
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePeptide conjugates with polyaromatic hydrocarbons can benefit the activity of catalytic RNAs†
Kevin J.
Sweeney
a,
Tommy
Le
a,
Micaella Z.
Jorge
 b,
Joan G.
Schellinger
b,
Luke J.
Leman
c and
Ulrich F.
Müller
*a
b,
Joan G.
Schellinger
b,
Luke J.
Leman
c and
Ulrich F.
Müller
*a
aDepartment of Chemistry and Biochemistry, University of California San Diego, La Jolla, CA 92093, USA. E-mail: ufmuller@ucsd.edu
bDepartment of Chemistry & Biochemistry, University of San Diego, San Diego, CA 92110, USA
cDepartment of Chemistry, The Scripps Research Institute, La Jolla, CA 92037, USA
First published on 12th September 2023
Abstract
Early stages of life likely employed catalytic RNAs (ribozymes) in many functions that are today filled by proteins. However, the earliest life forms must have emerged from heterogenous chemical mixtures, which included amino acids, short peptides, and many other compounds. Here we explored whether the presence of short peptides can help the emergence of catalytic RNAs. To do this, we conducted an in vitro selection for catalytic RNAs from randomized sequence in the presence of ten different peptides with a prebiotically plausible length of eight amino acids. This in vitro selection generated dozens of ribozymes, one of them with ∼900-fold higher activity in the presence of one specific peptide. Unexpectedly, the beneficial peptide had retained its N-terminal Fmoc protection group, and this group was required to benefit ribozyme activity. The same, or higher benefit resulted from peptide conjugates with prebiotically plausible polyaromatic hydrocarbons (PAHs) such as fluorene and naphthalene. This shows that PAH-peptide conjugates can act as potent cofactors to enhance ribozyme activity. The results are discussed in the context of the origin of life.
Introduction
The RNA world hypothesis states that an early stage in the evolution of life used RNA as genome, and as the only genome-encoded catalyst.1–5 However, the emergence of a self-replicating RNA system directly from a prebiotic environment would be unlikely if RNA oligomers containing only canonical nucleotides would have to originate directly from a chemically heterogeneous mixture.6 The concepts of chemical evolution and systems chemistry address this challenge: first, chemical evolution describes a process that shifts the spectrum of different chemical species over time, based on chemical characteristics such as chemical stability and reactivity in a dynamic environment.7,8 For example, canonical nucleosides seem to have been selected for high photochemical stability,9 and their N-glycosidic bond is hydrolytically more stable in canonical nucleosides than in alternative nucleosides.10 Other features must have been selected for advantages on the level of duplex formation: for example, the pKA and pKB values at the Watson–Crick face of canonical nucleosides are just right for efficient base pairing,11 and the conformation of ribose is optimal for positioning of the bases in base pairing.12 This selection advantage could have been mediated in part by the higher hydrolytic stability of double stranded RNA as compared to single stranded RNA.13 Second, systems chemistry studies the molecular interactions in a heterogeneous local environment, with a special focus on synergistic interactions.14 For example, the presence of alpha-hydroxy acids,15 or alpha-mercapto acids16 benefits the oligomerization of amino acids under wet–dry cycling conditions, and pyrimidine deoxynucleotides benefit the amidophosphorylation of purine deoxynucleotides.17 Finally, in a combination of chemical evolution and systems chemistry, pairs of compounds may mutually stabilize each other, and thereby enrich in the environment.18The mutual interactions of peptides and RNAs are promising to study regarding the origin of life because peptides likely existed in prebiotic environments that could give rise to RNAs: amino acids naturally arise under conditions that generate nucleotides,19–21 and amino acids are able to form peptides under wet–dry cycling conditions,22 especially in the presence of alpha-hydroxy acids,15 which likely existed together with amino acids.23 Prebiotic model reactions for peptide formation under these conditions give decent yields up to about 8-mers.15 Additionally, mutual RNA/peptide interactions could have been beneficial because these two polymers complement each other functionally, as seen in today's RNA/protein complexes: RNA has an advantage for the sequence specific recognition of nucleic acids, and polypeptides can use their larger chemical diversity to establish more efficient catalytic sites. A central question is whether, and how short peptides could have aided the emergence of catalytic RNAs (ribozymes). Previous studies found that polycationic proto-peptides increase the thermodynamic stability of duplex RNA,18 and that coacervates consisting of polyanionic and polycationic peptides can modulate the secondary structure of tRNA.24 The presence of an RNA-binding peptide can serve as switch for ribozyme activity,25 and polycationic peptides can enhance the function of existing ribozymes.26,27 However, to our knowledge no study has analyzed how short peptides could aid the emergence of catalytic RNAs from randomized sequence. This question can be addressed by in vitro selection procedures, to explore ligands for potential benefits.28–30 An in vitro selection of new ribozymes in the presence of peptides was used in this study to test whether peptides can help the emergence of catalytic RNAs.
As model system for a prebiotically relevant in vitro selection, we focused on a reaction with the prebiotically plausible energy source cyclic trimetaphosphate (cTmp).31–34 The desired ribozymes catalyze the nucleophilic attack of their own 5′-hydroxyl group on cTmp, thereby generating an RNA 5′-triphosphate. 5′-Triphosphates in the form of nucleoside 5′-triphosphates (NTPs) are the central energy currency in all known forms of life. Since an energetic driving force is essential for every life form, catalysts that convert the prebiotically plausible energy source cTmp into biochemically useful 5′-triphosphates can serve as models for a primitive energy metabolism in origins of life. Importantly, the described in vitro selection system generates a >1000-fold enrichment per selection cycle, can identify dozens to hundreds of active sequences in a single selection experiment, and identified few or no inactive sequences.34–37 Therefore, this in vitro selection procedure is an excellent model system to test whether peptides can benefit the emergence of ribozymes from random sequence.
Here we show the in vitro selection of self-triphosphorylating ribozymes in the presence of ten different octapeptides. After five rounds of selection the RNA library was dominated by active sequences. After high throughput sequencing analysis, the ribozyme sequence that appeared to benefit most from peptides was chosen for further analysis. In biochemical assays, this ribozyme showed 900 (±300)-fold higher activity with one specific peptide – but only when the peptide was linked to an N-terminal polyaromatic hydrocarbon (PAH) such as the Fmoc protection group, fluorene or naphthalene. The peptide-PAH conjugate showed very specific effects on the SHAPE probing signals, suggesting that the conjugate had a defined effect on the ribozyme's 3D structure. Together, this study showed unexpectedly that prebiotically generated peptides can strongly benefit ribozymes when conjugated to prebiotically available polyaromatic hydrocarbons.
Results
In vitro selection
To determine whether the presence of peptides can aid the emergence of catalytic RNAs we performed an in vitro selection of ribozymes in the presence of ten different octapeptides. The ten peptides were chosen to represent sets of amino acids that reflect different stages in the early evolution of life (Fig. 1A): peptides P1 to P6 are dominated by amino acids that are abundant in Miller-type reactions38 and were likely present in the earliest stages of life.39 Several of these peptides explored the addition of arginine and lysine: arginine precursors are generated in cyanosulfidic models for prebiotic reactions,19,20 and lysine has been identified in meteorites.40,41 In contemporary proteins, these two amino acids are the most common side chains in direct contact with RNA42 and were therefore promising to explore for their benefit to ribozyme function. Peptides P8, P9, and P10 incorporated amino acids that emerged later in evolution, including the aromatic amino acids whose synthesis may have required sophisticated enzymes. The length of the peptides was chosen so that they could have originated prebiotically due to wet–dry cycling in the presence of alpha-hydroxy carboxylic acids.15 The sequence of amino acids within each of the peptides was more or less arbitrary, to reflect some of the many possibilities. Additionally, all amino acids are the (biological) L-enantiomers, whereas prebiotic amino acids likely contained racemic mixtures. Of course, these ten peptides only serve as starting points to explore peptide benefits for ribozymes: if a specific peptide were found to be beneficial to ribozymes, it could be used as a foothold to explore the chemical space of related peptides that could have been useful in a prebiotic setting.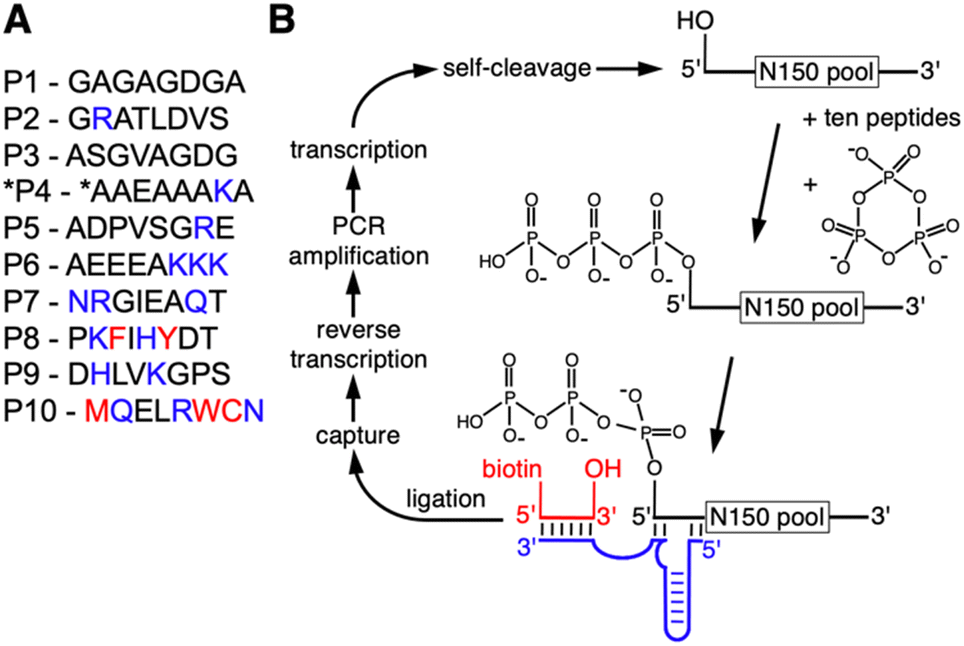 | ||
| Fig. 1 In vitro selection of ribozymes in the presence of ten octapeptides. (A) Ten octapeptides used for the in vitro selection. Amino acid colors describe their likely order in the emergence of life: black amino acids are generated in Miller-type prebiotic model reactions. Blue amino acids likely appeared at intermediate stages in evolution. Red amino acids, which includes the three aromatic amino acids, likely appeared latest. The asterisk at peptide P4 denotes the N-terminal Fmoc protection group, which was present in a portion of peptide *P4 due to an error in peptide purification. (B) Schematic for the in vitro selection procedure for self-triphosphorylating ribozymes in the presence of peptides. The RNA library (top) was prepared with a 5′-terminal hydroxyl group through the use of a self-cleaving hammerhead ribozyme. Upon incubation with the ten octapeptides and trimetaphosphate, catalytically active molecules converted their 5′-terminus to a 5′-triphosphate. Those molecules were covalently coupled to a biotinylated oligonucleotide (red) by using a ligase ribozyme (blue). The biotin modification allowed the capture of active molecules on streptavidine beads. Reverse transcription, PCR amplification, and transcription completed one selection round. | ||
The in vitro selection procedure for self-triphophorylation ribozymes was described earlier,34 and was modified for the current study to incorporate the presence of peptides during ribozyme reactions (Fig. 1B). The selection procedure started from a DNA library that contained a randomized sequence of 150 nucleotides, flanked by constant regions that served as primer binding sites, and preceded by the sequence of a hammerhead ribozyme that cleaved itself co-transcriptionally and thereby generated a 5′-hydroxyl at the library's 5′-terminus. The purified RNA library with 5′-hydroxyl termini was incubated with cTmp and the ten peptides simultaneously. After purification of the RNA library, those library molecules that catalyzed their own 5′-triphosphorylation were ligated to a short biotinylated DNA/RNA oligonucleotide, captured on magnetic streptavidin beads, reverse transcribed, and PCR amplified. The resulting DNA library was used for the next cycle of the selection procedure. The effective complexity of the library in the first round of selection was at least 7.8 × 1014. The library showed an increased fraction of self-triphosphorylated pool molecules after four rounds of selection, as judged by the required cycles of PCR after reverse transcription. To isolate the most active ribozymes, the selection pressure was increased starting in round six of the selection, by reducing the incubation time with cTmp and peptides, and reducing the cTmp concentration (Fig. S1†). The peptide concentration was kept constant at 0.5 mM for each peptide in all selection rounds. It was not reduced over the rounds because this would have penalized ribozymes that relied on peptides. A total of ten selection rounds were performed.
Identification of peptide-using ribozymes
Ribozyme sequences that were enriched during the selection experiment were identified by high throughput sequencing (HTS) analysis and clustering (Fig. 2A). To identify ribozyme clusters that depended in their activity on peptides, the library after selection round five was subjected to 12 parallel sub-rounds of selection: one sub-round contained no peptides, one sub-round contained all peptides, and ten sub-rounds contained each one individual peptide. Comparison of the HTS results from these selection sub-rounds identified individual ribozyme clusters that benefitted from specific peptides in their activity (Fig. 2B). Among the top 50 sequence clusters in selection round 5, the strongest effects on ribozyme enrichment stem from peptide *P4, and to a lesser extent from peptide P8. Interestingly, the two peptides *P4 and P8, together with P10, had the lowest solubility (less than 40 mg mL−1), while all other peptides were soluble at more than 60 mg mL−1. Therefore, both the positive effect on clusters C2, C4, C20, and 71 as well as the negative effects on many clusters (the grey lines showing two dips in Fig. 2B) may be caused in part by peptide aggregate formation. No sequence similarity between clusters 2, 4, 20, and 71 were found (Fig. S2†). Based on its strong signal with peptide *P4, ribozyme cluster 20 was chosen for further analysis. Sequence clusters that dominated the population in the later selection rounds 6–10 under increasing selection pressure (sequence clusters C7, C6, C13, and C74) did not depend on peptides for activity. This illustrated that self-triphosphorylation ribozymes are able to emerge in the absence or presence of peptides, and that some classes of ribozymes will strongly benefit from the presence of peptides. Future studies will show whether peptide-using ribozymes are more dominant when different peptides are used, or when different catalytic activities are selected. | ||
| Fig. 2 High throughput sequencing analysis of the selected sequences. (A) Enrichment of sequence clusters over the ten rounds of selection. Shown are the 25 most abundant sequence clusters that constituted at least 0.001% of the sequence reads in at least 5 selection cycles. Colored are four clusters that are also highlighted in Fig. 2B. Four additional clusters are labeled, which dominate the population of sequences at different stages of the selection. The black triangle illustrates the increasing selection pressure after selection round 5 (see Fig. S1†). (B) Effects of peptides on the activity of the same top 25 ribozyme clusters, based on HTS data. The activity is estimated from the frequency of a given cluster's sequences in the presence of a given peptide, as compared to its frequency in the absence of any peptide. By definition, the frequency is 1 for all clusters in the selection step without peptide (‘no pep.’). The response to the presence of only one specific peptide is shown to the right. A ratio of 1 denotes no peptide effect, a ratio larger than one suggests an increase in ribozyme activity by the peptide, and a ratio smaller than one a decrease in activity by the peptide. Data shown in color highlight the four clusters with increased activity in the presence of peptide *P4; the data for all other clusters are shown in grey. Labels below the graph show the number of each peptide (see Fig. 1A). | ||
To simplify the ribozyme/peptide model system, the ribozyme representing cluster 20 was truncated at its 3′-terminus while maintaining (or increasing) activity (Fig. S3 and S4†), generating a length of 120 nucleotides. In addition, the ribozyme was analyzed for the effect of five mutations that showed strong enrichment over selection rounds during HTS analysis (Fig. S5A†). Interestingly, all of these mutations were close to each other, at positions 91 to 95 in the ribozyme (U91C, C92U, G93A, G93U, G95A). After testing variants of the ribozyme with these individual mutations and their combinations, mutation G93U was chosen as the winner because its average activity was highest among all single- and double mutants of the five mutations, and 3.8-fold higher than the peak sequence of cluster 20 (Fig. S5B†). Both the peak sequence and the mutant G93U benefitted 4.3-fold from the combined presence of the ten peptides under selection conditions (each peptide at 0.5 mM). This 120 nt long ribozyme with the G93U mutation was chosen for further analysis, and named ‘ribozyme 20’ for simplicity.
Ribozyme 20 and its peptide interactions
To identify the optimal reaction conditions for ribozyme 20, the concentrations of MgCl2, cTmp, as well as the reaction pH and temperature were systematically varied. All assays were done in parallel in the absence and presence of the ten peptides to learn under which conditions the ribozyme activity may depend more or less on the peptides. When the concentrations of cTmp and MgCl2 were co-varied, the ribozyme showed highest activity at 200 mM MgCl2 and 100 mM cTmp (Fig. 3A). Activity in the absence of peptide 4 was barely detectable. The activity was highest at pH 8.3 (Fig. 3B), and at 25 °C (Fig. 3C).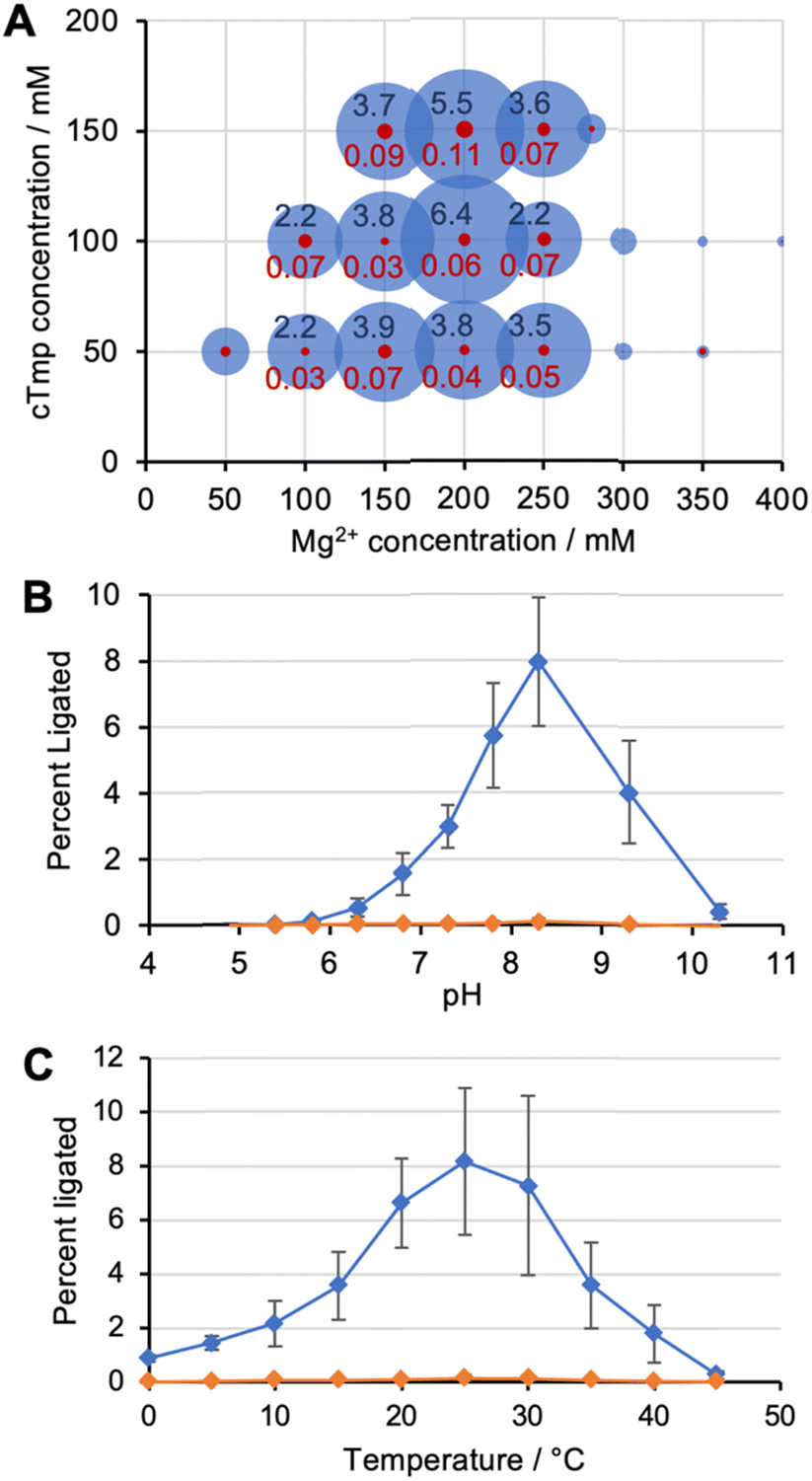 | ||
| Fig. 3 Optimization of reaction conditions for ribozyme 20. (A) Co-optimization of cTmp and Mg2+ concentrations. The size of the blue circles represents the activity with peptides; the size of the red circle represents the activity without peptides. The numbers in black and red are the percent of reacted ribozymes with, and without peptides, respectively. Peptides were used at 0.5 mM each, at a total of 5 mM. (B) Ribozyme activity as a function of the pH, in the presence of 200 mM Mg2+ and 100 mM cTmp. Blue symbols represent activity with peptides, orange symbols represent activity without peptides. (C) Ribozyme activity as function of temperature at 200 mM Mg2+, 100 mM cTmp, and pH 8.3. Blue symbols represent activity with peptides, orange symbols represent activity without peptides. Error bars are standard deviations from at least three experiments. | ||
When ribozyme 20 was tested under these optimized conditions with individual peptides (Fig. 4A), peptide *P4 resulted in strong activity whereas no other peptide resulted in activity above background. This confirmed the interpretation of the HTS data that peptide *P4 mediated a specific, and strong benefit to ribozyme 20. The absence of ribozyme activity with peptides P8 and P10 showed that the low peptide solubility displayed by peptides *P4, P8, and P10 was not sufficient to mediate ribozyme activity. Nevertheless, peptide aggregate formation appears to be important for the function of peptide *P4, as suggested by the following experiment: the dependence of ribozyme activity on the concentration of peptide *P4 gave a sigmoidal behavior with a half-maximum slightly above 1 mM peptide *P4, and saturation at 1.75 mM (Fig. 4B). The sigmoidal shape of the dependence suggested a cooperative effect of multiple peptide *P4 molecules. At 5 mM concentration, peptide *P4 increased the activity of ribozyme 20 by 900 +/− 300-fold at optimal conditions, which included 200 mM MgCl2, 100 mM cTmp, 50 mM Tris/HCl pH 8.3, for 3 hours at 25 °C. Because a low level of ribozyme activity was detectable without peptide *P4, the role of peptide *P4 was unlikely a contribution to catalysis and more likely the stabilization of the active ribozyme conformation. CD analysis of peptide P4 under reaction conditions (Fig. 4C) suggested that without RNA, the peptide *P4 existed at all tested peptide concentrations in random coil conformation.
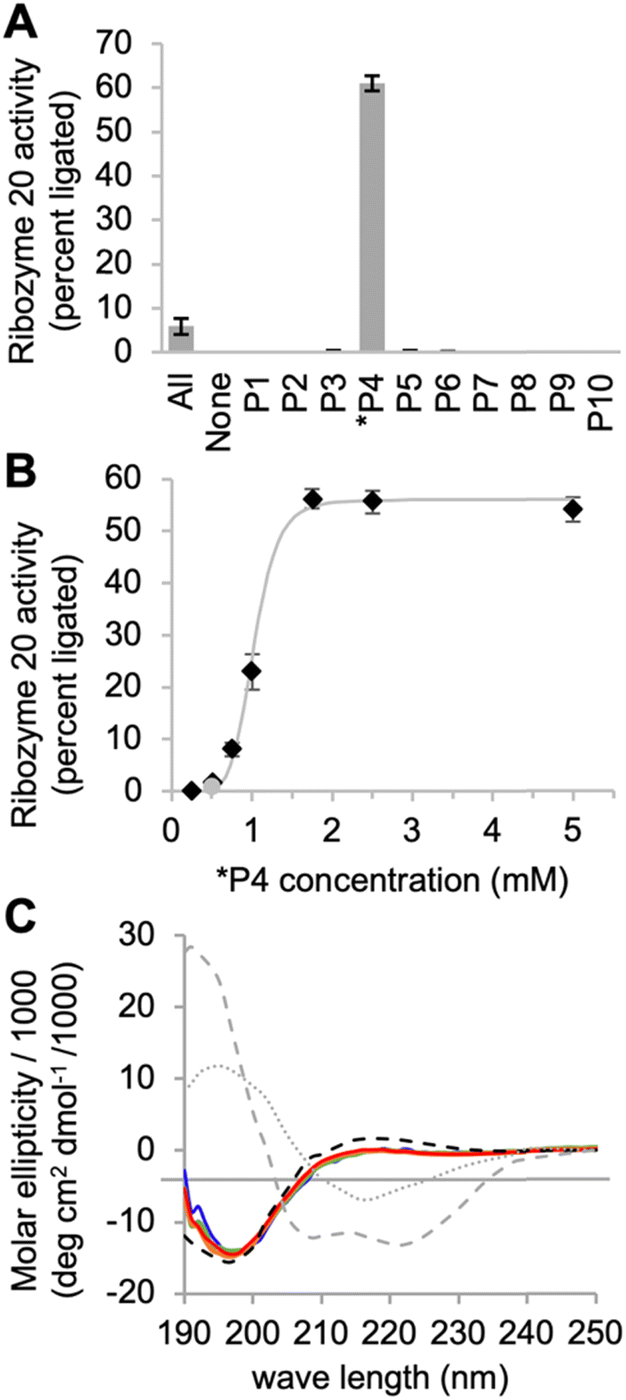 | ||
| Fig. 4 Interactions of ribozyme 20 with individual peptides. (A) Activity of ribozyme 20 in the presence of ten individual peptides, each at 5 mM, while the reaction with all peptides (‘All’) contains each peptide at 0.5 mM concentration. Error bars are standard deviations from three experiments. (B) Titration of peptide 4 into the activity assay of ribozyme 20. The grey line serves to illustrate the saturation at 1.75 mM peptide. The grey data point stems from the presence of all peptides, each at 0.5 mM. Error bars are standard deviations from three experiments. The grey line is a double exponential curve fit to illustrate the sigmoidal behavior. (C) CD spectra of peptide *P4 under the same ion conditions as the ribozyme reactions above. The peptide was tested at four different concentrations (blue: 0.5 mM; green: 1 mM; orange: 1.5 mM; red: 2 mM). The ideal CD spectra of possible peptide secondary structures43 are shown for alpha helix (grey, dashed), beta sheet (grey, dotted) and random coil (black, dashed). | ||
The secondary structure of ribozyme 20
To elucidate how peptide *P4 may modify the structure of ribozyme 20, the ribozyme secondary structure was analyzed by SHAPE probing44 (Fig. 5A). Differences in SHAPE reactivity without (left) and with (right) peptide 4 suggested four distinct peptide effects on the ribozyme structure (Fig. 5B). First, the decreased SHAPE reactivity in positions 7–9 suggested that the loop of helix H1, which is close to the catalytic site at nucleotide position 1, became more rigid in the presence of peptide *P4. Second, peptide *P4 appeared to modulate helix H3 by increasing the SHAPE reactivity of the upper part of the stem (58–61 and 68–72) and decreasing SHAPE reactivity in the loop positions 63 and 64. Third, the nucleotides 83–85 and 102–106 flanking helix H4 became more SHAPE reactive. The base complementary positions 68–70 and 102–104 became more SHAPE reactive upon addition of the peptide, suggesting that their pairing may prevent the ribozyme from accessing the catalytically active fold. Fourth, position 91 in the loop of the helix H4 became less SHAPE reactive. Since the loop of helix H4 contains all five mutations that improved ribozyme activity, its lower SHAPE reactivity may indicate the formation of the catalytic site. In total, 25 out of 105 analyzed positions in the ribozyme changed SHAPE reactivity outside experimental error. These results show that peptide *P4 modulated the secondary structure of ribozyme 20 but left most helical portions intact.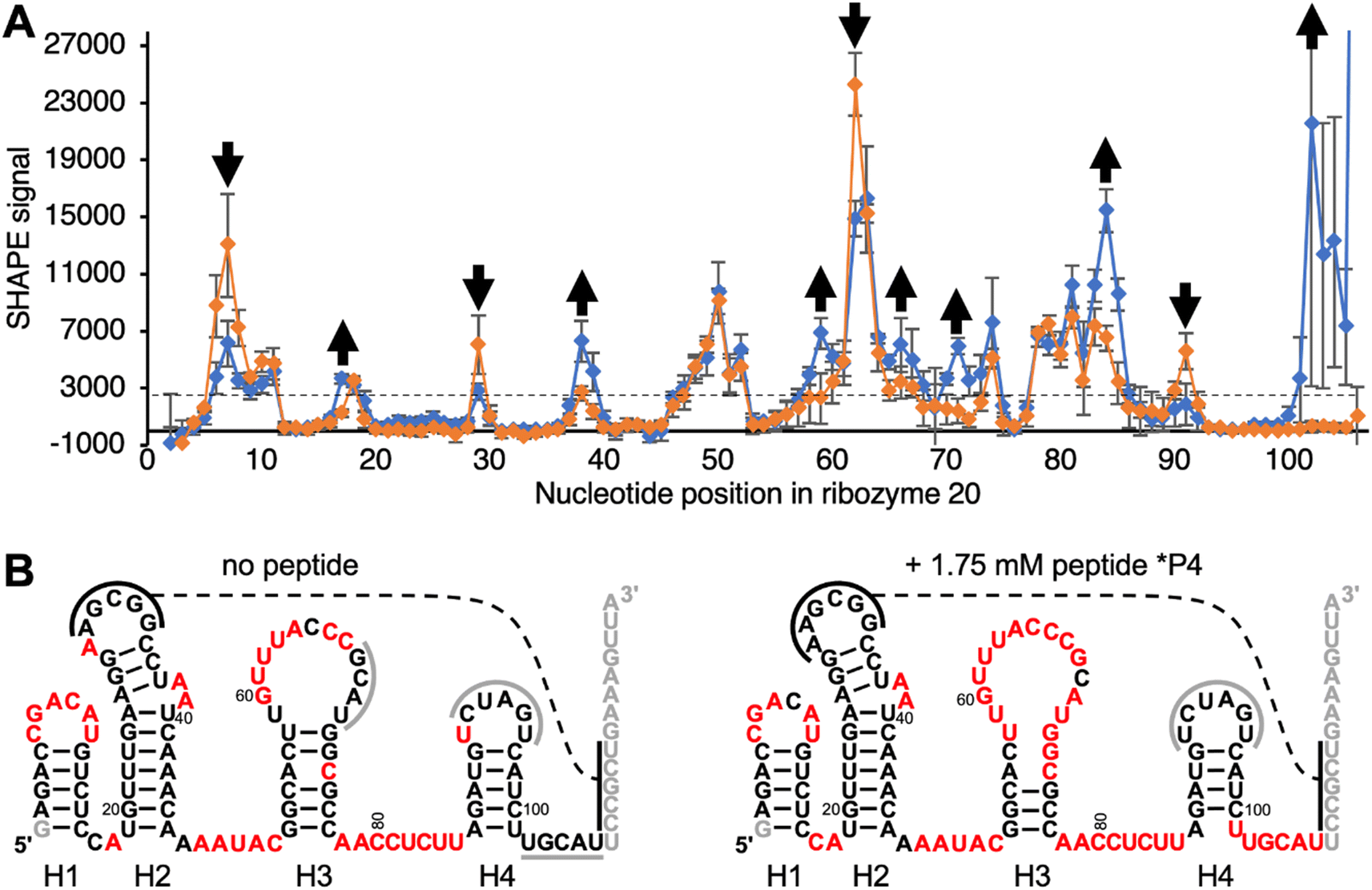 | ||
| Fig. 5 Secondary structure analysis of ribozyme 20 by SHAPE chemical probing in the absence and presence of peptide *P4. (A) The SHAPE reactivity is plotted as a function of nucleotide position, for the ribozyme without peptide (orange) and with 1.75 mM peptide *P4 (blue). The reactivity with the SHAPE reagent 1M7 was determined by terminating reverse transcription of 5′-[32P] radiolabeled primers, and PAGE analysis. Nucleotides downstream of position 106 were not analyzed because they were obscured by the reverse transcription primer. Error bars are standard deviations from triplicate experiments. For secondary structure prediction, the average signal intensity of 3000 was used as cutoff to discriminate between ‘reactive’ and ‘not reactive’ (dashed line). Positions where the presence of peptide *P4 increased, or decreased SHAPE reactivity outside of error are marked with arrows. (B) Predicted secondary structures without (left) and with 1.75 mM (right) peptide *P4, using constraints from SHAPE analysis. Positions in red denote a SHAPE signal above 3000 as shown in (A). The helices are numbered H1–H4 below the images. Black solid lines indicate a possible tertiary contact with five base pairs, and the dashed line denotes their possible interaction. Grey solid lines indicate protected loop regions and unknown interaction partners. Note that the ‘loop’ of helix H4, which contains the functionally important nucleotides 91–95 (Fig. S4†) is not SHAPE reactive. Grey nucleotide letters were not covered by SHAPE because they served to pair with the reverse transcription primer, or they were too close to the 5′-terminus. | ||
Polyaromatic hydrocarbon – peptide conjugates
During the analysis it turned out that a large fraction of peptide *P4 contained the N-terminal Fmoc-protection group, and that this group was required to mediate ribozyme activity (Fig. 6). Truncation of the peptide's C-terminus by one amino acid reduced the activity, and truncation to a dipeptide abolished activity, which showed that the peptide portion was required for function. While the Fmoc protection group shows similarity to the side chain of tryptophane, extending peptide 4 by tryptophane (or isoleucine, phenylalanine, diphenylalanine) did not mediate ribozyme activity (Fig. S6†). However, when the Fmoc group was replaced by the prebiotically plausible polyaromatic hydrocarbons (PAHs)45,46 fluorene or naphthalene, full activity of the ribozyme was restored (Fig. 6). This showed that PAH-peptide conjugates can benefit the emergence of ribozymes, with the PAH portion and the peptide portion required for activity. | ||
| Fig. 6 Effect of polyaromatic hydrocarbon (PAH) – peptide 4 conjugates on the activity of ribozyme 20. (A) Chemical structure of the conjugates tested for their benefit for ribozyme 20. From top to bottom: fluorenylmethoxycarbonyl-peptide 4 (Fmoc-P4), peptide 4 without modification (P4), Fmoc-P4 truncated at its C-terminus by one amino acid (Fmoc-P4_ct1), Fmoc-P4 truncated at its C-terminus by six amino acids (Fmoc-P4_ct6), fluorene acetic acid-P4 (Fluorene-P4), and naphthalene acetic acid-P4 (Naphthalene-P4). All peptides carried a C-terminal amide. (B) Activity assay of ribozyme 20 in the presence of the respective conjugate. Each self-triphosphorylation reaction contained 5 mM conjugate, 12.5% (v/v) of DMSO to improve the solubility of conjugates, 100 mM cTmp, 200 mM MgCl2, 50 mM Tris/HCl pH 8.3, and 5 μM ribozyme. Samples were analyzed at 7, 20, and 60 minutes triphosphorylation time, and 5′-triphosphorylated ribozymes were ligated to a short, radiolabeled oligonucleotide. Shown is a phosphorimage of [32P]-radiolabeled reaction products separated by denaturing polyacrylamide gel electrophoresis. Unreacted oligonucleotides are indicated, as well as gel-shifted oligonucleotides that indicate reactivity. (C) Quantitation of gel-shifted reaction products. Colored symbols denote fluorene-P4 (green circles), naphthalene-P4 (blue triangles), Fmoc-P4 (black squares), and Fmoc-P4 with a C-terminal truncation of 1 amino acid (grey diamonds). The symbols for compounds that did not mediate detectable activity are positioned on the X-axis, for the reactions without peptide, with peptide 4, and with Fmoc-P4 that was truncated at its C-terminus by six amino acids. Error bars denote the standard deviations from four to seven replicate experiments. | ||
Discussion
This study explored how peptides can aid the emergence of catalytic RNAs from random sequence, by performing an in vitro selection experiment for self-triphosphorylating ribozymes in the presence of ten different octapeptides. Dozens of ribozymes were identified, and one was studied in more detail. Ribozyme 20 increased about 900-fold in activity in the presence of Fmoc-protected peptide 4. While peptide 4 alone did not promote ribozyme activity, the peptide conjugate mediated full ribozyme activity when the Fmoc group was replaced by the prebiotically plausible polyaromatic hydrocarbons (PAHs) fluorene and naphthalene. This showed that conjugates between peptides and prebiotically plausible PAHs can increase the activity of ribozymes.Polyaromatic hydrocarbons (PAHs) are found abundantly in the insoluble organic matter of meteorites,46,47 interplanetary dust particles48 and comets.46 PAHs are also generated in prebiotic model reactions such as Fischer–Tropsch synthesis at temperatures above 350 °C.45 The synthesis of PAHs in interplanetary matter probably proceded via sugars generated by formose condensations, their dehydration, and cycloaddition reactions that yield PAHs.46,49 Importantly, the two PAHs naphthalene and fluorene are among the most abundant PAHs. PAHs exist in highly substituted form in meteorites,50 where the substitutions provide attachment points for conjugation with peptides. Together with the ease with which amino acids can be formed in prebiotic environments51 and peptides can form by dehydration in the presence of alpha-hydroxy acids15 and alpha-mercapto acids,16 this suggests that PAHs appeared in many of the same prebiotic environments as peptides, and peptide-PAH conjugates may have existed in these environments.
Polyaromatic hydrocarbons can facilitate prebiotically relevant catalysis with nucleic acids. The PAH proflavine is able to catalyze the ligation of chemically activated oligomers by at least 1000-fold, using the cooperative intercalation by at least three proflavine molecules to substrate and template strands.52 In contrast, no chemical activation groups are required for the oligomerization of nucleotides via nanoconfinement phenomena that decrease water activity in particle suspensions formed by the PAHs quinacridone and anthraquinone.53 The latter is especially attractive for the emergence of an RNA world from a prebiotic environment because it can overcome the problem that the polymerization of nucleotides into RNA is thermodynamically unfavorable in aqueous environments. These examples, together with our finding that PAH-peptide conjugates can strongly enhance ribozyme catalysis, show that PAHs could have fulfilled multiple different functions in a chemically diverse, prebiotic environment.
The ribozyme catalyzed triphosphorylation rate in the presence of the peptide 4-PAH conjugate is about 0.35 min−1, as judged by a single-exponential fit to the reaction mediated by the fluorene-peptide 4 conjugate at pH 8, 100 mM cTmp, and 200 mM MgCl2 (Fig. 6C). This is comparable to the ribozyme TPR1 (pH 8, 100 mM cTmp, 500 mM MgCl2), which was previously selected in the absence of peptides and showed a kOBS of 0.18 min−1 at pH 8, 100 mM cTmp, 500 mM MgCl2.34 The evolutionarily optimized cousin of TPR1, the seven-mutation variant TPR1e, reaches a kOBS of 5.7 min−1 under the same conditions. In contrast, ribozyme 51, which uses Yb3+ as catalytic cofactor (pH 7.3, 0.1 mM Yb3+, 1 mM cTmp, 500 mM KCl, 5 mM MgCl2 (ref. 54)), shows a kOBS of 1.2 min−1 without evolutionary optimization, which may be a result of the catalytic prowess of the Yb3+ ion. Future studies will elucidate the mechanism by which the PAH-peptide conjugate mediates the ∼900-fold increase in the activity of ribozyme 20.
The peptide portion of the peptide 4-PAH conjugate (AAEAAAKA) is dominated by alanine residues, and its glutamate and lysine are positioned ideally to form a salt bridge that stabilizes alpha-helices.46,49,55 The length of eight amino acids is too short to form an alpha-helix in free form, which was confirmed by CD analysis (Fig. 4C). However, it is possible that binding to the RNA stabilizes a specific peptide structure: previous studies on an N-terminal, arginine-rich peptide of the HTLV-Rex protein showed that binding to an RNA aptamer stabilized an alpha-helical conformation using only two hydrogen bonds between carbonyl oxygen and amide nitrogen of the i to i + 3 form.56 In general, peptide alpha-helices can bind into the major groove of RNA if the narrow major groove is widened by non-Watson–Crick interactions.57 Similar widening of the major groove can be accomplished by fraying near the end of a duplex, or disruptions by internal loops, especially if the two sides of the loop have different length.58 Because the narrow major groove is much richer in hydrogen-bonding partners than the openly accessible minor groove59 peptide 4 may be able to insert itself into major groove of helix 3 in ribozyme 20 to distort the helix, thereby destabilizing some secondary structure elements while stabilizing others. Future studies will reveal the conformation of the peptide 4 conjugate when bound to ribozyme 20.
The specificity of ribozyme 20 for peptide 4-PAH conjugates illustrates the evolutionary benefits of a primitive ribosome: by inserting specific functional groups (here a PAH) at specific positions (here the N-terminus), the encoded peptide synthesis of a primordial ribosome (which would execute the encoded insertion of specific amino acids) would have been able to increase the benefit of peptides.60–62 This transition could be driven also by the evolutionary benefit from the peptide's larger structural and functional repertoire compared to RNA.62 In today's biology, encoded peptides with specific sequences can play a chaperone-like role by forming a stable complex with functional RNA conformations.61 Because ribozyme 20 shows detectable activity even in the absence of the peptide 4-PAH conjugate (Fig. 4A) it seems that peptide 4-PAH does not participate in catalysis but stabilizes ribozyme 20 in a catalytically active conformation that is otherwise not well populated. Future studies on the ribozyme 20/peptide 4-PAH model system will be able to explore in mechanistic detail how peptide-PAH conjugates can benefit the function of catalytic RNAs.
Could the presence of short peptides have helped the emergence of ribozymes in early stages of life? The identification of the ribozyme 20/peptide 4-PAH conjugate suggests that PAH-peptide conjugates could have helped. Based on the HTS data (Fig. 3B), it appears that out of the top 25 ribozyme clusters, four show a similar peptide benefit profile as ribozyme 20 (ribozyme clusters 20, 71, 2, 4). A limitation of the current study for origin-of-life models was that only ten peptide sequences were included in the selection experiment, and all amino acids were in their (biological) L-form. Additionally, this study was confined to ribozymes that bind one negatively charged substrate (cTmp) and catalyze the nucleophilic attack of the ribozyme's 5′-hydroxyl group to a cTmp phosphorus, under a specific set of selection conditions. Future studies on different peptides, and different ribozymes will broaden our understanding how peptides and PAH-peptide conjugates could have helped the emergence of catalytic RNAs.
Experimental
Synthesis and purification of peptides for the in vitro selection
Peptides were synthesized on rink amide PS resin (0.77 mmol g−1, Novabiochem) and Rink Amide ChemMatrix (0.45 mmol g−1, Biotage) following the Fmoc/tBu strategy using a microwave-assisted peptide synthesizer (Alstra, Biotage). Side chain protecting groups for amino acids were as follows: Ser(tBu), His(Trt), Lys(Boc), Asp(OtBu), Glu(OtBu), Trp(Boc), Arg(Pbf), AsN(Trt), Thr(tBu), Gln(Trt), Tyr(tBu), Cys (Trt). Typical coupling reactions were performed at 0.200–0.300 mmol scale with 3.0 equiv. of Fmoc-protected amino acids, 2.94 equiv. of HBTU and 6.0 equiv. of NMM for 5 minutes at 75 °C. All reagents were pre-dissolved in DMF at 0.5 M. Fmoc deprotections were performed with 20% piperidine in DMF for 10 minutes at room temperature. Washing was performed after every deprotection and coupling step using DMF. For the syntheses of some sequences, the deprotection and coupling steps described above were either performed twice or with increased reagent equivalence to ensure reaction completion. Cleavage of the peptide sequences from the solid support with concomitant side chain deprotection was accomplished by placing the resin in fritted SPE tube and treating with TFA cleavage cocktail (∼20 mL g−1) containing 90![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5:2.5:2.5 TFA:dimethoxybenzene:H2O:TIS for 2 hours. For peptide sequences containing cysteine and methionine, EDT was added at 90:4:2.5:2.5:1.0 TFA:dimethoxybenzene:H2O:EDT:TIS. Cleaved peptides were then precipitated in cold ether, centrifuged, dissolved in methanol and reprecipitated in cold ether (3×).
:5:2.5:2.5 TFA:dimethoxybenzene:H2O:TIS for 2 hours. For peptide sequences containing cysteine and methionine, EDT was added at 90:4:2.5:2.5:1.0 TFA:dimethoxybenzene:H2O:EDT:TIS. Cleaved peptides were then precipitated in cold ether, centrifuged, dissolved in methanol and reprecipitated in cold ether (3×).
HPLC characterization and purification of peptides were carried out at room temperature on analytical (Jupiter C18 5 μm, 300 Å, 150 × 4.6 mm by Phenomenex, Torrance, CA) and semi-preparative columns (Aquasil C18 5 μm, 100 Å, 150 × 10 mm by Keystone Scientific Inc., Waltham, MA) with Prostar 325 Dual Wavelength UV-Vis Detector from Agilent Technologies with Varian pumps (Santa Clara, CA) with detection set at 225 and 406 nm. Peptides were eluted from column following a gradient using mobile phases A: 0.1% TFA in H20 and B: 0.1% TFA in CH3CN. MS analyses were obtained on a LTQ ESI-MS spectrometer (San Jose, CA). Solutions were prepared in either methanol or methanol/water (1% formic acid) with a flow rate of 10 μL min−1, spray voltage at 4.50 kV, capillary temperature at 300 °C, capillarity voltage at 7.00 V, tube lens at 135.00 V. Purified peptides were characterized by analytical HPLC (with purity typically greater than 90%) and MS (either by direct injection or LC-MS).
To remove a possible carryover of TFA, peptides were dissolved in a total volume of 2 to 5 mL 100 mM (NH4)HCO3, and frozen as a thin film on the inside of a glass bulb cooled in a dry ice/isopropanol bath. After desiccating the frozen solution to dryness in oil vacuum (∼2 mbar), the process was repeated once with 100 mM (NH4)HCO3, and once with water. The desiccated peptide was weighed, and dissolved in water to a stock concentration of 10 mM.
Synthesis and purification of peptides for peptide P4-PAH conjugates
Peptides were synthesized by using standard Fmoc chemistry with an Advanced Chemtech Apex 396 peptide synthesizer. A typical synthesis was performed on 0.12 mmol scale using 0.8 mmol g−1 Rink amide MBHA resin. Standard side chain protecting groups included Asp(tBu), Glu(OtBu), Asn(Trt), Gln(Trt), Tyr(tBu), Cys(Trt), Lys(Boc), Arg(Pbf), His(Trt), Ser(tBu), Thr(tBu), and Trp(Boc). Chain elongations were achieved using 1,3-diisopropylcarbodiimide (DIC) and Oxyma in dimethylformamide (DFM) with 60 minute coupling times. Fmoc deprotection was achieved by treating the resin twice with 25% piperidine in DMF for 10 minutes. The N-terminal Fmoc-protecting group did not fully cleave from peptide 4 (P4) under the standard deprotection conditions, so the Fmoc deprotection cycle was repeated for this peptide. 1-Naphthaleneacetic acid (Fisher) and fluorene-9-acetic acid (CombiBlocks) were coupled to the N-terminus using the typical chain elongation procedure. Peptides were cleaved from the resin with concomitant side chain deprotection by agitation in a solution of 95:2.5:2.5 TFA:triisopropylsilane (TIS):water for 2 hours. The peptide was precipitated with ether, centrifuged, and washed three additional times with ether. The crude peptides were purified by preparative reverse-phase (RP)-HPLC on a Vydac 218TP C18 or Thermo BioBasic C18 column. Purity was confirmed by analytical RP-HPLC and LC-mass spectrometry. Analytical RP-HPLC was performed using a Zorbax 300-SB C-18 column connected to a Hitachi D-7000 HPLC system. Binary gradients of solvent A (99% H2O, 0.9% acetonitrile, 0.1% TFA) and solvent B (90% acetonitrile, 9.9% H2O, 0.07% TFA) were employed for HPLC.
To remove a possible carryover of TFA, peptides were dissolved in a total volume of 2 to 5 mL 100 mM (NH4)HCO3, and frozen as a thin film on the inside of a glass bulb cooled in a dry ice/isopropanol bath. After desiccating the frozen solution to dryness in oil vacuum (∼2 mbar), the process was repeated once with 100 mM (NH4)HCO3, and once with water. The desiccated peptide was weighed, and dissolved in water to a stock concentration of 10 mM.
In vitro selection
The in vitro selection was performed essentially as described,34 with two important modifications:first, the RNA library was incubated with cTmp in the presence of a total of 5 mM peptides. The peptides had to be removed after this incubation to facilitate the ligation, and pull-down of successful library molecules. Second, the library 5′-terminus, ligase ribozyme, capture oligo, and library 3′-terminus were re-designed to avoid the selection of sequences from the previous study in our lab (Fig. 2).The DNA library consisted of a T7 RNA polymerase promoter, a hammerhead ribozyme, a 5′-constant region, 150 randomized positions, and a 3′-constant region. This double-stranded library was generated by PCR amplification from a custom-synthesized ultramerR DNA (IDTDNA) with the sequence 5′-TGCGATTACGTGTATA-N150-AGACATGTCGGTCTCGACTG-3′ (lower strand), the 5′-PCR primer 5′-AATTTAATACGACTCACTATAgggcggtctcctgacgagctaagcgaaactgcggaaacgcagtcGAGACCGACATGTCT-3′ and the 3′-PCR primer 5′-TGCGATTACGTGTATA-3′ where the italicized portion constitutes the T7 RNA polymerase promoter with 5′-terminal enhancer sequence,63 the lower case portion denotes the sequence of the 5′-terminal hammerhead ribozyme that is required to generate a 5′-terminal hydroxyl group, and the underlined portion is complementary to the ultramerR DNA. After transcription and PAGE purification the library RNA was incubated at a concentration of 200 nM with 50 mM Tris/HCl pH 8.3, 100 mM MgCl2, 50 mM trisodium-cTmp, and 3.3 mM NaOH (to compensate for the pH drop due to Mg2+ chelation by cTmp), and 0.5 mM of each of the ten peptides, in volumes of 10 mL for three hours at room temperature. After ethanol precipitation the large pellet was eluted with a total of 200 μL water, and the small, remaining pellet was dissolved in 0.5 mL water and extracted with 0.5 mL phenol equilibrated to 10 mM Tris/HCl pH 8.3, then with phenol/chloroform, and finally with chloroform. After ethanol precipitation the recovery was consistently about 80% of the inserted RNA.
The recovered RNA was ligated to the 3′-terminus of the biotinylated oligonucleotide 5′-bio-GTAGTGCTTCAArU-3′ using the R3C ligase ribozyme.64 The construct based on the R3C ligase ribozyme was designed to minimize interactions between the two arms of the ribozyme, and measuring the ligation efficiency of several constructs. The final construct showed a ligation efficiency of ∼40% on the N150 pool. The ligation products were captured on streptavidin coated magnetic beads (Promega), eluted with 96% formamide at 65 °C 5 min−1, and collected by ethanol precipitation. Recovered RNA was reverse transcribed with the 3′-PCR primer and Superscript III Reverse Transcriptase (Invitrogen). RT products were first PCR amplified with 5′-PCR primer 5′-TGCGATTACGTGTATA-3′ and 3′-PCR primer 5′-TGCGATTACGTGTATA-3′, then with the long 5′-PCR primer containing T7 promoter and hammerhead ribozyme to complete one round of selection. The number of PCR cycles in the first PCR that was necessary to see a clear band on an agarose gel was used to monitor the progress of the selection. The effective complexity of the library was at least 7.8 × 1015, as estimated by qPCR of a small sample of the initial library DNA oligonucleotide, and accounting for the losses during the selection procedure as described above. This estimate for the complexity is a lower limit because the amplification of the selection library used permissive PCR conditions, while qPCR was based on stringent PCR conditions.
HTS analysis
The selected libraries were prepared by attaching Illumina indexing sequences during PCR amplification. High-throughput sequencing was performed at the UCSD Institute for Genomic Medicine IGM on an Illumina MiSeq platform with 200 base pairs paired-end sequencing. The HTS data were received de-multiplexed (sorted by indices) in fastq format. The reads from opposite ends were merged using the merge function of usearch.65 After converting the fastq to fasta format, sequences were removed that did not contain the complete flanking regions, or that contained less than 135 nucleotides of the expected 150 nucleotides in the variable region. Using a python script, the number of instances for each unique sequence were counted. The unique sequences were then clustered based on the subpool from round 5, using the clustering function of usearch, with a cutoff of 75% nucleotide identity. For each cluster with over 30 total reads in round 5, the numbers of all sequences from that cluster were followed in every selection round and sub-round, with and without peptides.Self-triphosphorylation activity assays
To measure the self-triphosphorylation activity of different ribozymes and their variants, an assay was used in which short, radiolabeled oligonucleotides are ligated to triphosphorylated ribozymes, and where this ligation is detected in a gel shift assay.34 Because the radiolabeled oligonucleotide and the ribozymes were used at equimolar concentration the results report also the degree to which the ribozyme was triphosphorylated. The ribozymes were prepared by run-off transcription from PCR products containing the sequence of the T7 RNA polymerase promoter, the hammerhead ribozyme, and the triphosphorylation ribozyme. After transcription and co-transcriptional self-cleavage of the hammerhead ribozyme, the self-triphosphorylation ribozyme carrying a 5′-hydroxyl group was purified by denaturing PAGE. The 14-mer 5′-d(AGTAGTGCTTCAA)rU-3′ (identical to the biotinylated oligonucleotide as in the selection but without biotin) was 5′ radiolabeled using polynucleotide kinase (NEB) and γ-[32P] ATP (PerkinElmer), followed by PAGE purification. Under standard conditions, the self-triphophorylation ribozyme was incubated at 5 μM concentration with 50 mM Tris–HCl pH 8.3, 100 mM MgCl2, 50 mM trisodium trimetaphosphate, 3.33 mM NaOH, and peptides at 0 to 5 mM final concentration. The cTmp solution was made fresh. This mixture was incubated at ∼22 °C for 3 hours, after which time 2 μL were removed and added to 8 μL of a ligation pre-mix on ice. The final concentrations were 1 μM triphosphorylation ribozyme, 1 μM R3C ligase ribozyme, 1 μM unlabeled 14-mer, less than 10 nM of 5′-[32P] labeled 14-mer with 10000 cpm, 100 mM Tris-HCl pH∼8.5, 25 mM sodium-EDTA pH∼8.0 (a 5 mM excess over MgCl2), and 100 mM KCl. The mixture was heated to 65 °C and cooled at ∼0.1 °C s−1 to 30 °C to anneal the arms of the ligase ribozyme to the triphosphorylation ribozyme and the 14-mer. Upon reaching 30 °C, 10 μL of this mixture were added to 10 μL of 50 mM MgCl2, 4 mM spermidine, and 40% (w/v) PEG 8000. After incubation for 2 hours at 30 °C the reaction was stopped by ethanol precipitation. The products were separated on 7 M urea 10% PAGE for 45 minutes. Overnight exposures to a phosphorimaging screen were scanned on a Typhoon Phosphorimager (GE) imager, and the bands were quantified with the software Quantity One (Bio-Rad) using the rectangle method. The ‘fraction ligated’ was calculated as the [volume of ligated band]/[volume of ligated and unligated bands].
Because the fraction of ligated RNA depends not only on the self-triphosphorylation activity but also on the ability of the ribozyme to serve as substrate to the ligase ribozyme,34 a 5′-triphosphorylated version of ribozyme 20 (generated by transcription without a hammerhead ribozyme) was subjected to the ligation assay. The ligation yielded an average ‘fraction ligated’ of 68.6 ± 2.0%, under ligation conditions that simulated the triphosphorylation incubation under optimized conditions. There was no significant difference when this pre-incubation was performed in the absence, or presence of peptides. Therefore, the values of ‘fraction ligated’ under optimized conditions with the final ribozyme 20 construct can be converted to ‘triphosphorylated fraction of ribozyme’ when multiplying the ‘ligated fraction of radiolabeled primer’ by 1.46.
SHAPE probing
Shape probing was done on ribozymes carrying a 5′-hydroxyl group, and in the absence of cTmp. Ribozyme 20 was probed in 50 mM MgCl2, and 50 mM HEPES/KOH pH 8.0. Fourty-nine microliters of this solutions with 200 nM ribozyme was mixed with 1 μL of a solution containing 100 mM 1M7 (ref. 44) in dry DMSO or a control of DMSO, and incubated at room temperature for 3 minutes. After ethanol precipitation, the products were reverse transcribed using Superscript III Reverse Transcriptase (Invitrogen) and a 5′-[32P] radiolabeled primer complementary to the ribozyme 3′-terminus. RT products were heated in 750 mM NaOH for 5′/80 °C, ethanol precipitated, and separated on 7 M urea 10% polyacrylamide gels for 2–4 hours. Exposures of phosphorimager screens were scanned on a Typhoon Phosphorimager (GE). The bands were quantified using the rectangle method in Quantity One (Bio-Rad). The volumes of the bands in the ‘no 1M7’ band were subtracted from the volume of each corresponding band in the ‘with 1M7’ lane. Based on the reverse transcription primer 5′-GCTGGAGCTTAACT-3′ and the sequence of the full-length ribozyme, the bands were assigned to positions in the ribozyme, generating a SHAPE reactivity profile for each experiment. The profiles of replicate 2 and 3 were normalized by setting their strongest 10% signals to the same strength as the corresponding signals in first replicate. The averages and standard deviations of normalized signals at each position from three experiments were reported. The cutoffs chosen between ‘weak signals’ and ‘strong signals’ are indicated in Fig. 5A. The secondary structures are based on secondary structure predictions by mfold,66 constrained by the position of SHAPE reactive nucleotides.Conclusions
In summary, we report that conjugates between polyaromatic hydrocarbons (PAHs) and short peptides can serve as powerful cofactors to enhance ribozyme function. The proof-of-principle ribozyme was discovered from an in vitro selection of ribozymes from random sequence, in the presence of ten peptides, where one of the ten peptides contained an Fmoc protection group that could be replaced by the prebiotically plausible PAHs fluorene or naphtalene. Similar PAH-peptide conjugates might have been important for an RNA world scenario.Data availability
All data necessary for the reproduction of the described experiments are contained in the main manuscript and ESI.† For additional inquiries please contact the corresponding author.Author contributions
Conceptualization, methodology, funding acquisition, supervision, and manuscript writing by UFM. Synthesis and purification of peptides for the selection by MZJ and JGS. In vitro selection, HTS analysis, biochemical analysis, and data analysis by KJS. Analysis of peptides, synthesis and purification of PAH-peptide conjugates, and CD analysis by LJL. Analysis of PAH-peptide conjugate/ribozyme interactions by TL.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Russell Doolittle for help with the final purification of peptides. We thank Ram Krishnamurthy for helpful discussions. We thank Logan Norrell for pilot experiments. We thank NASA Exobiology grant NNX16AJ27G to UFM for funding of KJS, JS, MJ, UFM and NASA Exobiology grant NSSC22K1627 for funding TL, LL, UFM.Notes and references
-
A. Rich, Horizons in Biochemistry, New York, NY, 1962, pp. 103–126 Search PubMed
.
-
C. R. Woese, The molcular basis for genetic expression, Harper & Row, New York, 1967 Search PubMed
- F. H. C. Crick, J. Mol. Biol., 1968, 38, 367–379 CrossRef CAS PubMed
- L. E. Orgel, J. Mol. Biol., 1968, 38, 381–393 CrossRef CAS PubMed
- W. Gilbert, Nature, 1986, 319, 618 CrossRef
- G. F. Joyce, Nature, 2002, 418, 214–221 CrossRef CAS PubMed
- P. G. Higgs, J. Mol. Evol., 2017, 84, 225–235 CrossRef CAS PubMed
- R. Krishnamurthy and N. V. Hud, Chem. Rev., 2020, 120, 4613–4615 CrossRef CAS PubMed
- A. C. Rios and Y. Tor, Isr. J. Chem., 2013, 53, 469–483 CrossRef CAS PubMed
- A. C. Rios, H. T. Yu and Y. Tor, J. Phys. Org. Chem., 2015, 28, 173–180 CrossRef CAS PubMed
- R. Krishnamurthy, Acc. Chem. Res., 2012, 45, 2035–2044 CrossRef CAS PubMed
- R. Krishnamurthy, Synlett, 2014, 25, 1511–1517 CrossRef
- G. A. Soukup and R. R. Breaker, RNA, 1999, 5, 1308–1325 CrossRef CAS PubMed
- S. Islam and M. W. Powner, Chem, 2017, 2, 470–501 CAS
- J. G. Forsythe, S.-S. Yu, I. Mamajanov, M. A. Grover, M. Krishnamurthy, F. M. Fernandez and N. V. Hud, Angew Chem. Int. Ed. Engl., 2015, 54, 9871–9875 CrossRef CAS PubMed
- M. Frenkel-Pinter, M. Bouza, F. M. Fernández, L. J. Leman, L. D. Williams, N. V. Hud and A. Guzman, Thioesters Provide a Robust Path to Prebiotic Peptides, Nat. Commun., 2022, 13(1), 2569–2576 CrossRef CAS PubMed
- E. I. Jiménez, C. Gibard and R. Krishnamurthy, Angew. Chem., Int. Ed., 2021, 60, 10775–10783 CrossRef PubMed
- M. Frenkel-Pinter, J. W. Haynes, A. M. Mohyeldin, M. C, A. B. Sargon, A. S. Petrov, R. Krishnamurthy, N. V. Hud, L. D. Williams and L. J. Leman, Nat. Commun., 2020, 11, 3137 CrossRef CAS PubMed
- B. H. Patel, C. Percivalle, D. J. Ritson, C. D. Duffy and J. D. Sutherland, Nat. Chem., 2015, 7, 301–307 CrossRef CAS
- S. Islam, D.-K. Bučar and M. W. Powner, Nat. Chem., 2017, 9, 584–589 CrossRef CAS
- J. Xu, N. J. Green, C. Gibard, R. Krishnamurthy and J. D. Sutherland, Nat. Chem., 2019, 11, 457–462 CrossRef CAS PubMed
- N. Lahav, D. White and S. Chang, Science, 1978, 201, 67–69 CrossRef CAS PubMed
- E. T. Parker, H. J. Cleaves, J. L. Bada and F. M. Fernández, Rapid Commun. Mass Spectrom., 2016, 30, 2043–2051 CrossRef CAS PubMed
- F. P. Cakmak, S. Choi, M. O. Meyer, P. C. Bevilacqua and C. D. Keating, Nat. Commun., 2020, 11(1), 5949–5959 CrossRef CAS PubMed
- M. P. Robertson, RNA, 2004, 10, 114–127 CrossRef CAS PubMed
- S. Tagami, J. Attwater and P. Holliger, Nat. Chem., 2017, 9, 325–332 CrossRef CAS PubMed
- R. R. Poudyal, R. M. Guth-Metzler, A. J. Veenis, E. A. Frankel, C. D. Keating and P. C. Bevilacqua, Nat. Commun., 2019, 10, 490 CrossRef CAS PubMed
- D. Faulhammer and M. Famulok, Angew Chem. Int. Ed. Engl., 1996, 35, 2837–2841 CrossRef CAS
- J. D. Stephenson, M. Popović, T. F. Bristow and M. A. Ditzler, RNA, 2016, 22, 1893–1901 CrossRef CAS PubMed
- M. Popovic, P. S. Fliss and M. A. Ditzler, Nucleic Acids Res., 2015, 43(14), 7070–7082 CrossRef CAS PubMed
- M. A. Pasek, T. P. Kee, D. E. Bryant, A. A. Pavlov and J. I. Lunine, Angew Chem. Int. Ed. Engl., 2008, 47, 7918–7920 CrossRef CAS PubMed
- M. A. Pasek, J. P. Harnmeijer, R. Buick, M. Gull and Z. Atlas, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 10089–10094 CrossRef CAS PubMed
- B. Herschy, S. J. Chang, R. Blake, A. Lepland, H. Abbott-Lyon, J. Sampson, Z. Atlas, T. P. Kee and M. A. Pasek, Nat. Commun., 2018, 9, 1346 CrossRef PubMed
- J. E. Moretti and U. F. Muller, Nucleic Acids Res., 2014, 42, 4767–4778 CrossRef CAS
- G. F. Dolan, A. Akoopie and U. F. Muller, PLoS One, 2015, 10, e0142559 CrossRef
- A. Pressman, J. E. Moretti, G. W. Campbell, U. F. Muller and I. A. Chen, Nucleic Acids Res., 2017, 45, 8167–8179 CrossRef CAS PubMed
- J. T. Arriola and U. F. Müller, Nucleic Acids Res., 2020, 48, e116 CrossRef CAS PubMed
- E. T. Parker, H. J. Cleaves, J. P. Dworkin, D. P. Glavin, M. Callahan, A. Aubrey, A. Lazcano and J. L. Bada, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 5526–5531 CrossRef CAS PubMed
- E. N. Trifonov, J. Biomol. Struct. Dyn., 2004, 22, 1–11 CrossRef CAS
- C. D. Georgiou, Astrobiology, 2018, 18, 1479–1496 CrossRef CAS PubMed
- A. K. Cobb and R. E. Pudritz, Astrophys. J., 2014, 783, 12 CrossRef
- M. M. Hoffman, M. A. Khrapov, J. C. Cox, J. Yao, L. Tong and A. D. Ellington, Nucleic Acids Res., 2004, 32, D174–D181 CrossRef CAS PubMed
- N. J. Greenfield and G. D. Fasman, Biochemistry, 1969, 8, 4108–4116 CrossRef CAS PubMed
- S. A. Mortimer and K. M. Weeks, J. Am. Chem. Soc., 2007, 129, 4144–4145 CrossRef CAS PubMed
- A. I. Rushdi and B. R. Simoneit, Origins Life Evol. Biospheres, 2001, 31, 103–118 CrossRef CAS PubMed
- G. D. Cody, E. Heying, C. M. O. Alexander, L. R. Nittler, A. L. D. Kilcoyne, S. A. Sandford and R. M. Stroud, Proc. Natl. Acad. Sci. U.S.A., 2011, 108, 19171–19176 CrossRef CAS
- C. M. O. Alexander, M. Fogel, H. Yabuta and G. D. Cody, Geochim. Cosmochim. Acta, 2007, 71, 4380–4403 CrossRef CAS
- L. P. Keller, S. Messenger, G. J. Flynn, S. Clemett, S. Wirick and C. Jacobsen, Geochim. Cosmochim. Acta, 2004, 68, 2577–2589 CrossRef CAS
- G. Cooper, N. Kimmich, W. Belisle, J. Sarinana, K. Brabham and L. Garrel, Nature, 2001, 414, 879–883 CrossRef CAS PubMed
- G. D. Cody and C. M. O. ’D. Alexander, Geochim. Cosmochim. Acta, 2005, 69, 1085–1097 CrossRef CAS
- S. L. Miller, Science, 1953, 117, 528–529 CrossRef CAS PubMed
- S. S. Jain, F. A. Anet, C. J. Stahle and N. V. Hud, Angew Chem. Int. Ed. Engl., 2004, 43, 2004–2008 CrossRef CAS PubMed
- A. G. De Herrera, T. Markert and F. Trixler, Commun. Chem., 2023, 6, 69 CrossRef CAS PubMed
- K. J. Sweeney, X. Han and U. F. Müller, Nucleic Acids Res., 2023, 51, 7163–7173 CrossRef PubMed
- S. Marqusee and R. L. Baldwin, Proc. Natl. Acad. Sci., 1987, 84, 8898–8902 CrossRef CAS PubMed
- F. Jiang, A. Gorin, W. Hu, A. Majumdar, S. Baskerville, W. Xu, A. Ellington and D. J. Patel, Structure, 1999, 7, 1461–1472 CrossRef CAS PubMed
- J. L. Battiste, H. Mao, N. S. Rao, R. Tan, D. R. Muhandiram, L. E. Kay, A. D. Frankel and J. R. Williamson, Science, 1996, 273, 1547–1551 CrossRef CAS PubMed
- K. Weeks and D. Crothers, Science, 1993, 261, 1574–1577 CrossRef CAS PubMed
- N. C. Seeman, J. M. Rosenberg and A. Rich, Proc. Natl. Acad. Sci., 1976, 73, 804–808 CrossRef CAS PubMed
- E. Szathmary and J. Maynard Smith, J. Theor. Biol., 1997, 187, 555–571 CrossRef CAS PubMed
- A. M. Poole, D. C. Jeffares and D. Penny, J. Mol. Evol., 1998, 46, 1–17 CrossRef CAS PubMed
- H. F. Noller, RNA, 2004, 10, 1833–1837 CrossRef CAS PubMed
- G.-Q. Tang, R. P. Bandwar and S. S. Patel, J. Biol. Chem., 2005, 280, 40707–40713 CrossRef CAS PubMed
- J. Rogers and G. F. Joyce, Nature, 1999, 402, 323–325 CrossRef CAS PubMed
- R. C. Edgar, Bioinformatics, 2010, 26, 2460–2461 CrossRef CAS PubMed
- M. Zuker, Nucleic Acids Res., 2003, 31, 3406–3415 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc03540a |
| This journal is © The Royal Society of Chemistry 2023 |