqueous [Ru(bpy)3]2+ reference system, in which intersystem crossing is quantitative and the molar extinction coefficient of the GSB at 455 nm is known (Δε = −10100 M−1 s−1),85 the ISC-QYs of the four new PSs were calculated (Table 1). Expectedly, PS(I) and PS(II) showed the lowest ISC-QYs with 0.26 and 0.30, whereas PS(III) and PS(IV) had markedly higher ISC-QYs of 0.43 and 0.52, respectively.
To further characterize the S1 and T1 states, the radiative (kr(S1)) and non-radiative S1 decay rate constants (knr(S1)) to the electronic S0 ground state, as well the intersystem crossing rate constant (kISC) were calculated based on the known fluorescence quantum yield (ΦFL), the intersystem crossing quantum yield (ΦISC), and the singlet excited state lifetime (τS1). The key finding (summarized in Table 1) is that the ISC rate constant increases from 5.3 × 107 s−1 for PS(I) to 31 × 107 s−1 for PS(IV). The radiative singlet-excited state decay is comparatively little affected by the enhanced intersystem crossing (kr(S1) = 4.7 × 107 s−1 for PS(I), 4.3 × 107 s−1 for PS(II), 3.9 × 107 s−1 for PS(III), and 3.7 × 107 s−1 for PS(IV)). In contrast, the non-radiative singlet-excited state decay knr(S1) back to the ground state is accelerated from 1.0 × 108 s−1 for PS(I) to 2.5 × 108 s−1 for PS(IV). These findings are in line with the decreased singlet excited state lifetimes and fluorescence quantum yields when going from PS(I) & PS(II) to PS(III) & PS(IV), and with the exclusive observation of phosphorescence at 77 K in the case of PS(III) & PS(IV). Overall, these results validate our molecular design principles for obtaining all-organic combined singlet and triplet photosensitizers.
To further increase the ISC-QY, the external heavy atom effect was investigated by using heavy atom-containing salts or co-solvents.86–88 PS(I) was selected for this purpose, because it has the lowest inherent ISC-QY among the four investigated photosensitizers (Table 1). To quantify the ISC-QY, the same relative actinometry method as described above was employed. Tetra-n-butylammonium-bromide and -iodide (Table S1,† entries 2 & 3), chlorinated (Table S1,† entries 4 & 5), and brominated solvents (Table S1,† entries 6 & 7) lead either to insignificant increase of the ISC-QY or to fast decomposition upon pulsed excitation (see ESI† for details). However, CH2I2 (Table S1,† entry 8) provided to a two-fold improvement of the ISC-QY, confirming that the external heavy atom effect is applicable here to enhance intersystem crossing. The ISC-QY of PS(I) increases as a function of CH2I2 concentration and reaches a plateau at ca. 1 M (Fig. S2b†). However, this increase in ISC-QY is accompanied by a decrease of the triplet-excited state lifetime of anthracene from 71 μs to 3.6 μs as a result of accelerated reverse intersystem crossing on the acceptor molecule. Against this background, the exploitation of an external heavy atom effect was not further pursued in the ensuing photocatalytic and upconversion studies.
Photostability
Many applications in photoredox or energy transfer photocatalysis require long-term photostability. To assess the photorobustness, deaerated solutions of the individual photosensitizers with similar absorbance at 532 nm were irradiated with a 0.1 W cw laser at that wavelength. The fluorescence intensities of all four photosensitizers were monitored as a function of irradiation time following previously published methodologies.7,89–91 The decrease of the emission intensity during the irradiation time is illustrated in Fig. S4.† In neat solution without any substrates present, all four photosensitizers show decomposition within the first minutes. By determining the time, after which the luminescence intensity has decreased by 10%, the inherent photo-degradation quantum yields (Φdegr.) were obtained based on the known initial photosensitizer concentration and the number of absorbed photons during the respective irradiation time (see ESI† for details). PS(I) shows the highest photostability with a photodegradation quantum yield of 0.054% compared to 0.23% for PS(II), 0.36% for PS(III), and 0.13% for PS(IV), indicating that the iodo-substituted photosensitizers are more prone to photo-degradation. This finding likely reflects the higher extents of triplet-excited state formation in PS(I)–PS(III) due to enhanced intersystem crossing, since photo-degradation is often particularly prevalent from long-lived triplet-excited states.Under conditions, in which the lowest excited triplet state of the photosensitizers is rapidly quenched, in particular when 10 mM anthracene is present, all four photosensitizers are markedly more robust. Since the new isoacridone dyes exhibit strong fluorescence and only the triplet excited state is quenched by anthracene, the same methodology as described above was successfully applied for the photostability measurements. The resulting emission traces of all photosensitizers are in Fig. S4.† For all photosensitizers, the emission intensity showed no significant decrease as a function of irradiation time, indicating a much higher photorobustness under conditions of triplet–triplet energy transfer than in neat solution. An upper limit of ∼0.001% was estimated for the photo-degradation quantum yield (see ESI†) under TTET conditions, which corresponds to a photostability increase by more than a factor of 50 compared to the inherent photostability in neat solution. The main photo-decomposition pathway therefore involves the T1 state of the isoacridone dyes (3*PS), as illustrated in Fig. S3.† Given the remarkable photostabilities of all four dyes under TTET conditions, these newly developed photosensitizers seem promising for applications in energy-transfer catalysis and sTTA-UC.
Triplet–triplet energy transfer photocatalysis
To assess the potential photochemical reactivity of the newly developed photosensitizers, their singlet- and triplet-excited state redox potentials were estimated based on the respective excited state energies and ground state redox potentials (Table S4†). All four PS have ground state reduction potentials between −1.20 to −1.10 V vs. SCE and feature similar singlet-excited state energies of ∼2.3 eV and triplet energies of ∼1.9 eV (Table 1). This results in reduction potentials of ca. 1.2 V vs. SCE for the S1 state and ca. 0.8 V vs. SCE for the T1 state. Knowledge of these redox potentials and the triplet excited-state energies permits the identification of suitable biphotonic reactions, in which both a triplet–triplet energy transfer and a single electron transfer (SET) elementary reaction step is required (Fig. 4). Such combined reactivity can simultaneously exploit the strongly reducing character of the short-lived singlet-excited state (Fig. 4, purple part) and the energy donor capacity of the long-lived triplet-excited state (Fig. 4, orange part).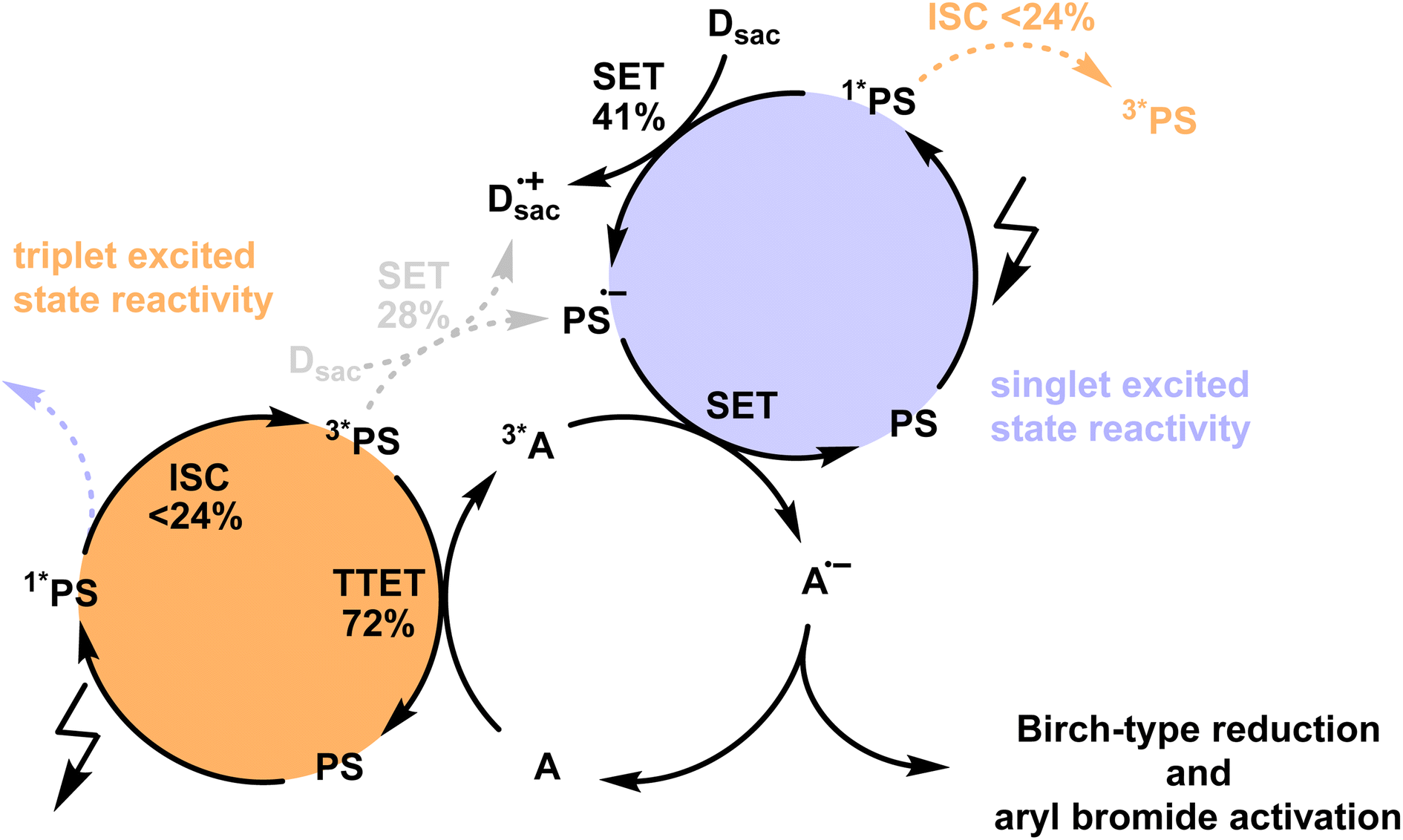 | ||
| Fig. 4 Postulated mechanism for Birch-type arene reduction and aryl bromide activation using the newly developed isoacridone photosensitizers (PS) with up to ∼24% intersystem crossing (ISC) efficiency under reaction conditions (see Section 4.3 of the ESI† for more information). The populated triplet excited state of the PS is mainly used for the triplet–triplet energy transfer (TTET) step (orange part) to the acceptor (A), whereas the singlet-excited state undergoes more efficient single electron transfer (SET, purple part) with a sacrificial electron donors (Dsac), owing to its stronger oxidizing properties with respect to the triplet excited state. The combination of TTET and SET on the same acceptor enables thermodynamically particularly challenging reactions, owing to the combined energy input from two photons. | ||
The previously investigated Birch-type photoreduction of anthracene derivatives is a good example of a biphotonic mechanism, which relies on the parallel operation of electron transfer and triplet–triplet energy transfer reaction pathways (Fig. 4).15 In the expectable mechanism, triplet–triplet energy transfer from the photosensitizer to the anthracene substrate (playing the role of the acceptor A in Fig. 4) occurs in parallel to a reductive singlet-excited state quenching by excess sacrificial electron donor, to produce the one-electron reduced form of the photosensitizer (PS˙−). Subsequent electron transfer from PS˙− to the triplet-excited anthracene substrate then forms anthracenyl radical anion (A˙− in Fig. 4), which, after a hydrogen atom transfer (HAT) and a proton transfer (PT) step (Fig. S8†), is converted to the Birch-type reduction product 9,10-dihydroanthracene. This mechanism has been previously put forward, and is assumed to be applicable to the new photosensitizer class investigated here (see ESI† Section 4.1 for further details).15 Based on the abovementioned triplet energies and excited-state redox potentials, both TTET and SET are thermodynamically viable with the new photosensitizers (see ESI†). The reduction of 9-phenylanthracene (Table 2, entry 1) was tested using PS(I), PS(III), and [Ir(dF(CF3)ppy)2(dtbpy)]+ as photosensitizers. After 18 hours of irradiation with an LED emitting in the range 465 nm–470 nm, isolated yields of 90% with PS(I), and 61% with PS(III) and 50% with [Ir(dF(CF3)ppy)2(dtbpy)]+ were obtained. With 9-methylanthracene (Table 2, entry 2) as substrate, isolated yields ranging from 53% to 66% were observed. When using anthracene (Table 2, entry 3), substrate conversions of 100% for [Ir(dF(CF3)ppy)2(dtbpy)]+, 91% for PS(I), and 70% for PS(III) were determined, but only 49% ([Ir(dF(CF3)ppy)2(dtbpy)]+), 35% (PS(I)), and 24% (PS(III)) of product were isolated. This large discrepancy between substrate conversion and product formation is attributable to anthracene dimerization.92
| Entry | Reaction | Isolated yields using | ||
|---|---|---|---|---|
| PS(I) | PS(III) | Ru or Ir-based PS | ||
| a The reactions were performed on 80.0 μmol scale in DMF (0.80 mL), under argon, during 18 h. b [Ir(dF(CF3)ppy)2(dtbpy)]PF6, with dF(CF3)ppy = 2-(2,4-difluorophenyl)-5-(trifluoromethyl)pyridine and dtbpy = 4,4′-di-tert-butyl-2,2′-bipyridine, was used as a PS. c The reactions were performed on 100 μmol scale in DMSO (0.50 mL). As the acceptor molecule, 9,10-diphenylanthracene was used. Anthracene and 9-phenylanthracene were also tested (see ESI for more details). d 4 mol% of PS(I) and DIPEA (4.0 eq.). e [Ru(bpy)3]Cl2 was used as a PS. | ||||
| 1a |
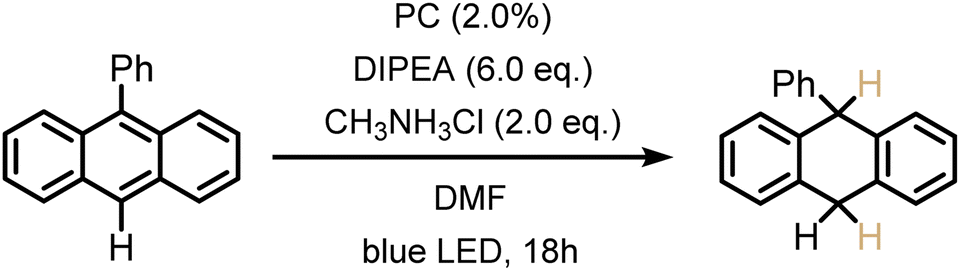
|
90% | 61% | 50%b |
| 2a |
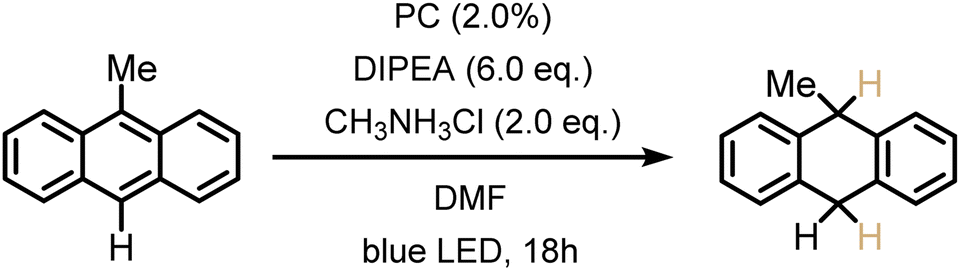
|
57% | 66% | 53%b |
| 3a |
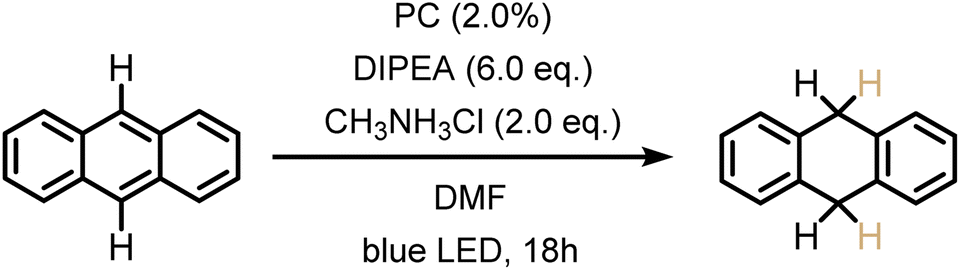
|
35% | 24% | 49%b |
| 4c |
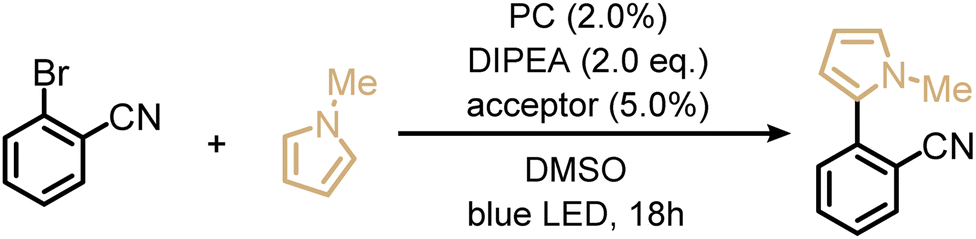
|
33%d | n/a | 31%e |
Next, we explored the utility of the isoacridone dyes for sensitization-initiated electron transfer, by focusing on the catalytic C–H arylation of an activated aryl bromide (Table 2, entry 4).14,16 Even though different mechanisms have been proposed in previous literature,93 two main reaction pathways seem particularly relevant (see ESI†).14,94 The first key option relies on a combination of SET and TTET processes of the PS to an acceptor molecule (Fig. 4 and S9a†),69 whereas the second key option relies solely on an energy transfer based sTTA-UC mechanism, for which only triplet–triplet energy transfer to an annihilator molecule is necessary (Fig. S9b†).14 In an attempt to distinguish between these two limiting cases of key mechanisms without performing in-depth transient absorption studies, the photocatalytic performance of PS(I) and PS(III), featuring markedly different ISC-QY of 0.26 and 0.43 but comparable redox potentials (Table S4†), was explored in the presence of anthracene as a triplet acceptor. The use of PS(I) resulted in an NMR-yield of 22% for the targeted C–C coupling product (Table S7,† entry 1), whereas PS(III) resulted in 20% NMR-yield (Table S7,† entry 5). The finding that the product yields are similar in both cases despite substantially different ISC-QYs could suggest that the overall photochemical reactivity is not exclusively governed by the long-lived triplet excited states, but instead involves also the respective singlet-excited states. This in turn would imply that the main reaction pathway is not sTTA-UC, but instead involves the classical Seni-ET mechanism (Fig. S9a†) relying on both SET (from the singlet-excited state) and energy transfer from the triplet excited state (TTET).
As alternatives to the anthracene co-catalyst used for the photoreaction in entry 4 of Table 2, 9-phenylanthracene (Table S7,† entry 10) and 9,10-diphenylanthracene (DPA, Table S7,† entry 11) were tested, because we anticipated less detrimental photodimerization than with unsubstituted anthracene. Both of the substituted triplet acceptors resulted in higher reaction yields (34% when using 9-phenylanthracene and 33% when using DPA) than simple anthracene (22%). Optimization of the photosensitizer, triplet acceptor, and electron donor concentrations (Table S7†), resulted in an isolated yield of 33% for the C–C coupling product.
As ISC and SET from the 1*PS are competing processes, and considering that the 3*PS may undergo both SET and TTET, we conducted a detailed mechanistic analysis for the sensitization-initiated electron transfer to support the viability of the proposed mechanistic interpretation. The reactivity of the 1*PS was considered by investigating the quenching efficiency (η) of the SET in the presence of a catalytically relevant electron donor concentration (Fig. S11†), resulting in a quenching efficiency of 54.8%. As SET reactions from singlet excited states often exhibit limited cage escape quantum yields (CE-QY), we selected one exemplary reaction system to investigate that aspect (see Fig. S12† for more details), resulting in a CE-QY of 0.75. This value indicates that only 75% of the SET transfer events from DIPEA to 1*PS(I) result in a productive pathway, forming the reduced PS(I)˙−. As a result, the SET between 1*PS(I) and DIPEA achieves a maximum overall efficiency of only 0.41 (=η × CE-QY = 0.54 × 0.75), as shown in Fig. 4. Since 54% of the 1*PS(I) undergo SET under these conditions, only 46% of the initial singlet excited state population can undergo ISC. With ISC-QY ranging from 0.52 to 0.26, the overall efficiency for the formation of the 3*PS ranges from 0.24 (=0.52 × 0.46) to 0.12 (=0.26 × 0.46), as shown in Fig. 4. The SET and TTET reaction rate constants from the 3*PS(I) were investigated using transient UV-vis absorption spectroscopy (Fig. S13†). With the catalytically relevant concentration of DIPEA as an electron donor (114 mM) and DPA as an energy acceptor (10 mM) the reaction rates were calculated (see details in Section 4.3 of the ESI†), resulting in a TTET efficiency of 72% compared to 28% SET efficiency (Fig. 4). These detailed mechanistic considerations (Section 4.3 of the ESI†) underpin the viability of the mechanistic interpretation given here.
Sensitized triplet–triplet annihilation upconversion
Given the relatively efficient intersystem crossing and triplet-excited state formation in some of our new photosensitizers, we anticipated that they could be amenable to sTTA-UC (Fig. 5a), which usually is performed with precious metal complexes exhibiting quantitative intersystem crossing.9,95–98 We identified (TMS)2pyr (Fig. 5b) as a suitable triplet acceptor and annihilator, since the triplet energy of unsubstituted pyrene (2.1 eV) is too high in comparison to the T1 energy of 1.9 eV of our photosensitizers. In polyaromatic hydrocarbons bearing alkyne substituents, the triplet state energies are often substantially lowered with respect to their unsubstituted analogues, which has been exploited for sTTA-UC in several previous cases.10,89,99,100 (TMS)2pyr fluoresces between 390 nm and 450 nm (Fig. S14†), in a spectral region in which all four isoacridone photosensitizers only absorb weakly (Fig. 2). The fluorescence quantum yield of the annihilator is high (0.90). The rate constants for triplet–triplet energy transfer (kTTET) from our photosensitizers to (TMS)2pyr were determined by transient UV-vis absorption spectroscopy using a Stern–Volmer-like approach (Fig. S16†), resulting in values of ∼7 ×108 M−1 s−1 for PS(III) and PS(IV), as well as values of ∼10 × 108 M−1 s−1 for PS(I) and PS(II). Based on excitation power dependent measurements of the decay of triplet-excited (TMS)2pyr by transient UV-vis absorption spectroscopy (Fig. S17†), a rate constant for triplet–triplet annihilation (TTA) of 3.6 × 109 M−1 s−1 was determined (see ESI†), similar to previously published annihilation rate constants.101,102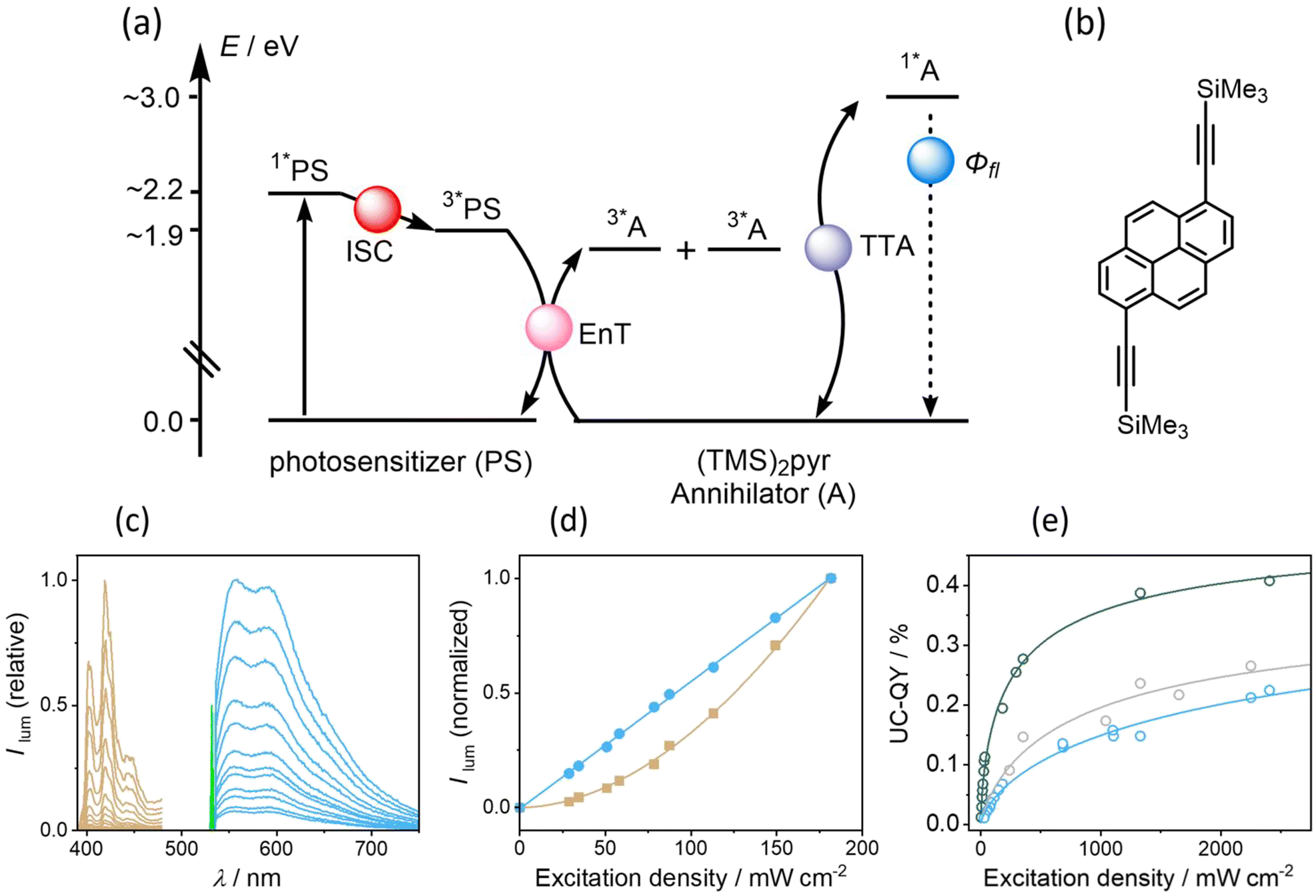 | ||
| Fig. 5 (a) Simplified energy level diagram for sensitized triplet–triplet annihilation upconversion (sTTA-UC) using the new photosensitizers and 1,6-bis((trimethylsilyl)ethynyl)pyrene as annihilator ((TMS)2pyr, panel (b)). (c) Excitation power dependent delayed upconversion fluorescence emitted by the annihilator (brown traces) and prompt photosensitizer fluorescence (blue traces), recorded from a solution containing 400 μM (TMS)2pyr and 20 μM PS(I) upon 532 nm cw laser excitation (marked by the green line). (d) Quadratic dependence of the integrated delayed upconversion fluorescence (brown squares) and linear dependence of prompt fluorescence emitted by PS(I) (blue circles) based on the data in (c). (e) Upconversion luminescence quantum yield (UC-QY) as a function of excitation power density, determined from solutions containing 400 μM (TMS)2pyr and 20 μM of the corresponding photosensitizer: PS(I), (blue), PS(III) (gray), PS(IV) (green). | ||
Upon excitation of 20 μM solutions of the four different photosensitizers with a 532 nm cw laser in the presence of 400 μM (TMS)2pyr, delayed (upconverted) fluorescence originating from the singlet-excited state of (TMS)2pyr peaking at 430 nm was detected (Fig. 5c). The pseudo anti-Stokes shift, as calculated by taking the energy difference between the excitation wavelength (532 nm = 2.2 eV) and the upconversion emission band maximum (430 nm = 2.9 eV), is 0.7 eV, similar to other upconversion systems made with organic photosensitizers.61,84,103–105 The excitation power dependence of the upconverted emission intensity is quadratic for all four investigated photosensitizers (Fig. 5d and S18†), confirming the expected biphotonic nature of the upconversion process. By contrast, the prompt photosensitizer fluorescence intensity depends linearly on the excitation power (Fig. 5d and S18†), as expected.
Given that the prompt fluorescence of the isoacridone photosensitizers remains unaffected by triplet–triplet annihilation, the upconversion quantum yields (ΦUC) were determined by comparing the ratios between the integrated prompt photosensitizer fluorescence and the delayed fluorescence emitted by the annihilator. Based on the available quantum yields for prompt photosensitizer fluorescence (ΦFL, see above), the upconversion quantum yields were then determined. At excitation power densities exceeding 700 mW cm−2, PS(II) showed poor photostability and consequently only the upconversion systems with PS(I), PS(III), and PS(VI) were fully explorable (Fig. 5e). Their upconversion quantum yields were estimated at an excitation power density of 1500 mW cm−2, at which photo-degradation does not play an important role on the timescale of our measurements. The obtained upconversion quantum yields correlate with the intersystem crossing quantum yields, manifesting in an increase of ΦUC along the series PS(I) (0.19%) < PS(III) (0.22%) < PS(IV) (0.38%).
With these ΦUC values, the observed upconversion serves as a promising starting point for a new class of purely organic UC-systems, which currently show UC-quantum yields typically below 5%61,84,99,100,103,106–113 to supplement purely organic UC-systems which mostly rely on BODIPY dyes50,60,61,108 or donor–acceptor cyanoarenes.95,99,100 Furthermore, the developed isoacridones represent a modular and tunable addition to the available range of organic sensitizers suitable for the TTA-UC. Based on a simplistic analysis (Section 5.5 of the ESI†), further improvement is expected mainly by adjusting the annihilator and not the photosensitizer properties, since the low TTA efficiency of the (TMS)2pyr annihilator is limiting the overall upconversion system efficiency more severely than any other sensitizer-related factor. Follow-up upconversion studies should therefore be geared at optimizing the annihilator rather than the isoacridone sensitizers.
Conclusion
Newly emerging biphotonic reactions targeting unusually challenging transformations such as Birch-type arene photoreductions or the activation of very inert substrates rely on a combination of photoinduced electron transfer and triplet–triplet energy transfer.12,15,16,94 This calls for new types of photosensitizers, which are able to promote both of these two elementary processes similarly well. In this context, the newly developed family of isoacridone photosensitizers fills an important gap by enabling photoinduced electron transfer from their lowest singlet-excited states and by facilitating triplet–triplet energy transfer from their lowest triplet-excited states. Importantly, in this photocatalysts family, both S1 and T1 are populated to similar extents, unlike in many other purely organic photosensitizers, in which only S1 is predominantly photoreactive, and unlike classical transition-metal complexes, which usually only react from triplet excited states. In contrast to compounds featuring thermally activated delayed fluorescence (TADF), the S1 and T1 states in the isoacridone dyes are not thermally equilibrated, but instead act independently of one another once they have been formed. Against this background, the simultaneous reactivity of S1 and T1 in our isoacridone dyes seems very attractive, particularly for the abovementioned increasingly important biphotonic reaction mechanisms. The anthracene derivative photoreductions and the C–H arylation reactions in Table 2 represent successful proof-of-concepts in this regard.Intersystem crossing is finely tuneable through chemical modification in our isoacridone dyes, which allows to balance the singlet- versus triplet-reactivity. In the present work, we have found that upconversion quantum yields correlate quantitatively with the intersystem crossing efficiencies. Such clear-cut relationships between these two basic quantities are rare.114 Most upconversion studies published to date rely on transition metal-based photosensitizers,8,9,72,73,96,97,115 in which intersystem crossing is not nearly as precisely tuneable as in the isoacridone family. Thus, the new photosensitizers permit unusally direct insight into fundamental aspects of photochemical triplet–triplet annihilation upconversion.
The high photostability of the isoacridone dyes under triplet energy transfer conditions is another attractive feature. Precious metal-based photosensitizers, including for example ruthenium(II) or iridium(III) complexes,116 can display similar inertness, but coordination compounds made from abundant first-row transition metal elements often suffer from limited photostability until now.26,27 In this context, the metal-free dyes investigated herein represent an attractive alternative. Despite important progress in the past few years with quantum yields for upconversion to the UV of up to ∼20%,31,32,35,60,99,105,106,109–114 the field of purely organic (metal-free) sensitizers with photoactive triplet excited states seems still underdeveloped.
Data availability
The data supporting this article has been uploaded as part of the ESI.† The data that support the findings of this study are openly available from Zenodo at https://doi.org/10.5281/zenodo.8380143.Author contributions
B. P. conceived the project, designed photochemical studies, conducted spectroscopic analyses, designed photocatalysis experiments, and performed mechanistic studies. V. H. conceived the project, designed and synthesized the PS, and conducted the catalysis. O. S. W. and C. S. contributed to project conception and provided guidance. B. P. and O. S. W. prepared the initial draft of the manuscript with inputs from all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
B. P. acknowledges a Ph. D. grant (number 14583224) by the National Research Fund, Luxembourg and V. H. support by the Alfred Werner Fund. Funding from the Swiss National Science Foundation through grant number 200020_207329 and the University of Basel is acknowledged.References
- M. Montalti, A. Credi, L. Prodi and T. M. Gandolfi, Handbook of photochemistry, CRC/Taylor & Francis, Boca Raton, FL, 3 edn, 2006 Search PubMed.
- M. Inokuti and F. Hirayama, J. Chem. Phys., 1965, 43, 1978–1989 CrossRef CAS.
- D. L. Dexter, J. Chem. Phys., 1953, 21, 836–850 CrossRef CAS.
- A. Kamkaew, S. H. Lim, H. B. Lee, L. V. Kiew, L. Y. Chung and K. Burgess, Chem. Soc. Rev., 2013, 42, 77–88 RSC.
- J. Grosskopf, T. Kratz, T. Rigotti and T. Bach, Chem. Rev., 2022, 122, 1626–1653 CrossRef CAS PubMed.
- T. Nevesely, M. Wienhold, J. J. Molloy and R. Gilmour, Chem. Rev., 2022, 122, 2650–2694 CrossRef CAS PubMed.
- L. Schmid, F. Glaser, R. Schaer and O. S. Wenger, J. Am. Chem. Soc., 2022, 144, 963–976 CrossRef CAS PubMed.
- B. Pfund, D. M. Steffen, M. R. Schreier, M. S. Bertrams, C. Ye, K. Börjesson, O. S. Wenger and C. Kerzig, J. Am. Chem. Soc., 2020, 142, 10468–10476 CrossRef CAS PubMed.
- T. N. Singh-Rachford and F. N. Castellano, Coord. Chem. Rev., 2010, 254, 2560–2573 CrossRef CAS.
- N. Harada, Y. Sasaki, M. Hosoyamada, N. Kimizuka and N. Yanai, Angew. Chem., Int. Ed., 2021, 60, 142–147 CrossRef CAS PubMed.
- J. Castellanos Soriano, T. J. B. Zähringer, J. C. Herrera-Luna, M. C. Jimenez, C. Kerzig and R. Pérez-Ruiz, Phys. Chem. Chem. Phys., 2023, 25, 12041–12049 RSC.
- F. Glaser, C. Kerzig and O. S. Wenger, Angew. Chem., Int. Ed., 2020, 59, 10266–10284 CrossRef CAS PubMed.
- F. Glaser and O. S. Wenger, JACS Au, 2022, 2, 1488–1503 CrossRef CAS PubMed.
- F. Glaser, C. Kerzig and O. S. Wenger, Chem. Sci., 2021, 12, 9922–9933 RSC.
- A. Chatterjee and B. König, Angew. Chem., Int. Ed., 2019, 58, 14289–14294 CrossRef CAS PubMed.
- I. Ghosh, R. S. Shaikh and B. König, Angew. Chem., Int. Ed., 2017, 56, 8544–8549 CrossRef CAS PubMed.
- D. Kim and T. S. Teets, Chem. Phys. Rev., 2022, 3, 021302 CrossRef CAS.
- L. Marzo, S. K. Pagire, O. Reiser and B. König, Angew. Chem., Int. Ed., 2018, 57, 10034–10072 CrossRef CAS PubMed.
- J. Twilton, C. Le, P. Zhang, M. H. Shaw, R. W. Evans and D. W. C. MacMillan, Nat. Rev. Chem., 2017, 1, 0052 CrossRef CAS.
- C. K. Prier, D. A. Rankic and D. W. MacMillan, Chem. Rev., 2013, 113, 5322–5363 CrossRef CAS PubMed.
- C. S. Wang, P. H. Dixneuf and J. F. Soule, Chem. Rev., 2018, 118, 7532–7585 CrossRef CAS PubMed.
- B. Zilate, C. Fischer and C. Sparr, Chem. Commun., 2020, 56, 1767–1775 RSC.
- M. R. Schreier, X. Guo, B. Pfund, Y. Okamoto, T. R. Ward, C. Kerzig and O. S. Wenger, Acc. Chem. Res., 2022, 55, 1290–1300 CrossRef CAS PubMed.
- D. Volz, M. Wallesch, C. Fléchon, M. Danz, A. Verma, J. M. Navarro, D. M. Zink, S. Bräse and T. Baumann, Green Chem., 2015, 17, 1988–2011 RSC.
- F. Glaser and O. S. Wenger, Coord. Chem. Rev., 2020, 405, 213129 CrossRef CAS.
- C. Wegeberg and O. S. Wenger, JACS Au, 2021, 1, 1860–1876 CrossRef CAS PubMed.
- N. Sinha and O. S. Wenger, J. Am. Chem. Soc., 2023, 145, 4903–4920 CrossRef CAS PubMed.
- O. S. Wenger, J. Am. Chem. Soc., 2018, 140, 13522–13533 CrossRef CAS PubMed.
- P. Chábera, L. Lindh, N. W. Rosemann, O. Prakash, J. Uhlig, A. Yartsev, K. Wärnmark, V. Sundström and P. Persson, Coord. Chem. Rev., 2021, 426, 213517 CrossRef.
- C. Förster and K. Heinze, Chem. Soc. Rev., 2020, 49, 1057–1070 RSC.
- E. Speckmeier, T. G. Fischer and K. Zeitler, J. Am. Chem. Soc., 2018, 140, 15353–15365 CrossRef CAS PubMed.
- E. Bassan, A. Gualandi, P. G. Cozzi and P. Ceroni, Chem. Sci., 2021, 12, 6607–6628 RSC.
- X.-F. Zhang, I. Zhang and L. Liu, Photochem. Photobiol., 2010, 86, 492–498 CrossRef CAS PubMed.
- L.-S. Cui, A. J. Gillett, S.-F. Zhang, H. Ye, Y. Liu, X.-K. Chen, Z.-S. Lin, E. W. Evans, W. K. Myers, T. K. Ronson, H. Nakanotani, S. Reineke, J.-L. Bredas, C. Adachi and R. H. Friend, Nat. Photon., 2020, 14, 636–642 CrossRef CAS.
- M. Y. Wong and E. Zysman-Colman, Adv. Mater., 2017, 29, 1605444 CrossRef PubMed.
- M. Jakobi and C. Sparr, Org. Process Res. Dev., 2022, 26, 2746–2760 CrossRef.
- J. Wellauer, D. Miladinov, T. Buchholz, J. Schutz, R. T. Stemmler, J. A. Medlock, W. Bonrath and C. Sparr, Chem. Eur. J., 2021, 27, 9748–9752 CrossRef CAS PubMed.
- A. Vega-Penaloza, J. Mateos, X. Companyo, M. Escudero-Casao and L. Dell'Amico, Angew. Chem., Int. Ed., 2021, 60, 1082–1097 CrossRef CAS PubMed.
- D. P. Hari and B. König, Chem. Commun., 2014, 50, 6688–6699 RSC.
- V. Srivastava and P. P. Singh, RSC Adv., 2017, 7, 31377–31392 RSC.
- G. Pandey, M. Karthikeyan and A. Murugan, J. Org. Chem., 1998, 63, 2867–2872 CrossRef CAS.
- C. W. Kee, K. F. Chin, M. W. Wong and C. H. Tan, Chem. Commun., 2014, 50, 8211–8214 RSC.
- W. Zhang, J. Gacs, I. Arends and F. Hollmann, ChemCatChem, 2017, 9, 3821–3826 CrossRef CAS PubMed.
- H. Schmaderer, P. Hilgers, R. Lechner and B. König, Adv. Synth. Catal., 2009, 351, 163–174 CrossRef CAS.
- J. Svoboda, H. Schmaderer and B. König, Chem. Eur. J., 2008, 14, 1854–1865 CrossRef CAS PubMed.
- S. Fukuzumi, K. Tanii and T. Tanaka, J. Chem. Soc. Chem. Commun., 1989, 816–818 RSC.
- E. Bassan, Y. Dai, D. Fazzi, A. Gualandi, P. G. Cozzi, F. Negri and P. Ceroni, Photochem. Photobiol. Sci., 2022, 21, 777–786 CrossRef CAS PubMed.
- K. Chen, W. Yang, Z. Wang, A. Iagatti, L. Bussotti, P. Foggi, W. Ji, J. Zhao and M. Di Donato, J. Phys. Chem. A, 2017, 121, 7550–7564 CrossRef CAS PubMed.
- J. Zhao, K. Xu, W. Yang, Z. Wang and F. Zhong, Chem. Soc. Rev., 2015, 44, 8904–8939 RSC.
- Z. Wang, J. Zhao, A. Barbon, A. Toffoletti, Y. Liu, Y. An, L. Xu, A. Karatay, H. G. Yaglioglu, E. A. Yildiz and M. Hayvali, J. Am. Chem. Soc., 2017, 139, 7831–7842 CrossRef CAS PubMed.
- L. Zhang, B. Pfund, O. S. Wenger and X. Hu, Angew. Chem., Int. Ed., 2022, 61, e202202649 CrossRef CAS PubMed.
- B. G. McCarthy, R. M. Pearson, C. H. Lim, S. M. Sartor, N. H. Damrauer and G. M. Miyake, J. Am. Chem. Soc., 2018, 140, 5088–5101 CrossRef CAS PubMed.
- R. M. Pearson, C. H. Lim, B. G. McCarthy, C. B. Musgrave and G. M. Miyake, J. Am. Chem. Soc., 2016, 138, 11399–11407 CrossRef CAS PubMed.
- H. Rao, C. H. Lim, J. Bonin, G. M. Miyake and M. Robert, J. Am. Chem. Soc., 2018, 140, 17830–17834 CrossRef CAS PubMed.
- S. M. Sartor, B. G. McCarthy, R. M. Pearson, G. M. Miyake and N. H. Damrauer, J. Am. Chem. Soc., 2018, 140, 4778–4781 CrossRef CAS.
- C. G. López-Calixto, M. Liras, V. A. de la Peña O'Shea and R. Pérez-Ruiz, Appl. Catal., 2018, 237, 18–23 CrossRef.
- G. Magagnano, A. Gualandi, M. Marchini, L. Mengozzi, P. Ceroni and P. G. Cozzi, Chem. Commun., 2017, 53, 1591–1594 RSC.
- D. Sasikumar, A. T. John, J. Sunny and M. Hariharan, Chem. Soc. Rev., 2020, 49, 6122–6140 RSC.
- T. Kowada, H. Maeda and K. Kikuchi, Chem. Soc. Rev., 2015, 44, 4953–4972 RSC.
- N. Kiseleva, M. A. Filatov, J. C. Fischer, M. Kaiser, M. Jakoby, D. Busko, I. A. Howard, B. S. Richards and A. Turshatov, Phys. Chem. Chem. Phys., 2022, 24, 3568–3578 RSC.
- W. Wu, H. Guo, W. Wu, S. Ji and J. Zhao, J. Org. Chem., 2011, 76, 7056–7064 CrossRef CAS PubMed.
- Y. Dong, P. Kumar, P. Maity, I. Kurganskii, S. Li, A. Elmali, J. Zhao, D. Escudero, H. Wu, A. Karatay, O. F. Mohammed and M. Fedin, Phys. Chem. Chem. Phys., 2021, 23, 8641–8652 RSC.
- S. Fukuzumi, H. Kotani, K. Ohkubo, S. Ogo, N. V. Tkachenko and H. Lemmetyinen, J. Am. Chem. Soc., 2004, 126, 1600–1601 CrossRef CAS PubMed.
- S. Fukuzumi, K. Ohkubo, T. Suenobu, K. Kato, M. Fujitsuka and O. Ito, J. Am. Chem. Soc., 2001, 35, 8459–8467 CrossRef PubMed.
- K. Suga, K. Ohkubo and S. Fukuzumi, J. Phys. Chem. A, 2006, 110, 3860–3867 CrossRef CAS PubMed.
- D. J. Wilger, J. M. Grandjean, T. R. Lammert and D. A. Nicewicz, Nat. Chem., 2014, 6, 720–726 CrossRef CAS PubMed.
- N. A. Romero, K. A. Margrey, N. E. Tay and D. A. Nicewic, Science, 2015, 349, 1326–1330 CrossRef CAS PubMed.
- A. Joshi-Pangu, F. Levesque, H. G. Roth, S. F. Oliver, L. C. Campeau, D. Nicewicz and D. A. DiRocco, J. Org. Chem., 2016, 81, 7244–7249 CrossRef CAS PubMed.
- A. R. White, L. Wang and D. A. Nicewicz, Synlett, 2019, 30, 827–832 CrossRef CAS.
- I. Ghosh, T. Ghosh, J. I. Bardagi and B. König, Science, 2014, 346, 725–728 CrossRef CAS.
- I. A. MacKenzie, L. Wang, N. P. R. Onuska, O. F. Williams, K. Begam, A. M. Moran, B. D. Dunietz and D. A. Nicewicz, Nature, 2020, 580, 76–80 CrossRef CAS PubMed.
- M. Uji, T. J. B. Zähringer, C. Kerzig and N. Yanai, Angew. Chem., Int. Ed., 2023, e202301506 CAS.
- L. Zeng, L. Huang, W. Lin, L. H. Jiang and G. Han, Nat. Commun., 2023, 14, 1102 CrossRef CAS PubMed.
- N. Yanai and N. Kimizuka, Angew. Chem., Int. Ed., 2020, 59, 10252–10264 CrossRef CAS PubMed.
- C. Fischer, C. Kerzig, B. Zilate, O. S. Wenger and C. Sparr, ACS Catal., 2019, 10, 210–215 CrossRef.
- K. Schmidt, S. Brovelli, V. Coropceanu, D. Beljonne, J. Cornil, C. Bazzini, T. Caronna, R. Tubino, F. Meinardi, Z. Shuai and J.-L. Brédas, J. Phys. Chem. A, 2007, 111, 10490–10499 CrossRef CAS PubMed.
- C. C. Kenry and B. Liu, Nat. Commun., 2019, 10, 2111 CrossRef PubMed.
- D. J. Wolstenholme, C. F. Matta and T. S. Cameron, J. Phys. Chem. A, 2007, 111, 8803–8813 CrossRef CAS PubMed.
- Z. Xu, C. Climent, C. M. Brown, D. Hean, C. J. Bardeen, D. Casanova and M. O. Wolf, Chem. Sci., 2020, 12, 188–195 RSC.
- V. Hutskalova and C. Sparr, Org. Lett., 2021, 23, 5143–5147 CrossRef CAS PubMed.
- C. Sparr and V. Hutskalova, Synlett, 2021, 33, 1180–1183 CrossRef.
- V. Hutskalova, A. Prescimone and C. Sparr, Helv. Chim. Acta, 2021, 104, e202100182 CrossRef.
- S. Guo, W. Wu, H. Guo and J. Zhao, J. Org. Chem., 2012, 77, 3933–3943 CrossRef CAS PubMed.
- H. C. Chen, C. Y. Hung, K. H. Wang, H. L. Chen, W. S. Fann, F. C. Chien, P. Chen, T. J. Chow, C. P. Hsu and S. S. Sun, Chem. Commun., 2009, 27, 4064–4066 RSC.
- P. Müller and K. Brettel, Photochem. Photobiol. Sci., 2012, 11, 632–636 CrossRef PubMed.
- L. Rodríguez, J. C. Lima, M. Ferrer, O. Rossell and M. Engeser, Inorg. Chim. Acta, 2012, 381, 195–202 CrossRef.
- K. Kikuchi, M. Hoshi, T. Niwa, Y. Takahashi and T. Miyashi, J. Phys. Chem., 1991, 95, 38–42 CrossRef CAS.
- J. Friedrich, G. Weinzierl and F. Dörr, Z. Naturforsch., 1976, 31 a, 748–753 CrossRef.
- J. A. Kübler, B. Pfund and O. S. Wenger, JACS Au, 2022, 2, 2367–2380 CrossRef PubMed.
- L. Schmid, C. Kerzig, A. Prescimone and O. S. Wenger, JACS Au, 2021, 1, 819–832 CrossRef CAS PubMed.
- J. B. Bilger, C. Kerzig, C. B. Larsen and O. S. Wenger, J. Am. Chem. Soc., 2021, 143, 1651–1663 CrossRef CAS PubMed.
- R. R. Islangulov and F. N. Castellano, Angew. Chem., Int. Ed., 2006, 45, 5957–5959 CrossRef CAS PubMed.
- M. Marchini, G. Bergamini, P. G. Cozzi, P. Ceroni and V. Balzani, Angew. Chem., Int. Ed., 2017, 56, 12820–12821 CrossRef CAS PubMed.
- M. S. Coles, G. Quach, J. E. Beves and E. G. Moore, Angew. Chem., Int. Ed., 2020, 59, 9522–9526 CrossRef CAS PubMed.
- S. Amemori, Y. Sasaki, N. Yanai and N. Kimizuka, J. Am. Chem. Soc., 2016, 138, 8702–8705 CrossRef CAS PubMed.
- B. D. Ravetz, A. B. Pun, E. M. Churchill, D. N. Congreve, T. Rovis and L. M. Campos, Nature, 2019, 565, 343–346 CrossRef CAS PubMed.
- P. Bharmoria, H. Bildirir and K. Moth-Poulsen, Chem. Soc. Rev., 2020, 49, 6529–6554 RSC.
- R. Perez-Ruiz, Top. Curr. Chem., 2022, 380, 23 CrossRef CAS PubMed.
- T. J. B. Zähringer, J. A. Moghtader, M. S. Bertrams, B. Roy, M. Uji, N. Yanai and C. Kerzig, Angew. Chem., Int. Ed., 2023, 62, e202215340 CrossRef PubMed.
- T. J. B. Zähringer, M.-S. Bertrams and C. Kerzig, J. Mater. Chem. C, 2022, 10, 4568–4573 RSC.
- F. Edhborg, A. Olesund and B. Albinsson, Photochem. Photobiol. Sci., 2022, 21, 1143–1158 CrossRef CAS PubMed.
- C. Kerzig and O. S. Wenger, Chem. Sci., 2018, 9, 6670–6678 RSC.
- T. N. Singh-Rachford and F. N. Castellano, J. Phys. Chem. A, 2009, 113, 5912–5917 CrossRef CAS PubMed.
- M. Majek, U. Faltermeier, B. Dick, R. Perez-Ruiz and A. Jacobi von Wangelin, Chem. Eur. J., 2015, 21, 15496–15501 CrossRef CAS PubMed.
- A. Olesund, J. Johnsson, F. Edhborg, S. Ghasemi, K. Moth-Poulsen and B. Albinsson, J. Am. Chem. Soc., 2022, 144, 3706–3716 CrossRef CAS PubMed.
- N. Yanai, M. Kozue, S. Amemori, R. Kabe, C. Adachi and N. Kimizuka, J. Mater. Chem. C, 2016, 4, 6447–6451 RSC.
- R. R. Islangulov, D. V. Kozlov and F. N. Castellano, Chem. Commun., 2005, 3776–3778 RSC.
- Y. Dong, B. Dick and J. Zhao, Org. Lett., 2020, 22, 5535–5539 CrossRef CAS PubMed.
- A. Mohan, E. Sebastian, M. Gudem and M. Hariharan, J. Phys. Chem. B, 2020, 124, 6867–6874 CrossRef CAS PubMed.
- M. Uji, N. Harada, N. Kimizuka, M. Saigo, K. Miyata, K. Onda and N. Yanai, J. Mater. Chem. C, 2022, 10, 4558–4562 RSC.
- M. Zheng, Y. Li, Y. Wei, L. Chen, S. Liu and X. Zhou, J. Phys. Chem. C, 2023, 127, 2846–2854 CrossRef CAS.
- W. Chen, F. Song, S. Tang, G. Hong, Y. Wu and X. Peng, Chem. Commun., 2019, 55, 4375–4378 RSC.
- J. K. Li, M. Y. Zhang, L. Zeng, L. Huang and X. Y. Wang, Angew. Chem., Int. Ed., 2023, 62, e202303093 CrossRef CAS PubMed.
- Z. Wang and J. Zhao, Org. Lett., 2017, 19, 4492–4495 CrossRef CAS PubMed.
- T. Schloemer, P. Narayanan, Q. Zhou, E. Belliveau, M. Seitz and D. N. Congreve, ACS Nano, 2023, 17, 3259–3288 CrossRef CAS PubMed.
- I. N. Mills, J. A. Porras and S. Bernhard, Acc. Chem. Res., 2018, 51, 352–364 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc02768f |
| ‡ These two authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |